
Klinické a genetické aspekty monogénovej obezity
Clinical and genetic aspects of monogenic obesity
High prevalence of obesity in all of age categories is currently one of the biggest problem in medicine. Identification of etiology of obesity can individualise an approach to the patient and it is essential for choosing a target management and therapy. Beside the largest group with polygenic inheritance are clinically important also patients with “syndromic obesity”, where obesity is only one of the signs and monogenic obesity, where obesity is the major clinical phenotype (patients with mutations in gene for leptin, leptine receptor, prohormone convertase 1, melanocortine receptor 4, brain‑derived neurotropic factor and tyrosin kinase receptor B). The monogenic obesity includes 3 – 4% of all patients with obesity. This review article brings newest insight on genetics, clinical manifestation, diagnostics and therapy of these diseases.
Key words:
prevalence of obesity – polygenic hereditability of obesity – monogenic obesity
Authors:
D. Lesayová 1,2; J. Staník 1,2; D. Gašperíková 1,3; I. Klimeš 1,3
Authors‘ workplace:
DIABGENE a Laboratórium diabetu a porúch metabolizmu ÚEE SAV Bratislava, Slovenská republika, vedúci prof. MU Dr. Iwar Klimeš, DrSc.
1; I. detská klinika Lekárskej fakulty UK a Dérerovej FNsP Bratislava, Slovenská republika, prednostka doc. MU Dr. Oľga Červeňová, CSc.
2; Molekulárno- medicínske centrum SAV Bratislava, Slovenská republika, riaditeľ MU Dr. Richard Imrich, PhD.
3
Published in:
Vnitř Lék 2010; 56(10): 1043-1049
Category:
Obesity 2010
Overview
Vysoký nárast prevalencie obezity vo všetkých vekových kategóriách predstavuje v súčasnosti jeden z veľkých medicínskych problémov. Poznanie skutočnej etiológie obezity umožňuje individualizáciu prístupu k pacientovi a je nevyhnutným predpokladom na zvolenie cieleného manažmentu a liečby pacienta. Popri najväčšej skupine s polygénovým typom dedičnosti sú počtom nezanedbateľní aj pacienti s tzv. „syndrómovou obezitou“, kde obezita je iba jedným z klinických príznakov pri danom syndróme a obezita vznikajúca na monogénovom podklade, kde obezita je hlavným a dominantným príznakom (pacienti s mutáciami pre leptín, leptínový receptor, pro‑opiomelanokortín, prohormón konvertázu 1, melanokortínový receptor 4, mozgový neurotropný faktor a tyrozín kinázový receptor B). Táto skupina predstavuje približne 3 – 4 % pacientov s obezitou. Predkladaný prehľadový článok prináša pohľad na najnovšie poznatky o genetike, klinických prejavoch, diagnostike a liečbe týchto ochorení.
Kľúčové slová:
prevalencia obezity – polygénový typ dedičnosti obezity – monogénová obezita
Nárast prevalencie obezity v nedávnej minulosti bol spájaný najmä s environmentálnymi, socioekonomickými a behaviorálnymi faktormi. V posledných rokoch sa však jednoznačne ukazuje, že telesný tuk a telesná hmotnosť, a teda aj „náchylnosť“ k vzniku obezity sú vo veľkej miere podmienené najmä genetickou výbavou jedinca [1,2]. Podľa najnovších štúdií sa predpokladá, že genetické faktory zodpovedajú až za 64–84 % výslednej hodnoty BMI [3–5].
Z hľadiska výskumu obezity a jej súvislosti s genetickou výbavou jedinca sú nepochybne najvýznamnejšie štúdie monozygotných dvojičiek a tzv. „adopčné štúdie“ vychádzajúce z faktu, že adoptované deti majú s adoptívnymi rodičmi spoločné iba environmentálne faktory, kým s biologickými rodičmi iba genetické faktory. Jedna z najväčších takýchto štúdií bola uskutočnená v Dánsku a dokázala štatisticky významnú koreláciu medzi BMI adoptovaných detí a ich biologickými rodičmi, ale iba minimálnu koreláciu medzi adoptovanými deťmi a ich adoptívnymi rodičmi [6].
V súčasnosti možno obezitu z genetického hľadiska klasifikovať ako:
- polygénovú (tzv. bežný typ obezity) – na jej vzniku sa zúčastňuje viacero génov, pričom tieto gény jednotlivo majú iba malý efekt na telesnú hmotnosť [7,8]. Kumulatívny efekt génov sa stáva signifikantným vtedy, keď je prítomná interakcia s faktormi prostredia (prejedanie sa, redukcia telesnej aktivity, hormonálne zmeny a socioekonomické faktory predisponujúce k ich fenotypovej expresii) [7–9].
