
APOBEC3A Is a Specific Inhibitor of the Early Phases of HIV-1 Infection in Myeloid Cells
Myeloid cells play numerous roles in HIV-1 pathogenesis serving as a vehicle for viral spread and as a viral reservoir. Yet, cells of this lineage generally resist HIV-1 infection when compared to cells of other lineages, a phenomenon particularly acute during the early phases of infection. Here, we explore the role of APOBEC3A on these steps. APOBEC3A is a member of the APOBEC3 family that is highly expressed in myeloid cells, but so far lacks a known antiviral effect against retroviruses. Using ectopic expression of APOBEC3A in established cell lines and specific silencing in primary macrophages and dendritic cells, we demonstrate that the pool of APOBEC3A in target cells inhibits the early phases of HIV-1 infection and the spread of replication-competent R5-tropic HIV-1, specifically in cells of myeloid origins. In these cells, APOBEC3A affects the amount of vDNA synthesized over the course of infection. The susceptibility to the antiviral effect of APOBEC3A is conserved among primate lentiviruses, although the viral protein Vpx coded by members of the SIVSM/HIV-2 lineage provides partial protection from APOBEC3A during infection. Our results indicate that APOBEC3A is a previously unrecognized antiviral factor that targets primate lentiviruses specifically in myeloid cells and that acts during the early phases of infection directly in target cells. The findings presented here open up new venues on the role of APOBEC3A during HIV infection and pathogenesis, on the role of the cellular context in the regulation of the antiviral activities of members of the APOBEC3 family and more generally on the natural functions of APOBEC3A.
Published in the journal:
APOBEC3A Is a Specific Inhibitor of the Early Phases of HIV-1 Infection in Myeloid Cells. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002221
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002221
Summary
Myeloid cells play numerous roles in HIV-1 pathogenesis serving as a vehicle for viral spread and as a viral reservoir. Yet, cells of this lineage generally resist HIV-1 infection when compared to cells of other lineages, a phenomenon particularly acute during the early phases of infection. Here, we explore the role of APOBEC3A on these steps. APOBEC3A is a member of the APOBEC3 family that is highly expressed in myeloid cells, but so far lacks a known antiviral effect against retroviruses. Using ectopic expression of APOBEC3A in established cell lines and specific silencing in primary macrophages and dendritic cells, we demonstrate that the pool of APOBEC3A in target cells inhibits the early phases of HIV-1 infection and the spread of replication-competent R5-tropic HIV-1, specifically in cells of myeloid origins. In these cells, APOBEC3A affects the amount of vDNA synthesized over the course of infection. The susceptibility to the antiviral effect of APOBEC3A is conserved among primate lentiviruses, although the viral protein Vpx coded by members of the SIVSM/HIV-2 lineage provides partial protection from APOBEC3A during infection. Our results indicate that APOBEC3A is a previously unrecognized antiviral factor that targets primate lentiviruses specifically in myeloid cells and that acts during the early phases of infection directly in target cells. The findings presented here open up new venues on the role of APOBEC3A during HIV infection and pathogenesis, on the role of the cellular context in the regulation of the antiviral activities of members of the APOBEC3 family and more generally on the natural functions of APOBEC3A.
Introduction
The apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 family (APOBEC3s) comprises 6 members of highly related cytidine deaminases [1]. The prototype of the family, APOBEC3G (or A3G), has been identified on the basis of its ability to inhibit HIV-1 infection in the absence of the Vif protein [2]. In this case, the antiviral effect of A3G is exerted via its incorporation into virion particles in virus-producing cells. This incorporation leads to the deamination of newly synthesized viral DNA during the subsequent cycle of infection [3]–[6], although a non-deaminase dependent mechanism of inhibition has also been described [7], [8]. In the presence of Vif, A3G is targeted to an E3-ubiquiting ligase complex and is thus degraded in virus-producing cells [9]–[17]. Even if this seems the major mechanism with which the virus protects itself, retroviruses can use a Vif-independent manner that does not involve the degradation of A3G, but results in the exclusion of the protein from assembling viral particles [18]–[21]. In the case of HIV-1, Vif has also been proposed to promote structural changes in A3G that negatively affect its ability to be incorporated into virion particles [22], [23].
While this host-pathogen struggle takes place in producing cells, the role of the pool of A3G molecules present in target cells and thus welcoming incoming viral particles remains unclear and in large part unexplored [24], [25]. The possibility that APOBEC3 members might exert an inhibitory effect directly on incoming viruses is interesting, because in these steps Vif is absent (or present only in trace amounts in virion particles), so that the virus would be denuded of protection against them [26], [27]. A3G has been reported to block the early phases of HIV-1 infection of quiescent T cells and DCs, but this activity has been recently ascribed to more general modifications of the activation status of quiescent lymphocytes, rather than to a specific effect of A3G silencing during the early phases of infection [24], [25].
All the members of the APOBEC3 family coded by primates (A through H) exhibit antiviral properties, although their potency may vary (for review see [28], [29] and [30] for the specific case of A3H). The exception to this rule is APOBEC3A (A3A) that is to date the only member of the APOBEC3 family whose ectopic expression in established cell lines (HeLa or HEK293T cells) bears no consequence for retroviruses [21], [31]. APOBEC3A is a single-domain cytidine deaminase, it is capable of editing exogenous and endogenous DNA upon expression in HeLa cells and has been shown to be responsible for the strong editing activity observed in primary monocytes [32]–[35]. A3A is capable of inhibiting the replication of LTR and non-LTR retroelements [36]–[39], of Parvoviruses [36] and has more recently been shown to moderately inhibit Alpharetroviruses (more specifically the Rous Sarcoma Virus, RSV, upon incorporation into virion particles, [40]). Increased expression of A3A, along with other APOBEC3 members, has been observed in keratinocytes and skin of precancerous cervical biopsies of human papillomavirus (HPV)-positive patients, presence that has been correlated to an increased evidence of HPV editing [41]. Although certain studies suggest that the editing activity of A3A may play a role in the host cell genome integrity and advance the hypothesis that this activity may lead or contribute to cancerogenesis [34], the inhibition of retroelements and of the adeno-associated virus 2 (AAV2) seems independent from the deaminase activity of A3A [36]–[39].
In the case of HIV-1, no firm evidence links A3A to an antiviral activity in either producing or receiving cells, although one study advanced the hypothesis that the increased replication of HIV-1 in differentiated macrophages over monocytes could be due to their lower content in A3A [42]. Together with a few recent articles indicating that A3A could be preferentially expressed in cells of the myeloid lineage, these reports call for a deeper analysis of the relationship existing between HIV-1 and myeloid cells [43], [44].
Primary circulating blood monocytes, along with differentiated macrophages and dendritic cells (DCs) are key cells in immune responses acting both as sentinels of danger signals, as well as instructors for other cells of the immune system (reviewed recently in [45]–[47]. These cells represent an important target for HIV-1 replication and have been shown to serve both as a vehicle for its dissemination through the body, as well as a viral reservoir. However, myeloid cells are collectively more resistant to HIV-1 infection than other cell types, as for example activated lymphocytes. This resistance varies according to the differentiation status and stimulation present and appears at different steps of the viral life cycle. Among these steps, a major impairment occurs during the early phases of infection, as a number of laboratories including ours have established [48]–[55]. The exact nature of this resistance is unclear and likely to be multifactorial. However, a number of recent evidences point to the existence of cellular restriction factors that affect incoming virus specifically in the peculiar environment of myeloid cells [50], [56]. In this respect, two very recent studies identified one such factor in SAMHD1, whose alternative name is dendritic cells derived interferon γ-induced protein (DCIP) [57] and the deficiency of which causes the Aicardi-Goutières syndrome, a disease in which interferon responses are deregulated [58]. Despite the fact that SAMHD1 is expressed in different cell types, it seems to restrict lentiviral infection specifically in myeloid cells, probably highlighting the need for a particular cellular context, or for specific cellular partners [59], [60].
Given that previous reports indicated that A3A was preferentially expressed in CD14-positive monocytes [42]–[44], we tried to determine whether the pool of A3A molecules present in target cells could inhibit de novo infection of myeloid cells by HIV-1.
To this end, we first determined that A3A is the sole member of the APOBEC3 family specifically expressed in different myeloid cells. Then, using either ectopic expression of A3A in established HeLa cells or specific silencing in primary macrophages and DCs, we determined that A3A does indeed inhibit the early phases of infection specifically in myeloid cells. Indeed, silencing of A3A not only increases the susceptibility of target cells to HIV-1 during single round infectivity assays, but also augments the spread of R5 tropic HIV-1 viruses. This antiviral effect is mediated by the pool of A3A present in target cells and is exerted through a decrease in viral DNA accumulation. The identification of TC editing in vDNA produced during HIV-1 infection of myeloid cells suggests that this antiviral mechanism may at least in part be exerted via deamination of the viral genome. The inhibition mediated by A3A targets also SIVMAC viruses, suggesting a conserved antiviral mechanism directed against primate lentiviruses. Finally, we provide evidence that Vpx, a protein coded by members of the SIVSM/HIV-2 lineage seems to provide partial protection against A3A by driving its degradation.
