
Brief insight into the in silico properties, structure–activity relationships and biotransformation of fruquintinib, an anticancer drug of a new generation containing a privileged benzofuran scaffold
Stručný pohľad na vlastnosti in silico, vzťahy štruktúra – –aktivita a biotransformáciu fruquintinibu, protinádorovo účinkujúceho liečiva novej generácie obsahujúceho privilegované benzofuránové zoskupenie
Súčasné trendy projekcie liečiv významne reflektujú tzv. privilegované zoskupenia ako základné (tzv. jadrové) štruktúrne fragmenty s rozhodujúcim vplyvom na afinitu k vhodne zvoleným biologickým cieľom, účinok, selektivitu aj toxikologické charakteristiky týchto liečiv a perspektívnych kandidátov na liečivá. Fruquintinib (1) je nový syntetický selektívny inhibítor izoforiem receptora vaskulárneho endotelového rastového faktora (z angl. vascular endothelial growth factor receptor; VEGFR), t. j. VEGFR-1, VEGFR-2 a VEGFR-3. Terapeutikum (1) obsahuje planárne bicyklické heteroaromatické jadro, v ktorom sú vhodne inkorporované dva atómy dusíka, základný (jadrový) bicyklický heteroaromatický kruh – privilegované (substituované) benzofuránové zoskupenie a skupinu pôsobiacu ako donor a akceptor väzby vodíkovým mostíkom (VVM), t. j. amidové funkčné zoskupenie. Fruquintinib (1) bol prvýkrát schválený v Číne pre liečbu metastázujúceho kolorektálneho karcinómu, závažného nádorového ochorenia s vysokou mortalitou. Táto prehľadová publikácia ponúkla stručný pohľad na tému privilegovaných štruktúr, ich niekoľkých parametrov, ktorých rozsah približuje tzv. liečivu podobné (drug-like) vlastnosti, farmakodynamické charakteristiky fruquintinibu (1) a rôzne in silico-deskriptory definujúce štruktúrne a fyzikálno-chemické vlastnosti tohto liečiva (molekulová hmotnosť, počet ťažkých atómov, počet aromatických tažkých atómov, frakcia C-atómov v sp3-hybridizovanom stave, počet akceptorov VVM, počet donorov VVM, celkový polárny povrch, molekulová refrakcia, molekulový objem aj parametre lipofility a rozpustnosti). Niektoré z týchto deskriptorov súviseli s farmakokinetikou aj distribúciou fruquintinibu (1) a navyše by mohli pomôcť predikovať jeho schopnosť pasívne prechádzať hematoencefalickou bariérou (HEB). V publikácii sa hodnotila aj eventuálna súvislosť medzi indukčným potenciálom liečiva (1) voči izoenzýmom cytochrómu P450 (CYP1A2 a CYP3A4) a jeho pasívnym transportom do centrálneho nervového systému via HEB. Stručne boli takisto načrtnuté súčasné klinické skúsenosti s fruquintinibom (1) a budúce liečebné možnosti tohto terapeutika.
Klíčová slova:
privilegované zoskupenie – fruquinti- nib – vlastnosti in silico – vzťahy štuktúra–aktivita – far- makokinetika
Authors:
Dominika Nádaská 1; Lucia Hudecova 2; Gustáv Kováč 2; Ivan Malík 1,2
Authors‘ workplace:
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University Bratislava, Slovak Republic
1; Institute of Chemistry, Clinical Biochemistry and Laboratory Medicine Faculty of Medicine, Slovak Medical University in Bratislava, Slovak Republic
2
Published in:
Čes. slov. Farm., 2023; 72, 267-276
Category:
Review Articles
doi:
https://doi.org/10.5817/CSF2023-6-267
Overview
Current trends in drug design notably consider so-called privileged scaffolds as the core structural fragments with decisive impact on affinity to properly chosen biological targets, potency, selectivity and toxicological characteristics of drugs and prospective drug candidates. Fruquintinib (1) is a novel synthetic selective inhibitor of vascular endothelial growth factor receptor (VEGFR) isoforms, i.e., VEGFR-1, VEGFR-2 and VEGFR-3. The therapeutic agent (1) consists of a flat bicyclic heteroaromatic ring, in which two nitrogens are suitablyincorporated, a core bicyclic heteroaromatic ring – privileged (substituted) benzofuran scaffold, and a pair of hydrogen bond (H-bond) donor and acceptor group, i.e., amide functional moiety. Fruquintinib (1) was first approved in China for the treatment of metastatic colorectal cancer, a severe malignant disease with a high mortality rate. The review article offered a brief insight into the topic of privileged structures, their drug - -like ranges of several parameters, pharmacodynamic characteristics of fruquintinib (1) and various in silico descriptors characterizing drug’s structural and physicochemical properties (molecular weight, number of heavy atoms, number of aromatic heavy atoms, fraction of sp3 C-atoms, number of H-bond acceptors, number of H-bond donors, total polar surface area, molar refractivity, molecular volume as well as parameters of lipophilicity and solubility). Some of these descriptors were related to pharmacokinetics and distribution of fruquintinib (1), and, in addition, might help predict its ability to cross passively the blood–brain barrier (BBB). Moreover, a possible connection between the induction potential on cytochrome P450 isoenzymes (CYP1A2 and CYP3A4) and passive transport of a given drug into the central nervous system via BBB was investigated. Current clinical experience and future directions regarding of fruquintinib (1) were also briefly outlined.
Keywords:
privileged scaffold – fruquintinib – in silico properties – structure–activity relationships – pharma- cokinetics
Current definition of the term privileged scaffold1) as well as its practical interpretation among medicinal chemists might "slightly" differ from the original perception of such scaffold coined by the research of Evans and his co-workers2). Theprivileged scaffold was first introduced as a single molecular framework capable to provide high-affinity ligands for more than one type of a receptor2). Following the more recent view and opinion of Zhao and Dietrich (2015), a chemical core structure can be regarded as a privileged scaffold if it is more probable that its derivatives can interact with several biological (protein) targets with high affinity and selectivity than other structures1).
Various structurally "simple" heterocycles containing O-, Sor N-atoms, such as morpholine, thiazole or suitablysubstituted triazine, incorporated in the structure of biologically active compounds, would be considered the privilegedscaffolds3–5) from a certain point of view. In fact, the given heterocyclic moieties meet the description according to Maclean et al. (2000) – privileged scaffolds are substructural features, which confer desirable (often drug-like) properties on compounds containing those features6). Other very well-known examples of privileged scaffolds7–11) are phenyl-substituted monocycles (biphenyls, N-arylpiperidines, N-arylpiperazines, 1,4-dihydropyridines or dihydropyrimidones), fused [7-6] ring systems (1,4-benzodiazepin-2-ones, 1,5-benzodiazepin-2-ones, 1,4-benzodiazepin-2,5-diones, pyrrolo[2,1-c][1,4]benzodiazepin-5,11-diones, 1,4-benzothiazepin-5-ones or 5,11-dihydrobenzo[e]pyrido[3,2-b][1,4]-diazepin-6-ones), fused [6-6] ring systems (benzopyrans, chromones, coumarins and pyranocoumarins or various quinoxalines / quinazolines) or fused [5-6] ring systems (indoles, benzimidazoles, azolo-1,2,4-triazines, azolopyrimidines, benzofurans or benzothiophenes).
