
Cationic Eudragit® Polymers as Excipients for Microparticles Prepared by Solvent Evaporation Method
Kationtové eudragitové polymery jako pomocné látky pro přípravu mikročástic metodou odpaření rozpouštědla
Jako potencionální polymery pro přípravu mikročástic metodou odpaření rozpouštědla se hodnotily tři kationtové akrylátové deriváty Eudragit® RL, Eudragit® RS a Eudragit® E 100. Prakticky nerozpustné léčivo mirtazapin a snadno rozpustné léčivo tramadol-hydrochlorid byly vybrány pro enkapsulaci jako modelová léčiva s extrémně rozdílnou rozpustností. Připravené mikrosféry byly podrobeny optické mikroskopii, analýze obsahu léčiva a disoluční zkoušce. Při použití Eudragitu® RL se nepodařilo vytvořit jakostní mikročástice, zatímco z Eudragitu® RS a Eudragitu® E 100 byly připraveny mikročástice optimálního tvaru. Vzorky s enkapsulovaným mirtazapinem vykazovaly prodloužené uvolňování léčiva, disoluční profily vzorků s obsahem tramadolu se blížily okamžitému uvolňování léčiva. Eudragit® RS poskytoval mikročástice s vyšší enkapsulační účinností, vyšším procentuálním obsahem léčiva v lékové formě a střední velikostí částic byla menší.
Klíčová slova:
mikročástice, odpaření rozpouštědla, prodloužené uvolňování léčiva, Eudragit®
Authors:
Jakub Vysloužil; Jana Bavoľárová; Martina Kejdušová; David Vetchý; Kateřina Dvořáčková
Authors‘ workplace:
University of Veterinary and Pharmaceutical Sciences Brno
; Department of Pharmaceutics, Faculty of Pharmacy
Published in:
Čes. slov. Farm., 2013; 62, 249-254
Category:
Original Articles
Overview
Three cationic acrylic polymers, i. e. Eudragit® RL, Eudragit® RS and Eudragit® E 100, were evaluated for the purpose of microparticles preparation by the solvent evaporation method. The practically insoluble drug mirtazapine and the freely soluble drug tramadol hydrochloride were selected for encapsulation as extreme limits of drug solubility. The prepared microspheres were analyzed by optical microscopy, drug content analysis and dissolution test. It was observed that Eudragit® RL did not provide microparticles while Eudragit® RS and Eudragit® E 100 yielded spherical microparticles. Samples prepared with mirtazapine showed sustained drug release whereas tramadol hydrochloride samples released the drug in a pattern similar to the immediate release profile. Eudragit® RS was found to be superior to Eudragit® E 100 in its encapsulation efficiency, drug loading and smaller mean size of microparticles.
Keywords:
microparticles, solvent evaporation, sustained drug release, Eudragit®
Introduction
Controlled drug delivery systems represent an effort of pharmaceutical technology to provide health care utilizing the most advanced therapeutic systems to achieve maximal compliance, convenience and treatment efficiency1). These systems are used for the drug release rate control or drug targeting to a specific site2). One of the integral branches of the drug delivery systems are microparticulate dosage forms. These highly developed delivery systems may possess several significant benefits, e.g. drug protection, dosage decrease and lower adverse effects or drug targeting3, 4). The most promising is the possibility of various release profiles, including sustained5) and pulsatile drug release6).
Several methods for microparticle preparation exist, including the solvent evaporation method7). Several modifications of this method were developed to enable encapsulation of a vast range of drugs8–10). The basic modification for drugs insoluble in water is their o/w (oil in water) modification11), consisting of several steps: 1. polymer dissolution and drug dissolution or dispersion in an organic solvent; 2. emulsification of the internal organic phase in a second, so called continuous (frequently aqueous) phase immiscible with the internal one; 3. solvent evaporation leading to the solid microspheres transformation; 4. microparticles collection and drying of the microspheres12). For water soluble drugs, a double emulsion method w/o/w is usually employed. The internal organic phase from the previous modification is here replaced with the primary emulsion w/o. The drug is dissolved in a small amount of water, while the polymer is dissolved in an organic solvent. The prepared primary emulsion is consequently mixed with the external water phase to form a double emulsion w/o/w (water in oil in water). The oil phase serves as a barrier against drug leakage13). The rest of the process remains identical to o/w modifications. A wide range of polymeric materials have been used for microparticle preparation, for example polylactic acid14), poly(lactic-co-glycolic acid)15), and poly(3-hydroxybutyrate)16).
