
Idiopatické zápalové myopatie u detí v Čechách a na Slovensku
Introduction:
Juvenile dermatomyositis (JDM) and juvenile polymyositis (JPM), both idiopathic inflammatory myopathies (IIM), are rare diseases with a potentially severe prognosis. Diagnosis and care for patients suffering from IIMs is concentrated in specialized centres.
Aim:
To retrospectively analyze a cohort of paediatric patients with JDM and JPM followed in two paediatric rheumatology centres in the Czech and Slovak Republics.
Methods:
Patients with JDM and JPM followed at the Centre for paediatric rheumatology and autoinflammatory diseases of the Department of Paediatrics and Adolescent Medicine, General University Hospital in Prague in the years 2005–2013 and at the Rhematology Clinic of the Department of Paediatrics, Comenius University Medical School and National Institute of Children’s Diseases in Bratislava in the years 2014–2018 were included. Demographic and clinical data were extracted from electronic and paper medical records. During routine follow-up visits, health and functional status were assessed using validated tools (MMT8, CMAS, VAS, CHAQ). Acquired data were analyzed retrospectively.
Results:
Altogether, 25 children with IIM were included. At the Czech centre, we identified 17 patients (12 female, 5 male) with IIM (15 JDM, 2 JPM), of whom 14 were still being followed-up at the time of inclusion. Eight pacients from the Slovak centre were included (all female, 7 JDM, 1 JPM), all of whom were followed up at the time of inclusion. In Czech patients, the diagnosis of IIM was established at the median age 7.7 (range 1.2–15.8) years, the Slovak patients were slightly younger (6.8; 1.1–15.0 years) at diagnosis. In the entire cohort, muscle (95%) and skin (88%, 100% for JDM) disease were most frequent, followed by constitutional (68%) and joint manifestations (28%). The concentration of at least one muscle enzyme (AST, ALT, LD or CPK) was elevated in 72% of patients, in up to 24% the elevation was only mild (up to 2–3 times the upper limit of normal). Magnetic resonance imaging of muscles (sensitivity 80%) and nailfold capillaroscopy (sensitivity 87%) proved to be useful diagnostic tools in our cohort. All patients (100%) were treated with glucocorticoids and the majority (96%) also with methotrexate with good results: muscle strength, as measured by standardized tests, improved significantly, quality of life was good and permanent damage was mild. However, long-term complications developed in a significant number of patients: lipoatrophy (n=8), contractures (n=8), calcinosis (n=10) and growth retardation (n=6). During follow-up (median time of follow up 3.3, range 0.2–12 years) there was no patient with a fatal outcome. Among Slovak patients, some cases with significant diagnostic delay were noted (median 0.5; range 0.1–9.0 vs. median 0.3; range 0.1–1.5 years in Czech patients).
Conclusion:
Demographic characteristics, clinical manifestation, treatment and outcomes in Czech and Slovak children with JDM and JPM were comparable to published cohorts. Despite adequate treatment, complications were frequent. Diagnostic delay in some cases may have contributed to their development.
Key words:
idiopathic inflammatory myopathies, juvenile dermatomyositis, juvenile polymyositis, clinical signs, treatment
Autoři:
T. Dallos 1,2; M. Vránová 1; D. Němcová 1; D. Mozolová 2; P. Doležalová 1
Působiště autorů:
Centrum dětské revmatologie a autoinflamatorních onemocnění, Klinika dětského a dorostového lékařství, Všeobecná fakultní nemocnice a 1. lékařská fakulta Univerzity Karlovy, Praha, Česká republika
1; Detská klinika, Lekárska fakulta Univerzity Komenského v Bratislave, Národný ústav detských chorôb, Bratislava, Slovenská republika
2
Vyšlo v časopise:
Čes-slov Pediat 2018; 73 (4): 198-208.
Kategorie:
Sympozium: dětská revmatologie
Souhrn
Úvod:
Juvenilná dermatomyozitída (JDM) a juvenilná polymyozitída (JPM) patria medzi idiopatické zápalové myopatie (IIM). Sú to zriedkavé a prognosticky závažné ochorenia, ich diagnostika a liečba sa sústreďuje v špecializovaných centrách.
Cieľ:
Retrospektívne analýzovať pacientov s IIM sledovaných v dvoch centrách pre detskú reumatológiu v Čechách a na Slovensku.
Metódy:
Z elektronickej a písomnej zdravotníckej dokumentácie sme získali demografické a klinické údaje pacientov s IIM sledovaných v rokoch 2005–2013 v Centre dětské revmatologie a autoinflamatorních onemocnění KDDL 1. LF UK a VFN v Prahe a pacientov sledovaných v Reumatologickej ambulancii DK LFUK a NÚDCH v Bratislave v rokoch 2014–2018. Pri rutinných kontrolách sme hodnotili ich aktuálny zdravotný a funkčný stav validovanými nástrojmi (MMT8, CMAS, VAS, CHAQ). Získané údaje sme retrospektívne analyzovali.
Výsledky:
Zaradili sme 25 detí s IIM. Na českom pracovisku sme identifikovali 17 pacientov (12 dievčat, 5 chlapcov, 15 JDM, 2 JPM). 14 bolo naďalej dispenzarizovaných. Všetci ôsmi slovenskí pacienti (všetko dievčatá, 7 JDM, 1 JPM) boli v čase zaradenia dispenzarizovaní. V českom súbore bola diagnóza potvrdená vo veku 7,7 (1,2–15,8) rokov, slovenskí pacienti boli málo mladší: 6,8 (1,1–15,0) rokov. V celom súbore boli svalové (95 %) a kožné (88 %, 100 % pri JDM) prejavy najčastejšie, nasledované systémovými (68 %) a kĺbovými príznakmi (28 %). Aspoň jeden svalový enzým (AST, ALT, LD alebo CK) bol zvýšený u 72 % pacientov, u 24 % len mierne (do 2–3 násobku normy). Ako prínosné sa ukázali magnetická rezonancia svalov (senzitivita 80 %) a kapilaroskopické vyšetrenie nechtového lôžka (senzitivita 87 %). Všetci pacienti užívali kortikoidy (100 %) a väčšina metotrexát (96 %) s dobrými výsledkami: svalová sila bola významne zlepšená, kvalita života bola vyhovujúca a trvalé poškodenie mierne. U nezanedbateľného počtu pacientov sa vyvinuli dlhodobé komplikácie: lipoatrofia (n = 8), kontraktúry (n = 8), kalcinóza (n = 10), znížený rast (n = 6). Počas sledovaného obdobia (medián 3,3 rokov, 0,2–12) žiadne dieťa nezomrelo. V slovenskom súbore sme zaznamenali deti s väčším oneskorením stanovenia diagnózy (medián 0,5; 0,1–9,0 rokov vs. medián 0,3; 0,1–1,5 u českých detí).
Záver:
Demografické charakteristiky, klinický obraz, používaná liečba ako aj jej výsledky u českých a slovenských detí s JDM alebo JPM sú porovnateľné s údajmi z literatúry. Napriek adekvátnej liečbe sú komplikácie časté. Na tom sa môže podieľať aj oneskorenie stanovenia diagnózy.