Bicyclic privileged structures could be defined by drug-like ranges of several parameters as follows12): 260.0 ≤ molecular weight (M.W.) ≤ 524.0, 0.9 ≤ lipophilicity descriptor generated in silico (ALogP) ≤ 5.4, 2 ≤ number ofheteroatoms (nhet; O, N, S or P) with one or more lone pairs, excluding atoms with positive formal charges, amide and pyrrole-type N-atoms, and aromatic Oand S-atoms in a bicyclic system ≤ 8, nhet (O, N, S or P)with one or more attached H-atoms ≤ 3, 21.0 Å2 ≤ polar surface area (calculated parameter using a 2D approximation; PSA) ≤ 128.6 Å2, 6.3 ≤ ratio of the PSA value divided by a total surface area (TPSA) value ≤ 34.2, 1 ≤ number of rotatable bonds (nrotb) ≤ 10, 2 ≤ number of rings ≤ 5, 1 ≤ number of chain assemblies ≤ 7, and atoms marked as EvenAtomStereo, OddAtomStereo or UnknownAtomStereo, and atoms that are internally perceived as having stereo and that are not marked as EvenAtomStereo or OddAtomStereo ≤ 4.
In addition, both number of hydrogen-bond (H-bond) donors and nrotb mainly modified the absorption of designedcompounds (drugs), while lipophilicity, flexibility, degree of branching and existence of some functional groups determined their fate in a metabolic process12, 13).
In fact, the design and development of biologically active compounds containing one or more privileged scaffolds has to be very precise. For example, a 2-aminothiazole (privileged) moiety is considered an integral part of various synthetic molecules providing notable pharmacodynamic effects. On the other hand, numerous pan-assay interference compounds contain a given chemotype suggesting potential risks of off-target activity if those molecules will be chosen for furtherdevelopment and optimization14).
Biologically active benzofuran derivatives – brief overview
Heterocyclic systems containing one or more O-atoms take up a central role as core components within a structure ofsynthetic or natural compounds showing diverse biological activities. These include anticancer, antimicrobial, antimycobacterial, antifungal, anti-inflammatory, analgesic, antioxidant, antiparasitic, antiviral, and antiseizure agents or the therapeutics for neuropsychiatric diseases, as published in various research and review papers15–21).
The structure of a benzofuran core consists of a fused benzene and furan ring. Following the frontier orbital theory, as the frontier electron populations of the parent benzo[b]furan are greater, the corresponding C-atoms are more reactive toward electrophiles22). Besides, a 2,3-dihydrobenzo[b]furan core is also considered a benzofuran unit. This framework can be foundin the structure23, 24) of natural molecules (ganodone, δ-viniferin, or ε-viniferin) as well as synthetic compounds – clinical drugs (citalopram or ramelteon).
Synthetic routes providing various (substituted) benzofurans were briefly mentioned in a review article25), for example, and several unconventional synthetic methodologies were comprehensively summarized and published26) as well. The introduction of proper substituents at specified positions of a benzofuran core leads to new derivatives with unique structural characteristics that may possess an excellent therapeutic value27, 28). The molecules containing a privileged benzofuran scaffold effectively interact with various biological targets and are considered powerful anticancer, antibacterial, antimycobacterial, antifungal, antiviral, anti-inflammatory, analgesic, antipyretic, antioxidant, anti-ulcer and anti-hyperlipidemic agents, insecticides, trypanocides as well as the compounds showing a beneficial impact in various phases of progressive neurodegenerative disorders such as Alzheimer’s disease25, 29–31).
Inhibition of vascular endothelial-derived growth factors by the activity of fruquintinib (1) as an important therapeuticstrategy to suppress tumor growth
Vascular endothelial growth factors (VEGFs), alternatively termed vascular permeability factors, play critical roles inangiogenesis, promoting cell survival as well as the growth and proliferation of endothelial cells. These VEGFs are classified into five isoforms, i.e., VEGF-A, VEGF-B, VEGF-C, VEGF-D, and VEGF-E, and placenta growth factor (abbreviation used: PlGF) as well. The VEGF-A is regarded as the most important factor commonly termed VEGF. Biological effects of given multifunctional peptides are mediated via canonical activation of several transmembrane VEGF receptor (VEGFR) subtypes,i.e., VEGFR-1 (also termed Flt-1 or fms-like Tyrosine Kinase 1), VEGFR-2 (KDR Kinase Domain Region / flk-1 or Fetal Liver Kinase 1) and VEGFR-3 (Flt-4), and neutropilins32–34). These VEGFRs belong to a family of receptor protein tyrosine kinases, whose upregulation has been observed in various tumors, both benign and malignant35).
Scientific literature survey offers numerous molecules effectively targeting VEGFRs, which are or would be clinically used to treat different types of cancer. The VEGFR inhibitory activity was observed for various compounds of natural origin, including epigallocatechin gallate, resveratrol, curcumin, wogonin, triptolide, or farnesiferol C, for example33). Many potent synthetic VEGFR inhibitors can potently inhibit VEGFR-1, VEGFR-2 and VEGFR-3. These molecules are classified as pan-VEGFR inhibitors such as axitinib, cediranib, dovitinib, motesanib, nintedanib, lenvatinib, pazopanib, regorafenib or tivozanib. Synthetic selective-VEGFR inhibitors, e.g., apatinib, brivanib, cabozantinib, foretinib, ponatinib, semaxanib, sorafenib, sunitinib or vandetanib, can suppress one or two of VEGFRs. Chemical structures of the small-molecule pan-VEGFR as well as selective-VEGFR inhibitors (characterized with M.W. < 900 – 1000 Da, among others) for targeted cancer therapy, their mechanism of action, biological targets and several data from their clinical trials can be found in the review papers33, 36, 37).
Fruquintinib (1), chemically 6-(6,7-dimethoxy - quinazolin-4-yloxy)-N,2-dimethylbenzofuran-3-carboxamide (CAS Registry Number: 1194506-26-7), is a novel, orally available drug containing a privileged benzofuran core. The molecule (1), also termed HMPL-013, potently and highly selectively inhibits desired biological targets, i.e., VEGFR-1, VEGFR-2, and VEGFR-3, for long term36). High selectivity to those receptors was observed when exploring a panel of more than 250 kinases, in which the synthetic compound (1) was found to potently inhibit VEGFR-1, VEGFR-2, and VEGFR-3 and provided weak to no inhibitory effect on all other kinases38).