Polyacrylate derivatives Eudragits® were predominantly developed as polymer coating materials with several benefits, including pH dependent drug release17). Nowadays, polyacrylate polymers products are tested as drug carriers forming matrix dosage forms18, 19). Presently, polyacrylate derivatives soluble in organic solvents represent potential polymers for microparticle preparation by the solvent evaporation method20). Eudragit® RL is a copolymer of ethyl acrylate, methyl acrylate and methacrylic acid ester. The polymer is insoluble in water and possess pH independent swelling. Quaternary ammonium groups of methacrylic acid ester in the form of salts give the polymer high water permeability. Eudragit® RS has a very similar composition with a lower frequency of quaternary ammonium groups. Eudragit® RS has thus a significantly lower permeability21). Eudragit® E 100 is composed of dimethylaminoethyl methacrylate, butyl methacrylate and methyl methacrylate. Designed as a protective polymer, Eudragit® E 100 is soluble up to pH 5.022).
The main objective of the preformulation experiment was to evaluate Eudragit® RL, Eudragit® RS and Eudragit® E 100 polyacrylate derivatives as potential polymers for microparticle preparation by the solvent evaporation method and to observe the effect of the polymer choice on microparticle properties.
Experimental section
Materials
Mirtazapine – (Zentiva, Prague, Czech Republic) was used as a poorly soluble model drug, tramadol hydrochloride (Zentiva, Prague, Czech Republic), Eudragit® RL , Eudragit® RS and Eudragit® E 100 (Evonik Industries AG, Essen, Germany) were used as materials for the polymer matrix formation. Dichloromethane – DM (Penta, Prague, Czech Republic) was used as an organic solvent and polyvinyl alcohol – PVA (Sigma Aldrich, St. Louis, USA) as an emulsifier. Buffer of pH 7.2 (sodium chloride, potassium chloride, calcium chloride, magnesium chloride, dodecahydrate sodium hydrogen phosphate, potassium dihydrogen phosphate – all by Merck KGaA, Darmstadt, Germany) and phosphate buffer of pH 6.8 (dodecahydrate sodium hydrogen phosphate, Merck KGaA, Darmstadt, Germany) were applied as dissolution media. All materials were of Ph. Eur. quality.
Microparticle preparation
Microparticles containing mirtazapin were prepared by the o/w modification. To form the internal phase of emulsion, 200 mg of the drug and 800 mg of a polymer were dissolved in 5 ml of dichloromethane. The mixture was stirred by a homogenizer Ultra-Turrax (T25 basic, IKA-Werke, Staufen, Germany) at 20 000 rpm for one minute to ensure complete polymer dissolution.
Microparticles containing tramadol hydrochloride were prepared by the w/o/w modification. To form the primary emulsion, 200 mg of the drug were dissolved in 2 ml of water and 800 mg of the polymer were dissolved in 5 ml of dichloromethane. Solutions were mixed together and the dispersion stirred by Ultra-Turrax (T25 basic, IKA-Werke, Staufen, Germany) at 20 000 rpm for one minute to form a fine emulsion.
All at once, the oil phase or the primary emulsion was emulsified into 800 ml of aqueous continuous phase, which contained 0.1% (w/w) PVA. The formed emulsion was stirred by a mechanical stirrer (Heidolph RZR 2021, Sigma Aldrich, St. Louis, USA) at 600 rpm for 1 hour to ensure a complete evaporation of the organic solvent. The prepared microparticles were collected on a fine mesh sieve (opening size 80 μμm), washed three times with purified water and dried at 25 °C in a cabinet drier (HORO – 048B, Dr. Hofmann GmbH, Ostfildern, Germany). The prepared samples were named in accordance with the polymer and drug type used. Characteristics of samples are shown in Table 1.