- syndrómovú – asociovanú s niektorými geneticky podmienenými syndrómami, vznikajúcimi v dôsledku malých genetických defektov alebo abnormalít chromozómov. Ide o 20 až30 ochorení, kde obezita je iba jedným z klinických príznakov [20]. Častá je asociácia s mentálnou retardáciou, dysmorfnými črtami a orgánovo špecifickými vývojovými abnormalitami (tzv. pleiotropné syndrómy) [10]. Do tejto skupiny patrí Praderov Williho syndróm [11], Bardetov Biedlov syndróm, Alströmov syndróm, Cohenov syndróm a iné.
- monogénovú – vznikajúcu na podklade mutácie v jednom z 11 v súčasnosti známych génov [12–14] (pričom sa neustále identifikujú ďalšie [1,15,16]) a kde obezita predstavuje hlavný klinický príznak. Jedinci s touto mutáciou majú normálny vývoj a nie sú u nich prítomné dysmorfické črty (na rozdiel od syndrómových obezít) [3]. Častá je asociácia s poruchami správania a endokrinnými poruchami [17].
Monogénová obezita
Monogénová obezita je podmienená mutáciou v jedinom z 11 doteraz známych génov [13,16]. Patofyziológia excesívneho ukladania tuku u monogénových obezít je pomerne dobre objasnená: v dôsledku mutácie dochádza k narušeniu leptín-melanokortínovej osi [18,19], ktorá zohráva veľmi dôležitú úlohu v neuronálnej kontrole pocitu sýtosti a hladu, teda udržiavaní energetickej homeostázy organizmu (obr. 1) [20,21].
![Úloha leptín-melanokortínovej osi pri regulácii telesnej hmotnosti [24].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/717929abb4f13431e2d3b1b547ab8197.jpg)
Na základe miesta porušenia leptín-melanokortínovej osi možno rozdeliť obezitu (Klasifikácia monogénovej obezity podľa Farooqiovej a Ichiharu [22,23]) na obezitu spôsobenú:
- mutáciou génu pre leptín a mutáciou leptínového receptora
- mutáciou POMC (proopiomelanokortín)
- mutáciou PC1 (prohormón konvertáza 1)
- mutáciou MC4R (melanokortínový receptor 4)
- mutáciou BDNF (mozgový neurotropný faktor) a jeho receptora TrkB (tyrozín kinázový receptor B)
Podľa odhadov monogénovo podmienená obezita sa vyskytuje u 3–4% všetkých obéznych pacientov [12,17]. V porovnaní s polygénovo podmienenou obezitou teda tvorí jej menšiu časť [16], ale jej diagnostika je dôležitá vzhľadom na špecifickú liečbu a manažment.
Genetický podklad monogénovej obezity
Gény, ktorých mutácie sa zúčastňujú na rozvoji monogénovej obezity, sa nachádzajú na autozómoch. Dedičnosť väčšiny týchto ochorení je autozómovo recesívna [25], pričom homozygoti (alebo zložení heterozygoti) majú plne rozvinutý klinický obraz s obezitou závažného stupňa vznikajúcou vo včasnom detstve [16]. Dedičnosť mutácií MC4R je autozómovo kodominantná [16,26], kde úplný klinický obraz je už pri mutáciách v heterozygotnej forme s tým, že homozygoti majú len väčšiu intenzitu príznakov. Mutácie génov majú väčšinou charakter bodových mutácií (zámena nukleotidov, delécie, inzercie) [27–29].
Klinické príznaky
Jednotlivé podtypy monogénovej obezity majú viaceré spoločné príznaky [3], a to: obezita ťažkého stupňa začínajúca v detstve, pozitívna rodinná anamnéza obezity a častá asociácia s endokrinopatiami. Klinická závažnosť a čas nástupu týchto príznakov závisí aj od mutovaného génu (tab. 1).
![Znaky monogénovo podmienených typov obezity. Modifikované podľa [10].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/80f0bb82dd5afea0039888da2957a449.jpg)
1. Mutácie génu pre leptín – deficit leptínu
Leptín (LEP) je proteín produkovaný tukovým tkanivom a pôsobí ako aferentný signál sýtosti, čím sa podieľa na regulácii telesnej hmotnosti [30]. Jeho hladina pozitívne koreluje s množstvom tukového tkaniva v organizme. Okrem toho ovplyvňuje aj ďalšie neuroendokrinné systémy asociované s fertilitou a pubertou [30,31].
Gén pre leptin je lokalizovaný na 7. chromozóme tzv. ob génu. Pre mutácie leptínového génu je charakteristický autozómovo recesívny typ dedičnosti s vyššou prevalenciou homozygotov v príbuzenských manželstvách.