The drug (1) received its first global approval in China for patients with metastatic colorectal cancer (mCRC) who havefailed at least two prior systemic antineoplastic therapies, including fluoropyrimidine, oxaliplatin, and irinotecan36). Fruquintinib (1) is the first Chinese original orally available small-molecule anticancer agent of a new generation approved by the National Medical Products Administration of China39) in 2018.
A brief view on particular phases of clinical development as well as the list of completed and ongoing clinical trialsinvolving fruquintinib (1) as the treatment for mCRC, (advanced) non-squamous non-small cell lung cancer, advancedgastric or gastroesophageal junction adenocarcinoma, and advanced solid tumors were summarized in40, 41). The current review paper focused primarily on in silico properties, structure–anti-VEGFR-2 inhibitory activity, and biotransformation pathways of this drug.
Fundamental physicochemical characteristics of fruquintinib (1) and structure–activity relationships in terms of interactions between a given ligand and vascular endothelial–derived growth factor receptor 2
The chemical structure of fruquintinib (1) can be "virtually" divided into several parts (Fig. 1) as follows:
i) flat bicyclic heteroaromatic ring containing two N-atoms (fragment A),
ii) core bicyclic heteroaromatic ring – a (substituted) benzofuran scaffold (B),
iii) pair of H-bond donor and acceptor groups consisting of an amide functional moiety (C).
In addition, the structure of other VEGFR-2 inhibitors also contains a monocyclic/bicyclic (hetero)aromatic ring, which can be unsubstituted (substituted with hydrogen, in fact) or substituted with one or more halogen atoms42), interacting with an allosteric hydrophobic pocket via numerous hydrophobic interactions.
Several physicochemical properties of the compound (1) were calculated by the authors of the current paper using aSwissADME applet43), a free web tool, in which a structure of fruquintinib (1) was converted to an appropriate digital notation, i.e., Simplified Molecular Input Line Entry System (SMILES). Based on generated SMILES, the obtained descriptors were as follows: M.W. = 393.39 g/mol (or in Da units), number of heavy atoms (nha) = 29, number of aromatic heavy atoms (nar-ha) = 19, fraction of sp3 C-atoms (Csp3) = 0.19, nrotb = 6, number of H-bond acceptors (nON) = 7, number of H-bond donors (nOHNH) = 1, TPSA = 95.7 Å2, molar refractivity (M.R.)= 106.8 Å2 and logarithm of a partition coefficient (log P) for an octan-1-ol/water partition system predicted by a Moriguchi44, 45) topological method (MLOGP) = 1.41.
In addition, the lipophilicity was also predicted by a fragment-based computational approach46) (CLOGP) = 3.03 with aPerkin Elmer’s ChemDraw ver. 19.0.0.22 (PerkinElmer Informatics, Waltham, MA, USA) software package47).
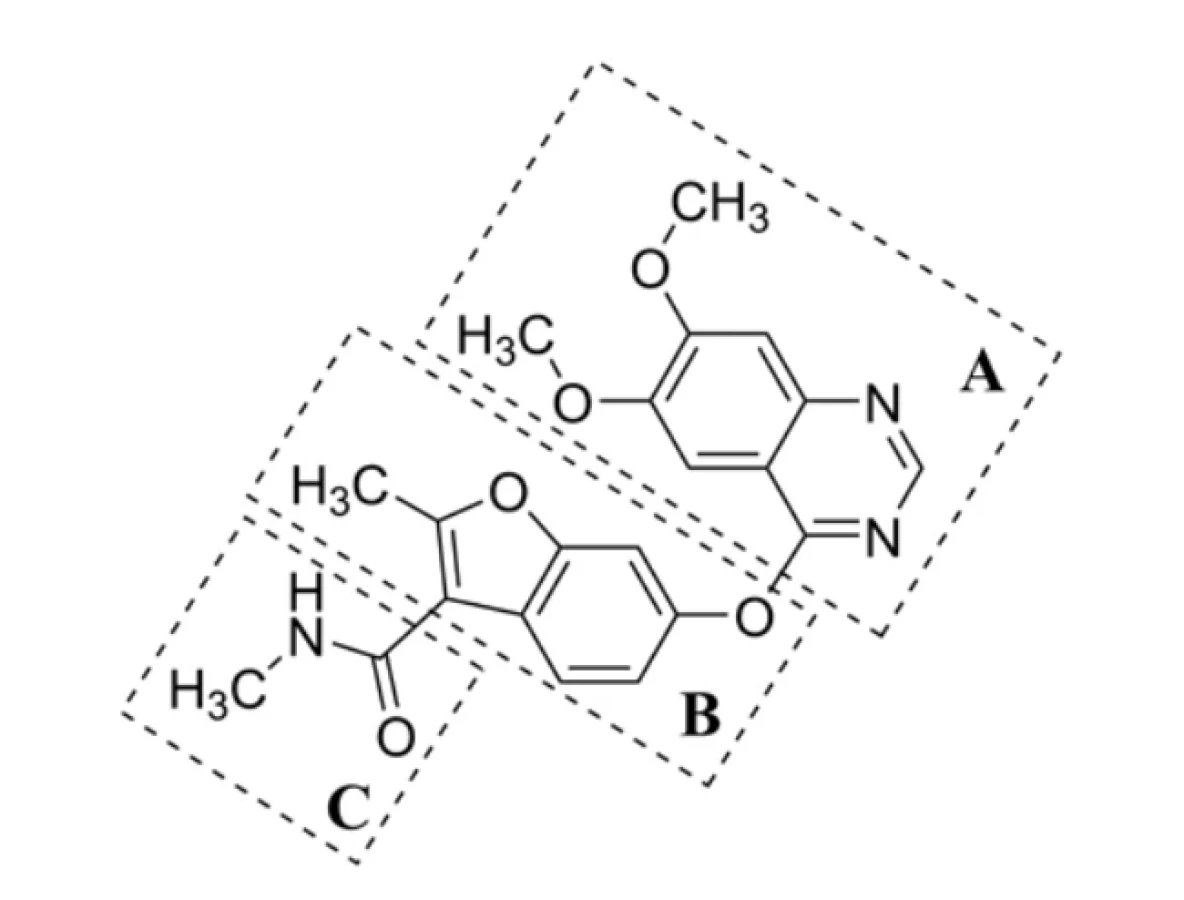
Fig. 1. Chemical structure of fruquintinib (1), an effective VEGFR-1, VEGFR-2 and VEGFR-3 inhibitor, virtually divided into three compartments A–C
Molecular volume (V, in A3 units) of fruquintinib (1), i.e., V = 341.3 A3, was also predicted using a MolinspirationCheminformatics48) property engine (Molispiration Cheminformatics, Slovenský Grob, Slovak Republic), in which the structure of (1) was converted to SMILES as in the case of the analysis by the SwissADME predictor.