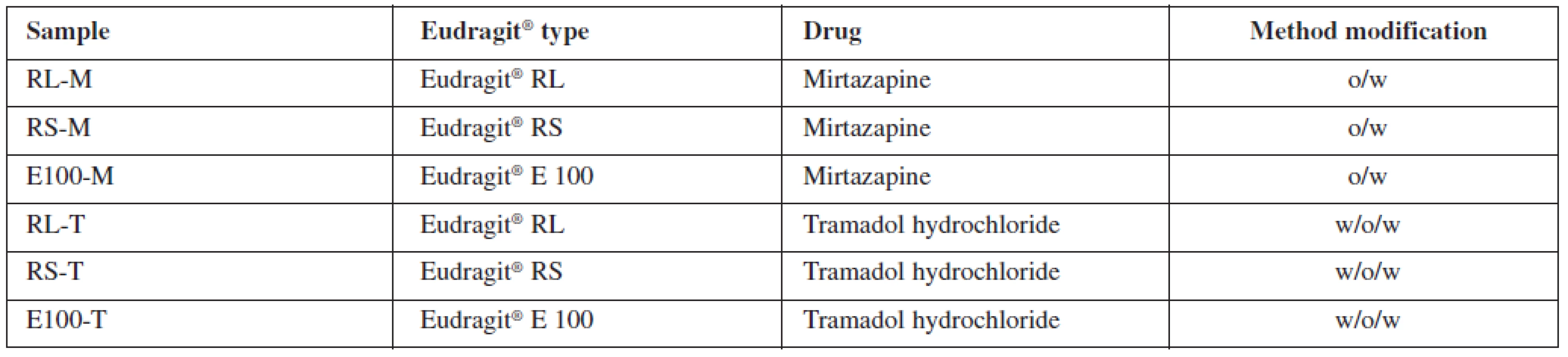
Microparticle characteristics
Drug content analysis
UV/Vis spectroscopy was employed for the drug content determination. An appropriate amount of dried microparticles was dissolved in 25 ml of dichloromethane. Absorbance of the samples was measured at 295 nm (mirtazapine) and 271 nm (tramadol hydrochloride) using a UV/Vis spectrometer (Lambda 25, Perkin Elmer, Waltham, USA). Encapsulation efficiency (EE) and drug load (DL) were calculated from the obtained values using the following equations23, 24):

where cs corresponds to the actual drug content and ct represents the theoretical drug load. The assay was carried out in triplicate. The results are expressed as mean values and standard deviations.

where w1 is the weight of a drug in microparticles and w2 is the gross weight of microparticles. The assay was carried out in triplicate and the results are stated as mean values and standard deviations.
Effectiveness of the process was also evaluated by the yield, calculated by the following equation25):

where w2 is the gross weight of microparticles and wt is the total weight of the drug and polymer used for microparticle preparation. Experiments were carried in triplicate.
Optical microscope analysis
200 microparticles of each sample were analyzed by a NIKON SMZ 1500 stereo microscope (Nikon, Tokyo, Japan) and a 72AUC02 USB camera (The Imaging Source, Bremen, Germany) in connection with the computer software NIS-Elements AR 4.0 (Nikon, Tokyo, Japan). Sphericity factor and mean size data were obtained. A photograph of every sample was taken.
In vitro release studies
In vitro drug release tests of drug-loaded microspheres were carried out using the Apparatus 1 method26) in an automatic dissolution device (SOTAX AT 7 On-Line System, Donau Lab, Zürich, Switzerland) at 75 rpm, kept at 37.0 ± 0.5 °C. Mirtazapin samples were tested in pH value of 7.2. Mirtazapin is practically insoluble in water and for this reason more than 24 hours sustained release was expected. Therefore, pH value of 7.2 was selected to simulate conditions for parenteral administration. On the other hand, tramadol hydrochloride is freely soluble in water which indicates much faster drug release. It is usually administered orally and therefore the conventional pH 6.8 dissolution test was performed with tramadol samples.