Results
A3A is the only member of the APOBEC3 family specifically expressed in primary blood cells of myeloid origins
A few reports have indicated that among circulating white blood cells, A3A was highly expressed in monocytes [42]–[44]. To determine more widely the pattern of expression of A3A and of the different members of the APOBEC3 family, quantitative RT-PCR was performed on quiescent or PHA-stimulated primary blood cells (PBLs, depleted of monocytes), monocytes, macrophages and dendritic cells (DCs) differentiated upon incubation of monocytes for 4 to 5 days with M-CSF and GM-CSF/IL4, respectively (Figure 1). Our analysis reveals that A3A is overexpressed by at least 100 fold in differentiated macrophages over PBLs, irrespectively of their activation status. This difference increases further in DCs and is the greatest in monocytes in which A3A is expressed over 5 logs more than in stimulated PBLs. The expression of the remaining APOBEC3 members did not display such cell type specific variations, with the exception of A3D/E that seems expressed at least 10 fold more in PBLs than in the myeloid cells tested. On the contrary, A3B and to a lower extent A3H are less expressed in non-stimulated monocytes than in stimulated PBLs, although their expression increased during differentiation into either macrophages or DCs. Overall, myeloid cells express consistent levels of all APOBEC3 members, but A3A is the only one whose expression is restricted to them. These results are in line with previous reports and extend them in the absolute quantification of the copy number of the different APOBEC3 members during the differentiation of circulating monocytes into macrophages and DCs. Of note, IFNα treatment increased the expression levels of A3A at both the mRNA and protein level, but the gradient of A3A expression did not change with respect to untreated cells: monocytes, DCs, macrophages and PBLs (from the highest to the lowest, Figure S1A and S1B). At the protein level, we confirm that A3A can be recognized after migration on an SDS-PAGE gel by the ApoC17 antibody initially raised against A3G [23] and Figure S2). In our hands, A3A can be detected as a doublet by WB. Detection of this doublet is however highly variable and unpredictable, probably influenced by the detection limit of A3A in a given experiment and by donor-to-donor variations. In any case, the antiviral effect of A3A seems not linked to the detection of an A3A doublet by WB.
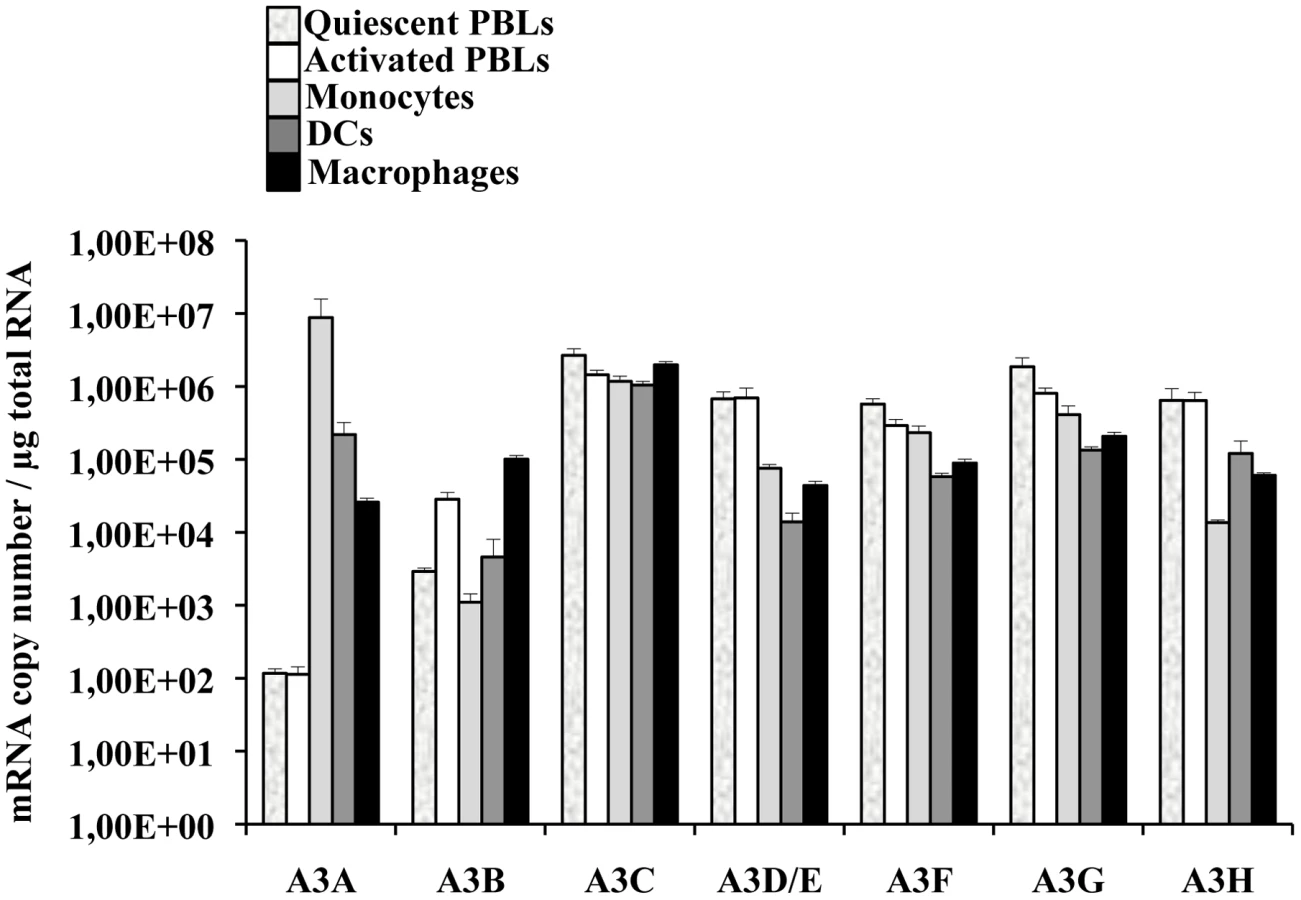
In conclusion, these results indicate that if myeloid cells express comparable levels of APOBEC3 members, A3A is the sole member of the APOBEC3 family whose expression is restricted to cells of myeloid origins, among cells of hematopoietic origins.
The expression levels of A3A specifically increase during HIV-1 spread in infected macrophages
To determine whether the levels of A3A were modulated during the course of HIV-1 infection, M-CSF-differentiated macrophages were infected with an R5 tropic HIV-1 virus (Yu2) and viral spread was assessed by determining the amount of virion-associated RT activity released in the cell supernatant (by exo-RT, Figure 2A). At day 7, when consistent viral spread had occurred, cell aliquots were lysed and the levels of APOBEC3 members analyzed at the mRNA level and, for A3A, also at the protein level by WB (Figures 2B and 2C, respectively). Spreading HIV-1 infection was associated to a consistent increase in A3A at both mRNA and protein levels. With the exception of A3B whose expression decreased during HIV-1 infection, the expression of the remaining members of the APOBEC3 family was not significantly modified. This increase was not due to the presence of type I interferon (IFNs) in the cultures, as assessed by the failure of supernatant derived from infected macrophages to inhibit the spread of a GFP-bearing Vesicular Stomatitis Virus in A549 cells (VSV, Figure S3). This assay is not only extremely sensitive, but allows the detection of the presence of all the subtypes of IFN α and β in the culture (in number of 13 and 2, respectively) [61], [62]. Although we cannot exclude the secretion of low levels of type I IFN and subsequent engagement of the IFN receptor, absence of IFN production during HIV-1 infection of dendritic cells has been recently reported [63]. Thus, although the expression of A3A can be induced by type I IFN [35], [43], [64], [65] and Figure S1), its expression during HIV-1 spreading infection of macrophages can also increase in a manner that is largely type I IFN-independent. To further support this argument, the expression of A3G, also modulated by type I IFN, is not significantly modified during infection. Thus, the upregulation of A3A observed here is likely to be part of a more complex response to viral infection that takes place in infected macrophages.