Homozygoti pre mutáciu leptínového génu sa rodia s normálnou pôrodnou hmotnosťou, avšak v dôsledku konštantnej hyperfágie od prvých dní života dochádza v prvých mesiacoch k rozvoju morbídnej obezity [32]. Pacienti mávajú pritom normálny rast a normálne hodnoty IGF-1 [33]. Súčasťou klinického obrazu deficitu leptínu je hyperinzulinémia (dôsledok zvýšeného množstva telesného tuku, pričom k manifestácii diabetu dochádza až v 3.–4. dekáde života [31,32,34]), hypotalamický hypotyreoidizmus, časté infekcie v detskom veku (v dôsledku zníženia počtu a poruchy funkcie T-lymfocytov [35]) a oneskorený nástup puberty s poruchou fertility (leptín v súčinnosti s hormonálnymi faktormi hrá dôležitú úlohu v inicializácii puberty). Špecifickým príznakom tejto mutácie sú dysfunkcie sympatikového nervového systému.
U týchto pacientov sa fenotyp môže vekom meniť a v niektorých prípadoch dochádza oneskorene k nástupu puberty a normalizácii imunitného systému a tyroidálnej osi [36].
U heterozygotov pre mutáciu v géne kódujúcom leptín je hladina leptínu signifikantne znížená, avšak táto stačí na zachovanie normálnej tyroidálnej funkcie, iniciáciu puberty a zachovanie plodnosti [37].
Terapia jedincov s deficitom leptínu je možná prostredníctvom ľudského rekombinantného leptínu podávaného vo forme subkutánnych injekcií [35]. Efekt leptínu sa neprejavuje iba významnou stratou telesnej hmotnosti so znížením hyperfágie v prvých mesiacoch podávania, ale aj poklesom hladín inzulínu, úprave glykémií, lipidového spektra [38] ako aj k zlepšeniu imunitnej funkcie organizmu [22,31]. Podávanie leptínu umožňuje tiež primeraný nástup puberty.
2. Mutácie leptínového receptoru
Gén pre leptínový receptor (LEPR) sa nachádza na chromozóme 1p31.
Receptory pre leptín sú široko distribuované v tele [39].
Klinické príznaky pri mutácii leptínového receptoru sú podobné ako pri samotnom deficite leptínu, líšia sa však tým, že pacienti majú znížený lineárny rast so zníženou bazálnou aj stimulovanou sekréciou rastového hormónu a nižšími hladinami IGF-1. Zaznamenaná bola aj emočná labilita, poruchy správania, nie však mentálna retardácia. Celkový stupeň obezity a klinických príznakov je nižší ako pri deficite leptínu [33].
Výskyt homozygotov a zložených heterozygotov pre mutácie leptínového receptora je relatívne vysoký; tvoria až 3% spomedzi detí s hyperfágiou a včasným rozvojom obezity [20,33].
Liečba u pacientov s mutáciou génu pre leptínový receptor spočíva v prevencii vzniku morbídnej obezity, diétnom manažmente a korekcii endokrinopatií [33,36]. Podávanie leptínu je vzhľadom na poškodenie receptoru neúčinné [20].
3. Mutácie v géne pre pro-opiomelanokortín
Gén pre proopiomelanokortín (POMC),skladajúci sa z 3 exónov, je lokalizovaný na chromozóme 2p23.3. Jeho produkt – pro-opiomelanokortín – je štiepený prohormón konvertázou 1 (PC1) a prohormón konvertázou 2 (PC2) na biologicky aktívne peptidy – v adenohypofýze na N terminálny peptid, ACTH, β-lipotropný hormón [40], v hypotalame je štiepený PC2 (v súčinnosti s PC1) na melanokortínové hormóny α,β,γ-MSH [41]. Melanokortíny sa navädzujú na MC3 alebo MC4 receptory, čím sa zapájajú do leptín-melanokortínovej osi a regulujú energetickú homeostázu organizmu [42].
U pacientov s týmto typom mutácie okrem rozvoja obezity v priebehu prvých rokov života dochádza v dôsledku deficitu ACTH už v neonatálnom období k rozvoju adrenálnej krízy. V súvislosti s nedostatkom MSH je pre ľudí postihnutých touto mutáciou takisto typická bledá pokožka a červené vlasy [43].
Okrem opísaných príznakov mávajú pacienti s POMC deficitom aj zvýšenú hladinu TSH a hranične nízky voľný tyroxín, pričom TRH stimulačný test je v norme [27]. Pacienti mávajú aj zvýšenú náchylnosť k infekciám.