Klúčové slova:
idiopatické zápalové myopatie, juvenilná dermatomyozitída, juvenilná polymyozitída, klinický obraz, liečba
ÚVOD
Idiopatické zápalové myopatie (IIM) tvoria heterogénnu skupinu zriedkavých zápalových ochorení svalu, z ktorých sa 16–20 % prejaví už v detskom veku. U detí sú najčastejšie juvenilná dermatomyozitída (JDM) a zriedkavejšia juvenilná polymyozitída (JPM). Aj incidencia JDM je však veľmi nízka (0,19–0,32/100 000 detí) [1, 2] a predstavuje najväčšiu prekážku pre pokroky v diagnostike a liečbe tohto raritného ochorenia.
Klinický obraz JDM popísalo viacero autorov už v 19. sto-ročí. JDM je výsledkom zápalu (vaskulárneho, perivaskulárneho a perimyziálneho) s charakteristickými prejavmi na koži (heliotropný exantém, Gottronove papuly, malárny raš, ulcerácie), svaloch (symetrická proximálna svalová slabosť) a niekedy na slizniciach (postihnutie ďasien, vaskulitída mezenteriálnych ciev). Generalizovaná lipoatrofia a dystrofická kalcinóza podkožia sú najčastejšie dlhodobé komplikácie neskoro diagnostikovaného alebo dlhodobo aktívneho ochorenia u detí [3]. Postihnutie myokardu a intersticiálna pneumopatia sa v detskom veku, na rozdiel od dermatomyozitídy dospelých, považujú za zriedkavé, i keď novšie práce poukazujú na známky pľúcneho postihnutia až u 26–37 % pacientov s JDM [4]. Diagnostické kritériá pre dermatomyozitídu dospelých podľa Bohana a Petera [5] pochádzajú zo 70-tych rokov minulého storočia. Hoci neboli validované pre detský vek, používajú sa aj na stanovenie diagnózy JDM u detí. JPM je charakteristická podobným obrazom svalovej slabosti, i keď na podklade odlišného histologického nálezu v svale a najmä bez postihnutia kože.
Mortalita JDM dosahovala pred 50-tymi rokmi minulého storočia až 30 % [6, 7]. Po zavedení intenzívnej a dlhodobej liečby glukokortikoidmi a imunosupresívami poklesla na 1–2 % a v súčasnosti má adekvátne liečená JDM už celkovo lepšiu prognózu prežívania s nižším stupňom funkčného postihnutia ako dermatomyozitída u dospelých. Prognóza JDM však závisí od včasnej diagnózy a intenzívnej a dlhodobej liečby [3].
Cieľom tejto práce bolo retrospektívne kriticky zhodnotiť diagnostické a terapeutické postupy u pacientov s idiopatickou zápalovou myopatiou dispenzarizovaných v Centre dětské revmatologie a autoinflamatorních onemocnění pri KDDL 1. LF UK a VFN v Prahe a v Reumatologickej ambulancii DK LFUK a NÚDCH v Bratislave. Sekundárnym cieľom bolo zhodnotiť prínos vykonaných vyšetrení pre stanovenie diagnózy zápalovej myopatie.
METÓDY
V roku 2013 resp. 2018 sme v elektronickom archíve hospitalizovaných pacientov vyhľadali pacientov s kódmi MKCH začínajúcimi M33 od roku 2005, kedy bol zavedený nemocničný informačný systém na oboch pracoviskách. V databáze Centra pro dětskou revmatologii a autoinflamatorní onemocnění pri KDDL 1. LF UK a VFN v Prahe sme identifikovali 25 pacientov. Z nich sme z ďalšej analýzy vylúčili 5 detí, u ktorých sa ďalšími vyšetreniami IIM nepotvrdila. U ďalších 2 detí nebola dostupná dokumentácia v dostatočnom rozsahu, ale započítali sme ich do výpočtu incidencie. Jedno dieťa bolo vyšetrené len raz a pochádzalo zo Slovenska, nebolo sledované v analyzovanom slovenskom centre, a preto nebolo zaradené do žiadnej z analýz. Rovnako sme postupovali v roku 2018 pri analýze pacientov dispenzarizovaných v Reumatologickej ambulancii Detskej kliniky LFUK a NÚDCH v Bratislave a do súboru sme zaradili 8 pacientov.
Z elektronickej a písomnej dokumentácie pacientov sme extrahovali demografické dáta, údaje o klinických a laboratórnych prejavoch na začiatku a v ďalšom priebehu ochorenia, o type a načasovaní diagnostických metód a o liečebnom postupe. Od stále sledovaných pacientov sme získali aktuálne anamnestické údaje, fyzikálny nález a pomocou validovaných nástrojov sme hodnotili svalovú silu (svalové testy Childhood Myositis Assessment Scale – CMAS a Manual Muscle Test 8 – MMT8), trvalé následky (muscle damage index – sMDI) ako aj funkčný stav a kvalitu života (CHAQ).
Základnú štatistickú analýzu demografických parametrov sme uskutočnili v programe MS Excel. Vzhľadom na malý počet zaradených pacientov s týmito raritnými ochoreniami sme nepovažovali štatistické porovnanie oboch súborov za adekvátne. Demografické údaje preto prezentujeme ako medián a rozsah (minimum – maximum). Ostatné údaje v záujme prehľadnosti prezentujeme v absolútnych aj relatívnych hodnotách (%). Údaje z oboch súborov prezentujeme v tabuľkách 1–4.
Pokúsili sme sa o odhad incidencie IIM v Českej republike, pričom sme vychádzali z dostupných demografických údajov Statistického úřadu ČR a predpokladu, že všetky deti s chronickou idiopatickou myozitídou diagnostikovanou v sledovanom období, ktoré pochádzali približne z polovice českej detskej populácie, boli vyšetrené aj na KDDL VFN v Prahe. Pre absenciu údajov o výskyte JDM a JPM v iných centrách sme odhad incidencie na Slovensku neuskutočnili.
VÝSLEDKY
Odhad incidencie v Českej republike
Od januára 2005 do novembra 2013 sme v Centre pro dětskou revmatologii a autoinflamatorní onemocnění KDDL 1. LF UK a VFN v Prahe sledovali 19 pacientov s idiopatickou zápalovou myopatiou. V uvedenom období bolo novo diagnostikovaných 13 pacientov. Incidenciu idiopatických zápalových myopatií sme odhadli na 1,4/1 milión/rok. Predpokladáme, že výskyt JDM a JPM v Slovenskej republike je podobný, i keď v rokoch 2014–2016 bolo na analyzovanom pracovisku novo diagnostikovaných až 5 nových pacientov s JDM a 1 pacient s JPM, čo bolo ale pravdepodobne spôsobené významným oneskorením stanovenia diagnózy až u 3 z nich.
Súbor pacientov
Pre potreby nášho vyhodnotenia bola na KDDL 1. LF UK a VFN v Prahe dostupná kompletná dokumentácia 17 pacientov. V čase retrospektívnej analýzy bol medián veku pacientov 13,2 rokov. Jeden pacient bol už sledovaný reumatológom pre dospelých a 2 pacienti v inom centre, z toho jedna pacientka v zahraničí. Z vyhodnotených 17 pacientov bolo v čase ukončenia analýzy 14 naďalej v sledovaní centra minimálne 4 mesiace a maximálne 12 rokov (medián 4 roky) (tab. 1). Na DK LFUK a NÚDCH v Bratislave sme analyzovali 8 dispenzarizovaných pa-cientov, ktorých medián veku v čase zaradenia do súboru bol 10,7 rokov a boli v sledovaní pracoviska 1,5–6,3 rokov (medián 3,3 roku).