The compound (1) was "moderately soluble" based on a generated log S value describing solubility (S) according to amethod of Ali and his co-workers49). The log S parameter was found in a range43, 49) of –6 < log S < –4, as the calculation procedure showed. Fragmental method integrated within a Filter-IT program ver. 1.0.2 (Silicos-IT, Wijnegem, Antwerpen, Belgium) assigned the category "poorly soluble"43, 50). Thus, the classification group for (1) was defined with the log S value belonging to an interval of –10 < log S < –6.
Some of these parameters were used to provide a more detailed "theoretical" view on the compound’s ability of being passively absorbed per os as well as its capability to pass the blood–brain barrier (BBB) into the central nervous system (CNS) by a passive mechanism.
The VEGFR-2, as a type of membrane-bound receptor tyrosine kinases, regulates the process of vasculogenesis and angiogenesis51). The inhibitors of a given 210 to 230 kDa glycoprotein can be roughly classified into three types52), i.e.,adenosine-5´-triphosphate (ATP) competitive inhibitors, which are able to bind at a pocket reserved for the accommodation ofan adenine ring of ATP (type I), inhibitors interacting specifically to the inactive (DFG-out) conformation of VEGFR-2 (type II), and inhibitors, which covalently interact to the active site of this receptor at a hinge region in order to prevent the ATP binding at a catalytic domain (type III).
Molecular docking of the VEGFR-2 inhibitors approved by the U.S. Food and Drug Administration indicated the occupation ofmainly four regions of the glycoprotein, i.e., hydrophobic region I, hydrophobic region II, H-bond rich region (DFG domain), andhinge region. Thus, those ligands are regarded as indirectly competitive inhibitors42). The N-atom within an A fragment,which was opposite to an O-aryl group (Figure 1), interacted at the hinge region of VEGFR-2, forming H-bonds with anamino acid (A.A.) residuum (Cys919). The interaction was essentially required for the inhibitory activity of (1). The central B ring interacted with another A.A. residuum (Lys868) within a hydrophobic region I. In addition, the H-bond-rich regionwas engaged by an amide group within a C compartment of fruquintinib (1), forming H-bonds with specific AAs42), i.e.,Glu885 (interaction with an NH-moiety of a drug) and Asp1046 (CO-group).
Several in silico predictions connected with pharmacokinetics and distribution of fruquintinib (1)
When the drug molecule was characterized by M.W. > 500 Da, CLOGP > 5.00 (or Moriguchi`s MLOGP > 4.15), nON > 10, nOHNH > 5, nrotb > 10 and PSA > 140.0 Å2, i.e., if its (nON + nOHNH) > 12, it would not be sufficiently absorbed passively from a gastrointestinal tract when beingadministered per os44–46, 53, 54). In fact, the values of these descriptors calculated for fruquintinib (1) supported its oral administration55).
Kelder and his co-workers56) found out that non-CNS drugs transported passively and transcellularly needed PSA (TPSA) ≤ 120.0 Å2, whereas the drugs can be targeted to the CNS with PSA (TPSA) < 60.0–70.0 Å2. On the other hand, van deWaterbeemd with his scientific team57) suggested a cut-off limit of PSA (TPSA) for CNS penetration to ≤ 90.0 Å2 and M.W. < 450 Da. Levin58) proposed the M.W. value cut-off ≤ 400 Da. Hansch and Leo59) found out that BBB penetration was optimal when the value of a log P descriptor for a CNS-active drug was within an interval of 1.5–2.7. In addition, the small-molecule drugs with V = 740.0–970.0 Å3 could passively permeate via BBB60). Following the criteria published in56, 57, 59, 60), fruquintinib (1) would not be passively transported via BBB into CNS.
Moreover, the research61) summarized several essential attributes of successful CNS-active drugs as follows: M.W. < 450 Da, CLOGP < 5.00 (currently investigated anticancer compound (1) passed both criteria), nON < 7 (did not meet),nOHNH < 3 (passed), nrotb < 8 (passed), H-bonds < 8 (did not meet) and PSA (TPSA) < 60.0–70.0 Å2 (did not meet), respectively. If the difference CLOGP – (number of nitrogens + number of oxygens) was > 0.00, then the compound had a high probability of entering the CNS.
The predicted solubility of (1) in aqueous environment49) was 2.98 μg/ml; however, the CNS-active agents should becharacterized with solubility > 60.00 μg/ml61).
In general, the compounds possessing a tertiary N-atom show a higher degree of brain permeation. The predicted value of an acid-base dissociation constant (pKa) parameter62) for the molecule (1) was 14.99. The research63) regarded pKa = 4–10 as an optimal interval for passive permeation of a drug through BBB; the study61) was slightly more rigorous and indicated a neutral or basic molecule with pKa = 7.5–10.5 (avoiding acids) as a suitable CNS-active drug.
Considering the values of all given descriptors and the total number of N - and O-atoms56, 57, 59–61) within the structure offruquintinib (1), it might be concluded that this anticancer drug would not be passively transported via BBB into the CNS. In fact,its distributions in adipose, adrenal, and kidney were slightly higher than or almost equivalent to the corresponding plasma levels.The lowest distribution was observed in the brain, testis, and bone marrow when experimental animal models (rats) were used55).
Biotransformation of fruquintinib (1)
Regarding fruquintinib’s (1) metabolism, three major oxidative metabolites, i.e., a metabolite M1 (2), M2 (3), and a series of M3 (4) metabolites, were identified in liver microsomes of several animal models (mouse, rat, dog, and monkey) and humans (Figure 2). Oxidation (M1) and oxidative O-demethylation (M2) within phase I of biotransformation, followed by O-glucuronidation (phase II) are the major in vivo metabolic pathways55).
Two cytochrome P450 (CYP) enzymes are responsible for oxidation and oxidative O-demethylation in vitro of (1), leading to M1 (2) and M2 (3), respectively. These enzymes are well-known CYP3A4, a main enzyme biotransforming (1), and CYP2D6. The oxidation in vitro of a fundamental structural core, as schematically drawn for the metabolites from a series M3 (4) of a parent compound (1), was possible via the activity of CYP3A4, CYP2D6 as well as CYP2C19 (Fig. 2). Itwas predicted that fruquintinib (1) might show favorable human pharmacokinetic properties and low efficacious dose55). The compound (1) provided no induction potential on CYP1A2 and CYP3A4 in human hepatocytes and probably did not induce some transporters, like P-glycoprotein (P-gp), after multi-dosing55).