Heterozygotné mutácie nemusia nutne viesť ku vzniku vyššie opísaného fenotypu a deficitu kortizolu, skôr predisponujú k obezite pri tzv. „obezitogénnom prostredí“ [27].
Terapia. Postihnutí jedinci veľmi dobre reagujú na suplementáciu kortikoidov. Nakoľko sa predpokladá, že β-MSH je najdôležitejší ligand MC4R, realistickou možnosťou budúcnosti sú analógy β-MSH [16] alebo samotný proopiomelanokortín, podávaný periférne a prechádzajúci do centrálneho nervového systému [36].
4. Mutácie v géne kódujúcom prohormón konvertázu 1
Prohormón konvertáza 1 (PC1) štiepi prohormóny (POMC, proglukagón, proinzulín) na aktívne hormóny [41,44]. Predpokladá sa, že PC1 sa okrem hypofýzy a hypotalamu nachádza aj v enteroendokrinných bunkách a hrá zásadnú úlohu v absorbčnej schopnosti tenkého čreva [45].
Dominantným príznakom u väčšiny pacientov s deficitom PC1 je ťažká refraktérna neonatálna hnačka malabsorbčného typu [46] – teda skôr neprospievanie v prvom roku života. Neskôr popri obezite dominuje hypokortizolémia a veľmi typická postprandiálna hypoglykémia, hypoinzulinémia so zvýšenými hladinami proinzulínu v plazme [10,47].
Pacienti mávajú aj hypotalamický hypogonadizmus a sú infertilní [48].
5. Mutácie génu pre melanokortínový receptor 4
Ľudský melanokortínový receptor 4 (MCR4) je kódovaný génom skladajúcim sa z jedného exónu, lokalizovaným na chromozóme 18q22 [49,50]. Funkcia MC4 receptoru spočíva v ovplyvňovaní centra sýtosti v hypotalame, vo vytváraní správnej odpovede organizmu na príjem vysoko tukovej stravy s udržiavaním pocitu sýtosti, zvyšovaním metabolizmu a termogenézy [51,52]. Zúčastňuje sa aj na regulácii kardiovaskulárneho systému, glukózovej a lipidovej homeostázy, reprodukčných a sexuálnych funkciách, ako aj vnímania bolesti a strachu [53].
Pacienti s mutácou MC4R majú normálnu pôrodnú hmotnosť, ale v dôsledku hyperfágie sa rozvíja obezitav prvom roku života [54–56]. V priebehu detstva majú akcelerovaný rast (zvyčajne nad 97. percentil) [57,58], pričom sa zvyšuje tuková aj netuková hmota ako aj denzita kostí [54]. Býva prítomný hyperinzulinizmus, ale väčšina pacientov nemá porušenú toleranciu glukózy [54]. Pubertálny vývoj a fertilita je v norme [59] a vývojové, intelektuálne, behaviorálne problémy ani dysmorfné črty sa nevyskytujú [55]. Rozdielom oproti iným typom obezity je, že nosiči mutácií MC4R nemávajú hypertenziu [60]. MC4R sa totiž podieľa aj na aktivácii sympatikového nervového systému, ktorý je vo veľkej miere zodpovedný za rozvoj hypertenzie [61]. Pacienti s homozygotnou formou mutácie majú závažnejší klinický prejav než heterozygoti [54].
Prevalencia. Mutácia MC4R predstavuje najčastejšie sa vyskytujúcu formu monogénovej obezity. V Európe je najvyššia prevalencia popisovaná v Anglicku, kde sa mutácia vyskytuje u 5,8 % obéznych detí, v ostatných krajinách Európy sa prevalencia pohybuje medzi 0,5% (Taliansko) a 2,6% (Francúzko). V českej populácii je popisovaná prevalencia MC4R u obézných detí a adolescentov 2,4 % [62].
Liečba. V poslednom čase sa venovalo množstvo výskumných tímov vytvoreniu efektívnych látok pôsobiacich na báze agonistov melanokortínového receptoru 3 a 4 [53,63]. Všeobecne akceptovaným endogénnym agonistom MC4 receptoru bol doteraz α-MSH (α-melanocyty-stimulujúci hormón), preto sa väčšina výskumov upriamila na vývoj vhodnej terapeutickej látky s touto funkciou. Až neskôr sa ukázalo, že túto funkciu lepšie spĺňa β-MSH (tzv. peptid 6), ktorý predstavuje potentného agonistu MC3 a MC4 receptorov s redukovanou aktivitou na MC1 receptor a inaktivitou voči MC5 receptoru. Ukázalo sa, že subkutánna alebo intracerebrálna aplikácia β-MSH u potkanov jednoznačne spôsobuje pokles hmotnosti, úpravu energetickej rovnováhy a zvýšenie spotreby tuku – avšak u potkanov, kde obezita bola spôsobená diétou, nie u homozygotných mutácií MC4 receptoru. To súčasne ale predstavuje pravdepodobne určitú šancu pre heterozygotov tejto mutácie a takisto je možné, že tieto látky sa budú ďalej rozvíjať a v budúcnosti sa môžu používať u ľudí s polygénovým typom obezity („tzv. bežná obezita“).