The samples for the dissolution test were weighted with respect to their encapsulation efficiency. An equivalent quantity of microparticles was weighted to achieve a 15 mg amount of mirtazapine. 1000 ml of pH 7.2 buffer was used as the dissolution medium. Samples were analyzed using an UV spectrophotometer (Lambda 25, Perkin Elmer, St. Louis, USA) at 295 nm.
A relevant quantity of tramadol hydrochloride samples was weighted to obtain a 20 mg amount of the drug. 500 ml of pH 6.8 buffer served as the dissolution medium. Samples were measured at 271 nm. In vitro drug release was observed. The results were expressed as mean values and standard deviations.
The similarity factor f2 was originally designed for comparison of originals and generics dissolution profiles27). It can be also used for the determination of possible dependence. The similarity factor f2 value ranged between 0 and 100. If f2 ≥ 50, drug release profiles were more than 90 % similar. If f2 < 50, release profiles are not similar and the observed influence of formulation or process variables is considered as significant.

Ri – drug amount (%) released at time interval i; reference sample, Ti – drug amount (%) released at time interval i; tested sample, n – total number of samplings.
Results and discussion
During the drying phase, Eudragit® RS and Eudragit® E 100 was observed to yield spherical particles. On the other hand, Eudragit® RL did not form microparticles under the determined conditions. The RL-M sample resulted only in a formless gel substance and the RL-T sample formed irregular microparticle deformities. Eudragit® RL microparticles were previously prepared only in non-aqueous systems28–30). In aqueous systems, Eudragit® RL was used just as a component of polymer blends31). With respect to a higher permeability of Eudragit® RL and the use of the aqueous PVA solution as the external phase of the system, the microparticle preparation could be heavily disrupted. Because of poor results, Eudragit® RL samples were not included in further evaluations.
Encapsulation process
Table 2 shows the drug content analysis results. Overall, Eudragit® RS as a drug carrier provided better results than Eudragit® E 100. Insoluble mirtazapine loaded with RS-M microparticles had a higher encapsulation efficiency (49.1 ± 4.27%), greater drug loading (14.5 ± 1.26%) and yield (67.9%) than its relevant Eudragit® E 100 counterpart (EE 33.3 ± 0.29%, DL 15.2 ± 0.14%, yield 43.7%). Similar results were also obtained for the tramadol hydrochloride samples. The sample RS-T was characterized by significantly higher values of the observed parameters (EE 27.8 ± 0.8%, DL 8.3 ± 0.21%, yield 67.1%) than the E100-T sample (EE 1.58 ± 0.1%, DL 1.4 ± 0.17%, yield 29.8%). While Eudragit® RS was designed as a polymer for controlled drug release32), Eudragit® E 100 was primarily developed as a protective coating polymer33) which can serve as a drug carrier under certain conditions (generally pH higher than 5, for oral administration protection against low stomach pH is necessary)34). However, it is not suggested as a matrix forming material for sustained drug release. Having a different chemical structure from Eudragit® RS resulting in higher permeability and swellability, and under the conditions connected with the usage of conventional o/w and w/o/w methods, the leakage of freely soluble drug is too high to obtain respectable results. Therefore, the observed superiority of Eudragit® RS for this purpose is not surprising. Mirtazapine and tramadol hydrochloride samples are not comparable as different modifications of a solvent evaporation method were used for the microparticles preparation.