In fact, these pharmacokinetic features, if taken "separately", did not provide the light to clearly
answer the question of whether fruquintinib (1) might be passively transported via BBB into the CNS or not. The reason wasthat a successful CNS-active drug must have no significant CYP2D6 metabolism. In the case of (1), the meeting this criterion was quite questionable, as the research55) indicated. Furthermore, the CNS-active drug must be a nonpotent CYP3A4inducer61) – the compound (1) passed55) that requirement – and must have no to low affinity in vivo to the P-gp transporter61, 64). Focusing on such affinity studies, however, no relevant in vivo (using experimental animal models) or clinical data were available.
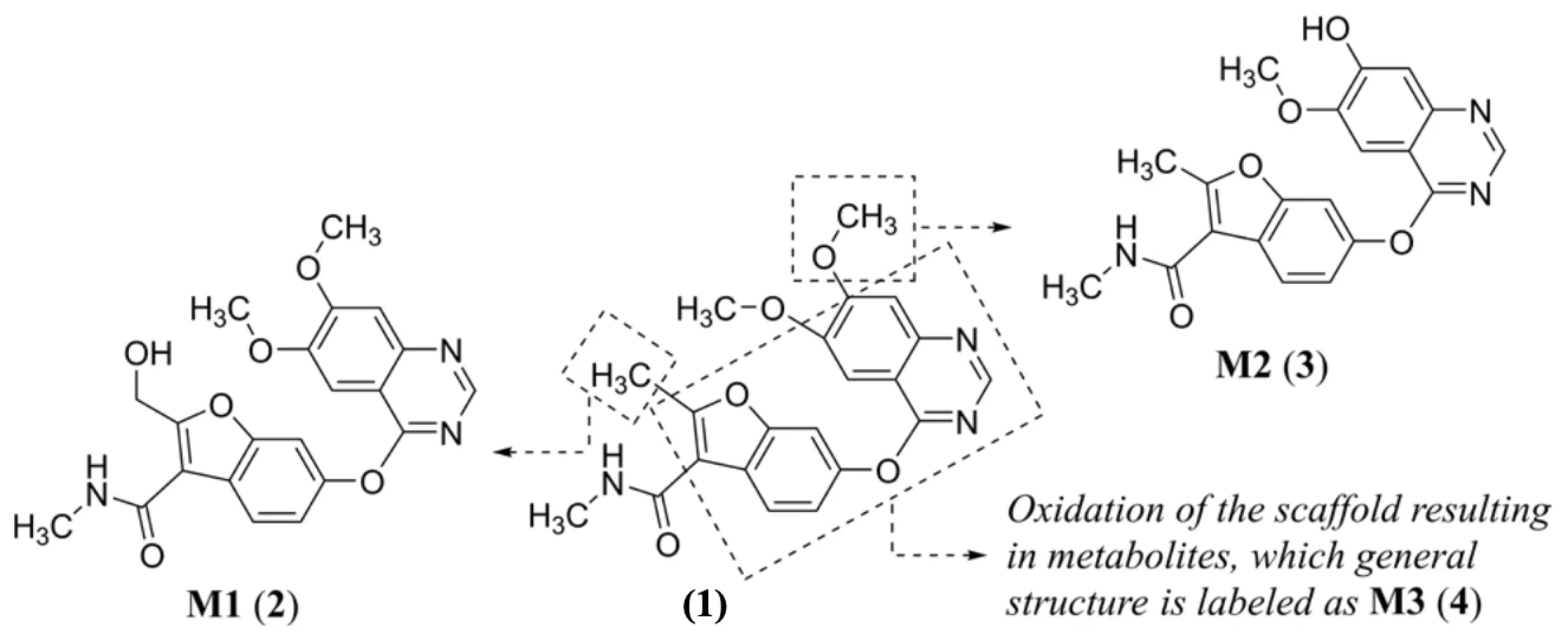
Fig. 2. Proposed biotransformation in vitro55) of fruquintinib (1) with indication of main metabolites M1 (2)–M3 (4)
Current clinical experience and future directions
From a clinical point of view, beneficial therapeutic interventions against some (severe) cancers might be done employing fruquintinib (1), as the conclusions from various clinical trials indicated. For example, the administration of a given compound after local radiotherapy may be an effective option in the treatment of specific populations suffering from mCRC with high microsatellite instability and v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS) exon 2 p. G12D mutation65).
Combining fruquintinib (1) and a suitable immune checkpoint inhibitor, i.e., programmed cell death protein 1(programmed cell death receptor-1; PD-1) / PD-ligand 1 (PD-L1) antibody, might improve the efficacy compared tomonotherapy when treating various cancers in preclinical and clinical studies. Sintilimab, as a fully human IgG4 monoclonal antibody, binds to a programmed cell PD-1 in order to block the interaction between PD-1 and its ligands (PD-L1 and PD-L2) and consequently help to restore the endogenous antitumour T-cell response66). The clinical trial studies67, 68) regarding fruquintinib (1) plus sintilimab and fruquintinib (1) plus toripalimab, a recombinant, humanized PD-1 monoclonal antibody69), respectively, showed promising clinical activity of these combinations in patients with mismatch repair-proficient mCRC. In addition, fruquintinib (1) was reported for the first time to have favorable efficacy and a convenient safety profile as an optional treatment modality for patients with advanced bone and soft tissue sarcoma who failed in multi-line therapies70).
The drug (1) also showed promising efficacy and acceptable toxicity as second or further-line therapy in advanced or metastatic biliary tract cancer71), a quite rare but aggressive disease.
On the other hand, fruquintinib (1) as a relatively new drug brought not only clinical benefits but also several adverse reactions as the first case of fruquintinib (1)-associated aortic dissection was recently reported72). The reactions might be suppressed by administering a pH-triggered size-converted nano-drug delivery system to co-deliver fruquintinib (1) togetherwith other chemotherapeutic agents, such as doxorubicin. The clinical use of such a nanoparticle system could simultaneously achieve rapid tumor tissue enrichment and high efficiency tumor tissue penetration providing excellent antitumor effects of both anticancer therapeutics as well as effective inhibition of tumor growth and metastasis73).