Najčastejší typ IIM v celom súbore (n = 25) bola JDM (n = 22), nasledovaná JPM (n = 3); iné typy IIM sme nezaznamenali. Medzi postihnutými bolo viac dievčat (n = 20, 80 %), pričom na Slovensku to boli výlučne dievčatá. Ochorenie sa najčastejšie prejavilo v nižšom školskom veku (medián 6,5; 1,1–15 rokov), ale až u 9 detí (36 %) vo veku do 5 rokov. U väčšiny detí bola diagnóza potvrdená do 3 mesiacov od manifestácie ochorenia. Najmarkantnejší rozdiel medzi dvoma súbormi bol práve v dĺžke času od manifestácie prvých príznakov do stanovenia diagnózy a teda začiatku liečby, keďže v rokoch 2014–2016 bola na Slovensku diagnóza u 3 z 5 novozistených prípadov JDM stanovená s výrazným oneskorením (8 mesiacov, 2 a 9 ro-kov). Príčiny tohto oneskorenia diagnózy sme retrospektívne analyzovali.
Klinické príznaky
V celom súbore 25 pacientov s IIM bol myopatický syndróm s proximálnou svalovou slabosťou objektivizovaný svalovými testami (MMT8, CMAS) najčastejším klinickým prejavom (95 %), ale nebol prítomný u všetkých pacientov v čase stanovenia diagnózy. Typická myopatická chôdza a Gowersov príznak (šplhanie) sa vyskytli len u 52 % resp. 36 % pacientov. Menej častá bola pohmatová citlivosť svalov (28 %), myalgie ale udávalo až 64 % pacientov. Kontraktúry a svalová hypotrofia, ktoré sa všeobecne považujú za neskoré príznaky myozitídy, boli už v čase stanovenia diagnózy prítomné u 28 %, resp. 16 % pacien-tov. V ďalšom priebehu sa len u 1 pacienta vyvinuli kontraktúry resp. svalová hypotrofia. Až u 36 % pacientov sa vyskytol niektorý z prejavov postihnutia viscerálnych svalov ako rinolália, dysfónia, dysfágia prípadne až aspirácia (tab. 2). Z ďalších menej častých muskuloskeletálnych príznakov sa prejavili artralgie resp. artritída (28 %), pričom u niektorých pacientov bola artritída klinicky dominantným nálezom. Zo systémových prejavov bola častá najmä únava (68 %), menej časté boli nechutenstvo, pokles hmotnosti a zvýšená telesná teplota (36 %, 24 % resp. 16 %).

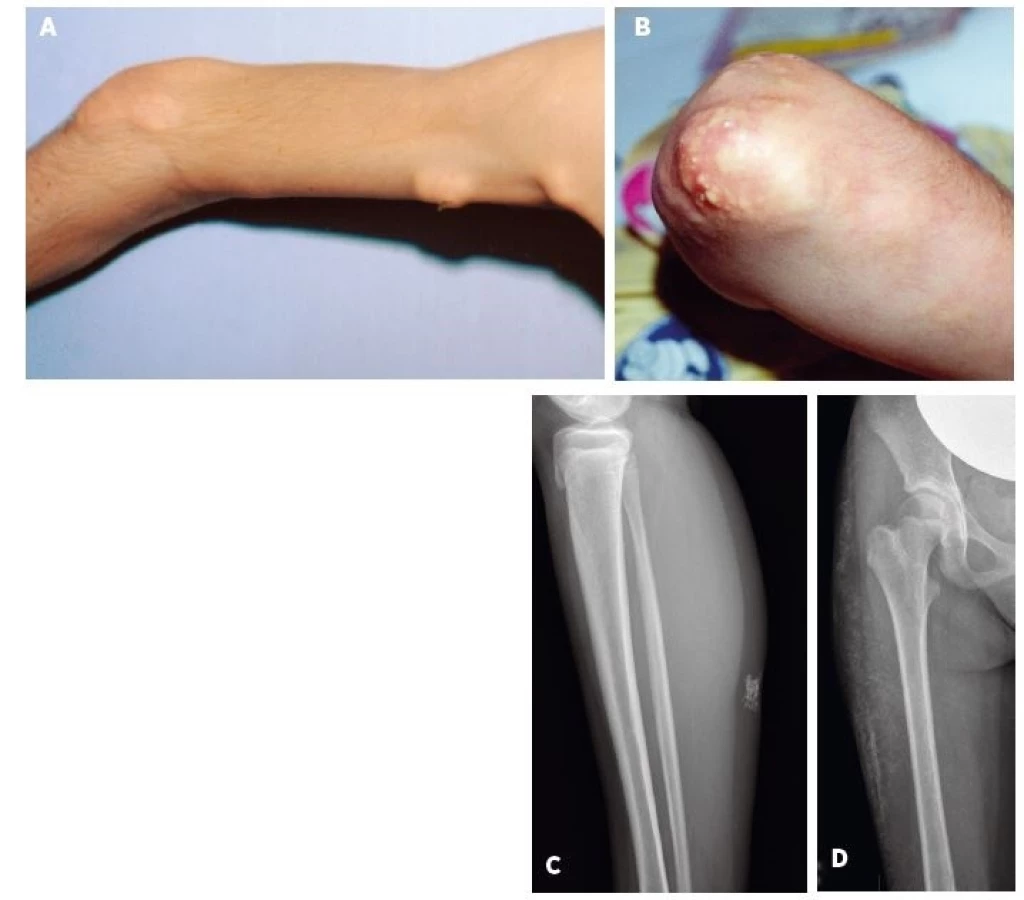
Z definície JPM vyplýva, že nemá kožné prejavy, tak ako to bolo aj v prípade 3 pacientov s JPM v našom súbore. Kožné prejavy sme preto hodnotili výlučne u 22 pacientov s JDM. U týchto bol najčastejší nález Gottronových papúl (86 %) nad veľkými (80 %) a o niečo menej aj malými kĺbmi (73 %). Častým príznakom bol aj malárny raš (motýľový exantém) u 72 % resp. heliotropný exantém u 68 % pacientov. Každý pacient s JDM (n = 23, 100 %) mal aspoň jeden z typických kožných prejavov JDM. U viac ako polovice pacientov boli klinicky zjavné aj prejavy kapilaritídy ako začervenanie nechtového lôžka, teleangiektázie na očných viečkach a angiektázie na slizniciach ďasien. U jedného pacienta s ťažkým priebehom JDM bola prítomná generalizovaná dermatitída a u 3 pacientov kožné ulcerácie. Príklady typických kožných nálezov pri JDM sú na obrázku 1. Kalcinóza, považovaná za komplikáciu chronicky aktívnej JDM, bola už v čase diagnózy prítomná u 4 pacientov (18 %). V ďalšom priebehu sa vyvinula až u ďalších 9 pacientov a postihovala teda celkovo 59 % pacientov s JDM. U 2 slovenských detí sa podarilo intenzifikáciou liečby (intravenózne imunoglobulíny, navýšenie dávky metotrexátu) progresiu kalcinózy zvrátiť, ale u jednej pacientky s oneskoreným stanovením diagnózy (2 roky) má kalcinóza napriek agresívnej liečbe charakter tzv. calcinosis universalis s progresívnym priebehom (obr. 2D). Súčasne sa u nej vyvíja aj generalizovaná lipoatrofia a už v čase stanovenia diagnózy boli prítomné kontraktúry.