Optical microscope analysis
Microparticle mean size and sphericity factor values are presented in Table 2. The photographs of the prepared microparticles are shown in Figure 1. In general, every prepared sample consisted of spherical particles (Fig. 1); the sphericity factor ranged between 0.86–0.96. The RS-T sample was characterized by the highest sphericity (0.96), the lowest value of the observed parameter was found in the sample E100-M (0.86), which could be attributed to the presence of deformed polymer filaments and sticks (Fig. 1c), which formed along with the particles. Eudragit® RS samples provided particles with a smaller mean size (RS-M – 198.75 μμm; RS-T – 345.28 μμm) in comparison with their equivalent Eudragit® E 100 counterparts (E100-M – 237.89 μμm; E100-T – 484.66 μμm). The larger mean size of tramadol hydrochloride samples was probably reflection of their different preparation modification.

Drug release behavior
Drug dissolution profiles of mirtazapine samples are shown in Figure 2. The E100-M sample showed sustained release of the drug26) and during 72 hours of the dissolution time 99 % of the drug was released. The RS-M sample was characterized by a remarkably slower drug release with only 12 % of the drug released within 72 hours. A significant difference between the release profiles was confirmed by similarity factor analysis. The similarity factor f2 between the samples RS-M and E100-M totalled 14.34, which meant that the release curves were not similar. Both samples offered sustained drug release in days, which could predetermine the dosage forms based on this Eudragit® polymer type for parenteral administration of new drug molecules with varying solubility. In view of their non-biodegradability35) and non-absorbability36), the Eudragit® dosage forms would have to be surgically removed upon its exhaustion37) from a patient’s body.

Figure 3 depicts dissolution profiles of tramadol hydrochloride microparticle samples. The E100-T sample released 94 % of tramadol hydrochloride within three hours, while the RS-T sample released 102 % during the same time. Both samples, i.e. RS-T and E100-T, released slightly less than 80 % of the drug in 45 minutes and therefore did not meet the conditions for dosage forms with immediate drug release26). The similarity factor f2 totalled 51.14 and both profiles could be described as similar.

Conclusion
Eudragit® RS and Eudragit® E 100 microparticles were successfully prepared, while Eudragit® RL did not provide microparticles at all. Microparticles prepared from Eudragit® RS had a greater encapsulation efficiency, higher drug loading and a smaller mean size in comparison with Eudragit® E 100. With respect to a wide range of dissolution profiles, drug solubility seems to be one of the main factors affecting the dissolution behavior of Eudragit® RS microparticles. The selection of a suitable drug candidate for encapsulation is based on its solubility and the required dissolution profile. Eudragit® E 100 microparticles, intended for administration under conditions characterized by pH higher than 5, showed such a low encapsulation efficiency of tramadol hydrochloride that this polymer would be appropriate only for encapsulation of poorly soluble drugs. The next choice for preparation of Eudragit® RL microparticles could be the use of it in a polymer blend or selection of a different modification of the solvent evaporation method.
Conflict of interest: none.
Received 13 September 2013
Accepted 29 October 2013
Mgr. Jakub Vysloužil (∗) • J. Bavoľárová • M. Kejdušová • K. Dvořáčková • D. Vetchý
Department of Pharmaceutics, Faculty of Pharmacy
University of Veterinary and Pharmaceutical Sciences Brno
Palackého 1/3, 612 42 Brno, Czech Republic
e-mail: jakub.vyslouzil@gmail.com
Sources
1. Kumar M. N. V. R. Nano and microparticles as controlled drug delivery devices. J Pharm Pharm Sci 2000; 3, 234–258.
2. Vasir J. K., Tambwekar K., Garg S. Bioadhesive microspheres as a controlled drug delivery system. Int J Pharm 2003; 255, 13–32.
3. Tran V. T., Benoît J. P., Venier-Julienne M. C. Why and how to prepare biodegradable, monodispersed, polymeric microparticles in the field of pharmacy? Int J Pharm 2011; 407, 1–11.
4. Gelperina S., Kisich K., Iseman M. D., Heifets L. The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. Am J Respir Crit Care 2005; 172, 1487–1490.
5. Kim S. J., Hahn S. K., Kim M. J., Kim D. H., Lee Y. P. Development of a novel sustained release formulation of recombinant human growth hormone using sodium hyaluronate microparticles. J Control Release 2005; 104, 323–335.