Napriek tomu, že v súčasnosti teda nie je možnosť kauzálnej liečby u ľudí postihnutých touto mutáciou – skorá diagnostika je veľmi dôležitá, nakoľko pri tomto type obezity je nutná agresívna symptomatická liečba vo forme režimových opatrení, keďže pacienti nemajú navodený pocit sýtosti po jedle pre nefunkčnú leptín-melanokortínovú os, je nutná dôsledná limitácia prísunu potravy [64] už od skorého detstva, aby sa tak predišlo vzniku morbídnej obezity a jej komplikácií [65].
6. Mutácia mozgového neurotropného faktora a tyrozín kinázového receptora (brain derived neurotrophic factor – BDNF, tyrosine kinase receptor B – TRKB)
BDNF je neurálny rastový faktor, ktorý sa naväzuje na tyrozín kinázový receptor B, reguluje vývoj neurónov a moduluje plasticitu synáps, čo zohráva dôležitú úlohu v pamäťových funkciách a učení sa [66]. Expresia BDNF v hypotalame je regulovaná nutričnym statusom [67].
Pacienti s mutáciou v BDNF/TRkB mávajú retardáciu psychomotorického vývoja, poruchu krátkodobej pamäte a poruchy správania, emočnú labilitu a porušené vnímanie bolesti [68].
Význam DNA diagnostiky monogénovej obezity
Poznanie skutočnej etiológie obezity (obr. 2) je dôležité nielen z klasifikačného hľadiska, ale umožňuje poskytnutie viacerých dôležitých informácií pre pacienta ako aj pre ošetrujúceho lekára.
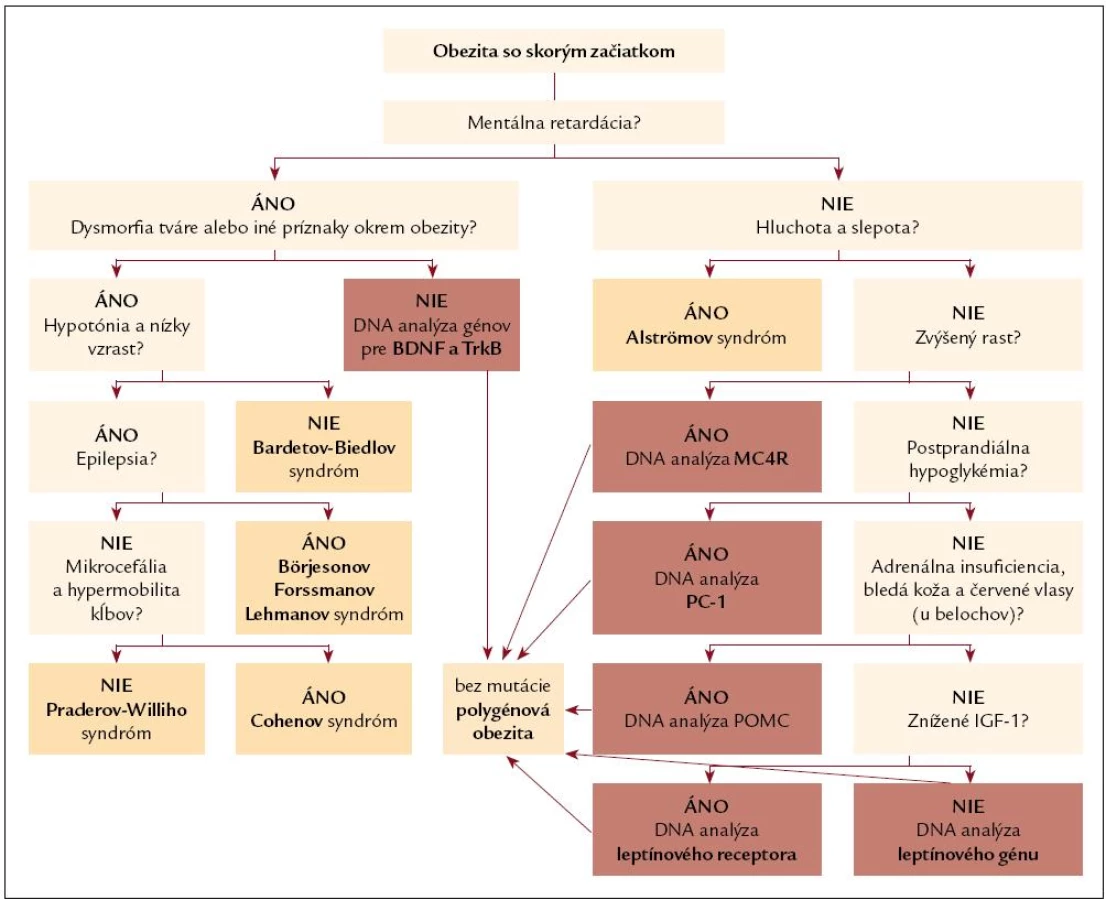
Najlákavejšou je možnosť kauzálnej liečby, ktorá v súčasnosti prichádza do úvahy pri mutáciách génu pre leptín. Ostatné formy monogénovej obezity zatiaľ možnosť farmakologickej liečby nemajú, ale v blízkej budúcnosti je možné predpokladať ich objavenie sa. V každom prípade je nepochybne dôležitý včasný záchyt takéhoto pacienta s možnosťou edukácie rodičov a pacienta, správnych režimových opatrení a tým pádom prevencii vzniku morbídnej obezity a jej komplikácií [65].