Conclusions
Colorectal cancer (CRC) belongs to a group of the most predominant malignancies with a high mortality rate globally. Approximately a quarter of CRC patients presented metastatic disease (mCRC) at diagnosis, while almost half of them will develop metastases. The treatment paradigm for CRC/mCRC is nowadays moving towards a tailored approach based on clinical and molecular characteristics. Fruquintinib (1), an orally bioavailable innovative small-molecule tyrosine kinase inhibitor highly selectively targeting VEGFR-1, VEGFR-2, and VEGFR-3, is successfully utilized clinically for the treatment of patients suffering from mCRC. The structural arrangement of the molecule (1) favors its interactions with particular VEGFR subtypes. Several in silico descriptors listed in this paper supported the "decision" to administer fruquintinib (1)per os, and, in addition, they could indicate the compound’s (in)ability to be passively transported via BBB into CNS. Accordingly, CNS side effects of a given molecule (1) were expected to be quite low. The antiangiogenic agent (1) containing a privileged benzofuran scaffold could significantly improve major efficacy parameters, including response rate,progression-free survival, and overall survival. Moreover, fruquintinib (1) might be successfully involved in combinationtherapy focusing on the treatment of not only rat sarcoma (RAS) oncogene-mutated or chemo-refractory mCRC but also (locally) advanced gastric cancer, non-small cell lung cancer, (locally) advanced rectal cancer, advanced pancreatic canceror esophageal squamous cell carcinoma in order to prolong survival and improve the quality of patients` life.
Acknowledgments
We would like to thank the Czech and Slovak Pharmacy Journal (Česká a slovenská farmacie) for the opportunity to publish this scientific article.
Conflict of interest: none.
Sources
- Zhao H., Dietrich J. Privileged scaffolds in lead generation. Expert Opin. Drug. Discov. 2015; 10, 781–790. doi:10.1517/17460441.2015.1041496
- Evans B. E., Rittle K. E., Bock M. G., DiPardo R. M., Freidinger R. M., Whitter W. L., Lundell G. F., Veber D. F., Anderson P. S., Chang R. S. L., Lotti V. J., Cerino D. J., Chen T. B., Kling P. J., Kunkel K. A., Springer J. P., Hirshfield J. Methods for drug discovery: development of potent,selective, orally effective cholecystin antagonists. J. Med. Chem. 1988; 31, 2235–2246. doi: 10.1021/ jm00120a002
- Kourounakis A. P., Xanthopoulos D., Tzara A. Morpholine as a privileged structure: A review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 2020; 40, 709–752. doi: 10.1002/med.21634
- Datusalia A. K., Khatik G. L. Thiazole heterocycle: A privileged scaffold for drug design and discovery. Curr. Drug Discov. Technol.2018; 15, 162. doi: 10.2174/157016381503180620153423
- Gharat R., Prabhu A., Khambete M. P. Potential of triazines in Alzheimer’s disease: A versatile privileged scaffold. Arch. Pharm. (Weinheim) 2022; 355, art. no. e2100388 (12 pp.). doi: 10.1002/ardp.202100388
- Maclean D., Baldwin J. J., Ivanov V. T., Kato Y., Shaw A., Schenider P., Gordon E. M. Glossary of terms used in combinatorial chemistry (technical report). J. Comb. Chem. 2000; 2, 562–578. doi: 10.1021/cc000071u
- Horton D. A., Bourne G. T., Smythe M. L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures.Chem. Rev. 2003; 103, 893–930. doi: 10.1021/cr020033s
- Costantino L., Barlocco D. Privileged structures as leads in medicinal chemistry. Curr. Med. Chem. 2006; 13, 6585. doi: 10.2174/092986706775197999
- Rusinov V. L., Charushin V. N., Chupakhin O. N. Biologically active azolo-1,2,4-triazines and azolopyrimidines. Russ. Chem. Bull. 2018; 67, 573–599. doi: 10.1007/ s11172-018-2113-8
- Voinkov E. K., Drokin R. A., Fedotov V. V., Butorin I. I., Savateev K. V., Lyapustin D. N., Gazizov D. A., Gorbunov E. B., Slepukhin P. A., Gerasimova N. A., Evstigneeva N. P., Zilberberg N. V., Kungurov N. V., Ulomsky E. N., Rusinov V. L. Azolo[5,1-c][1,2,4]triazines and azoloazapurines: Synthesis, antimicrobial activity and in silico studies. ChemistrySelect 2022; 7, art. no. e202104253 (8 pp.). doi: 10.1002/slct.202104253
- Savateev K. V., Ulomsky E. N., Butorin I. I., Charushin V. N., Rusinov V. L., Chupakhin O. N. Azoloazines as A2a receptor antagonists.Structure–activity relationship. Russ. Chem. Rev. 2018; 87, 636–669. doi: 10.1070/RCR4792
- Han Ch., Zhang J., Zheng M., Xiao Y., Li Y., Liu G. An integrated drug-likeness study for bicyclic privileged structures: from physicochemical properties to in vitro ADME properties. Mol. Divers. 2011; 15, 857–876. doi: 10.1007/s11030-011-9317-2
- Hubatsch I., Ragnarsson E. G. E., Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007; 2, 2111–2119. doi: 10.1038/nprot.2007.303
- Jakopin Ž. 2-Aminothiazoles in drug discovery: Privileged structures or toxicophores? Chem. Biol. Interact. 2020; 330, art. no. 109244 (8 pp.). doi: 10.1016/j. cbi.2020.109244
- Atmaram U. A., Roopan S. M. Biological activity of oxadiazole and thiadiazole derivatives. Appl. Microbiol. Biotechnol. 2022; 106, 3489–3505. doi: 10.1007/s00253-022-11969-0
- He M., Fan M., Peng Z., Wang G. An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur. J. Med. Chem. 2021; 221, art. no. 113546 (29 pp.). doi: 10.1016/j.ejmech.2021.113546
- Rakesh K. P., Shantharam C. S., Sridhara M. B., Manukumar H. M., Qin H.-L. Benzisoxazole: a privileged scaffold for medicinal chemistry. Med. Chem. Commun. 2017; 8, 2023–2039. doi: 10.1039/c7md00449d
- Saroha B., Kumar G., Kumari M., Kaur R., Raghav N., Sharma P. K., Kumar N., Kumar S. A decennary update on diverse heterocycles and their intermediates as privileged scaffolds for cathepsin B inhibition. Int. J. Biol. Macromol. 2022; 222 (Part B),2270–2308. doi: 10.1016/j. ijbiomac.2022.10.017
- Avula S. K., Das B., Csuk R., Al-Harrasi A. Naturally occurring O-heterocycles as anticancer agents. Anticancer Agents Med.Chem. 2022; 22, 3208–3218. doi: 10.2174/1871520621666211108091444