A) heliotropný exantém lokalizovaný periorbitálne a malárny raš (motýľový exantém), B) Gottronove papuly nad veľkými
kĺbmi, C) symetricky rozložené Gottronove papuly nad malými kĺbmi.
Fig. 1. Chracteristic skin findings in patiens with recent-onset JDM.
A) periorbital heliotropic rash and malar („butterfly“) rash, B) Gottron’s papules on extensor surfaces of large (elbow
and knee) joints, C) symmetrical Gottron’s papules over small joints of hands.

A) heliotropný exantém lokalizovaný periorbitálne a malárny raš (motýľový exantém), B) Gottronove papuly nad veľkými kĺbmi, C) symetricky rozložené Gottronove papuly nad malými kĺbmi.
Fig. 1. Chracteristic skin findings in patiens with recent-onset JDM.
A) periorbital heliotropic rash and malar („butterfly“) rash, B) Gottron’s papules on extensor surfaces of large (elbow and knee) joints, C) symmetrical Gottron’s papules over small joints of hands.
Zastúpenie väčšiny klinických príznakov bolo v oboch súboroch pacientov podobné. V slovenskom súbore bolo vyššie zastúpenie postihnutia viscerálnych svalov. Zaujímavé je úplné chýbanie postihnutia svalov (klinicky, laboratórne aj na magnetickej rezonanci – MRI) u 1 slovenskej pacientky s tzv. juvenilnou dermatomyositis sine myositis, u ktorej bola diagnóza potvrdená až po 9 rokoch trvania ochorenia. Atypický bol aj priebeh u českej pacientky, ktorá bola liečená pre juvenilnú idiopatickú artritídu a až nález dystrofickej kalcinózy a minimálnej neobratnosti viedol k indikácii MRI svalov a potvrdeniu diagnózy JDM. Pozoruhodný bol aj výskyt polyartritídy u slovenskej pacientky s JPM.
Laboratórne a pomocné vyšetrenia
I keď mierne zvýšenie sedimentácie erytrocytov (do 30 mm/h) sme zaznamenali u 38 % pacientov, u prevažnej väčšiny pacientov nebolo CRP významne zvýšené. V krvnom obraze sa nevyskytovali odchýlky od normy. U jednej pacientky sa po 2 rokoch trvania ochorenia prejavila akútna imúnna trombocytopenická purpura, ktorá bola zvládnutá liečbou vysoko dávkovanými intravenóznymi imunoglobulínmi. U 65 % pacientov, u ktorých bola uskutočnená imunofenotypizácia periférnych lymfocytov (n = 16), sme pri diagnóze ochorenia zistili zvýšené zastúpenie CD19+ B-lymfocytov (>22 %) a ich podiel klesal s klesajúcou klinickou aktivitou ochorenia pod liečbou. Aktivita von Willebrandovho faktora (vWF) bola, ako prejav poškodenia endotelu pri vaskulitíde (>150 %), zvýšená u 53 % vyšetrených pacientov (n = 9). U jednej pacientky s potvrdenou von Willebrandovou chorobou boli hodnoty vWF patologicky nízke (tab. 3).

+ pacient s von Willebrandovou chorobou (n = 1), **pacienti s deficitom IgA (<0,05 g/l) (n = 2)

Aktivita aspoň jedného svalového enzýmu bola aspoň mierne zvýšená u väčšiny pacientov (n = 18, 72 %), i keď sa ojedinele vyskytli aj extrémne hodnoty najmä u pacientov s JPM. Najčastejšie boli zvýšené hodnoty LD (67 %), nasledované AST (63 %), CK (46 %) a ALT (42 %). Až u 4 pacientov (16 %) sme v úvode ochorenia nezaznamenali zvýšenie ani jedného zo 4 vyšetrovaných svalových enzýmov, u ďalších (n = 7, 28 %) nebolo zvýšené AST ani ALT a u takmer štvrtiny (n = 6, 24 %) bolo zvýšenie hraničné (menej ako 2- až 3-násobok hornej hranice normy). V ďalšom priebehu ochorenia sa v čase relapsov správali hodnoty CK ako najdynamickejší parameter (údaje neuvedené). Koncentrácie myoglobínu sme stavnovili len u jednej slovenskej pacientky s JPM, u českých pacientov (9 JDM, 1 JPM) boli v čase diagnózy zvýšené u všetkých vyšetrených pacientov, i keď u troch z nich bolo toto zvýšenie len hraničné. U všetkých pacientov so zvýšenou koncentráciou myoglobínu boli súčasne významne zvýšené aj aktivity svalových enzýmov.
Hladiny imunoglobulínov nevykazovali významné zvýšenie nad vekovo špecifické normy. Dvaja pacienti splnili kritériá deficitu IgA. Antinukleárne protilátky boli pozitívne asi u 44 % pacientov a nevykazovali žiadnu špecificitu. Protilátky špecifické pre myozitídu boli zachytené len u jedného českého pacienta (anti-Jo1). U jednej slovenskej pacientky sme identifikovali protilátku asociovanú s myozitídou anti-NXP2. Táto sa spája vývojom dystrofickej kalcinózy, ktorú sme identifikovali aj u tejto pacientky, i keď zatiaľ len v diskrétnej forme.
U 22 pacientov s JDM a u jednej pacientky s JPM bol k dispozícii kapilaroskopický nález nechtového lôžka a u 87 % z nich vykazoval aspoň jednu z charakteristických známok postihnutia kapilár. Najčastejšími nálezmi boli znížená hustota (65 %), dilatácia (65 %) a tortuozita (50 %) kapilár a hemorágie (50 %). Ďalej sa vyskytlo „kríčkovanie“ kapilár (37 %), megakapiláry (25 %) a trombotizácia (12 %) či elongácia (12 %) kapilár. U všetkých 8 slovenských pacientov s JDM bola kapilaroskopia nechtových lôžok uskutočnená pomocou dermatoskopu (typ Heine 20) a všetci pacienti mali pri tomto vyšetrení typický nález kapilaritídy. Najmä u pacientky s JDM sine myositis bol tento nález rozhodujúci pre stanovenie správnej diagnózy. Pod imunosupresívnou liečbou došlo k úplnej alebo aspoň čiastočnej úprave kapilaroskopického nálezu u väčšiny pacientov.
Výkonnosť diagnostických kritérií pre JDM
Výkonnosť diagnostických kritérií pre JDM sme analyzovali na 15 pacientoch z českého súboru. Diagnostické kritériá pre JDM (podľa Bohana a Petera, 1985) splnilo len 60 % pacientov s JDM. Ak bol v čase diagnózy prítomný myopatický syndróm, charakteristické kožné zmeny aj zvýšenie svalových enzýmov, všetky významne prispeli k splneniu diagnostických kritérií u všetkých pacientov. U niektorých českých pacientov bolo v priebehu vyšetrovacieho procesu indikované elektromyografické (n = 7) alebo bioptické (n = 8) vyšetrenie svalu, ktoré v 83 % resp. 87,5 % vyšetrení preukázali typické zmeny pri zápalovom postihnutí svalu a teda boli prínosné pre diagnózu podľa štandardných kritérií. Z pomocných vyšetrení bolo v celom súbore najčastejšie indikované MRI vyšetrenie svalov, ktoré preukázalo charakteristické zmeny (zvýšený signál svalu, perifasciálny edém, edém podkožia, svalová atrofia, tuková degenerácia) u celkovo 80 % vyšetrených pacientov. Pri rozšírení diagnostických kritérií o MRI nález stúpol počet českých pacientov, ktorí tieto modifikované kritériá splnili na 80 % a prínos elektromyografického a bioptického vyšetrenia klesol na 16 % resp. 0 %. V slovenskom súbore bolo histologické vyšetrenie svalu indikované len u jednej pacientky s JPM, u ktorej bol nález na MRI fyziologický. Keďže slovenskí pacienti s JDM mali typický nález kapilarity na nechtových lôžkach a obraz myozitídy na MRI vyšetrení svalu (okrem pacientky bez myozitídy), svalová biopsia nebola indikovaná u žiadneho z nich.