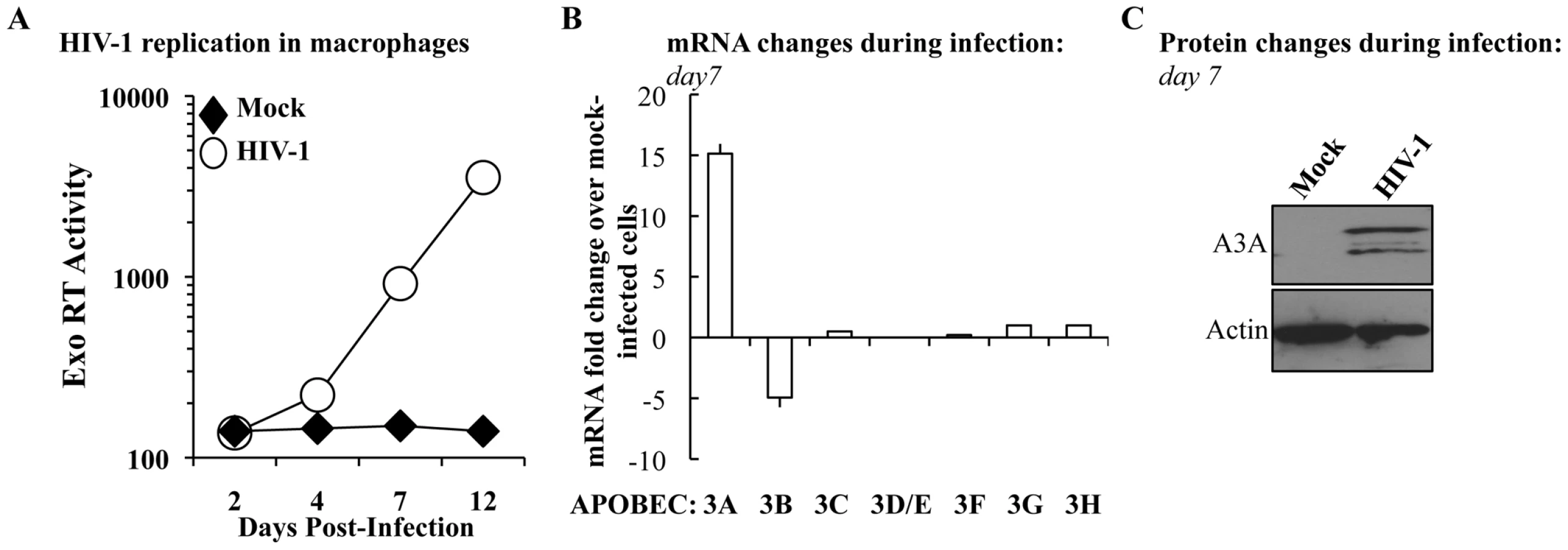
A3A inhibits the early phases of HIV-1 infection specifically in myeloid cells
The main described anti-retroviral function of APOBEC3 members involves the pool of APOBEC3s molecules present in virus-producing cells. On the contrary, the role of the pool of APOBEC3 molecules present in target cells has been explored only for A3G and remains highly debated [24], [25]. Among the members of this family, A3A is to date the sole member devoid of known inhibitory activity against HIV-1 when overexpressed in established cell lines [21], [31]. In agreement with these previous results, single cycle infection of HeLa cells overexpressing an HA-tagged version of A3A (that is editing-competent, data not shown) did not significantly modulate the susceptibility of target cells to infection with both complete or minimal HIV-1 vectors (i.e. containing or lacking non-structural viral proteins, Figure 3A). Given that no differences were observed between complete and minimal vectors in the experiments described below, the formers were routinely used for their slight but consistent higher infectivity (data not shown and [66]).
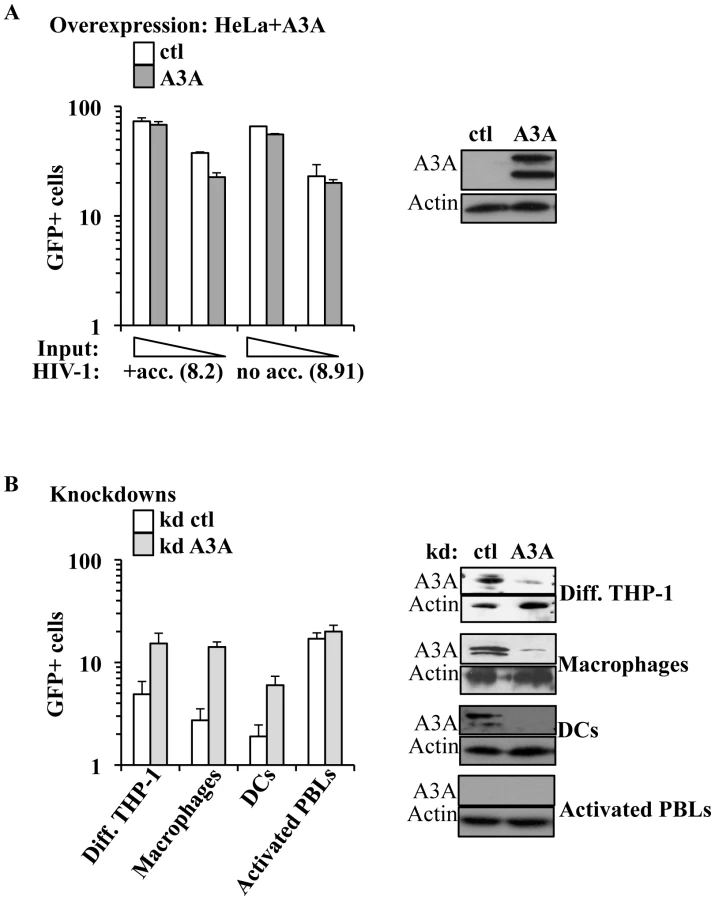
Contrarily to what observed in HeLa cells, silencing of A3A increased the susceptibility of target myeloid cells to HIV-1 infection (Figure 3B). Silencing was established using either miR-shRNAs expressed upon HIV-1-vectors mediated gene transduction [67] or liposome-mediated transfection of siRNAs. Silenced cells were then challenged with GFP-coding HIV-1 vectors. Given that both silencing methods yielded similar results, the formers were routinely used, as they allowed for more potent silencing. Knockdown of A3A increased the susceptibility of all cells of myeloid origins tested to the challenge with HIV-1 by an average of 5 to 7 fold: THP-1 differentiated into macrophage-like cells upon incubation with PMA (Diff. THP-1), as well as macrophages and DCs (Figure 3B). As expected, knockdown of A3A exerted no effect on the infection of PHA-stimulated PBLs (in the presence or absence of IFNα, Figure 3B and Figure S4). This result was expected as A3A is barely expressed in both quiescent and activated PBLs even after IFNα treatment, if compared to myeloid cells. The absence of an effect of the A3A knockdown on the susceptibility of PBLs to HIV-1 infection suggests the lack of non-specific effects (i.e. the A3A target must be present for the observed phenotype). To further control the specificity of our knockdowns, quantitative RT-PCR analysis was carried out on mRNA preparations derived from miR-shRNA-silenced macrophages for all the members of the APOBEC3 family (Figure S5A). This analysis indicated a specific decrease in A3A mRNA, but not in the mRNAs of other members of the family. As further proof of the specificity of the A3A knockdowns, A3A was silenced in macrophages upon liposome-mediated transfection of siRNAs. These siRNAs where chosen to target sequences within the A3A mRNA that were distinct from those targeted by the miR-shRNAs. Again, knockdown of A3A yielded an increase in the infectivity of HIV-1 similar to the one observed for miR-shRNAs (Figure S5B).
Overall, these experiments reveal that A3A plays a specific antiviral role during the early phases of HIV-1 infection in cells of myeloid origins in which it is naturally highly expressed.
A3A inhibits spread of HIV-1 in primary macrophages
To determine whether the antiviral effect of A3A could be observed also during replicative infection, silenced macrophages were challenged with R5 tropic HIV-1 (Yu2) at MOI of 0,05 and viral spread monitored by exo-RT activity (Figure 4). Even in this case, the specific silencing of A3A (shown in the WB side panel) increased viral spread in the culture. Although this result does not exclude an effect of A3A in virus-producing cells (the most classically described antiviral effect of APOBEC3s), this increase does correlate with the one observed during the early phases of infection in single round infectivity assays, indicating that A3A inhibits viral spread in macrophages. In light of the data presented in Figure 2B indicating an increase of A3A during spreading infection of HIV-1, we can hypothesize that the effect of silencing of A3A is mitigated by the overexpression of A3A observed during spreading HIV-1 infection. Similar experiments were attempted in DCs, but were abandoned due to the high mortality of silenced DCs upon viral challenge. This higher mortality is independent from the identity of the silenced gene, as it is observed in control and A3A knockdowns alike and does not seem specific for HIV-1.