- Pairas G. N., Perperopoulou F., Tsoungas P. G., Varvounis G. The isoxazole ring and its N-oxide: A privileged core structure in neuropsychiatric therapeutics. ChemMedChem. 2017; 12, 408–419. doi: 10.1002/ cmdc.201700023
- Wang Xi., Wang Xu., Zhao Y., Zhang X. Two previously undescribed benzofuran derivatives from the flowers of Callistephuschinensis. Phytochem. Lett. 2022; 51, 145–148. doi: 10.1016/j.phytol.2022.08.012
- Abu-Hashem A. A., Hussein H. A. R., Aly A. S., Gouda M. A. Reactivity of benzofuran derivatives. Synth. Commun. 2014; 44, 2899–2920. doi: 10.1080/00397911.2014.907425
- Abbas A. A., Dawood K. M. Anticancer therapeutic potential of benzofuran scaffolds. RSC Adv. 2023; 13, 11096–11120. doi: 10.1039/d3ra01383a
- Fuloria Sh., Sekar M., Khattulanuar F. S., Gan S. H., Rani N. N. I. M., Ravi S., Subramaniyan V., Jeyabalan S., Begum M. Y., Chidambaram K., Sathasivam K. V., Safi Sh. Z., Wu Y. S., Nordin R., Maziz M. N. H., Kumarasamy V., Lum P. T., Fuloria N. K.Chemistry, biosynthesis and pharmacology of viniferin: Potential resveratrol-derived molecules for new drug discovery, development and therapy. Molecules 2022; 27, art. no. 5072 (33 pp.). doi: 10.3390/molecules27165072
- Khanam H., Shamsuzzaman. Bioactive benzofuran derivatives: A review. Eur. J. Med. Chem. 2015; 97, 483–504. doi:10.1016/j.ejmech.2014.11.039
- Chiummiento L., D’Orsi R., Funicello M., Lupattelli P. Last decade of unconventional methodologies for the synthesis of substituted benzofurans. Molecules 2020; 25, art. no. 2327 (52 pp.). doi: 10.3390/molecules25102327
- Modell A. E., Blosser S. L., Arora P. S. Systematic targeting of protein–protein interactions. Trends Pharmacol. Sci. 2016; 37, 702–713. doi: 10.1016/j.tips.2016.05.008
- Farhat J., Alzyoud L., Alwahsh M., Al-Omari B. Structure–activity relationship of benzofuran derivatives with potential anticancer activity. Cancers (Basel) 2022; 14, art. no. 2196 (22 pp.). doi: 10.3390/cancers14092196
- Dawood K. M. Benzofuran derivatives: a patent review. Expert Opin. Ther. Pat. 2013; 23, 1133–1156. doi:10.1517/13543776.2013.801455
- Xu Zh., Zhao Sh., Lv Z., Feng L., Wang Y., Zhang F., Bai L., Deng J. Benzofuran derivatives and their anti-tubercular, antibacterial activities. Eur. J. Med. Chem. 2019; 162, 266–276. doi: 10.1016/j.ejmech.2018.11.025
- Nevagi R. J., Dighe S. N., Dighe S. N. Biological and medicinal significance of benzofuran. Eur. J. Med. Chem. 2015; 97, 561–581. doi: 10.1016/j.ejmech.2014.10.085
- Ahmad A., Nawaz M. I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell. Biochem. 2022; 123, 1938–1965. doi: 10.1002/jcb.30344
- Malekan M., Ebrahimzadeh M. A. Vascular endothelial growth factor receptors [VEGFR] as target in breast cancer treatment: Current status in preclinical and clinical studies and future directions. Curr. Top. Med. Chem. 2022; 22, 891–920. doi: 10.2174/1568026622666220308161710
- Olsson A.-K., Dimberg A., Kreuger J., Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006; 7, 359–371. doi: 10.1038/nrm1911
- Mabeta P., Steenkamp V. The VEGF/VEGFR axis revisited: Implications for cancer therapy. Int. J. Mol. Sci. 2022; 23, art. no. 15585 (14 pp.). doi: 10.3390/ijms232415585
- Zhang Y., Zou J.-Y., Wang Zh., Wang Y. Fruquintinib: a novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manag. Res. 2019; 11, 7787–7803. doi: 10.2147/CMAR.S215533
- Li X., Zhou J., Wang X., Li Ch., Ma Z., Wan Q., Peng F. New advances in the research of clinical treatment and novel anticanceragents in tumor angiogenesis. Biomed. Pharmacother. 2023; 163, art. no. 114806 (16 pp.). doi: 10.1016/j.biopha.2023.114806
- Sun Q., Zhou J., Zhang Zh., Guo M., Liang J., Zhou F., Long J., Zhang W., Yin F., Cai H., Yang H., Zhang W., Gu Y., Ni L., Sai Y., Cui Y., Zhang M., Hong M., Sun J., Yang Zh., Qing W., Su W., Ren Y. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol. Ther. 2014; 15, 1635–1645. doi:10.4161/15384047.2014.964087
- Chen Zh., Jiang L. The clinical application of fruquintinib on colorectal cancer. Expert Rev. Clin. Pharmacol. 2019; 12, 713–721. doi:10.1080/17512433.2019.1630272
- Shirley M. Fruquintinib: First global approval. Drugs 2018; 78, 1757–1761. doi: 10.1007/s40265-018-0998-z
- Lavacchi D., Roviello G., Guidolin A., Romano S., Venturini J., Caliman E., Vannini A., Giommoni E., Pellegrini E., Brugia M., Pillozzi S., Antonuzzo L. Evaluation of fruquintinib in the continuum of care of patients with colorectal cancer. Int. J. Mol. Sci. 2023; 24, art. no. 5840 (12 pp.). doi: 10.3390/ijms24065840
- Modi S. J., Kulkarni V. K. Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: Binding site analysis of type II, ’DFG-out’ inhibitors. J. Biomol. Struct. Dyn. 2022; 40, 5712–5727. doi: 10.1080/07391102.2021.1872417
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017; 7, art. no. 42717 (13 pp.). doi: 10.1038/ srep42717
- Moriguchi I., Hirono Sh., Liu Q., Nakagome I., Matsushita Y. Simple method of calculating octanol / water partition coefficient.Chem. Pharm. Bull. 1992; 40, 127–130. doi: 10.1248/cpb.40.127
- Moriguchi I., Hirono Sh., Nakagome I., Hirano H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994; 42, 976–978. doi: 10.1248/cpb.42.976
- Lipinski Ch. A., Lombardo F., Dominy D. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001; 46, 3–26. doi: 10.1016/s0169-409x(00)00129-0
- PerkinElmer. https://www.perkinelmer.com/analytical-and-enterprise-solutions.html (accessed on: September 24, 2023)
- Molinspiration Cheminformatics. https://www.molinspiration.com/cgi-bin/properties (accessed on: September 24, 2023)
- Ali J., Camilleri P., Brown M. B., Hutt A. J., Kirton S. B. In silico prediction of aqueous solubility using simple QSPR models: theimportance of phenol and phenol-like moieties. J. Chem. Inf. Model. 2012; 52, 2950–2957. doi: 10.1021/ci300447c
- Silicos-IT. https://www.silicos-it.be/ (accessed on: September 24, 2023)