LIEČBA
Všetci pacienti s JDM aj JPM boli liečení systémovo podávanými kortikoidmi, pričom miernejší klinický priebeh umožnil výlučné perorálne podávanie u 16 % pacientov. Všetci pacienti boli liečení aspoň jedným imunosupresívom zo skupiny chorobu modifikujúcich antireumatík (DMARDs), a to predovšetkým metotrexátom (96 %) a cyklosporínom A (48 %), pričom metotrexát bol použitý ako liek prvej voľby u prevažnej väčšiny z nich (80 %). Niektorí pacienti užívali antimalarikum (36 %) predovšetkým na kožné prejavy pri JDM. Sulfasalazín bol prvým imunosupresívnym liekom u 1 pacientky, u ktorej v začiatku ochorenia dominovali kĺbové prejavy ochorenia a diagnóza JDM bola stanovená neskôr. Neúčinnosť uvedenej štandardnej terapie si vyžiadala liečbu vysoko dávkovanými imunoglobulínmi (n = 4 v ČR, n = 3 v SR), cyklofosfamidom (n = 1 v ČR), mykofenolátom mofetilu (n = 1 v SR) a inhibítorom TNFα (n = 1 v ČR). Bisfosfonáty sme použili na liečbu osteoporózy resp. dystrofickej kalcinózy u 4 českých pacientov a 1 slovenskej pacientky.
Výsledky liečby
V priebehu nášho sledovania nedošlo k žiadnemu úmrtiu. Na konci sledovania bolo 35 % českých pacientov a 87 % slovenských pacientov v stave klinickej a laboratórnej inaktivity, väčšina z nich pod trvajúcou liečbou. Aktivitu ochorenia a jeho následky sme pomocou validovaných nástrojov bližšie analyzovali v českom súbore: pacienti mali vyhovujúcu svalovú silu (MMT8 medián 76; 27–80, maximum 80 bodov) a svalovú výdrž (CMAS medián 42; 13–52, maximum 52 bodov) a priaznivo hodnotili aktivitu ochorenia na vizuálnej analógovej škále (VAS medián 0; 0–70, najvyššia aktivita 100) a svoj funkčný stav (CHAQ medián 0; 0–2,5, maximum funkčného obmedzenia 3,0).
Z dlhodobých komplikácií sa vyvinuli kontraktúry (41 %), kalcinóza (59 %), znížený rast (29 %) a osteoporóza (29 %). Tieto čiastočne odzrkadľuje aj skóre trvalého poškodenia sMDI, ktoré ale dosiahlo nízke hodnoty (medián 1,5; 0–6, maximum 35). Lokalizovaná alebo generalizovaná lipoatrofia sa vyvinula u 41,1 % pacientov a u niektorých detí bola sprevádzaná charakteristickými metabolickými komplikáciami (dyslipoproteinémia, inzulínová rezistencia). Viaceré z uvedených nežiaducich účinkov (osteoporóza, dyslipoproteinémia, znížený rast) ako aj ďalšie komplikácie (cushingoidný habitus, glaukóm, katarakta, infekcie) pravdepodobne súviseli s dlhodobou systémovou kortikoterapiou. V čase ukončenia zberu údajov pre potreby tejto analýzy v celom súbore kortikoidy užívalo až 19 pacientov (76 %), z nich 13 (t.j. 68 %) viac ako 2 roky a 6 (t.j. 31 %) viac ako 4 roky.
DISKUSIA
Idiopatické zápalové myopatie patria k zriedkavým ochoreniam detského veku s incidenciou 1,9 a 2,6–4,1/1 milión detí vo Veľkej Británii resp. USA [1, 2]. Náš hrubý odhad incidencie v Českej republike 1,4/1 milión detí je na dolnej hranici rozhrania incidencie v uvedených krajinách. Podľa definície WHO, ale aj v podmienkach špecializovaného reumatologického centra, ide o raritné ochorenia (orphan diseaase), ktoré sú zriedkavé aj medzi ostatnými reumatickými ochoreniami detského veku.
Podobne ako sa uvádza v literatúre, aj v našom súbore prevažujú dievčatá a ochorenie sa väčšinou manifestovalo v mladšom školskom veku [8]. Nízky vek manifestácie do 3.–5. roku života, ktorý sme pozorovali u 36 % detí, sa v minulosti považoval za nepriaznivý prognostický faktor. Závažnejší priebeh či horšiu odpoveď na liečbu sme u 9 pacientov v tejto vekovej kategórii nezaznamenali. Toto pozorovanie je v súlade s novšími retrospektívnymi analýzami väčších súborov, ktoré síce poukázali na častejší výskyt nekompletného klinického obrazu JDM [9], prípadne vyšší výskyt ulcerácií [10] v tejto vekovej kategórii, ale nepotvrdili horšiu dlhodobú prognózu týchto detí [9, 10]. Z prognostického hľadiska je pre individuálneho pacienta dôležitejšie včasné stanovenie diagnózy a začatie intenzívnej liečby [3]. U väčšiny českých pacientov bola diagnóza IIM stanovená do 3–4 mesiacov od prvých klinických príznakov ochorenia. Avšak u niektorých detí to bolo dlhšie resp. až výrazne dlhšie. Čas stanovenia diagnózy predstavoval najmarkantnejší rozdiel medzi dvoma súbormi. Medzi slovenskými deťmi s JDM sa vyskytli až 3 deti, u ktorých bola diagnóza JDM stanovená s významným oneskorením 8 mesiacov resp. až 2–9 rokov, kým v českom súbore bolo len jedno dieťa, u ktorého bola správna diagnóza stanovená po 18 mesiacoch trvania ochorenia.
Až 40 % pacientov nesplnilo v čase stanovenia diagnózy JDM kritériá pre definitívnu diagnózu podľa Bohana a Petera [5]. Preto sme v snahe identifikovať faktory, ktoré mohli ovplyvniť nesplnenie týchto kritérií a teda aj oneskorené stanovenie diagnózy, vyhodnotili klinické príznaky pri manifestácii ochorenia. Zo všetkých príznakov bola najčastejšia proximálna svalová slabosť (95 %), s ktorou pravdepodobne súvisel aj najčastejší systémový prejav – únava a znížená výkonnosť. Prítomnosť svalovej slabosti bola objektivizovaná štandardnými svalovými testami. Avšak jej „klasické“ prejavy ako je myopatická chôdza (52 %) a Gowersov príznak (36 %) zďaleka neboli prítomné u všetkých pacientov so svalovou slabosťou. Na druhej strane, aj naša skúsenosť ukazuje, že ani chýbanie objektívne zistiteľnej svalovej slabosti nevylučuje zápalové postihnutie svalu. Naopak, aj kontraktúry a hypotrofia svalov, ktoré sa považujú za neskorý dôsledok neliečeného ochorenia, sa môžu vyskytnúť už v čase stanovenie diagnózy. K výraznému oneskoreniu stanovenia správnej diagnózy u slovenských pacientov mohlo prispieť viacero skutočností: úplné chýbanie postihnutia svalov (n = 1), slabo vyjadrený myopatický syndróm (n = 1) a chýbanie významného zvýšenia svalových enzýmov (n = 3).