A3A impairs the accumulation of vDNA during the early phases of infection, but does not affect the kinetics of accumulation of infectious vDNA
To identify the step affected by the depletion of A3A in target cells, silenced macrophages were lysed 24 hours post infection and the accumulation of vDNA analyzed by qPCR. The use of an HIV-1 based miR-shRNA vector precluded the analysis of all the viral DNA products synthesized over the course of infection. However, the presence of GFP in the challenge HIV-1 vector enabled the analysis of vDNA products obtained after the minus strand strong stop, which represent one of the major restrictive step during the infection of myeloid cells. Under these conditions, silencing of A3A increased the accumulation of vDNA by 4 to 5 fold (Figure 5A), in agreement with the increase observed in infectivity. While the analysis of vDNA products by PCR yields quantitative data on their accumulation as a whole, this technique does not reveal the kinetic behavior of truly infectious vDNA. Infectious vDNA constitutes a minor fraction of the total vDNA produced during infection [68], a phenomenon that is exacerbated in primary cells more resistant to infection. Thus, determining the behavior of these genomes represents an important additional parameter during the early phases of infection. To determine the kinetic behavior of infectious vDNA and more specifically to determine how fast it was completed during infection, we employed a technique we had previously described [48]. In this setup, infectious viral genomes are defined as those capable of expressing the viral-coded GFP reporter and their accumulation over time is determined by arresting the reverse transcription process at different times post infection through the addition of the RT inhibitor Nevirapine. The percentage of GFP-positive cells at each time point is then determined by flow cytometry 3 days post infection and values are graphed after normalization to control infections carried out in the absence of RT inhibitors. This assay yields a kinetic measurement of the speed at which completion of infectious vDNA is carried out. In this respect, the assay does not yield information on the total amount of vDNA synthesized during infection as in Fig. 5A, but describes another parameter which is the speed at which infectious vDNA is produced. When control and A3A-silenced macrophages were thus analyzed, no significant changes were observed in the kinetics of reverse transcription of HIV-1 infectious genomes (Figure 5B). These results indicate that A3A does not modify the kinetics of reverse transcription, but affects the overall amount of vDNA synthesized. This decrease is of the same amplitude of the effect observed in viral infectivity.
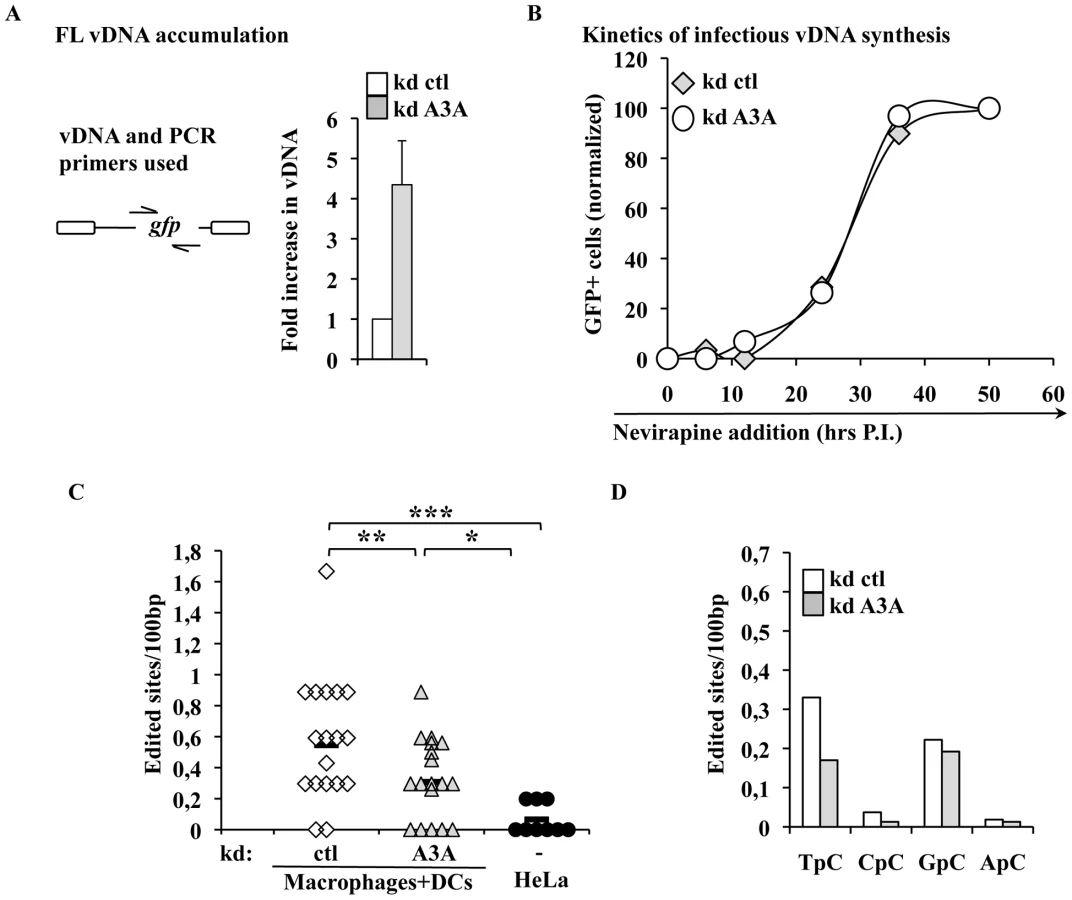
The intracellular levels of A3A modulate the levels of TC editing found in vDNA produced during infection of myeloid cells
Inhibition of AAV or retroelements by A3A seems to follow a deaminase-independent mechanism. To determine whether this was the case here, vDNA products accumulated during infection of macrophages were amplified, cloned and sequenced in search of potential cytidine editing. HIV-1 vDNA produced in macrophages exhibited a significantly higher proportion of edited cytidine with respect to vDNA synthesized in HeLa cells (Figure 5C). This deamination signature depended at least in part from A3A, since the number of edited sites decreased upon silencing of A3A. When the dinucleotide motifs surrounding the edited cytidine were examined, a minority was found to be CC (described as specific of A3G), while more than a half were TC (described to be preferentially recognized by the remaining A3 members) (Figure 5D). Cytidine deamination of the latter TC motif depended from A3A as their proportion specifically decreased upon A3A silencing. Overall, these results indicate that a small yet detectable cytidine editing signature is present in vDNA produced during the infection of myeloid cells, a portion of which is clearly influenced by the intracellular levels of A3A.
The antiviral effect of the A3A pool present in target cells is conserved among primate lentiviruses
To determine whether A3A is a specific inhibitor of HIV-1, or if it exerts a conserved antiviral function directed against primate lentiviruses, A3A or control-silenced differentiated THP-1 cells and DCs were challenged with SIVMAC vectors. Given that we have previously determined that the non-structural protein Vpx plays an important role during the infection of myeloid cells, Vpx-positive and deficient SIVMAC viruses were compared side by side (Figure 6A). Silencing of A3A resulted in an increase in the infectivity of WT SIVMAC viruses in both differentiated THP-1 cells and DCs, indicating that A3A targets not only HIV-1, but more generally primate lentiviruses. A positive effect of A3A silencing was also observed in the infectivity of SIVMAC vectors devoid of Vpx, that display a strong defect during the infection of myeloid cells. In differentiated THP-1, the infectivity of SIVMAC Vpx-deficient viruses approached –albeit not equaled- that of the WT in control silenced cells. In DCs, in which SIVMAC Vpx-deficient viruses display a drastic infectivity defect, silencing of A3A allowed the detection of infection clearly above the background, although not to the same levels of WT SIVMAC. These results suggest that the depletion of A3A may contribute to relieve the defect of Vpx-minus viruses. This result is of interest for two reasons. First because we and others have previously determined that Vpx is required for the completion of the early phases of infection specifically in myeloid cells in a manner that may involve the degradation of an unknown cellular restriction factor [50], [56], [67], [69]–[71]. Second because A3A has been previously shown to interact with SIVMAC Vpx upon overexpression in HeLa cells [72]. Thus, we decided to pursue the characterization of the relationship between Vpx and A3A further.