6. Thomasin C., Corradin G., Men Y., Merkle H. P., Gander B. Tetanus toxoid and synthetic malaria antigen containing poly (lactide)/poly (lactide-co-glycolide) microspheres: importance of polymer degradation and antigen release for immune response. J Control Release 1996; 41, 131–145.
7. Jyothi N. V. N., Prasanna P. M., Sakarkar S. N., Prabha K. S., Ramaiah P. S., Srawan G. Y. Microencapsulation techniques, factors influencing encapsulation efficiency. J Microencapsul 2010; 27, 187–197.
8. Tuncay M., Çaliş S., Kaş H. S., Ercan M. T., Peksoy I., Hincal A. A. Diclofenac sodium incorporated PLGA (50 : 50) microspheres: formulation considerations and in vitro/in vivo evaluation. Int J Pharm 2000; 195, 179–188.
9. Kirby D. J., Kaur R., Perrie Y. Formulation and Characterisation of PLGA Microspheres as Vaccine Adjuvants. In Flower D. R., Perrie Y. eds. Immunomic Discovery of Adjuvants and Candidate Subunit Vaccines Springer, 1st ed. New York: Springer 2013.
10. Zakeri-Milani P., Loveymi B. D., Jelvehgari M., Valizadeh H. The characteristics and improved intestinal permeability of vancomycin PLGA-nanoparticles as colloidal drug delivery system. Colloids Surf B Biointerfaces 2013; 103, 174–181.
11. Beck L. R., Cowsar D. R., Lewis D. H., Gibson J. W., Flowers C. E. New long-acting injectable microcapsule contraceptive system. Am J Obstet Gynecol 1979; 135, 419–426.
12. Li M., Rouaud O., Poncelet D. Microencapsulation by solvent evaporation: State of the art for process engineering approaches. Int J Pharm 2008; 363, 26–39.
13. Ogawa Y., Yamamoto M., Okada H., Yashiki T., Shimamoto T. A new technique to efficiently entrap leuprolide acetate into microcapsules of polylactic acid or copoly (lactic/glycolic) acid. Chem Pharm Bull 1988; 36, 1095–1103.
14. Bodmeier R., McGinity J. W. Solvent selection in the preparation of poly (DL-lactide) microspheres prepared by the solvent evaporation method. Int J Pharm 1988; 43, 179–186.
15. Patel R. S., Cho D. Y., Tian C., Chang A., Estrellas K. M., Lavin D., Furtado S., Mathiowitz E. Doxycycline delivery from PLGA microspheres prepared by a modified solvent removal method. J Microencapsul 2012; 29, 344–352.
16. Hui J., Yu X. J., Zhang Y., Hu F. Q. Characterization of Anti-Cancer Drug Materials Loaded Poly (3-hydroxybutyrate - co-3-hydroxyhexanoate) Microspheres for Drug Delivery System in Biochemical Material System. Adv Mat Res 2012; 600, 137–143.
17. Ashford M., Fell J. T., Attwood D., Woodhead P. J. An in vitro investigation into the suitability of pH-dependent polymers for colonic targeting. Int J Pharm. 1993; 91, 241-245.
18. Rasool F., Ahmad M., Murtaza G., Khan H. M. S., Khan S. A. Pharmacokinetic Studies on Metoprolol-Eudragit Matrix Tablets and Bioequivalence Consideration with Mepressor®. Tropical Journal of Pharmaceutical Research 2012; 11, 281–287.
19. Dave V. S., Fahmy R. M., Bensley D., Hoag S. W. Eudragit® RS PO/RL PO as rate-controlling matrix-formers via roller compaction: Influence of formulation and process variables on functional attributes of granules and tablets. Drug Dev Ind Pharm 2012; 38, 1240–1253.
20. Cortesi R., Ravani L., Menegatti E., Esposito E., Ronconi F. Eudragit® microparticles for the release of budesonide: A comparative study. Indian J Pharm Sci 2012; 74, 415–421.