Febrilný priebeh ochorenia sme pozorovali len u 4 pacientov, pričom išlo o najzriedkavejší systémový prejav. Častejší bol pokles hmotnosti a zmena nálady. V akútnej fáze sa vyskytlo aj postihnutie viscerálnych svalov (dysfónia, dysfágia), ktoré sa môžu, tak ako u 2 pacientov, komplikovať aspiráciou. Túto možnosť je potrebné predvídať a aspirácii predchádzať zavedením nazoduodenálnej sondy alebo totálnej parenterálnej výživy.
Kožné prejavy sa vyskytujú pri JDM a nie pri JPM. Pri ich typickom vzhľade, súčasnom myopatickom syndróme a charakteristickom laboratórnom obraze ich prítomnosť umožňuje zdržanlivý prístup k ďalším, najmä invazívnym vyšetreniam. Najčastejšie sú Gottronove papuly v charakteristických lokalizáciách nad extenzorovými plochami veľkých a malých kĺbov (v našom súbore 86 %). Je ich preto možné jednoducho rozpoznať, problémy však môže spôsobiť podobná lokalizácia a vzhľad so psoriatickými eflorescenciami. Heliotropný exantém je síce menej častý (v našom súbore 68 %), ale špecifický príznak. Podobný výskyt (v našom súbore 72 %) má malárny raš, ktorý je identický s motýľovým exantémom pri systémovom lupus erythematosus. Predpokladáme, že významnému oneskoreniu diagnózy JDM najmä u niektorých slovenských pacientov prispelo práve nesprávne hodnotenie kožného nálezu ako psoriáza alebo subakútny lupus erythematosus ešte pred prvým vyšetrením na reumatologickom pracovisku. U jedného z týchto detí boli atrofické Gottronove papuly nesprávne hodnotené ako vitilligo, pre ktoré dieťa podstúpilo aj fototerapiu, teda metódu, ktorá je potenciálne škodlivá u tohto fotosenzitívneho ochorenia. U viac ako tretiny pacientov sme už pri manifestácii ochorenia pozorovali klinicky zjavné prejavy postihnutia kapilár (začervenanie nechtových lôžok, teleangiektázie na očných viečkach a sliznici alveolárnych výbežkov). Tieto sú priamym prejavom vaskulitídy pri JDM, a preto aj prejavom aktivity ochorenia [11].
Napriek tomu, že JDM a JPM patria medzi zápalové systémové ochorenia spojiva, aj naša skúsenosť dokazuje, že humorálna aktivita pri nich nebýva zvýšená. Laboratórne parametre, ktoré odzrkadľujú poškodenie svalu (svalové enzýmy, myoglobín), bývajú často, ale nie univerzálne zvýšené, a to ani vo floridnom štádiu novo manifestovaného ochorenia. Súčasne je nutné konštatovať, že ich hodnoty nemusia navzájom spoľahlivo korelovať. Platí preto, že ani normálny laboratórny nález nevylučuje zápalové postihnutie svalu pri JDM alebo JPM. V našom súbore sa von Willebrandov faktor, ktorý sa uvoľňuje z poškodeného endotelu, ukázal ako pomerne citlivý indikátor prítomnosti vaskulitídy pri JDM. I keď jeho korelácia s aktivitou ochorenia je spoľahlivá, podľa literatúry nemá väčší prínos ako monitoring aktivity svalových enzýmov [12] a nevie predpovedať relaps ochorenia. Dysregulácia imunitného systému v podobe zvýšeného zastúpenia B-lymfocytov bola prítomná u 2/3 všetkých vyšetrených pacientov. V ďalšom priebehu sme indikovali imunofenotypizáciu periférnych lymfocytov v relapse resp. remisii u 8 pacientov, u ktorých sme pozorovali vzostup resp. pokles ich zastúpenia. Pri JDM môže ísť o potenciálny marker aktivity ochorenia [13].
Celkovo možno konštatovať, že pacienti, u ktorých došlo k významnému oneskoreniu diagnózy, sa manifestovali netypickým klinickým (absencia významnej svalovej slabosti, dominantná artritída, absencia kožných prejavov pri JPM) alebo laboratórnym obrazom (absencia zvýšenia svalových enzýmov). Na rozdiel od charakteristického klinického obrazu u dieťaťa s myopatickým syndrómom, typickým kožným nálezom a zvýšenou aktivitou svalových enzýmov, ktorý nenecháva pochybnosti o diagnóze JDM, práve skupina pacientov s netypickou manifestáciou môže profitovať zo správne indikovaných pomocných vyšetrení.
K pomocným vyšetreniam, ktoré sú súčasťou diagnostických kritérií, patrí elektromyografia a histologické vyšetrenie svalu [14, 15]. Aj naše skúsenosti potvrdzujú ich vysokú výťažnosť (80 %). Napriek tomu sme ich indikovali len u menej ako 50 % českých a u 1 zo slovenských pacientov (pacient s JPM). Nižší počet svalových biopsií v slovenskom súbore, ktorý bol vyhodnotený o 5 rokov neskôr, odzrkadľuje všeobecný trend k minimalizácii týchto invazívnych vyšetrení. Biopsia svalov sa v súčasnosti odporúča pri chýbaní typických kožných prejavov a mala by sa vykonávať cielene z postihnutého svalu, ktorý je možné identifikovať pomocou MRI svalov. V nie-ktorých prácach sa ukázalo, že na základe histologického nálezu spolu s prítomnosťou autoprotilátok špecifických alebo asociovaných s myozitídou je možné predpovedať prognózu detí s JDM [16].
Neinvazívne metódy, ktoré potvrdzujú zápalové postihnutie svalu (MRI) resp. kapilaritídu (kapilaroskopia nechtového lôžka), predstavujú najmä v detskej reumatológii veľmi prínosnú alternatívu k štandardným pomocným vyšetreniam, a to aj napriek tomu, že zatiaľ nie sú súčasťou platných diagnostických kritérií. MRI svalov sa aj v našich centrách stalo najčastejšie využívanou pomocnou vyšetrovacou metódou (80 % pacientov) s pozitívnym nálezom u 80 % z nich, a teda porovnateľnou výťažnosťou ako iné pomocné metódy (obr. 3). Táto dostupná a neinvazívna metóda [17] sa v súčasnosti podľa medzinárodného prieskumu uznáva ako štvrté najprínosnejšie diagnostické kritérium pre JDM hneď po náleze proximálnej svalovej slabosti, typických kožných zmenách a zvýšených svalových enzýmoch [18]. Potenciál MRI svalov ako ďalšieho diagnostického kritéria pre JDM demonštrujú aj naše skúsenosti – význam invazívnych vyšetrení (EMG a biopsia) pre diagnózu JDM by významne poklesol, ak by bol nález na MRI ďalším plnohodnotným diagnostickým kritériom pre JDM. Je však nutné si uvedomiť, že zmeny v MRI obraze neodlíšia iné typy svalového postihnutia (vrátane pre detský vek vysoko relevantných svalových dystrofií), a preto nie sú pre zápalové postihnutie svalu špecifické. Na druhej strane môže zobrazenie svalového postihnutia prispieť k zvýšeniu výťažnosti cielenej svalovej biopsie alebo elektromyografického vyšetrenia, ak sú tieto potrebné [19]. I keď celotelové MRI prináša určité výhody pri diagnostike JDM [20], vyšetrenie obmedzené na proximálne svaly stehna, má v bežnej klinickej praxi porovnateľnú výpovednú hodnotu [21].