DNA diagnostika umožňuje aj genetické poradenstvo, nakoľko ide o ochorenia s rodinným výskytom (často z príbuzenských manželstiev).
Pri splnení kritérií (tab. 2) sa prosím obráťte na www.diabgene.sk, kde nájdete aj dotazník monogénovej obezity a formulár súhlasu s DNA analýzou. Kontaktný e-mail: daniela.gasperikova@savba.sk.

Poďakování
Táto publikácia vznikla vďaka podpore v rámci operačného programu Výskum a vývoj pre projekt: Transfer poznatkov genetického endokrinologického výskumu do klinickej praxe (26240220051), spolufinancovaný zo zdrojov Európskeho fondu regionálneho rozvoja.
prof. MU Dr. Iwar Klimeš, DrSc.
www.sav.sk
e-mail: iwar.klimes@savba.sk
Sources
1. Farooqi S. Obesity genes ‑ it’s all about the parents! Cell Metab 2009; 9 : 487 – 488.
2. Farooqi S, O’Rahilly S. Genetics of obesity in humans. Endocr Rev 2006; 27 : 710 – 718.
3. Farooqi S, O’Rahilly S. New advances in the genetics of early onset obesity. Int J Obes (Lond) 2005; 29 : 1149 – 1152.
4. Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 1997; 27 : 325 – 351.
5. Meyre D, Lecoeur C, Delplanque J et al. A genome ‑ wide scan for childhood obesity‑associated traits in French families shows significant linkage on chromosome 6q22.31 – q23.2. Diabetes 2004; 53 : 803 – 811.
6. Stunkard AJ, Sørensen TI, Hanis C et al. An adoption study of human obesity. N Engl J Med 1986; 314 : 193 – 198.
7. Willer CJ, Speliotes EK, Loos RJ et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41 : 25 – 34.
8. Hinney A, Vogel CI, Hebebrand J. From monogenic to polygenic obesity: recent advances. Eur Child Adolesc Psychiatry 2010; 19 : 297 – 310.
9. Hetherington MM, Cecil JE. Gene ‑ environment interactions in obesity. Forum Nutr 2010; 63 : 195 – 203.
10. O’Rahilly S, Farooqi S. Genetics of obesity. Philos Trans R Soc Lond B Biol Sci 2006; 361 : 1095 – 1105.
11. Santos VM, Henrique de Paula F, Osterne EM et al. Morbid obesity in an adolescent with Prader ‑ Willi syndrome. Rev Med Chil 2009; 137 : 264 – 268.
12. Ranadive SA, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am 2008; 37 : 733 – 751.
13. Tolson KP, Gemelli T, Gautron L et al. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J Neurosci 2010; 30 : 3803 – 3812.
14. Perrone L, Marzuillo P, Grandone A et al. Chromosome 16p11.2 deletions: another piece in the genetic puzzle of childhood obesity. Ital J Pediatr 2010; 36 : 43.
15. de Smith AJ, Purmann C, Walters RG et al. A deletion of the HBII ‑ 85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 2009; 18 : 3257 – 3265.
16. Farooqi S, O’Rahilly S. Mutations in ligands and receptors of the leptin‑melanocortin pathway that lead to obesity. Nat Clin Pract Endocrinol Metab 2008; 4 : 569 – 577.