- Wang X., Bove A. M., Simone G., Ma B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020; 8, art. no. 599281 (12 pp.). doi: 10.3389/fcell.2020.599281
- Peng F.-W., Liu D.-K., Zhang Q.-W., Xu Y.-G., Shi L. VE-GFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin. Ther. Pat. 2017; 27, 987–1004. doi: 10.1080/13543776.2017.1344215
- Lipinski Ch. A. Leadand drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004; 1, 337–341. doi: 10.1016/j.ddtec.2004.11.007
- Veber D. F., Johnson S. R., Cheng H.-Y., Smith B. R., Ward K. W., Kopple K. D. Molecular properties that influence the oralbioavailability of drug candidates. J. Med. Chem. 2002; 45, 2615–2623. doi: 10.1021/jm020017n
- Gu Y., Wang J., Li K., Zhang L., Ren H., Guo L., Sai Y., Zhang W., Su W. Preclinical pharmacokinetics and disposition of a novel selective VEGFR inhibitor fruquintinib (HMPL-013) and the prediction of its human pharmacokinetics. Cancer Chemother.Pharmacol. 2014; 74, 95–115. doi: 10.1007/s00280-014-2471-3
- Kelder J., Grootenhuis P. D. J., Bayada D. M., Delbressine L. P. C., Ploemen J.-P. Polar molecular surface as a dominatingdeterminant for oral absorption and brain penetration of drugs. Pharm. Res. 1999; 16, 1514–1519. doi: 10.1023/A:1015040217741
- van de Waterbeemd H., Camenish G., Folkers G., Chretien J. R., Raevsky O. A. Estimation of blood–brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998; 6, 151–156. doi: 10.3109/10611869808997889
- Levin V. A. Relationship of octanol / water partition coefficient and molecular weight to rat brain capillary permeability. J. Med. Chem.1980; 23, 682–684. doi: 10.1021/ jm00180a022
- Hansch C., Leo A. J. Substituent constant for correlation analysis in chemistry and biology. New York: Wiley 1979. doi:10.1002/jps.2600690938
- Ghose A. K., Herbertz T., Hudkins R. L., Dorsey B. D., Mallamo J. P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012; 3, 50–68. doi: 10.1021/cn200100h
- Pajouhesh H., Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005; 2, 541–553. doi: 10.1602/neurorx.2.4.541
- de Klerk D. J., Honeywell R. J., Jansen G., Peters G. J. Transporter and lysosomal mediated (multi)drug resistance to tyrosine kinase inhibitors and potential strategies to overcome resistance. Cancers (Basel) 2018; 10, art. no. 503 (27 pp.). doi: 10.3390/cancers10120503
- Fischer H., Gottschlich R., Seelig A. Blood–brain barrier permeation: Molecular parameters governing passive diffusion. J. Membr. Biol. 1998; 165, 201–211. doi: 10.1007/ s002329900434
- Raub T. J., Lutzke B. S., Andrus P. K., Sawada G. A., Staton B. A. Early preclinical evaluation of brain exposure in support of hitidentification and lead optimization. In: Borchardt R. T., Kerns E. H., Hageman M. J., Thakker D. R., Stevens J. L. (eds.) Optimizing the"Drug-Like" Properties of Leads in Drug Discovery. Biotechnology: Pharmaceutical Aspects, Vol. IV. New York: Springer 2006; 355–410. doi: 10.1007/978-0-387-44961-6_16
- Wang R., Cong D., Bai Y., Zhang W. Case report: long-term sustained remission in a case of metastatic colon cancer with high microsatellite instability and KRAS exon 2 p.G12D mutation treated with fruquintinib after local radiotherapy: a case report and literature review. Front. Pharmacol. 2023; 14, art. no. 1207369 (8 pp.). doi: 10.3389/fphar.2023.1207369
- Hoy S. M. Sintilimab: First global approval. Drugs 2019; 79, 341–346. doi: 10.1007/s40265-019-1066-z
- Guo Y., Zhang W., Ying J., Zhang Y., Pan Y., Qiu W., Fan Q., Xu Q., Ma Y., Wang G., Guo J., Su W., Fan S., Tan P., Wang Y., Luo Y., Zhou H., Li J. Phase 1b/2 trial of fruquintinib plus sintilimab in treating advanced solid tumours: The dose-escalation and metastatic colorectal cancer cohort in the dose-expansion phases. Eur. J. Cancer 2023; 181, 26–37. doi: 10.1016/j.ejca.2022.12.004
- Ma Sh., Chen R., Duan L., Li Ch., Yang T., Wang J., Zhao D. Efficacy and safety of toripalimab with fruquintinib in the third-line treatment of refractory advanced metastatic colorectal cancer: results of a single-arm, single-center, prospective, phase II clinical study. J. Gastrointest. Oncol. 2023; 14, 1052–1063. doi: 10.21037/jgo-23-108
- Keam S. J. Toripalimab: First global approval. Drugs 2019; 79, 573–578. doi: 10.1007/s40265-019-01076-2
- Ding X., Liu Y., Zhang Y., Liang J., Li Q., Hu H., Zhou Y. Efficacy and safety of fruquintinib as thirdor further-line therapy for patients with advanced bone and soft tissue sarcoma: a multicenter retrospective study. Anticancer Drugs 2023; 34, 877–882. doi: 10.1097/ CAD.0000000000001482
- Zhang P., Yang Y., Gou H., Li Q. Phase II study of fruquintinib as secondor further-line therapy for patients with advanced biliary tract cancer. J. Clin. Oncol. 2023; 41(Suppl), art. no. e16161 (1 pp.). doi: 10.1200/ JCO.2023.41.16_suppl.e16161
- Deng Y.-Y., Chen Y.-W., Wang M.-X., Zhu P.-F., Pan Sh.-Y., Jiang D.-Y., Chen Zh.-L., Yang L. Acute aortic dissection caused by fruquintinib for metastatic colorectal cancer–a case report and literature review. Transl. Cancer Res. 2023; 12, 177–185. doi: 10.21037/tcr-22-1872
- Zhang N., Xin X., Feng N., Wu D., Zhang J., Yu T., Jiang Q., Gao M., Yang H., Zhao S., Tian Q., Zhang Zh. Combiningfruquintinib and doxorubicin in size-converted nano-drug carriers for tumor therapy. ACS Biomater. Sci. Eng. 2022; 8, 1907–1920. doi:10.1021/acsbiomaterials.1c01606
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2023 Issue 6
Most read in this issue
- Analysis of medication administration in relation to food and beverages in inpatients
- Potential impact on mental health in patients with treatment-resistant schizophrenia – clozapine augmentation with long-acting parenteral antipsychotics: a case series
- Ethical aspects of conducting clinical trials of human medicinal products
- Brief insight into the in silico properties, structure–activity relationships and biotransformation of fruquintinib, an anticancer drug of a new generation containing a privileged benzofuran scaffold