A) fyziologický nález u pacientky s amyopatickou formou JDM, B) patologický nález u pacientky s floridnou JDM, C) regresia patologických zmien u rovnakej pacientky po liečbe.
Fig. 3. MRI of lower girdle and thigh muscles.
A) physiological findings in amyopathic JDM, B) pahtological findings in active JDM (edema of muscle and subcutis),
C) reversal of pathological findings in the same patient after treatment.

Kapilaroskopicky viditeľné zmeny nechtového lôžka sú síce taktiež nešpecifické, ale zato typické pre JDM [11]. Aj u našich pacientov sa u viac ako 90 % podarilo týmto jednoduchým a dostupným vyšetrením identifikovať známky kapilaritídy. Toto vyšetrenie má aj prognostický význam, keďže rozsah kapilaroskopických zmien koreluje so závažnosťou myopatie a aktivitou vaskulopatie pri JDM [12]. U všetkých slovenských pacientov bol na tento účel použitý kvalitný dermatoskop, ktorý sa ukázal ako jednoduchý a spoľahlivý nástroj na detekciu kapilaritidy [22].
Napriek ustupujúcemu trendu využívania dlhodobej kortikoterapie napr. pri liečbe juvenilnej idiopatickej artritídy, ostávajú kortikoidy základným pilierom liečby JDM a JPM. Tomu zodpovedá aj ich využitie u 100 % našich pacientov, väčšinou aj formou i.v. pulzov. Dlhodobá kortikoterapia je spojená so zvýšeným výskytom potenciálne závažných komplikácií (glaukóm, katarakta, osteoporóza, infekcie) u nezanedbateľnej časti pacientov, ktoré je nutné cielene monitorovať (oftalmologická dispenzarizácia, denzitometria). K rýchlemu rozvoju osteoporózy prispieva aj znížená pohyblivosť dieťaťa spôsobená svalovou slabosťou. V bazálnej liečbe sa uplatňuje najmä metotrexát (15–20 mg/m2/týždeň, najvhodnejšie subkutánne), ktorý sa ukázal ako rovnako účinný, ale menej toxický ako cyklosporín A [23]. Liečba vysokodávkovanými imunoglobulínmi (2 g/kg á 1 mesiac minimálne 6 mesiacov) je považovaná za štandardnú pri ťažkých formách ochorenia s nedostatočnou odpoveďou na konvenčnú liečbu kortikoidmi a metotrexátom [24] i keď ešte stále v režime „off-label“. Až 23 % českých a 37 % slovenských pacientov takúto liečbu vyžadovalo a väčšina z nich z tejto liečby aj profitovala. V slovenskom súbore táto liečba pomohla navodiť regresiu dystrofickej kalcinózy u 2 pacientov. Aj naše skúsenosti teda podporujú použitie tejto nákladnej liečby u pacientov s reraktérnym priebehom JDM. K novším terapeutickým prístupom patrí využitie biologických liečiv u refraktérnych foriem (1 pacient) a Na-tiosulfátu i.v. alebo lokálne na liečbu kalcinózy [25, 26]. Lokálna aplikácia Na-tiosulfátu u dvoch slovenských pacientiek s JDM nemala presvedčivý efekt.
Rutinné používanie aktuálnych medzinárodných diagnostických a terapeutických štandardov [27] v našich centrách reflektujú aj výsledky liečby porovnateľné s publikovanými údajmi z iných krajín. Tieto sú charakterizované významným poklesom mortality oproti obdobiu pred zavedením kortikoterapie [28], ale aj pretrvávajúcou významnou morbiditou [29] a trvalými následkami samotného ochorenia a liečby [30]. Svalová sila, aktivita ochorenia a kvalita života hodnotené štandardizovanými metódami mali tendenciu sa upraviť do uspokojivých hodnôt. Napriek tomu je pomerne častý výskyt dlhodobých komplikácií (lipoatrofia, kalcinóza, svalová atrofia, hyperlipoproteinémia), i keď miera celkového trvalého poškodenia je pomerne nízka a najmä pacienti hodnotia svoju kvalitu života priaznivo (VAS, CHAQ). Lipodystrofia bola u 1 pacienta sprevádzaná metabolickými zmenami charakteristickými pre metabolický syndróm, ktoré je nutné očakávať najmä u pacientov s protrahovaným priebehom aktívneho ochorenia a kalcinózou [31].
Výpovede našej práce majú niekoľko dôležitých obmedzení: i) vzhľadom na zriedkavosť idiopatických zápalových myopatií, a teda malý súbor pacientov, je detailnejšie štatistické vyhodnotenie problematické, ii) retrospektívna povaha našej štúdie je zodpovedná za to, že niektoré vyšetrenia neboli vykonané u všetkých pacientov, alebo niektoré výsledky neboli v čase zberu dát už k dispozícii, iii) pomerne dlhé obdobie sledovania bolo spojené s individuálnym prístupom ku každému pacientovi podľa v tom čase aktuálnych odporúčaní a výsledkov štúdií, iv) všade, kde to bolo prípustné, sme vzhľadom na malý počet jedincov údaje pre JDM a JPM vyhodnocovali spoločne, v) český a slovenský súbor boli vyhodnotené s odstupom takmer 5 rokov. Napriek takto vzniknutej heterogenite súboru však môžeme konštatovať, že získané údaje predstavujú reprezentatívnu vzorku českých a slovenských detí liečených pre IIM od roku 2005 a odzrkadľujú aktuálny prístup k diagnostike a liečbe týchto ochorení v Českej a Slovenskej republike. Charakteristiky našich pacientov ako aj výsledky ich liečby zodpovedajú publikovaným výsledkom z iných zahraničných pracovísk a dostupným údajom z väčších, prevažne medzinárodných, súborov [27].
ZÁVER
Súhrnne je možné konštatovať, že výskyt a klinický obraz idiopatických zápalových myopatií detského veku v našom regióne je porovnateľný s údajmi z iných krajín. Aj v našich podmienkach sa MRI svalov stalo významnou pomocnou diagnostickou metódou s vysokou výťažnosťou, ktorá umožňuje obmedziť invazívne vyšetrenia (EMG a biopsia) a má potenciál tieto nahradiť aj ako diagnostické kritérium. K zlepšeniu diagnostiky JDM by mohli prispieť aj nové klasifikačné kritériá pre IIM, i keď ich hlavnou úlohou je najmä správne klasifikovať pacientov pre vedecké účely [32]. Dlhodobá kortikoterapia v kombinácii s metotrexátom predstavuje štandardnú liečbu, ktorá aj u našich pacientov účinne znížila mortalitu, morbiditu a výskyt komplikácií, i keď za cenu pomerne vysokého výskytu nežiaducich účinkov.