17. Farooqi S, O’Rahilly S. Monogenic obesity in humans. Annu Rev Med 2005; 56 : 443 – 458.
18. Hochberg I, Hochberg Z. Hypothalamic obesity. Endocr Dev 2010; 17 : 185 – 196.
19. Bochukova EG, Huang N, Keogh J et al. Large, rare chromosomal deletions associated with severe early ‑ onset obesity. Nature 2010; 463 : 666 – 670.
20. Lee YS. The role of leptin‑melanocortin system and human weight regulation: lessons from experiments of nature. Ann Acad Med Singapore 2009; 38 : 34 – 41.
21. Coll AP, Farooqi S, O’Rahilly S. The hormonal control of food intake. Cell 2007; 129 : 251 – 262.
22. Farooqi S. Monogenic human obesity. Front Horm Res 2008; 36 : 1 – 11.
23. Ichihara S, Yamada Y. Genetic factors for human obesity. Cell Mol Life Sci 2008; 65 : 1086 – 1098.
24. Krude H, Grüters A. Implications of proopiomelanocortin (POMC) mutations in humans: the POMC deficiency syndrome. Trends Endocrinol Metab 2000; 11 : 15 – 22.
25. Kimber W, Peelman F, Prieur X et al. Functional characterization of naturally occurring pathogenic mutations in the human leptin receptor. Endocrinology 2008; 149 : 6043 – 6052.
26. Tan K, Pogozheva ID, Yeo GS et al. Functional characterization and structural modeling of obesity associated mutations in the melanocortin 4 receptor. Endocrinology 2009; 150 : 114 – 125.
27. Creemers JW, Lee YS, Oliver RL et al. Mutations in the amino‑terminal region of proopiomelanocortin (POMC) in patients with early ‑ onset obesity impair POMC sorting to the regulated secretory pathway. J Clin Endocrinol Metab 2008; 93 : 4494 – 4499.
28. Tan K, Pogozheva ID, Yeo GS et al. Functional characterization and structural modeling of obesity‑associated mutations in the Melanocortin 4 Receptor. Endocrinology 2009; 150 : 114 – 125.
29. Kimber W, Peelman F, Prieur X et al. Functional characterisation of naturally occurring pathogenic mutations in the human leptin receptor. Endocrinology 2008; 149 : 6043 – 6052.
30. Farooqi IS, Bullmore E, Keogh J et al. Leptin regulates striatal regions and human eating behavior. Science 2007; 317 : 1355.
31. Farooqi S, O’Rahilly S. Leptin: a pivotal regulator of human energy homeostasis. Am J Clin Nutr 2009; 89 : 980S – 984S.
32. Farooqi S, Jebb SA, Langmack G et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 1999; 341 : 879 – 884.
33. Farooqi IS, Wangensteen T, Collins S et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med 2007; 356 : 237 – 247.
34. Farooqi S. Insights from the genetics of severe childhood obesity. Horm Res 2007; 68 (Suppl 5): 5 – 7.
35. Farooqi IS, Matarese G, Lord GM et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/ metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 2002; 110 : 1093 – 1103.
36. Oswal A, Yeo GS. The leptin melanocortin pathway and the control of body weight: lessons from human and murine genetics. Obes Rev 2007; 8 : 293 – 306.
37. Farooqi IS. Monogenic human obesity syndromes. Prog Brain Res 2006; 153 : 119 – 125.
38. Prieur X, Tung YC, Griffin JL et al. Leptin regulates peripheral lipid metabolism primarily through central effects on food intake. Endocrinology 2008; 149 : 5432 – 5439.
39. Guerra B, Santana A, Fuentes T et al. Leptin receptors in human skeletal muscle. J Appl Physiol 2007; 102 : 1786 – 1792.
40. Chan LF, Webb TR, Chung TT et al. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc Natl Acad Sci USA 2009; 106 : 6146 – 6151.
41. Bicknell AB. The tissue ‑ specific processing of pro‑opiomelanocortin. J Neuroendocrinol 2008; 20 : 692 – 699.
42. Coll AP, Farooqi IS, Challis BG et al. Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab 2004; 89 : 2557 – 2562.
43. Krude H, Biebermann H, Luck W et al. Severe early ‑ onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998; 19 : 155 – 157.
44. Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin‑releasing hormone and proopiomelanocortin. Endocrinology 2007; 148 : 4191 – 4200.
45. Bataille D. Pro‑protein convertases in intermediary metabolism: islet hormones, brain/ gut hormones and integrated physiology. J Mol Med 2007; 85 : 673 – 684.
46. Jackson RS, Creemers JW, Farooqi IS et al. Small‑intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest 2003; 112 : 1550 – 1560.