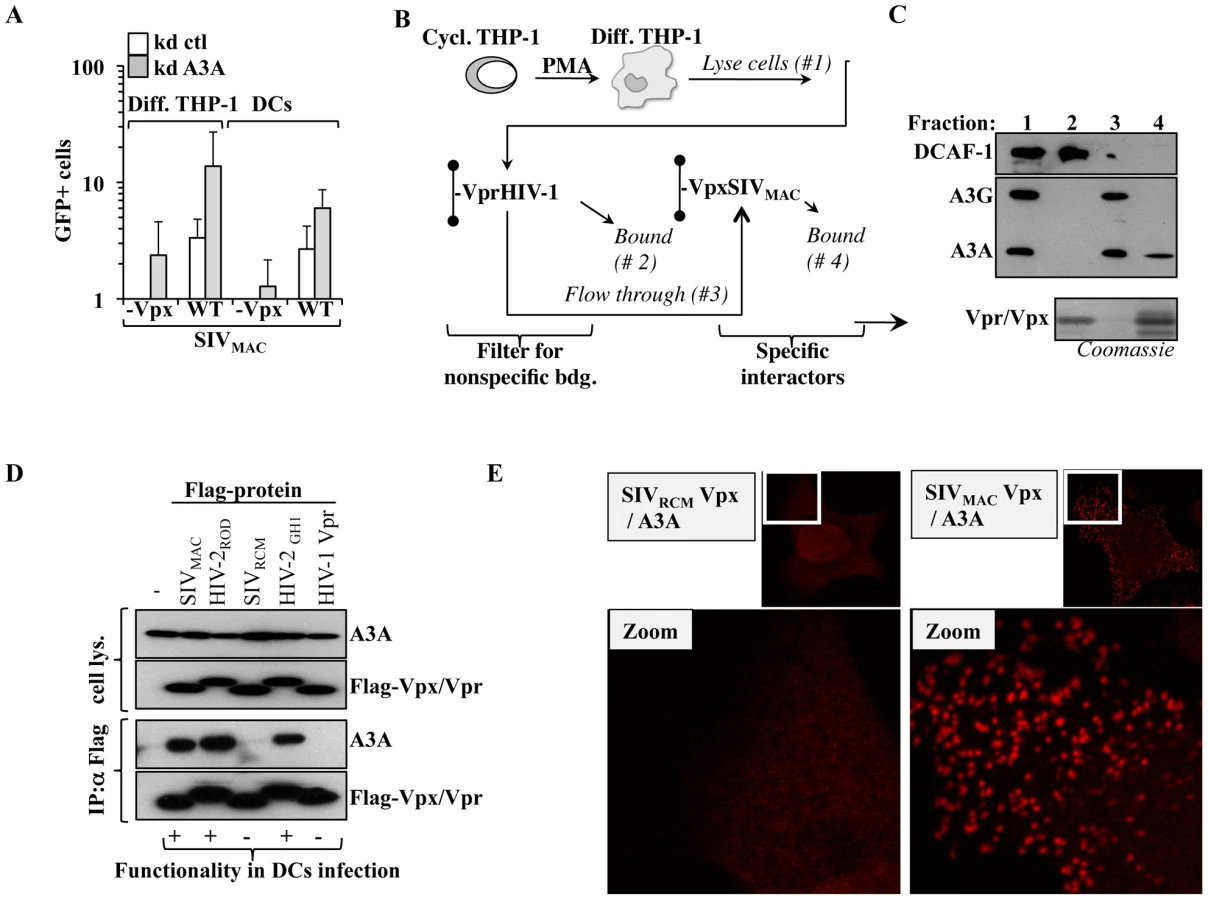
SIVMAC Vpx associates functionally to A3A
His-tagged SIVMAC Vpx and HIV-1 Vpr were purified on nichel columns and used to pull down endogenous A3A expressed in PMA-treated THP-1 cells. We have already determined that although these proteins are structurally related, only SIVMAC Vpx exerts a strong positive effect during the early phases of infection of myeloid cells [50]. HIV-1 Vpr was used to filter the lysate obtained from differentiated THP-1 of non specific interactors, prior to binding to a second column containing SIVMAC Vpx (Figure 6B). The different fractions were then subjected to WB analysis for the indicated proteins (Figure 6C). The DDB1 and CUL4-associated factor 1 (DCAF1) adaptor was examined, as it has been shown to associate to both HIV-1 Vpr and Vpx proteins derived from almost all SIV and HIV-2 strains tested [56], [67], [69], [73]–[78], although the relevance of this binding for the functions of Vpx remains controversial [67], [69], [70]. As expected, DCAF1 was almost completely retained by the HIV-1 Vpr column, thus validating our filtration scheme. On the contrary, A3G and A3A, recognized by the same antibody and distinguishable by size upon migration in an SDS-PAGE gel, were not. Upon binding of the flow-through to the SIVMAC Vpx column, only A3A was retained indicating that it bound endogenous A3A but not A3G in differentiated THP-1 cells. To characterize this interaction further, HEK293T cells were transfected with several Flag-tagged Vpr and Vpx proteins of known functionality during the early phases of lentiviral infection in primary myeloid cells along with a construct coding HA-Tagged A3A (Figure 6D). Cells were lysed and co-immunoprecipitations were carried out using anti-Flag antibody-coated beads. Under these conditions, A3A co-immunoprecipitated only with Vpx proteins derived from SIVMAC, HIV-2ROD and HIV-2GH1, but not with SIVRCM Vpx nor with HIV-1 Vpr. Thus, A3A associates with proteins that exert a positive function during the early phases of infection of myeloid cells, but does not associate to non-functional ones.
To provide further evidence of the A3A-Vpx interaction with an independent technique, DuoLink experiments were carried out. In DuoLink-based immunofluorescence, the spatial proximity between two proteins (≤40 nm) is required for the generation of a fluorescent signal that is amplified in situ and that appears in the form of a dot upon image analysis. Very simplistically, instead than being directly coupled to a fluorophore, secondary antibodies are coupled to 2 distinct single-stranded oligonucleotides that, when in close proximity, interact with a substrate oligonucleotide that serves as the basis for fluorescence development. Thus, the DuoLink technique does not show the intracellular distribution of two proteins, but reveals if and where they might interact in the cell (Figure 6E). Under these conditions, a diffused background signal was obtained in HeLa cells stained solely with 2ndary antibodies or upon transfection of A3A with the non-functional SIVRCM Vpx (in our experience, this diffuse staining is the normal background obtained with DuoLink). On the contrary, a clear positive signal in the form of dots was obtained when A3A was co-transfected with SIVMAC Vpx. These dots were mostly localized in the cytoplasm of transfected cells, supporting the notion that A3A and Vpx interact in this intracellular location and corroborating our previous findings indicating that Vpx is significantly -although not exclusively- localized in the cytoplasm of expressing cells. Of note, the distribution of Vpx and A3A did not change when these proteins were expressed alone or together, as determined by standard confocal microscopy, indicating that this interaction does not induce a massive relocalization of either proteins (not shown).
All together, these results indicate that A3A associates functionally with Vpx.
SIVMAC Vpx specifically degrades A3A in HeLa cells and in primary DCs
A previous report indicated that SIVMAC Vpx could degrade A3A [72]. To determine whether this was the case, A3A and A3G were co-transfected along with HIV-1 Vif and SIVMAC Vpx in HeLa cells (Figure 7A). As expected, HIV-1 Vif decreased the steady state levels of A3G and A3A, although this effect was less pronounced for A3A. On the contrary, SIVMAC Vpx affected only A3A, but not A3G. When the stability of A3A was determined, a clear decrease in its stability was observed upon co-expression of SIVMAC Vpx (Figure 7B). This degradation seems to involve a proteasome dependent mechanism since degradation of A3A by Vpx is significantly blocked upon incubation with the proteasome inhibitor MG132 (Figure 7C).

To determine whether Vpx was also able to degrade endogenous A3A in myeloid cells, SIVRCM and SIVMAC Vpx proteins were expressed in DCs after lentiviral-mediated gene transduction. Briefly, HIV-1 vectors coding the 2 Vpx proteins were produced in 293T cells, normalized by protein content and used to transduce DCs. Under these conditions, Vpx was not incorporated into HIV-1 particles and thus did not affect the infectivity of these vectors in DCs (data not shown). When DCs were analyzed 4 days after transduction, a clear decrease in the levels of A3A was observed in DCs expressing SIVMAC Vpx, but not in DCs expressing SIVRCM Vpx. This decrease was specific for A3A, as the levels of A3G were unaffected by the expression of Vpx proteins.
Overall, these results indicate that SIVMAC Vpx may partially protect SIVMAC viruses from the negative effects of A3A by inducing its degradation during the early phases of infection.
Discussion
In the present study we reveal a novel role for A3A in controlling HIV-1 replication specifically in myeloid cells. Cells belonging to this lineage express all members of the APOBEC3 family, but A3A is the only one restricted to myeloid cells among the white blood cells targeted by HIV-1 in vivo and is also the sole member whose expression augments during spreading infection of primary macrophages. The results presented here indicate that the specific silencing of A3A in differentiated THP-1 cells (that mimic macrophages upon differentiation), primary macrophages and DCs increases their susceptibility to de novo HIV-1 infection, while on the contrary silencing in primary PBLs or ectopic expression of A3A in established cell lines exerts no effect on these phases. Thus, these results indicate that the antiviral effect of A3A is common to cells of myeloid origins. We have been so far unable to silence A3A in monocytes in a robust manner that also preserves their non-stimulated phenotype, so that the effect of A3A on HIV-1 could not be explored. However, in light of its extremely high expression, we may surmise that A3A hampers HIV-1 in non-stimulated monocytes, as suggested previously [42].
Overall, our findings are in agreement with past studies indicating that A3A does not exert an antiviral activity in HeLa or HEK293T cells [21], [31], and extend them by revealing that this property is explicit only in cells in which A3A is naturally expressed, i.e. myeloid cells.