Včasná diagnóza a liečba ostávajú rozhodujúcim faktorom pre dosiahnutie priaznivého terapeutického výsledku, a preto by aj deti s netypickou manifestáciou ochorenia mali byť v čo najkratšom čase odosielané do centier so skúsenosťami s touto skupinou ochorení. Naše skúsenosti potvrdzujú, že problémom ostáva rozpoznanie JDM odborníkmi – nereumatológmi. Kapilaroskopické vyšetrenie nechtových lôžok má vysokú výťažnosť a u detí sa dá spoľahlivo vykonávať aj štandardným dermatoskopom [22]. Umožňuje tak aj dermatológovi odlíšiť JDM od iných, podstatne častejších dermatologických diagnóz, včas vysloviť podozrenie a tak zlepšiť prognózu detí s týmto závažným systémovým ochorením [33].
Poďakovanie
Študijný pobyt MUDr. T. Dallosa, PhD., v Centre dětské revmatologie KDDL VFN v Prahe podporila Európska liga proti reumatizmu (EULAR) a nadačný fond Pro Sophia.
MUDr. Tomáš Dallos, PhD.
Detská klinika LFUK
Národný ústav detských chorôb
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: dallos@dfnsp.sk
Zdroje
1. Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis, 1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum 2003; 49 (3): 300–305.
2. Symmons DP, Sills JA, Davis SM. The incidence of juvenile dermatomyositis: results from a nation-wide study. Br J Rheumatol 1995; 34 (8): 732–736.
3. Pachman LM, Abbott K, Sinacore JM, et al. Duration of illness is an important variable for untreated children with juvenile dermatomyositis. J Pediatr 2006; 148 (2): 247–253.
4. Sanner H, Aaløkken TM, Gran JT, et al. Pulmonary outcome in juvenile dermatomyositis: a case-control study. Ann Rheum Dis 2011; 70 (1): 86–91.
5. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975; 292: 344–347.
6. Bitnum S, Daeschner CW, Travis LB, et al. Dermatomyositis. J Pediatr 1964; 74: 101–131.
7. Huber A, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep 2005; 7 (6): 441–446.
8. Pachman LM, Hayford JR, Chung A, et al. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol 1998; 25 (6): 1198–1204.
9. Patwardhan A, Rennebohm R, Dvorchik I, et al. Is juvenile dermatomyositis a different disease in children up to three years of age at onset than in children above three years at onset? A retrospective review of 23 years of a single center‘s experience. Pediatr Rheumatol Online J 2012; 10 (1): 34.
10. Martin N, Krol P, Smith S, et al. Juvenile Dermatomyositis Research Group (JDRG). Comparison of children with onset of juvenile dermatomyositis symptoms before or after their fifth birthday in a UK and Ireland juvenile dermatomyositis cohort study. Arthritis Care Res (Hoboken) 2012; 64 (11): 1665–1672.
11. Doležalová P, Young SP, Bacon PA, et al. Nailfold capillary microscopy in healthy children and in childhood rheumatic diseases: a prospective single blind observational study. Ann Rheum Dis 2003; 62 (5): 444–449.
12. Guzmán J, Petty RE, Malleson PN. Monitoring disease activity in juvenile dermatomyositis: the role of von Willebrand factor and muscle enzymes. J Rheumatol 1994; 21 (4): 739–743.
13. O‘Gorman MR, Corrochano V, Roleck J, et al. Flow cytometric analyses of the lymphocyte subsets in peripheral blood of children with untreated active juvenile dermatomyositis. Clin Diagn Lab Immunol 1995; 2 (2): 205–208.
14. Cibulčík F, Hančinová V, Hergottová A, et al. Elektromyografia v diagnostike zápalových ochorení svalov. Neurológia 2007; 2 (Suppl 1): 16.
15. Špalek P. Dermatomyozitída – patogenéza, klinický obraz, diagnostické kritériá a liečby. Neurológia 2007; 2 (3): 161–168.
16. Deakin CT, Yasin SA, Simou S, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol 2016; 68 (11): 2806–2816.
17. Maillard SM, Jones R, Owens C, et al. Quantitative assessment of MRI T2 relaxation time of thigh muscles in juvenile dermatomyositis. Rheumatology (Oxford) 2004; 43 (5): 603–608.
18. Brown VE, Pilkington CA, Feldman BM, et al. Network for Juvenile Dermatomyositis, Paediatric Rheumatology European Society (PReS). An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford) 2006; 45 (8): 990–993.
19. Tuen VC, Zingula SN, Moir C, et al. MRI guided wire localization muscle biopsy in a child with juvenile dermatomyositis. Pediatr Rheumatol Online J 2013; 11 (1): 15.
20. Malattia C, Damasio MB, Madeo A, et al. Whole-body MRI in the assessment of disease activity in juvenile dermatomyositis. Ann Rheum Dis 2014; 73 (6): 1083–1090.
21. Madeo A, Damasio MB, Dellepiane M, et al. Whole-body versus localized magnetic resonance imaging in the assessment of juvenile dermatomyositis, abstrakt PREsS-FINAL-2129. Pediatr Rheumatol 2013; 11 (Suppl 2): 141.
22. Bergman R, Sharony L, Schapira D, et al. The handheld dermatoscope as a nail-fold capillaroscopic instrument. Arch Dermatol 2003; 139 (8): 1027–1030.
23. Ruperto N, Pistorio A, Oliveira S, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet 2016; 387 (10019): 671–678.
24. Lam CG, Manlhiot C, Pullenayegum EM, et al. Efficacy of intravenous Ig therapy in juvenile dermatomyositis. Ann Rheum Dis 2011; 70 (12): 2089–2094.
25. Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr 2012; 160 (3): 520.
26. Pagnini I, Simonini G, Giani T, et al. Sodium thiosulphate for the treatment of juvenile dermatomyositis complicated by calcinosis. abstrakt PREsS-FINAL-2015. Pediat Rheumatol 2013; 11 (Suppl 2): 26.
27. Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017; 76 (2): 329–340.
28. Taborda AL, Azevedo P, Isenberg DA. Retrospective analysis of the outcome of patients with idiopathic inflammatory myopathy. A long-term follow up. Clin Exp Rheumatol 2014; 32 (2): 188–193.
29. Ravelli A, Trail L, Ferrari C, et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res (Hoboken) 2010; 62 (1): 63–72.
30. Rider LG, Lachenbruch PA, Monroe JB, et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum 2000; 60 (11): 3425–3435.
31. Bingham A, Mamyrova G, Rother KI, et al. Childhood Myositis Heterogeneity Study Group. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore) 2008; 87 (2): 70–86.
32. Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheumatol 2017; 69 (12): 2271–2282.
33. Dallos T, Mozolová D, Lukáč J, et al. Ako môže dermatológ zlepšiť prognózu detí s juvenilnou dermatomyozitídou? Pediatr praxi 2017; 18 (3): 112–116.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2018 Číslo 4
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Využití hodnoticích skóre a objektivních nástrojů při léčbě astmatu
- Nech brouka žít… Ať žije astma!
Najčítanejšie v tomto čísle
- Uveitida asociovaná s juvenilní idiopatickou artritidou
- Idiopatické zápalové myopatie u detí v Čechách a na Slovensku
- Péče o pacienty s autoinflamatorními onemocněními: Česko-slovenská adaptace překladu evropských doporučení SHARE*
- Před 110 lety se narodil vynikající pediatr prof. MUDr. Antonín Mores