21. Duarte A. R. C., Roy C., Vega-González A., Duarte C. M., Subra-Paternault P. Preparation of acetazolamide composite microparticles by supercritical anti-solvent techniques. Int J Pharm 2007; 332, 132–139.
22. http://eudragit.evonik.com/product/eudragit/en/products-services/ eudragit-products/protective-formulations/e100/Pages/default.aspx
23. Saravanan M., Bhaskar K., Maharajan G., Pillai K. S. Ultrasonically controlled release and targeted delivery of diclofenac sodium via gelatin magnetic microspheres. Int J Pharm 2004; 283, 71–82.
24. Wang S. B., Chen A. Z., Weng L. J., Chen M. Y., Xie X. L. Effect of Drug‐loading Methods on Drug Load, Encapsulation Efficiency and Release Properties of Alginate/Poly‐L‐Arginine/Chitosan Ternary Complex Microcapsules. Macromol Biosci 2004; 4, 27–30.
25. Haznedar S., Dortunc B. Preparation and in vitro evaluation of Eudragit microspheres containing acetazolamide. Int J Pharm 2004; 269, 131–140.
26. European Pharmacopoeia Online http://online6.edqm.eu/ep708/
27. Vetchý D., Vetchá M., Rabišková M., Gryczová E., Bartošíková L. Comparison in vitro felodipine release rate from the original versus generic product with controlled release of the drug. Medicina (Kaunas) 2007; 43, 326–331.
28. Singh V., Chaudhary A. K. Preparation of Eudragit E100 microspheres by modified solvent evaporation method. Acta Pol Pharm 2011; 68, 975–980.
29. Basu S. K., Adhiyaman R. Preparation and Characterization of Nitrendipine-loaded Eudragit RL 100 Microspheres Prepared by an Emulsion-Solvent Evaporation Method. Tropical Journal of Pharmaceutical Research 2008; 7, 1033–1041.
30. Alhnan M. A., Basit A. W. Engineering polymer blend microparticles: An investigation into the influence of polymer blend distribution and interaction. Eur J Pharm Sci 2011; 42, 30–36.
31. El-Kamel A. H., Sokar M. S., Al Gamal S. S., Naggar V. F. Preparation and evaluation of ketoprofen floating oral delivery system. Int J Pharm. 2001; 220, 13–21.
32. Khatun M., Islam S. M. A., Akter P., Quadir M. A., Reza M. S. Controlled release of naproxen sodium from Eudragit RS 100 transdermal film. Dhaka University Journal of Pharmaceutical Sciences 2004; 3, 1–2.
33. Cerea M., Zheng W., Young C. R., McGinity J. W. A novel powder coating process for attaining taste masking and moisture protective films applied to tablets. Int J Pharm 2004; 279, 127–139.
34. Guzman M. L., Manzo R. H., Olivera M. E. Eudragit E100 as a Drug Carrier: The Remarkable Affinity of Phosphate Ester for Dimethylamine. Mol Pharmaceutics 2012; 9, 2424–2433.
35. Damgé C., Maincent P., Ubrich N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Control Release 2007; 117, 163–170.
36. Hanamura T. Experimental study of acrylic resin coated gentamicin tablets for local antibiotic therapy. Nihon Seikeigeka Gakkai zasshi 1984; 58, 555–565.
37. Stallmann H. P., Faber C., Bronckers A. L., Amerongen A. V. N., Wuisman P. I. In vitro gentamicin release from commercially available calcium-phosphate bone substitutes influence of carrier type on duration of the release profile. BMC Musculoskelet Disord 2006; 7, 1–8.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2013 Issue 6
Most read in this issue
- A view on providing care in the field of medicines in Slovakia – the pharmacist and the patient
- Alkalimetric titrations of salts of organic bases in the Pharmacopoeia
- Influence of quaternary ammonium salt on liberation of drug with antiseptic effect
- Physiological factors with impact on the drug behaviour in the gastrointestinal tract