While the restricted expression of A3A indicates a tight transcriptional regulation, the lack of antiviral activity upon ectopic expression in HeLa cells suggests that the mere expression of A3A is not sufficient to confer an antiviral state to recipient cells. This observation suggests that A3A may be regulated also at the post-translational level through modifications that affect its activity, localization, association to co-factors, processivity or all these things together. This cell type dependent regulation is not unprecedented among members of the APOBEC3 family, as an as yet unidentified inhibitory activity has been recently proposed to regulate the editing activity of A3G in T cells [79]. In the case of A3A, these different levels of control may be extremely important to regulate its activity and cytotoxicity. In our hands, the stable expression of A3A is particularly cytotoxic (data not shown), an effect that is likely linked to its ability to edit the cellular genome and to induce a DNA damage response that is followed by cell cycle arrest in A3A-overexpressing cell lines [32], [34], [35]. However, myeloid cells express constitutively high levels of A3A and must have devised manners to protect themselves from these negative effects. Since all the myeloid cells tested here are non-dividing, it is possible that this state makes them less susceptible to editing of the cellular genome or to a DNA damage response than cycling cells. Alternatively, and not mutually exclusive with the hypotheses mentioned above, a specific post-translational regulation might protect myeloid cells. Such modifications, and/or the association with other cellular components may strongly influence the ability of A3A to recognize its target for example by shaping the single-stranded DNA docking groove of A3A, as identified in [80].
The results presented in this study indicate that the pool of A3A present in target cells is directly capable of inhibiting incoming viruses. This possibility had been raised before in quiescent T cells and DCs for A3G, although a recent report tempered enthusiasm over the function of APOBEC3 members during these phases [25]. Our results demonstrate that the specific knockdown of A3A positively impacts the early phases of HIV-1 infection, as clearly observed using vectors capable of a single round of infection. An antiviral effect at this step is conceptually interesting because Vif, that normally inhibits APOBEC3s during the phases of virus production, is absent in virion particles [26], [27]. As a consequence, incoming virus may be largely deprived of one of its major protection against APOBEC3 members present in recipient cells. Indeed, as a further proof that Vif cannot influence the early phases of infection, use of minimal HIV-1 vectors deficient in Vif, did not modify the results obtained here (data not shown). At present, we ignore whether this inhibition during the early phases of infection is specific to the combination A3A-myeloid cells, or if it is more generally present in other primary cells. Despite the fact that a number of studies have characterized the lack of effect of APOBEC3 members during the early phases of infection, rare are those that examined this question in the natural targets of HIV-1 replication. In light of the results presented here, we believe this issue deserves further investigation.
A3A inhibits HIV-1 during reverse transcription, the step that a number of laboratories including ours have determined as a major restriction during the infection of primary myeloid cells [48]–[55]. The process of reverse transcription is extremely sensitive to the intracellular environment and multiple factors have been shown to affect it (core-stability, viral protein processing, dNTPs levels, possibly intracellular trafficking and so forth). So, in principle, A3A could inhibit this process by affecting any of these parameters either more potently than other APOBEC3 members or else more specifically in myeloid cells in which these steps take place at a slower pace with respect to other cell types. Viral DNA produced upon infection of primary myeloid cells presents cytidine editing in higher levels than the one synthesized in HeLa cells. More than 50% of edited cytidines are present within a TC dinucleotide recognized by all members of the APOBEC3 family with the exception of A3G, while most of the remaining edited sites are within a GC dinucleotide that does not seem to be specific for APOBEC3s. Only a minority of edited cytidine is present in a CC dinucleotide context specific to A3G (10 fold less than TC). Among the different sites, the levels of TC editing are specifically influenced by the intracellular levels of A3A, indicating it as a major responsible for TC editing in myeloid cells. The amount of cytidine editing observed here is small and unlikely to account for the antiviral effect of A3A observed here. However, this low amount of editing may represent only the detectable fraction of the total edited vDNA, as edited vDNA may be efficiently removed or repaired in myeloid cells. Given that detection of this small amount of vDNA editing is challenging, previous reports indicating its absence upon A3A inhibition of LINE-1 retrotransposons and AAV2 may mean that either editing is in these cases lower than what observed here, or else that A3A can exert its antiviral effect independently from deamination [36]–[39].
Although our results reveal that A3A plays a role directly in target cells, the finding that A3A is a cell type specific restriction factor suggests that A3A may also exert the most classical of the antiviral activities of APOBEC3s, namely the one of being incorporated into HIV-1 virions and then deaminate newly synthesized vDNA. In light of the exquisite cell type dependent regulation of the activity of A3A this possibility is highly likely and in this case HIV-1 Vif that does degrade A3A, although less efficiently than A3G, may protect the virus against A3A as it does against other APOBEC3s.
The features of HIV-1 inhibition by A3A seem essentially conserved among primate lentiviruses, as the depletion of A3A increases also the infectivity of SIVMAC. However, we have noticed that Vpx-deficient SIVMAC viruses are more responsive to A3A depletion than WT ones, a phenotype that is more marked in differentiated THP-1 cells, although less so in DCs. This may relate to the more drastic infectivity defect of Vpx-deficient SIVMAC viruses in the latters. Vpx is to date the sole non-structural viral protein coded by primate lentiviruses capable of affecting the early phases of infection specifically in myeloid cells [50]. In the past, we have hypothesized that this could be due to the block of a restriction factor specifically expressed in these cells [50], [56]. In our hands, Vpx proteins interact functionally with A3A using three different methods: binding of endogenous A3A obtained from the lysate of differentiated THP-1 cells, co-IP after co-expression and DuoLink immunolocalisation that requires the physical proximity of two proteins for a positive signal to develop (less than 40 nm). This interaction leads to the degradation of A3A, but not of A3G, in HeLa cells and more importantly in primary DCs. Thus, our results are in agreement and extend a past study indicating that Vpx functionally interacts and degrades A3A after co-expression in established HeLa cells [72]. In the past, we have shown that SIVMAC Vpx modifies the susceptibility of myeloid cells to lentiviral infection with both cognate and non-cognate viruses (as HIV-1) [50], [81]. Under these conditions, the major effect of Vpx on the early phases of HIV-1 infection was exerted on the speed at which viral DNA accumulated, a phenomenon that we had hypothesized to protect vDNA from the negative effects of a particularly hostile intracellular milieu [50]. Given that knockdown of A3A does not affect the kinetics of vDNA formation following HIV-1 infection, we believe that A3A may not be the sole effector counteracted by Vpx (Figure S6). However, the fact that A3A silencing increases in a more dramatic manner the infectivity of Vpx-deficient viruses, as well as the fact that A3A is degraded upon expression of Vpx in HeLa cells and DCs indicates that A3A is an important target of Vpx during the infection of myeloid cells. Two recent studies identified SAMHD1 as a cellular factor that is specifically bound and degraded by Vpx [59] [60]. Contrarily to A3A, SAMHD1 is expressed in cells other than myeloid cells, as its isolation from HEK 293T cells indicates [59]. However, its antiviral effect is apparent mainly in myeloid cells, arguing for the need of a particular environment or for specific cellular co-factors. In light of the results presented here, A3A whose expression is truly restricted to the myeloid lineage (among cells of hematopoietic origin) may be a co-factor in the antiviral action of SAMHD1 or conversely, SAMHD1 may be an important cofactor in the action of A3A. The fact that Vpx seems to degrade two factors that hamper the early phases of infection specifically in myeloid cells evokes the possibility that they may act on the same pathway.
In conclusion, our results reveal a novel role for A3A in inhibiting the de novo infection of myeloid cells with HIV-1 and more generally with primate lentiviruses. In light of the extreme importance of myeloid cells during HIV-1 pathogenesis, these results call for a more ample evaluation of the impact of A3A during HIV-1 pathogenesis in cells that play a key instructive role in immune system responses.
Materials and Methods
Cell culture, cytokines and antibodies
293T and HeLa cells were maintained in Dulbecco's Eagle Modified Medium (DMEM) supplemented with 10% of fetal calf serum (FCS) and 100 U of penicillin/streptomycin. The THP-1 monocytic cell line was maintained in RPMI1640 medium supplemented with 10% FCS, HEPES 10 mM, 0,05 mM β-mercaptoethanol and 100 U of penicillin/streptomycin. THP-1 cells were differentiated for 24 hours in presence of 100 ng/ml of phorbol-12-myristate-13-acetate (PMA, SIGMA) to induce their differentiation into macrophage-like cells. Primary blood human monocytes and peripheral blood lymphocytes (PBLs) were purified from the blood of healthy donors obtained at the blood bank of Lyon (EFS-Lyon), as described before [67], yielding cell populations of purity greater than 95%. Monocytes were differentiated into macrophages or immature dendritic cells (DCs) upon incubation for 4 to 5 days in RPMI1640 complete medium supplemented with 100 ng/ml of MCSF or 100 ng/ml of GM-CSF/IL-4, respectively (AbCys). PBLs were either left in the absence of stimulus (quiescent) or stimulated with phytohemmagglutinin (PHA, SIGMA at 1 µg/ml) and IL2 (150 U/ml, AIDS Reagents and Reference Program of the NIH) for 24 hrs prior to infection. When indicated cyclohexamide (CHX, SIGMA) was added at a concentration of 100 µg/ml to arrest cell translation and MG132 (SIGMA) was added for 8 hrs at 10 µg/ml to block proteasomal degradation. When indicated IFNα2A (Tebu-Bio) was used at a final concentration of 1000 U for 24 hrs prior to infection. Anti-Flag, anti-HA, anti-actin and anti-tubulin monoclonal antibodies were purchased from SIGMA while, anti-HIV-1 Vif and anti-A3G Apo-C17 antibodies that recognize A3G and also A3A (recognized for its smaller size) were obtained from the AIDS Reagents and Reference Program of the NIH.