47. O’Rahilly S, Farooqi IS. Human obesity as a heritable disorder of the central control of energy balance. Int J Obes (Lond) 2008; 32 (Suppl 7): S55 – S61.
48. Farooqi IS, Volders K, Stanhope R et al. Hyperphagia and early ‑ onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/ 3. J Clin Endocrinol Metab 2007; 92 : 3369 – 3373.
49. Sundaramurthy D, Campbell DA, Leek JP et al. Assignment of the melanocortin 4 receptor (MC4R) gene to human chromosome band 18q22 by in situ hybridisation and radiation hybrid mapping. Cytogenet Cell Genet 1998; 82 : 97 – 98.
50. Alfieri A, Pasanisi F, Salzano S et al. Functional analysis of melanocortin‑4 - receptor mutants identified in severely obese subjects living in Southern Italy. Gene 2010; 457 : 35 – 41.
51. Chai B, Li JY, Zhang W et al. Melanocortin‑4 receptor activation inhibits c ‑ Jun N‑terminal kinase activity and promotes insulin signaling. Peptides 2009; 30 : 1098 – 1104.
52. Masuzaki H, Tanaka T, Ebihara K et al. Hypothalamic melanocortin signaling and leptin resistance – perspective of therapeutic application for obesity ‑ diabetes syndrome. Peptides 2009; 30 : 1383 – 1386.
53. Tao YX. The melanocortin‑4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev 2010; 31 : 506 – 543.
54. Farooqi IS, Keogh JM, Yeo GS et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003; 348 : 1085 – 1095.
55. Lee YS, Poh LK, Kek BL et al. Novel melanocortin 4 receptor gene mutations in severely obese children. Clin Endocrinol (Oxf) 2008; 68 : 529 – 535.
56. Petry CJ, López ‑ Bermejo A, Díaz M et al. Association between a common variant near MC4R and change in body mass index develops by two weeks of age. Horm Res Paediatr 2010; 73 : 275 – 280.
57. Rettenbacher E, Tarnow P, Brumm H et al. A novel non‑synonymous mutation in the melanocortin‑4 receptor gene (MC4R) in a 2‑year ‑ old Austrian girl with extreme obesity. Exp Clin Endocrinol Diabetes 2007; 115 : 7 – 12.
58. Wangensteen T, Kolsgaard ML, Mattingsdal M et al. Mutations in the melanocortin 4 receptor (MC4R) gene in obese patients in Norway. Exp Clin Endocrinol Diabetes 2009; 117 : 266 – 273.
59. Stutzmann F, Tan K, Vatin V et al. Prevalence of melanocortin‑4 receptor deficiency in Europeans and their age ‑ dependent penetrance in multigenerational pedigrees. Diabetes 2008; 57 : 2511 – 2518.
60. Maier T, Hoyer J. Modulation of blood pressure by central melanocortinergic pathways. Nephrol Dial Transplant 2010; 25 : 674 – 677.
61. Sayk F, Heutling D, Dodt C et al. Sympathetic function in human carriers of melanocortin‑4 receptor gene mutations. J Clin Endocrinol Metab 2010; 95 : 1998 – 2002.
62. Hainerova I, Larsen LH, Holst B et al. Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis. J Clin Endocrinol Metab 2007; 92 : 3689 – 3696.
63. Szewczyk JR, Laudeman CP, SammondDM et al. A concise synthesis of 1,4 - dihy-dro‑[1,4]diazepine ‑ 5,7 - dione, a novel 7-TMreceptor ligand core structure with melano-cortin receptor agonist activity. Bioorg Med Chem 2010; 18 : 1822 – 1833.
64. Reinehr T, Hebebrand J, Friedel S et al. Lifestyle intervention in obese children with variations in the melanocortin 4 receptor gene. Obesity (Silver Spring) 2009; 17 : 382 – 389.
65. Loos RJ, Rankinen T, Tremblay A et al. Melanocortin‑4 receptor gene and physical activity in the Quebec Family Study. Int J Obes (Lond) 2005; 29 : 420 – 428.
66. Price RD, Milne SA, Sharkey J et al. Advances in small molecules promoting neurotrophic function. Pharmacol Ther 2007; 115 : 292 – 306.
67. Dhillo WS. Appetite regulation: an overview. Thyroid 2007; 17 : 433 – 445.
68. Gray J, Yeo GS, Cox JJ et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain‑derived neurotrophic factor (BDNF) gene. Diabetes 2006; 55 : 3366 – 3371.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 10
Most read in this issue
- Genetika obezity
- Hormony tukové tkáně
- Prevence obezity
- Výskyt obezity a jejích komplikací v České republice