To determine the amount of type I interferons released in the supernatants of infected macrophages, we adopted a well established method that allows the simultaneous detection of multiple α and β IFN subtypes thanks to their ability to functionally inhibit the infection of target A549 cells with a GFP-bearing VSV. VSV is replicative virus that is extremely susceptible to the presence of type I interferons. Briefly, supernatant dilutions harvested from macrophage cultures were incubated with A549 cells prior to challenge with GFP-bearing VSV. The percentage of GFP-infected cells is assessed 24 hrs later by flow cytometry and the amount of type I IFN (U/ml) obtained against a standard curve. Plasmacytoid DCs (pDCs obtained by positive selection, Miltenyi) purified from the blood of healthy donors were used as control.
DNA constructs and viral production
The plasmids coding HA-tagged A3A and A3G were a kind gift of Dr. Kenzo Tokunaga (National Institute of Infectious Diseases, Tokyo, Japan). Flag-tagged Vpx/Vpr coding plasmids were described before in [67]. The plasmid coding HIV-1 Vif was obtained from the AIDS Reagents and Reference Program of the NIH.
The HIV-1 and SIVMAC251 GFP-coding lentiviral vectors and their production has been described before [67]. Briefly, these single round infectivity vectors were produced by co-transfection of HEK293T cells with 3 plasmids coding: the packaging proteins Gag-Pro-Pol and viral non structural proteins (unless otherwise specified), a mini viral genome bearing a CMV-driven eGFP reporter and the Vesicular Stomatitis Virus G envelope (VSVg) conferring ample cellular tropism to viral vectors. Virions released in the supernatant of transfected cells were then purified through a 25% sucrose cushion, resuspended and their infection titers determined onto HeLa cells or by protein content against standards of known infectivity (exo-RT activity).
Replication competent R5-tropic HIV-1 virus (Yu2) was produced by transient transfection of HEK293T cells then used to infect target cells.
miR30-shRNA hairpins were designed using the Open Biosystem Website facility and cloned in the context of an HIV-1 lentiviral construct allowing their stable expression upon viral transduction (the targeted sequences are listed in Table 1, [67]). In this case, HIV-1 vectors bearing a miR30-shRNA expression cassette were produced as mentioned above and quantified by exo-RT over standards of known infectious titer. For A3A silencing, a mixture of the 3 targeting miR30-shRNAs was used in the transfection during the step of virus production and the overall DNA ratio of packaging to vector constructs was kept constant between specific and control miR30-shRNA coding vectors.
Infections
Infections were carried out for 2 hrs on 105 cells prior to extensive cells washing. The percentage of infected cells was monitored 3 to 4 days post infection by flow cytometry. For miR-shRNA lentiviral vector-mediated silencing, infections were carried out on 5.106 monocytes for 24 hrs prior to the addition of fresh media. Cells were then differentiated for 5 days in macrophages or DCs upon incubation with the appropriate cytokine cocktail. Cells were challenged with GFP-coding vectors four days afterwards. A multiplicity of infection (MOI) comprised between 3 and 5 was used for both silencing and challenging vectors.
Replicative infections were carried out as described above at MOI of 0,05 except that a fraction of the supernatant was stored and replaced with fresh media every 2–4 days. Viral spread through the culture was monitored by exo-RT. When used to reveal deamination of produced vDNA, virions were treated twice with 20 U/ml RNase free DNase RQ1 (Promega) for 2 hours at 37°C, and DNA extracted from infected cells was treated overnight at 37°C with DpnI. vDNA obtained after PCR amplification was cloned and sequenced.
Transfection of siRNAs
M-SCF-differentiated macrophages were transfected with a total of 100 pmol of siRNAs (Darmacon) using Lipofectamine2000, according to the manufacturer's protocol (Invitrogen). After 6 hours, fresh media was added and cells were analyzed 48 hours post transfection by Western blot or challenged.
Quantitative real time PCRs
RNA was extracted on silica columns (NucleoSpin RNA XS; Macherey-Nagel) and reverse transcribed after DNAse treatment, using the SuperScript II Reverse Transcriptase (Invitrogen). Quantitative PCR was performed on a StepOnePlus Real-time PCR system (Applied Biosystems) using the FastStart Universal SYBR Green Master mix (Roche Diagnostics). Primers specific for each member of the APOBEC3 family genes and house-keeping genes (TBP, HPRT1 and RPL13A) had been previously described [44]. The level of expression of these genes does not vary among the different cell types and conditions used here. Results are expressed in mRNA copies per µg of total RNA.
For the analysis of vDNA produced upon infection, cells were lyzed 24 hours post infection and DNA amplified using primers specific for the GFP sequence carried by the challenge virus (see Table 1). Control infections were carried out in the presence of RT inhibitors (Nevirapine and AZT, at 20 µg/ml, obtained through the AIDS Reagents and Reference Program of the NIH). Values were first normalized for the amount of cellular DNA (actin) then subtracted for the values obtained for each sample in control infections performed in the presence of RT inhibitors. Experiments were discarded if the latter represented values higher than 1/10 of the ones obtained in the absence of RT inhibitors.
Co-Immunoprecipitations
PMA-differentiated THP-1 cells were lysed in SD buffer 150 mM (50 mM Tris/HCl pH 7.4, 150 mM NaCl, 0,5% Triton-X100) in presence of a cocktail of protease inhibitors (Roche). The lysate was passed through a nichel column with immobilized HIV-1 Vpr protein, then on a second column containing SIVMAC Vpx. Columns were washed thrice with SD buffer 400 mM (50 mM Tris/HCl pH 7.4, 400 mM NaCl, 0,5% Triton-X100) prior to analysis by WB. Transfections of HEK293T cells were carried out in 6 well plates using a 1∶1 ratio of the indicated Vpx/Vpr and APOBEC3 proteins. Thirty-six hours post transfection, cells were lyzed in SD buffer 150 mM, then immunoprecipitated by addition of anti-Flag antibody-coated beads (Sigma) for 3 hours at +4°C. Beads were washed thrice with SD buffer 400 mM prior to WB analysis.
DuoLink immunofluorescence
HeLa cells were seeded on coverlips and transfected with 300 ng of DNA using JetPei, according to the manufacturer's protocol (Polyplus). Twenty-four hours post transfection, cells were washed twice in PBS and fixed in 3% Paraformaldehyde in PBS (PFA). Free aldehydes were quenched by the addition of 50 mM NH4Cl in PBS before an additional wash in PBS. Cells were then permeabilized in 0,2% Triton-X100 for 5 min and saturated for 45 min in 0,2% gelatin from cold water skin fish (SIGMA) before immunostaining with DuoLink kit (Eurogentec), following the manufacturer's protocol. Image acquisition was carried out on an Axiovert 100 M Zeiss LSM 510 confocal microscope.
Accession numbers
APOBEC3A: Ensembl:ENSG00000128383; APOBEC3B: Ensembl:ENSG00000179750; APOBEC3C: Ensembl:ENSG00000244509; APOBEC3D/E: Ensembl:ENSG00000243811; APOBEC3F: Ensembl:ENSG00000128394; APOBEC3G: Ensembl:ENSG00000239713; APOBEC3H: Ensembl:ENSG00000100298.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AlbinJSHarrisRS 2010 Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev Mol Med 12 e4
2. SheehyAMGaddisNCChoiJDMalimMH 2002 Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418 646 650
3. ZhangHYangBPomerantzRJZhangCArunachalamSC 2003 The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424 94 98
4. MangeatBTurelliPCaronGFriedliMPerrinL 2003 Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424 99 103
5. LecossierDBouchonnetFClavelFHanceAJ 2003 Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300 1112
6. HarrisRSBishopKNSheehyAMCraigHMPetersen-MahrtSK 2003 DNA deamination mediates innate immunity to retroviral infection. Cell 113 803 809
7. BishopKNVermaMKimEYWolinskySMMalimMH 2008 APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 4 e1000231
8. NewmanENHolmesRKCraigHMKleinKCLingappaJR 2005 Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol 15 166 170
9. KaoSKhanMAMiyagiEPlishkaRBuckler-WhiteA 2003 The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol 77 11398 11407
10. MehleAStrackBAncutaPZhangCMcPikeM 2004 Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem 279 7792 7798
11. MehleAGoncalvesJSanta-MartaMMcPikeMGabuzdaD 2004 Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev 18 2861 2866
12. YuXYuYLiuBLuoKKongW 2003 Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302 1056 1060
13. MarinMRoseKMKozakSLKabatD 2003 HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9 1398 1403
14. ConticelloSGHarrisRSNeubergerMS 2003 The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol 13 2009 2013
15. BergeronJRHuthoffHVeselkovDABeavilRLSimpsonPJ 2010 The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog 6 e1000925
16. ShirakawaKTakaori-KondoAKobayashiMTomonagaMIzumiT 2006 Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology 344 263 266
17. SheehyAMGaddisNCMalimMH 2003 The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9 1404 1407
18. RulliSJJrMirroJHillSALloydPGorelickRJ 2008 Interactions of murine APOBEC3 and human APOBEC3G with murine leukemia viruses. J Virol 82 6566 6575
19. BogerdHPTallmadgeRLOaksJLCarpenterSCullenBR 2008 Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J Virol 82 11889 11901
20. OhsugiTKoitoA 2007 Human T cell leukemia virus type I is resistant to the antiviral effects of APOBEC3. J Virol Methods 139 93 96
21. DoehleBPSchaferAWiegandHLBogerdHPCullenBR 2005 Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J Virol 79 8201 8207
22. Goila-GaurRKhanMAMiyagiEKaoSOpiS 2008 HIV-1 Vif promotes the formation of high molecular mass APOBEC3G complexes. Virology 372 136 146
23. KaoSMiyagiEKhanMATakeuchiHOpiS 2004 Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1 27
24. PionMGranelli-PipernoAMangeatBStalderRCorreaR 2006 APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med 203 2887 2893
25. KamataMNagaokaYChenIS 2009 Reassessing the role of APOBEC3G in human immunodeficiency virus type 1 infection of quiescent CD4+ T-cells. PLoS Pathog 5 e1000342
26. DettenhoferMYuXF 1999 Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J Virol 73 1460 1467
27. SimonJHMillerDLFouchierRAMalimMH 1998 Virion incorporation of human immunodeficiency virus type-1 Vif is determined by intracellular expression level and may not be necessary for function. Virology 248 182 187
28. MalimMH 2009 APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci 364 675 687
29. ChiuYLGreeneWC 2008 The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol 26 317 353
30. OhAinleMKernsJALiMMMalikHSEmermanM 2008 Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4 249 259
31. BishopKNHolmesRKSheehyAMDavidsonNOChoSJ 2004 Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14 1392 1396
32. LandrySNarvaizaILinfestyDCWeitzmanMD 2011 APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep 12 444 50
33. ThielenBKMcNevinJPMcElrathMJHuntBVKleinKC 2010 Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J Biol Chem 285 27753 27766
34. SuspeneRAynaudMMGuetardDHenryMEckhoffG 2011 Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci U S A 108 4858 4863
35. StengleinMDBurnsMBLiMLengyelJHarrisRS 2010 APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol 17 222 229
36. ChenHLilleyCEYuQLeeDVChouJ 2006 APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol 16 480 485
37. BogerdHPWiegandHLHulmeAEGarcia-PerezJLO'SheaKS 2006 Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A 103 8780 8785
38. BogerdHPWiegandHLDoehleBPLuedersKKCullenBR 2006 APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res 34 89 95
39. MuckenfussHHamdorfMHeldUPerkovicMLowerJ 2006 APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem 281 22161 22172
40. WiegandHLCullenBR 2007 Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J Virol 81 13694 13699
41. VartanianJPGuetardDHenryMWain-HobsonS 2008 Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320 230 233
42. PengGGreenwell-WildTNaresSJinWLeiKJ 2007 Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110 393 400
43. KoningFANewmanENKimEYKunstmanKJWolinskySM 2009 Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol 83 9474 9485
44. RefslandEWStengleinMDShindoKAlbinJSBrownWL 2010 Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 38 4274 4284
45. HeathWRCarboneFR 2009 Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol 10 1237 1244
46. PiguetVSteinmanRM 2007 The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol 28 503 510
47. MartinezFOSicaAMantovaniALocatiM 2008 Macrophage activation and polarization. Front Biosci 13 453 461
48. ArfiVLienardJNguyenXNBergerGRigalD 2009 Characterization of the behavior of functional viral genomes during the early steps of human immunodeficiency virus type 1 infection. J Virol 83 7524 7535
49. ArfiVRiviereLJarrosson-WuillemeLGoujonCRigalD 2008 Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J Virol 82 6557 6565
50. GoujonCRiviereLJarrosson-WuillemeLBernaudJRigalD 2007 SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology 4 2
51. TriquesKStevensonM 2004 Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J Virol 78 5523 5527
52. SonzaSMaerzADeaconNMeangerJMillsJ 1996 Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J Virol 70 3863 3869
53. O'BrienWANamaziAKalhorHMaoSHZackJA 1994 Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J Virol 68 1258 1263
54. NeilSMartinFIkedaYCollinsM 2001 Postentry restriction to human immunodeficiency virus-based vector transduction in human monocytes. J Virol 75 5448 5456
55. CollinMGordonS 1994 The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology 200 114 120
56. SharovaNWuYZhuXStranskaRKaushikR 2008 Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog 4 e1000057
57. LafuseWPBrownDCastleLZwillingBS 1995 Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J Leukoc Biol 57 477 483
58. RiceGIBondJAsipuABrunetteRLManfieldIW 2009 Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41 829 832
59. HreckaKHaoCGierszewskaMSwansonSKKesik-BrodackaM 2011 Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474 658 661
60. LaguetteNSobhianBCasartelliNRingeardMChable-BessiaC 2011 SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474 654 657
61. RoseJMCrowleyCKFleischmannWRJr 1985 Evidence that IFN-alph/beta induces two antiviral states active against different viruses. J Gen Virol 66 Pt 5 1153 1158
62. FamillettiPCRubinsteinSPestkaS 1981 A convenient and rapid cytopathic effect inhibition assay for interferon. Methods Enzymol 78 387 394
63. HarmanANLaiJTurvilleSSamarajiwaSGrayL 2011 HIV infection of dendritic cells subverts the interferon induction pathway via IRF1 and inhibits type 1 interferon production. Blood 118 298 308
64. PengGLeiKJJinWGreenwell-WildTWahlSM 2006 Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med 203 41 46
65. BonvinMAchermannFGreeveIStrokaDKeoghA 2006 Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43 1364 1374
66. GoujonCJarrosson-WuillemeLBernaudJRigalDDarlixJL 2003 Heterologous human immunodeficiency virus type 1 lentiviral vectors packaging a simian immunodeficiency virus-derived genome display a specific postentry transduction defect in dendritic cells. J Virol 77 9295 9304
67. GoujonCArfiVPertelTLubanJLienardJ 2008 Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol 82 12335 12345
68. ThomasJAOttDEGorelickRJ 2007 Efficiency of human immunodeficiency virus type 1 postentry infection processes: evidence against disproportionate numbers of defective virions. J Virol 81 4367 4370
69. SrivastavaSSwansonSKManelNFlorensLWashburnMP 2008 Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog 4 e1000059
70. BergamaschiAAyindeDDavidALe RouzicEMorelM 2009 The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J Virol 83 4854 4860
71. KaushikRZhuXStranskaRWuYStevensonM 2009 A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe 6 68 80
72. BergerAMunkCSchweizerMCichutekKSchuleS 2010 Interaction of Vpx and apolipoprotein B mRNA-editing catalytic polypeptide 3 family member A (APOBEC3A) correlates with efficient lentivirus infection of monocytes. J Biol Chem 285 12248 12254
73. WenXDuusKMFriedrichTDde NoronhaCM 2007 The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J Biol Chem 282 27046 27057
74. McCallCMMiliani de MarvalPLChastainPD2ndJacksonSCHeYJ 2008 Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol Cell Biol 28 5621 5633
75. TanLEhrlichEYuXF 2007 DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J Virol 81 10822 10830
76. Le RouzicEBelaidouniNEstrabaudEMorelMRainJC 2007 HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6 182 188
77. HreckaKGierszewskaMSrivastavaSKozaczkiewiczLSwansonSK 2007 Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci U S A 104 11778 11783
78. BelzileJPDuisitGRougeauNMercierJFinziA 2007 HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog 3 e85
79. ThielenBKKleinKCWalkerLWRieckMBucknerJH 2007 T cells contain an RNase-insensitive inhibitor of APOBEC3G deaminase activity. PLoS Pathog 3 1320 1334
80. BulliardYNarvaizaIBerteroAPeddiSRohrigUF 2011 Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol 85 1765 1776
81. GoujonCJarrosson-WuillemeLBernaudJRigalDDarlixJL 2006 With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther 13 991 994
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence