
Human Cytomegalovirus Drives Epigenetic Imprinting of the Locus in NKG2C Natural Killer Cells
Upon viral infection, the innate interferon (IFN)-γ producing Natural Killer (NK) cells provide fast, but short-term protection, while adaptive T cells confer delayed, but long-lasting immunity. Once acquired, effector properties remain stably imprinted in the T cell memory progeny. Recently, it was shown that human cytomegalovirus (HCMV) infection can shape the human NK cell repertoire and drive the generation and maintenance of NK cell expansions, which express the activating receptor CD94/NKG2C and have been described as memory-like NK cells. However, the molecular mechanisms underlying NK cell adaptive properties driven by HCMV infection have not been completely defined. In this study, we identify epigenetic imprinting of the IFNG locus as selective hallmark and crucial mechanism driving strong and stable IFN-γ expression in HCMV-specific NK cell expansions, thus providing a molecular basis for the regulation of adaptive features in innate cells.
Published in the journal:
Human Cytomegalovirus Drives Epigenetic Imprinting of the Locus in NKG2C Natural Killer Cells. PLoS Pathog 10(10): e32767. doi:10.1371/journal.ppat.1004441
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004441
Summary
Upon viral infection, the innate interferon (IFN)-γ producing Natural Killer (NK) cells provide fast, but short-term protection, while adaptive T cells confer delayed, but long-lasting immunity. Once acquired, effector properties remain stably imprinted in the T cell memory progeny. Recently, it was shown that human cytomegalovirus (HCMV) infection can shape the human NK cell repertoire and drive the generation and maintenance of NK cell expansions, which express the activating receptor CD94/NKG2C and have been described as memory-like NK cells. However, the molecular mechanisms underlying NK cell adaptive properties driven by HCMV infection have not been completely defined. In this study, we identify epigenetic imprinting of the IFNG locus as selective hallmark and crucial mechanism driving strong and stable IFN-γ expression in HCMV-specific NK cell expansions, thus providing a molecular basis for the regulation of adaptive features in innate cells.
Introduction
In order to successfully fight infections caused by intracellular pathogens, interferon (IFN)-γ is expressed during an immune response primarily by T cell lineages and natural killer (NK) cells. While NK cells display constitutive IFNG promoter activity and express IFN-γ at early maturation stages [1], the expression by CD8+ and CD4+ T cells is restricted to differentiated effector/memory cells. In particular, naïve CD4+ T cells must undergo a differentiation process towards type 1 T helper cells (TH1), in order to acquire the ability to stably express IFN-γ [2], [3]. A key mechanism stabilizing TH1-lineage commitment is epigenetic imprinting of the IFNG locus, which leads to heritable DNA and histone modifications of cis-elements, such as the promoter and several conserved non-coding sequences (CNS). The IFNG cis-elements that show the most stringent TH1-specific hypersensitivity include the promoter and the activation-induced proximal upstream element CNS1, which is located 6 kb or 4 kb upstream of the mouse Ifng and human IFNG promoter, respectively. These regulatory regions display binding sites for T-bet, STAT4, NF-κB, and NFAT. Once in an open configuration, both regions function as crucial enhancers of IFNG transcriptional activity in TH1 cells, especially in response to TCR stimulation, due to the presence of binding sites for NFAT, which is activated after engagement of the TCR but not of the cytokine receptors in T cells [3]–[8]. Moreover, effector/memory but not naïve CD8+ T cells were shown to display an open configuration in these two IFNG regions [9], [10]. Although epigenetic analysis of the IFNG locus in total NK cells revealed an open configuration in some regions analyzed [1], [9], [11]–[13], the IFNG promoter undergoes partial demethylation during NK cell differentiation [14]. Thus, a more detailed analysis of additional IFNG regulatory regions in NK cell subsets is of great importance.
Though being part of the innate immune system, defined subsets of NK cells display adaptive features [15]. NK cells with memory-like properties have been described during mouse cytomegalovirus (MCMV) infection [16], after hapten-induced contact hypersensitivity [17], as well as after cytokine (IL-15+IL-12+IL-18) priming [18]–[20]. Primary MCMV infection of C57BL/6 mice induces the clonal expansion of Ly49H+ NK cells, which can directly interact with the m157 protein of MCMV [21]. Ly49H+ memory-like NK cells persist for several months and, upon rechallenge, undergo secondary expansion and display enhanced effector functions [16]. The mechanisms underlying the generation of memory-like features in NK cells are a field of intense research [22]–[27]. For instance, it has been shown that proliferation of MCMV-specific Ly49H+ NK cells relies on IL-12R and STAT4 signaling and PLZP-mediated antagonism of Blimp-1 [22], [24]. However, it is not clear whether this phenomenon might be associated with imprinting of the IFNG locus, similar to memory CD8+ T cells and CD4+ TH1 cells. Human CMV (HCMV) infection also plays an important role in shaping the human NK cell repertoire. In HCMV seropositive (HCMV+) individuals, CD94/NKG2C+, further referred to as NKG2C+, NK cells are present at higher frequency compared to HCMV seronegative (HCMV−) ones [28] and can be expanded in vitro in the presence of HCMV-infected fibroblasts [29]. In the course of HCMV reactivation after solid organ or hematopoietic cell transplantation, NKG2C+ NK cells from certain donors undergo extensive proliferation in vivo [30]–[33]. These NKG2C+ expansions are characterized by bright expression of NKG2C (NKG2Chi) and by a terminally differentiated phenotype, as suggested by the preferential expression of CD57 [31], [34]. The specific mechanisms how HCMV might mediate “priming” and expansion of NKG2Chi NK cells is still not completely clear. Both NKG2C and its inhibitory counterpart CD94/NKG2A, further referred to as NKG2A, recognize the non-classical HLA class I molecule HLA-E [35]–[37], whose surface expression is stabilized by peptides derived from the leader sequence of other HLA class I molecules in a transporter associated with antigen processing (TAP)-dependent fashion [38], [39]. Interestingly, a peptide derived from the gpUL40 protein of HCMV strains and clinical isolates is able to stabilize HLA-E in a TAP-independent manner and enables recognition by both NKG2A and NKG2C [40], [41]. HCMV exploits several strategies to induce down-regulation of MHC class I molecules in infected cells, for instance by blocking TAP [42]. Thus, while classical MHC class I molecules are down-regulated, gpUL40 induces surface expression of HLA-E on infected cells [40]. As the inhibitory receptor NKG2A generally displays a higher affinity for HLA-E compared to NKG2C [41], [43], gpUL40-induced up-regulation of HLA-E may represent an escape route for HCMV against NK cell mediated elimination of infected cells. However, while NKG2C is often co-expressed with NKG2A in HCMV− individuals, NKG2Chi expansions in HCMV+ individuals typically lack NKG2A, while expressing killer cell immunoglobulin-like receptor (KIR) specific for self-MHC (sKIR), especially KIR2DL1 or KIR2DL2/3, and LIR-1 [28], [33], [34], [44]. Importantly, NKG2Chi expansions persist over time, are highly competent IFN-γ producers and exhibit even higher IFN-γ expression upon HCMV reactivation [30]–[32]. For all these reasons, it has been suggested that NKG2Chi NK cell expansions found in HCMV infected individuals might also represent NK cells with memory-like properties [15], [31], [32].
With the aim to analyze whether epigenetic regulation of the IFNG locus could contribute to determine high IFN-γ competence and stability in NK cells described to display memory-like properties, we here showed that only HCMV-specific NKG2Chi expansions and cytokine-primed NK cells displayed an open configuration at the IFNG CNS1, which acted as important enhancer of IFNG transcriptional activity in response to NKG2C and activating receptors (actR) engagement.
Results
Epigenetic remodeling of the CNS1 is an exclusive marker of NKG2Chi expansions and cytokine-primed NK cells
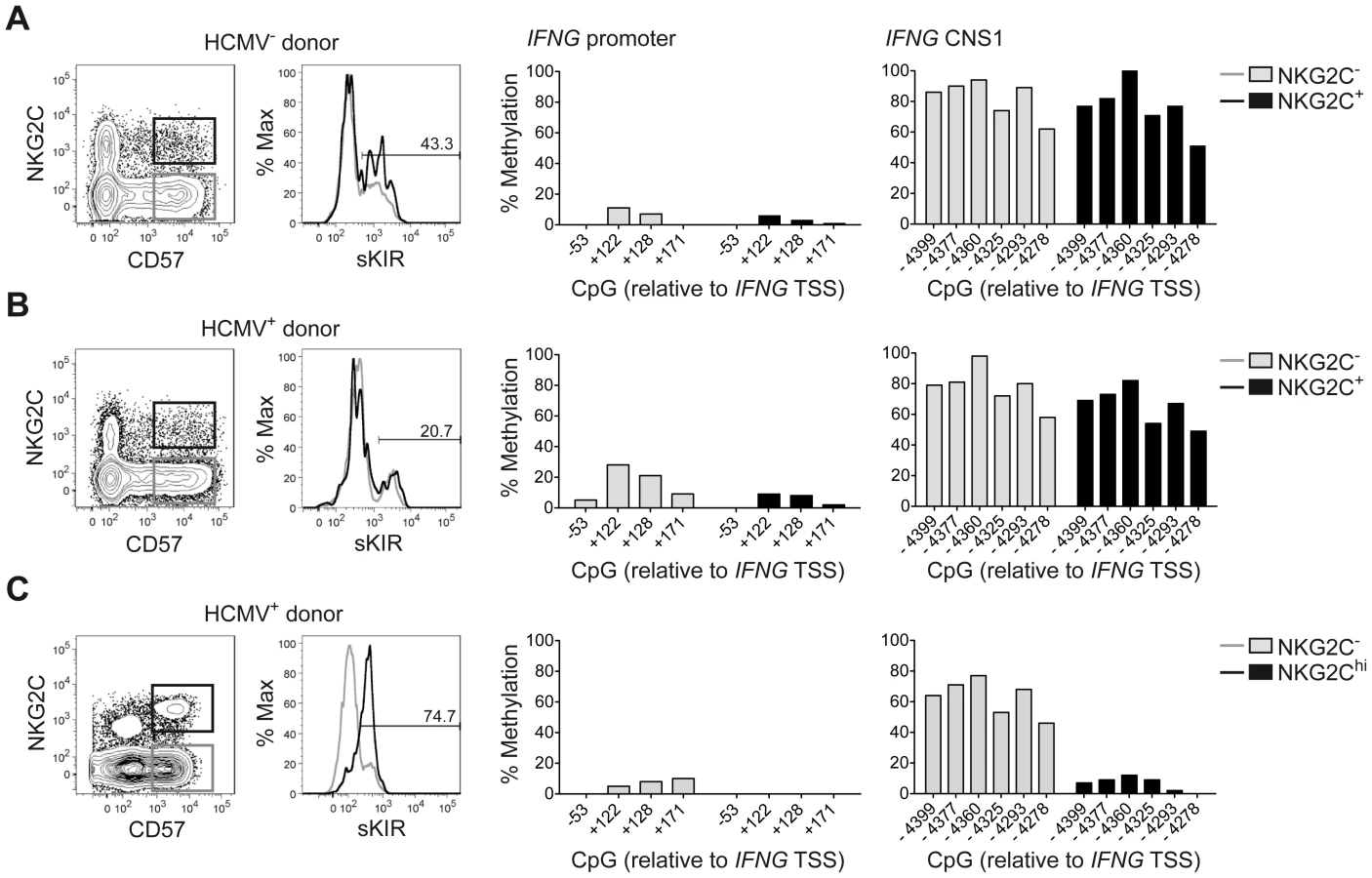
Among the described regulatory regions conserved between the human and mouse IFNG/Ifng locus (Figure 1A), we analyzed the methylation status of selected CpG residues present in the frame of the promoter and the CNS1, which are important enhancers of IFNG transcriptional activity and represent epigenetic hallmarks of TH1 differentiation [3], [4]. While the promoter was largely demethylated in NK cells and TH1 cells (Figure 1B), CNS1 was completely methylated in NK cells, resembling the configuration of naïve CD4+ T cells, rather than of TH1 cells (Figure 1C). Previous reports have described that after HCMV infection or priming with IL-15+IL-12+IL-18, NK cells can display memory-like properties and the ability to stably produce high amounts of IFN-γ after rechallenge [19], [30], [31]. Considering the parallels to memory TH1 cell formation, we analyzed the methylation status of the CNS1 in cytokine-primed NK cells and in HCMV-specific NK cell expansions. Cytokine-primed NK cells generated in vitro in the presence of IL-15+IL-12+IL-18 displayed complete demethylation of the CNS1. Conversely, only partial and progressive opening of both regions could be observed in the presence of IL-15+IL-12, IL-15+IL-18 or IL-15 alone. Substituting IL-18 by IL-1 almost led to similar results (Figure 1D). Next, we analyzed the methylation status of the IFNG CNS1 in NKG2C+ and NKG2C− NK cells isolated from HCMV− (Figure 2A) or HCMV+ (Figure 2B) individuals as well as in NKG2C+ NK cell expansions, selectively occurring in some HCMV+ individuals (Figure 2C). These NKG2C+ expansions are characterized by preferential expression of CD57 and strong enrichment in at least one sKIR (Figure 2C). As CD57+ NKG2C+ expansions display higher surface expression of NKG2C (Figure S1), they are further referred to as NKG2Chi NK cells [31], [44]. For a fair comparison of all NK cell subsets, CpG methylation analysis was performed after sorting CD56dim CD57+ NK cells. CD56dim CD57+ NK cells displayed an open configuration of the IFNG promoter independent of NKG2C expression or HCMV status (Figures 2A, 2B and 2C). NKG2C− NK cells derived from HCMV+ or HCMV− individuals as well as NKG2C+ NK cells from HCMV− individuals consistently displayed a closed configuration of the CNS1 (Figures 2A, 2B and 2C). Importantly, among HCMV+ individuals, an open configuration of the CNS1 occurred exclusively in donors displaying a distinctive expansion of NKG2Chi sKIR+ NK cells (Figure 2C). Conversely, if no expansion was present, NKG2C+ NK cells from HCMV+ individuals displayed a closed configuration of the CNS1, similar to the NKG2C− subsets (Figure 2B). Altogether, these data show that among NK cells only HCMV-specific NKG2Chi expansions and cytokine-primed NK cells, which were described as memory-like NK cells [15], [19], [30], [31], share the ability to undergo remodeling of the CNS1 with TH1 cells.
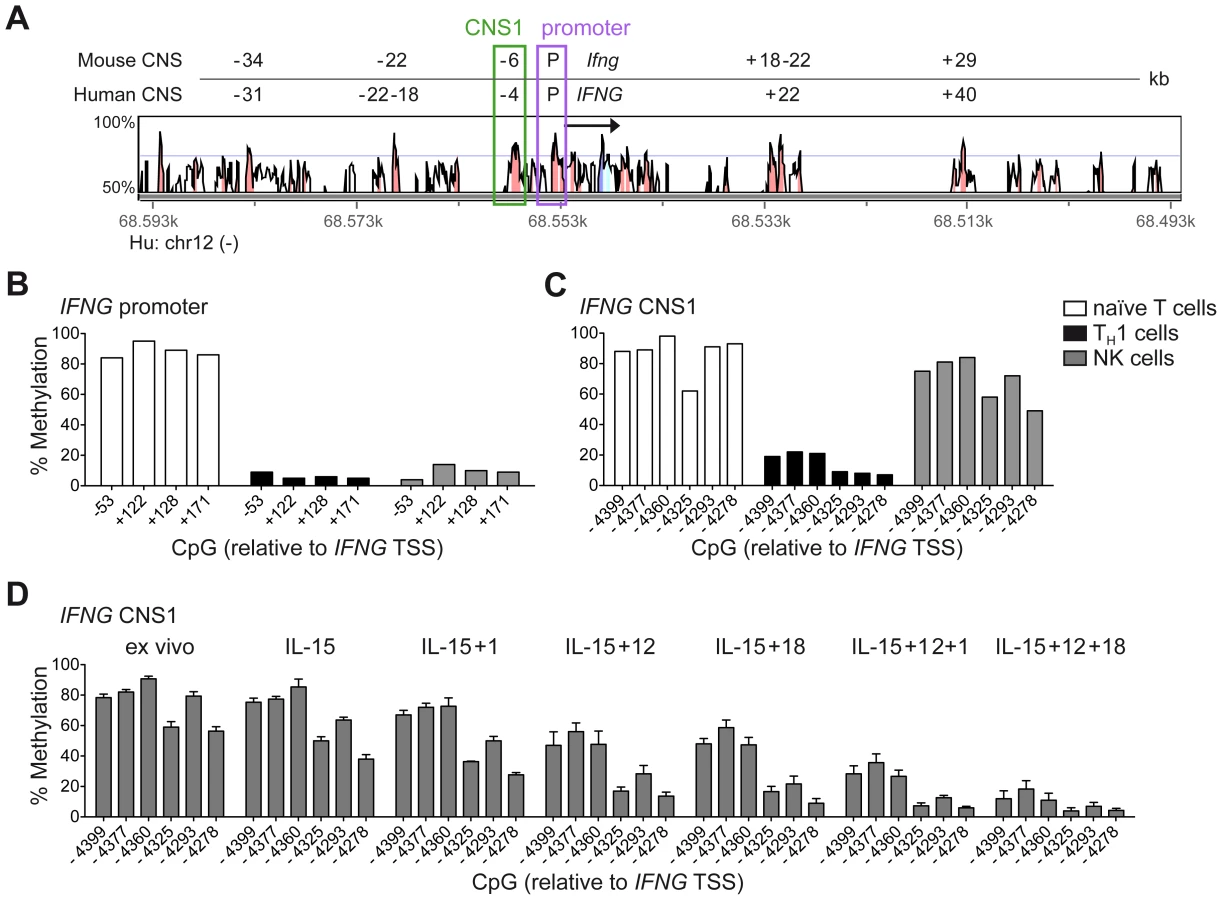
CNS1 demethylation occurs in NKG2Chi NK cell expansions, independent of sKIR or CD57 expression and remains stably imprinted in the progeny
One major phenotypic difference between CD56dim CD57+ NKG2Chi cell expansions and CD56dim CD57+ NKG2C+ cells from HCMV+ donors is the high enrichment in expression of at least one sKIR, which defines educated NK cells. Thus, we next asked whether CNS1 demethylation was a peculiar feature of sKIR+ educated NK cells, rather than of NKG2Chi cells. Among NKG2Chi NK cells, CNS1 was accessible not only in CD57+ sKIR+ cells, but also in CD57+ sKIR− cells (Figure 3A). Surprisingly, among NKG2C+ cells, CNS1 was accessible even in CD57− sKIR− cells (Figure 3B). We finally aimed to determine whether the open configuration of the CNS1 was stably imprinted in NKG2Chi NK cell expansions, by analyzing the same HCMV+ individual after one year. The size and phenotype of the NKG2Chi NK cell pool remained constant over time (Figure 3C), in line with previous data [44]. Importantly, CD57+ NKG2Chi but not CD57+ NKG2C− NK cells still displayed a completely open configuration at both the IFNG promoter and CNS1 (Figures 3C and S2), indicating that the IFNG locus remains stably imprinted in NKG2Chi NK cell progeny, similar to memory TH1 cells.

NKG2Chi NK cell expansions share a global epigenetic signature with TH1 and CD8+ memory T cells
In order to understand whether IFNG CNS1 demethylation was part of a broader epigenetic remodeling occurring in NKG2Chi expansions from HCMV+ individuals, we performed RRBS-based global methylation analysis of CD57+ NKG2Chi, CD57+ NKG2C− and CD57− NKG2C− NK cells in comparison with CD8+ memory and CD4+ TH1 cells as well as with CD8+ or CD4+ naïve T cells isolated from the same HCMV+ individuals. CD8+ memory and CD4+ TH1 cells, CD8+ or CD4+ naïve T cells, and NK cells constitute three different clusters. Interestingly, NKG2Chi NK cells distinctively shared the methylation profile of a large set of CpG sites with CD8+ memory or CD4+ TH1 cells. Conversely, CD57− NKG2C− NK cells were more similar to naïve CD8+ or CD4+ T cells, with CD57+ NKG2C− NK cells displaying a methylation profile intermediate to the two other NK cell subsets (Figure 4A). The NK cell subsets show a decreasing gradient of similarities to naïve CD8+ and CD4+ T cells from CD57− NKG2C− via CD57+ NKG2C− to CD57+ NKG2Chi, as judged by the Euclidian distance, and a complementary, increasing gradient of similarities to CD8+ memory and CD4+ TH1 cells (Figure 4B). This observation was further confirmed by employing an unsupervised strategy and performing principal component analysis (PCA) (Figure 4C). Most of the variation among the different lymphocyte subsets was captured by the first principal component (PC1). CD8+ and CD4+ naïve T cells were at one extreme, while CD8+ memory together with TH1 cells were at the other extreme of the PC1-axis, suggesting that PC1 defined the direction of lymphocyte differentiation, which was characterized by a global epigenetic remodeling (Figure 4B). NK cell subsets distributed along the PC1, with CD57− NKG2C− cells displaying the highest PC1 score, similar to naïve T cells, while CD57+ NKG2Chi NK cells displayed the lowest PC1 score. T cell and NK cell lineage-defining epigenetic modifications were likely described by the second principal component (PC2). Analyzing the methylation pattern of other genes of interest, which are shared between NK cells and TH1 or CD8+ memory T cells, such as TBX21 (T-bet), EOMES (Eomesodermin), and PRF1 (perforin), we observed that all NK cell subsets displayed similar or even more pronounced demethylation pattern at several CpG regions compared to TH1 or CD8+ memory T cells (Figure 4D). TNF was mainly demethylated in both CD57+ NKG2Chi and CD57+ NKG2C− NK cell subsets, similar to TH1 or CD8+ memory T cells. Recently, it was shown that PRDM1 (Blimp-1) and ZBTB32 (also known as PLZP, ROG, FAZF and TZFP), which are not expressed in naïve T cells but are up-regulated in differentiated T cells [45]–[47], regulate the proliferative burst of Ly49H+ memory NK cells during MCMV infection [24]. Interestingly, some CpGs of the PRDM1 and ZBTB32 genes were consistently demethylated in NKG2Chi NK cells and TH1 or memory CD8+ T cells, compared to other NK cell subsets or naïve T cells. Altogether, these data suggest that CNS1 demethylation in NKG2Chi expansions from HCMV+ individuals is part of a broader epigenetic remodeling, which is partially shared by TH1 and memory CD8+ T cells.

NKG2C is a main trigger for IFN-γ production in NKG2Chi NK cells
NKG2Chi NK cells are described as potent IFN-γ producers [30]. However, in line with their phenotype of CD57+ terminally differentiated cells, they respond less efficiently to IL-12+IL-18 stimulation, compared to CD57− NK cells, due to the lower expression of IL12RB2 and IL-18R (Figure S3A and S3B), in line with previous data [34]. Moreover, although terminally differentiated CD57+ sKIR+ NK cells are generally more proficient in producing IFN-γ in response to activating receptor triggering [14], several actR, such as NKp46 and NKp30 are prominently down-regulated in NKG2Chi NK cells, in line with previous findings [28], [44] and their engagement led to only poor IFN-γ production (Figure S3C and S3E). As NKG2C triggering has been shown to induce effector functions in NKG2C+ NK cells from HCMV+ donors [31], [34], we hypothesized that NKG2C engagement alone or in combination with 2B4 could be a major trigger of IFN-γ expression in expanded NKG2Chi NK cells. Since NKG2C could not be stained after cross-linking, IFN-γ expression was first analyzed after gating on CD57+ NKG2A− sKIR+ NK cells, of which the large majority expressed NKG2C only in HCMV+ donors displaying an expansion (Figure S3D). Triggering of NKG2C alone was able to induce high IFN-γ production in CD57+ NKG2A− sKIR+ NK cells only in HCMV+ individuals displaying NKG2Chi expansions and this effect was even more pronounced when NKG2C and 2B4 were engaged in combination. Conversely, very low IFN-γ expression could be detected in CD57+ NKG2A− sKIR+ NK cells from HCMV+ individuals with or without NKG2Chi expansions when 2B4 alone was engaged (Figures 5A and 5B). Next, we tested whether a similar effect could be observed when NK cells were stimulated via the physiological engagement of the 2B4 ligand CD48 and the NKG2C ligand HLA-E, by using the CD48+ cell line LCL.221 (221) or 221-AEH [39], which expressed high levels of HLA-E on their surface, in contrast to 221 (Figure 5C). In this system, analysis of IFN-γ expression among CD57+ NKG2A− sKIR+ cells could be performed directly by gating on NKG2Chi or NKG2C− cells from HCMV+ donors as well as on NKG2C+ cells from HCMV− donors. In line with the data obtained with antibody cross-linking, only NKG2Chi expansions proficiently produced IFN-γ in response to 221-AEH (Figure 5D and 5E). Conversely, stimulation with 221 resulted in only low IFN-γ expression in all subsets analyzed. Thus, these data show that engagement of NKG2C alone or together with 2B4 represents a major trigger of IFN-γ production in NKG2Chi NK cells.
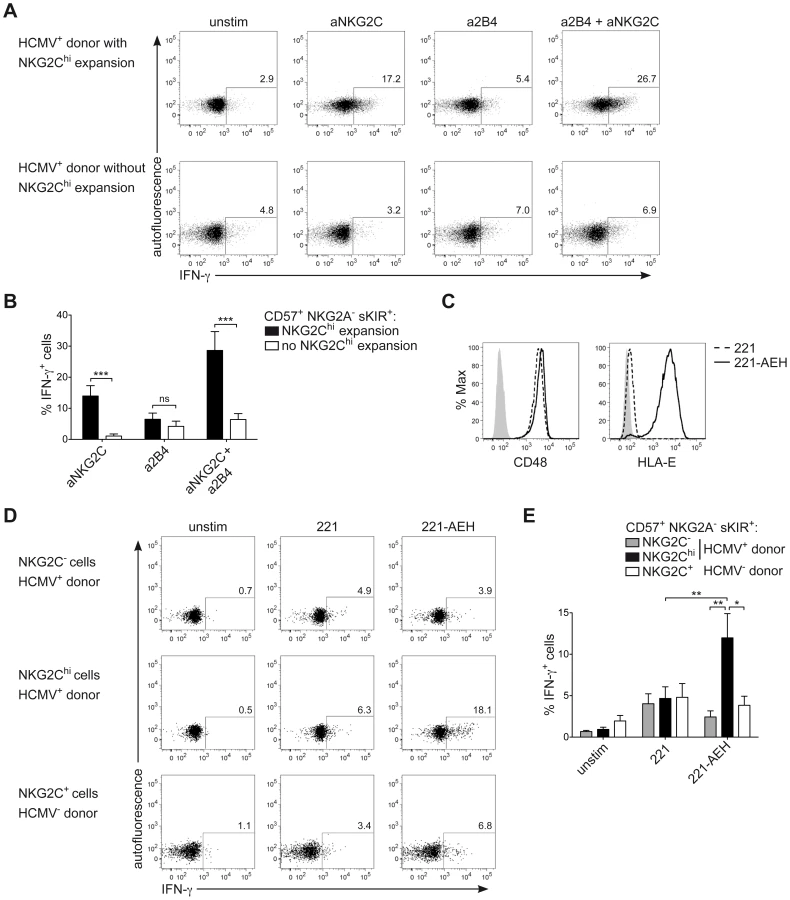
CNS1 accessibility enhances IFNG transcriptional activity induced by NKG2C and 2B4 engagement
Next, we aimed to understand whether CNS1, which was selectively demethylated in NKG2Chi NK cells, would be able to enhance IFNG transcriptional activity induced by engagement of NKG2C alone or in combination with 2B4. To perform luciferase reporter assays, we took advantage of the NK cell line NKL. NKL expressed 2B4 but only intermediate levels of NKG2C (Figure 6A), and only co-engagement of NKG2C and 2B4, but not of NKG2C alone, efficiently induced IFN-γ expression in NKL (Figure 6B). In line with these data, IFNG transcriptional activity in NKL, measured after transfection with the luciferase reporter vector pGL3 containing the IFNG promoter (IFNGp) (−571 to +71), could be induced only after co-engagement of 2B4 and NKG2C (Figure 6C). Next, we tested whether CNS1 was able to enhance IFNG transcriptional activity induced by the engagement of NKG2C and 2B4 and whether selective methylation would suppress it. To this aim, we cloned the minimal IFNGp (−49 to +71), not containing any CpG site, into the CpG-free luciferase reporter vector pCpGL [48], in combination or without the CNS1 sequence containing the CpG sites of interest. The construct was in vitro treated or not with CpG-methyltransferase (M.SssI), which selectively methylated the CNS1 CpG sites. CNS1 induced a dramatic enhancement of IFNG transcriptional activity in response to co-engagement of 2B4 and NKG2C, which was completely abolished by in vitro methylation of the CNS1 construct (Figure 6D). In further support of the concept that DNA methylation plays an important role in IFN-γ expression in NK cells as in TH1 cells, we treated NKL with the DNA methyltransferase inhibitor 5-azacytidine (AZA). Addition of AZA resulted in partial CNS1 demethylation and consistently enhanced expression of IFN-γ induced by NKG2C alone or by NKG2C and 2B4 co-stimulation (Figure S4A and S4B). Altogether, this data show that accessibility of CNS1 is crucial to enhance IFNG transcriptional activity in response to NKG2C engagement.

Discussion
Our present study showed that epigenetic remodeling of the CNS1 region in the IFNG locus is a hallmark of NKG2Chi NK cells expanded in HCMV+ individuals as well as cytokine-primed NK cells, which have been previously described as memory-like NK cells [15], [19], [31], [32] and share an open configuration of this region with TH1 cells. CNS1 is a crucial enhancer of IFNG transcriptional activity in TH1 cells, especially in response to TCR triggering [4], [8]. In line with its important role as TCR-dependent enhancer in TH1 cells, we showed that accessibility of CNS1 enhanced IFNG transcriptional activity of NK cells upon triggering of actR, especially NKG2C. Whereas resting NK cells generally need simultaneous engagement of multiple actR [49], the sole engagement of NKG2C was sufficient to induce IFN-γ production in expanded NKG2Chi cells, while 2B4 mainly functions as coreceptor [50]. We propose that CNS1 accessibility contributes to the robust IFN-γ expression in response to NKG2C engagement as one important mechanism. Epigenetic remodeling of the IFNG locus, together with high expression of NKG2C, might help NK cells to compensate for other actR deficiencies. Several characteristics displayed by NKG2Chi NK cells, such as their in vivo expansion, persistence in time and high effector functions after HCMV reactivation support the concept that these cells display adaptive properties [15]. Indeed, our genome wide analysis of DNA methylation showed that NKG2Chi NK cell expansions show remarkably lower epigenetic distance to TH1 and CD8+ memory T cells as compared to the other NK cell subsets analyzed. This observation further strengthens the idea that common epigenomic features might be functional hallmarks of memory-like NK cells and memory T cells.
It is still not clear to which extent some of the adaptive features of NKG2Chi expansions depend on low-level viral reactivation events occurring in healthy HCMV+ individuals [51]. In line with this interpretation, it was shown that during allogeneic hematopoietic stem cell transplantation from HCMV+ donor to HCMV+ recipient, NKG2C+ NK cells also expand in the absence of clinically detectable HCMV viremia [32]. Similarly, clinically silent reactivation of HCMV or of other herpesviruses also stimulates HCMV-specific T cells and dramatically shapes the T cell repertoire [52], [53].
Which signals are driving CNS1 demethylation exclusively in expanded NKG2Chi NK cells? Co-culture with 221-AEH cells, which express the NKG2C ligand HLA-E, or with HCMV-infected fibroblasts leads to expansion of NKG2C+ NK cells [29], [44]. Although engagement of NKG2C might contribute to induce CNS1 opening in these cells, we do not have evidences for that. On the other hand, we show that IL-12+IL-15+IL-18 stimulation is sufficient to drive CNS1 demethylation in NK cells. Priming by pro-inflammatory cytokines, which largely contribute to anti-viral defense [54], might not only induce CNS1 opening but also contribute to select NKG2C+ cells with proliferative and/or survival advantage [34], [55], thus enabling the persistence of a progeny, which has undergone epigenetic remodeling of the CNS1, similar to memory TH1 cells. Further supporting a central role of pro-inflammatory cytokines in this process, it was shown that generation of MCMV-specific Ly49H+ memory-like NK cells is strongly dependent on IL-12R/STAT4 signaling [22] and that induction of PLZP, required for the proliferative burst of Ly49H+ NK cells, is accomplished by IL-12 and IL-18 but not by Ly49H engagement [24]. A preferential expansion/survival in response to inflammatory cytokines might also partially explain why NKG2C+ NK cells from HCMV+ individuals could expand in the course of other viral infections, such as with Hantavirus, Chikungunya or Hepatitis C virus [34], [55], [56]. This unique ability of NKG2Chi NK cells to expand and become activated in different infections closely resembles what has been observed for HCMV-specific T cell responses. It was shown that the broad HCMV-specific T cell compartment not only controls viral reactivation, but can also alter the outcome of unrelated infections [57], a phenomenon known as heterologous immunity [58]. Herpesvirus-specific CD8+ T cells, especially HCMV- and EBV-specific ones, are frequently activated in the setting of other infections, among which Hantavirus and Hepatitis virus have been reported [57], [59]. This phenomenon does not rely exclusively on T cell receptor cross-reactivity but also acts through antigen-independent bystander mechanisms, such as cytokines, in particular IL-15 [51], [57]. Similarly, during Hantavirus and Hepatitis virus infections proinflammatory cytokines might promote the expansion of NKG2Chi NK cells, which were previously primed during HCMV infection. Interestingly, we also showed that, among CD56dim NKG2Chi NK cells, CNS1 demethylation is not confined to the CD57+, but also present in the CD57− subset, which is highly responsive and proliferates in response to cytokines [44], [60]. Thus, our and other data [44] suggest that CD57− NKG2Chi cells, rather than their CD57+ counterpart, might represent memory-like precursors replenishing and stably maintaining the peripheral pool of NKG2Chi NK cells.
In summary our data identified epigenetic imprinting in the CNS1 region of the IFNG locus as a hallmark and mechanism underlying adaptive features and IFN-γ memory in NK cells. We also showed that a broader epigenetic remodeling accompanies these adaptive features demonstrating that both inflammatory and pathogen cues induce epigenetic remodeling of NK cells. These two observations extend our view of the molecular control caused by inflammation in innate immune cells [61], [62] and furthermore highlight adaptive epigenetic similarities among the two branches of the immune system.
Materials and Methods
Ethics statement
Leukocyte concentrates were obtained from adult healthy individuals (DRK, Berlin, Germany) after written informed consent and approval by the local ethics committees on human studies (Charité Berlin, Germany).
Cell isolation and flow cytometry
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll Hypaque density gradient centrifugation from leukocyte concentrates obtained from healthy individuals (DRK, Berlin, Germany). NK cells, CD4+ and CD8+ T cells were enriched by magnetic cell sorting using CD56, CD4 or CD3 microbeads respectively (Miltenyi Biotec), followed by flow cytometric sorting (FACS) using FACS Aria II (BD Biosciences). NK cell subsets were FACS sorted as viable CD3− CD56+ (Figure 1) or as CD3− CD56dim CD57+/− (CD62L+/−) sKIR+/− NKG2C+/−, hereby showing purity over 95%. sKIR corresponds to KIR2DL1+ in HLA-C2+ or KIR2DL3+ in HLA-C1+ donors, respectively. Naïve CD4+ T cells were FACS sorted as CD3+ CD4+ CD45RA+ CCR7+ and TH1 cells as CD3+ CD4+ CD45RA− CCR7− CCR5+ IL-18Ra+ [63], [64]. Naïve CD8+ T cells were FACS sorted as CD3+ CD8+ CD45RA+ CCR7+ cells and CD8+ memory cells as CD3+ CD8+ CD45RO+ cells. T cell subsets were FACS sorted to over 95% purity. For flow cytometric (FC) analysis, PBMC were stained with mAbs against the following molecules: CD56 PE-Cy7 (NCAM16.2), CD3 APC-H7 (SK27), CD3 V500 (UCHT1), CD4 V450 (RPA-T4), CCR5 APC-Cy7 (2D7/CCR5) (BD Biosciences), CD56 BV605 (HCD56), CD62L PerCP-Cy5.5 (Dreg56), CD57 Pacific Blue (HCD57), CD45RA PE-Cy7 (HI100), CCR7 PerCP-Cy5.5 (G043H7), KIR3DL1 PE (DX9), HLA-E PE (3D12) (Biolegend), KIR2DL1 APC (143211), KIR2DL3 FITC (180701), NKG2C PE (134591), IL-18Ra PE (70625), NKG2C (134522) in-house coupled to biotin (R&D Systems), 2B4 biotin (C1.7) (eBioscience). KIR2DL1/S1 (11PB6) and KIR2DL2/L3/S2 (GL183), kindly provided by D. Pende and A. Moretta, Genova, Italy, were self-conjugated to Cy5 or Alexa Fluor 700. CD8 Cy5 (GN11/134D7) and CD45RO FITC (UCHL1) were self-conjugated. CD48 (CO202) was kindly provided by A. Moretta, Genova, Italy. Secondary staining of biotinylated mAbs was performed with Streptavidin-BV421 (Biolegend), and of unconjugated IgM mAbs with aIgM PE (RMM-1, Biolegend). Viable cells were detected using Live/Dead (LD) fixable dead cell stains (Invitrogen). Flow cytometric (FC) analysis was performed at BD FACS Fortessa employing FACSDiva Software (BD Biosciences). Obtained data was analyzed with FlowJo software (Tree Star).
Cell culture, stimulation and intracellular staining
FACS sorted NK cell subsets were maintained in RPMI-1640 (Gibco BRL) (100 U/mL penicillin and 0.1 mg/ml streptomycin added) supplemented with 10% human AB serum or 10% FCS (Lonza). To cross-link distinct actR, biotinylated mAbs specific for 2B4 (C1.7, eBioscience), NKp30 (AF29-4D12), NKp46 (9E2) (Miltenyi Biotec) and purified NKG2C (134522, R&D Systems) (in-house coupled to biotin) were conjugated to anti-biotin MACSiBead particles (all Miltenyi Biotec). For NK cell stimulation, beads were used at a bead∶cell ratio of 5∶1 and co-incubated with NK cells for 16 hours. Alternatively NK cells were stimulated with 50 ng/ml IL-12 and 50 ng/ml IL-18 for 16 hours. 10 µg/ml Brefeldin A (Sigma) was added for the time of stimulation. The target cell line LCL.221 or 221-AEH [39] was used for stimulation at a NK cell∶target cell ratio of 10∶1 for 6 hours, adding 10 µg/ml Brefeldin A and Golgistop (BD Biosciences) after 1 hour. 221 and 221-AEH were maintained in complete RPMI medium supplemented with 10% FCS and 721.221-AEH in the presence of hygromycin B (Invitrogen) at 250 µg/ml. After stimulation, cells were fixed with 1.6% paraformaldehyde (Electron Microscopy Services) and stained intracellularly in the presence of 0.5% saponin (Sigma-Aldrich). Intracellular cytokines were stained using the following mAbs: IFN-γ APC (B27) (BD Biosciences) or IFN-γ PE-Cy7 (B27) (Biolegend). To generate cytokine-induced memory-like NK cells, FACS sorted and CFSE-labeled (500 nM, Invitrogen) NK cells were incubated with 20 ng/ml IL-1β, 10 ng/ml IL-12, 20 ng/ml IL-15 (Miltenyi Biotec) and/or 20 ng/ml IL-18 (R&D systems) for 5 days. The NK cell line NKL (ATCC) was cultured in complete RPMI medium supplemented with 10% FCS in the presence of 100 U/ml IL-2 (Miltenyi Biotec). To test the impact of 5-Azacytidine (AZA), NKL cells were cultured for 5 days in the presence of 5 µM AZA (Sigma).
Quantitative RT-PCR (qPCR)
RNA was isolated from FACS sorted NK cell subsets using NucleoSpin RNAII (Macherey Nagel) and reverse transcribed using Reverse Transcription Reagents. mRNA levels were analyzed by qPCR using a StepOnePlus real-time PCR system and TaqMan gene expression assays according to manufacturer's instructions (all Applied Biosystems): Hs00155486_m1 IL12RB2, 4333764F GAPDH. mRNA content was normalized to GAPDH expression and mean relative gene expression was determined using the ΔΔCT method.
CpG methylation analysis
Genomic DNA of ex vivo FACS sorted NK and T cell subsets was extracted using the QIAamp DNA Mini Kit and bisulfite conversion was performed by using the EpiTect Bisulfite Kit (all purchased from Qiagen). Regions of interest in the IFNG locus were amplified using primers as described before [9], and subsequently pyrosequenced by Varionostic GmbH, Germany.
Reduced Representation Bisulfite Sequencing (RRBS)
Genomic DNA of ex vivo FACS sorted CD3− CD56dim CD57+/− NKG2C+/− NK cell subsets, naïve CD4+ T cells, TH1 cells as well as naïve and memory CD8+ T cells from two donors was extracted using the QIAamp DNA Mini Kit (Qiagen). Libraries for RRBS were prepared according to the protocol previously described by Boyle et al [65]. For the MspI digestion 160 ng DNA was used. After end-repair and A-tailing, sample specific adaptors, with a unique sequence barcode for each sample, were ligated and purified by Agencourt AMPure XP beads (Beckman Coulter). The adaptor ligand mix was amplified by PCR (12 cycles) and purified by AMPure beads. All barcoded DNA samples were pooled into one library per donor, with respectively 7 DNA samples. The multiplexed libraries were amplified by PCR (12 cycles), purified by AMPure beads and sequenced (∼2.5 lanes per library) on the Illumina HiSeq 2500 platform in a 100 bp single read configuration. Since RRBS libraries exhibit low complexity base composition at the first three bases (due to the MspI digestion all fragments start with “CGG”) a tailored sequencing recipe with three initial dark cycles (designed with the help of Illumina) was used for the sequencing run. Raw reads were trimmed in RRBS-mode for adapter contamination and low-quality tails (Phred <20) with the Cutadapt wrapper Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) [66] for RRBS and mapped with GSNAP [67] to the human genome (hs37d5). After the initial alignment, Bis-SNP [68] was applied to realign the reads overlapping known insertions/deletions as defined by dbSNP v138, to recalibrate quality scores and finally to call methylation levels. This was done with default parameters, except a maximal allowed coverage of 1000. The methylation calls were filtered with RnBeads [69], requiring a minimum coverage of 5 reads in each sample. A genome wide comparison was performed by principle component analysis (PCA) of mean methylation levels from two donors for each cell-type and site. The PCA revealed a clear separation of CD4+ TH1 and CD8+ T memory cells, naïve CD4+ and CD8+ T cells, and NK cells by the first two components, with explained proportions of variance of 48.15%, 23.51%, 11.03%, 6.78%, 5.64% and 4.90% for PC1 to PC6. Moreover, 1,000 most variant methylation sites of 30,000 randomly chosen sites, localized within the gene bodies and promotor regions (−2 kb TSS) were used for clustering analysis and visualization of the methylation profiles (Beta-values of methylation sites) and similarities between cell subsets (Euclidian distance). This strategy was chosen to account for a representative proportion of genome wide methylation and to avoid a potential bias towards outstanding differences between particular cell types. The clustering was performed with the WARD-linkage criterion and Euclidian distance as similarity metric.
Luciferase reporter assay
DNA constructs containing regulatory sequences of the human IFNG locus (CNS1 and promoter) were generated with the Expand High Fidelity PCR System (Roche) and cloned upstream of the Firefly luciferase (Luc) gene into the basic pGL3 reporter vector (Promega). For in vitro methylation assays, constructs were cloned into the CpG-free luciferase vector pCpGL (kindly provided by M. Rehli) [48]. pCpGL vectors were methylated in vitro by M.SssI according to manufacturer's recommendations (New England Biolabs), followed by purification with the NucleoSpin Extract II kit (Macherey Nagel). Transfection of rested (deprived of IL-2 for 24 hours) NKL cells was performed by nucleofection using the Amaxa Human NK cell Nucleofector Kit (Lonza). 3×106 cells were transfected with 5 µg pGL3/pCpGL vector and 0.25 µg pRL-TK Renilla luciferase reporter vector (Promega), as internal control. After transfection, cells were rested for 1 hour followed by stimulation via NKG2C and/or 2B4 cross-linking as described before. Subsequently, cells were collected for measurement of luciferase activity, which was performed using the Dual Luciferase Reporter Assay system according to manufacturer's instructions (Promega) and analyzed with an Orion L luminometer (Titertek Berthold). Relative luciferase units (RLU) were normalized to Renilla luciferase and to empty vectors pGL3 or pCpGL, respectively. A list of all primers synthesized upon request (TIB Molbiol Berlin) is provided in Supporting Information.
Statistical analysis
Wilcoxon signed rank two-tailed and Mann-Whitney test were used for statistical analysis of data sets consisting of at least six independent experiments (*p<0.05, **p<0.01, ***p<0.001), using GraphPad Prism (GraphPad Software).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. StetsonDB, MohrsM, ReinhardtRL, BaronJL, WangZE, et al. (2003) Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 198: 1069–1076.
2. MurphyKM, ReinerSL (2002) The lineage decisions of helper T cells. Nat Rev Immunol 2: 933–944.
3. WilsonCB, RowellE, SekimataM (2009) Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 9: 91–105.
4. BalasubramaniA, MukasaR, HattonRD, WeaverCT (2010) Regulation of the Ifng locus in the context of T-lineage specification and plasticity. Immunol Rev 238: 216–232.
5. LeeDU, AvniO, ChenL, RaoA (2004) A distal enhancer in the interferon-gamma (IFN-gamma) locus revealed by genome sequence comparison. J Biol Chem 279: 4802–4810.
6. ShnyrevaM, WeaverWM, BlanchetteM, TaylorSL, TompaM, et al. (2004) Evolutionarily conserved sequence elements that positively regulate IFN-gamma expression in T cells. Proc Natl Acad Sci U S A 101: 12622–12627.
7. MacianF (2005) NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5: 472–484.
8. SchoenbornJR, DorschnerMO, SekimataM, SanterDM, ShnyrevaM, et al. (2007) Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol 8: 732–742.
9. DongJ, ChangHD, IvascuC, QianY, RezaiS, et al. (2013) Loss of methylation at the IFNG promoter and CNS-1 is associated with the development of functional IFN-gamma memory in human CD4(+) T lymphocytes. Eur J Immunol 43: 793–804.
10. FitzpatrickDR, ShirleyKM, McDonaldLE, Bielefeldt-OhmannH, KayGF, et al. (1998) Distinct methylation of the interferon gamma (IFN-gamma) and interleukin 3 (IL-3) genes in newly activated primary CD8+ T lymphocytes: regional IFN-gamma promoter demethylation and mRNA expression are heritable in CD44(high)CD8+ T cells. J Exp Med 188: 103–117.
11. ChangS, AuneTM (2005) Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc Natl Acad Sci U S A 102: 17095–17100.
12. TatoCM, MartinsGA, HighFA, DiCioccioCB, ReinerSL, et al. (2004) Cutting Edge: Innate production of IFN-gamma by NK cells is independent of epigenetic modification of the IFN-gamma promoter. J Immunol 173: 1514–1517.
13. HattonRD, HarringtonLE, LutherRJ, WakefieldT, JanowskiKM, et al. (2006) A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25: 717–729.
14. Luetke-EverslohM, CicekBB, SiracusaF, ThomJT, HamannA, et al. (2014) NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol 44: 2074–2084.
15. RolleA, PollmannJ, CerwenkaA (2013) Memory of infections: an emerging role for natural killer cells. PLoS Pathog 9: e1003548.
16. SunJC, BeilkeJN, LanierLL (2009) Adaptive immune features of natural killer cells. Nature 457: 557–561.
17. O'LearyJG, GoodarziM, DraytonDL, von AndrianUH (2006) T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol 7: 507–516.
18. CooperMA, ElliottJM, KeyelPA, YangL, CarreroJA, et al. (2009) Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A 106: 1915–1919.
19. RomeeR, SchneiderSE, LeongJW, ChaseJM, KeppelCR, et al. (2012) Cytokine activation induces human memory-like NK cells. Blood 120: 4751–4760.
20. NiJ, MillerM, StojanovicA, GarbiN, CerwenkaA (2012) Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med 209: 2351–2365.
21. DokunAO, KimS, SmithHR, KangHS, ChuDT, et al. (2001) Specific and nonspecific NK cell activation during virus infection. Nat Immunol 2: 951–956.
22. SunJC, MaderaS, BezmanNA, BeilkeJN, KaplanMH, et al. (2012) Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med 209: 947–954.
23. NabekuraT, KanayaM, ShibuyaA, FuG, GascoigneNR, et al. (2014) Costimulatory molecule DNAM-1 is essential for optimal differentiation of memory natural killer cells during mouse cytomegalovirus infection. Immunity 40: 225–234.
24. BeaulieuAM, ZawislakCL, NakayamaT, SunJC (2014) The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol 15: 546–553.
25. Min-OoG, BezmanNA, MaderaS, SunJC, LanierLL (2014) Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J Exp Med 211: 1289–1296.
26. FirthMA, MaderaS, BeaulieuAM, GasteigerG, CastilloEF, et al. (2013) Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J Exp Med 210: 2981–2990.
27. ZawislakCL, BeaulieuAM, LoebGB, KaroJ, CannerD, et al. (2013) Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proc Natl Acad Sci U S A 110: 6967–6972.
28. GumaM, AnguloA, VilchesC, Gomez-LozanoN, MalatsN, et al. (2004) Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 104: 3664–3671.
29. GumaM, BudtM, SaezA, BrckaloT, HengelH, et al. (2006) Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 107: 3624–3631.
30. FoleyB, CooleyS, VernerisMR, PittM, CurtsingerJ, et al. (2012) Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 119: 2665–2674.
31. Lopez-VergesS, MilushJM, SchwartzBS, PandoMJ, JarjouraJ, et al. (2011) Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A 108: 14725–14732.
32. FoleyB, CooleyS, VernerisMR, CurtsingerJ, LuoX, et al. (2012) Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol 189: 5082–5088.
33. KuijpersTW, BaarsPA, DantinC, van den BurgM, van LierRA, et al. (2008) Human NK cells can control CMV infection in the absence of T cells. Blood 112: 914–915.
34. BeziatV, DalgardO, AsselahT, HalfonP, BedossaP, et al. (2012) CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol 42: 447–457.
35. BraudVM, AllanDS, O'CallaghanCA, SoderstromK, D'AndreaA, et al. (1998) HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391: 795–799.
36. BorregoF, UlbrechtM, WeissEH, ColiganJE, BrooksAG (1998) Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med 187: 813–818.
37. LeeN, LlanoM, CarreteroM, IshitaniA, NavarroF, et al. (1998) HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A 95: 5199–5204.
38. BraudV, JonesEY, McMichaelA (1997) The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol 27: 1164–1169.
39. LeeN, GoodlettDR, IshitaniA, MarquardtH, GeraghtyDE (1998) HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol 160: 4951–4960.
40. TomasecP, BraudVM, RickardsC, PowellMB, McSharryBP, et al. (2000) Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287: 1031.
41. HeatleySL, PietraG, LinJ, WidjajaJM, HarpurCM, et al. (2013) Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J Biol Chem 288: 8679–8690.
42. PloeghHL (1998) Viral strategies of immune evasion. Science 280: 248–253.
43. Vales-GomezM, ReyburnHT, ErskineRA, Lopez-BotetM, StromingerJL (1999) Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J 18: 4250–4260.
44. BeziatV, LiuLL, MalmbergJA, IvarssonMA, SohlbergE, et al. (2013) NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121: 2678–2688.
45. KalliesA, NuttSL (2007) Terminal differentiation of lymphocytes depends on Blimp-1. Curr Opin Immunol 19: 156–162.
46. OmoriM, YamashitaM, InamiM, Ukai-TadenumaM, KimuraM, et al. (2003) CD8 T cell-specific downregulation of histone hyperacetylation and gene activation of the IL-4 gene locus by ROG, repressor of GATA. Immunity 19: 281–294.
47. MiawSC, ChoiA, YuE, KishikawaH, HoIC (2000) ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity 12: 323–333.
48. KlugM, RehliM (2006) Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1: 127–130.
49. BrycesonYT, MarchME, LjunggrenHG, LongEO (2006) Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107: 159–166.
50. SivoriS, ParoliniS, FalcoM, MarcenaroE, BiassoniR, et al. (2000) 2B4 functions as a co-receptor in human NK cell activation. Eur J Immunol 30: 787–793.
51. WhiteDW, Suzanne BeardR, BartonES (2012) Immune modulation during latent herpesvirus infection. Immunol Rev 245: 189–208.
52. SylwesterAW, MitchellBL, EdgarJB, TaorminaC, PelteC, et al. (2005) Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202: 673–685.
53. MossP, KhanN (2004) CD8(+) T-cell immunity to cytomegalovirus. Hum Immunol 65: 456–464.
54. BironCA, NguyenKB, PienGC, CousensLP, Salazar-MatherTP (1999) Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol 17: 189–220.
55. BjorkstromNK, LindgrenT, StoltzM, FauriatC, BraunM, et al. (2011) Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med 208: 13–21.
56. PetitdemangeC, BecquartP, WauquierN, BeziatV, DebreP, et al. (2011) Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog 7: e1002268.
57. SandalovaE, LaccabueD, BoniC, TanAT, FinkK, et al. (2010) Contribution of herpesvirus specific CD8 T cells to anti-viral T cell response in humans. PLoS Pathog 6: e1001051.
58. WelshRM, CheJW, BrehmMA, SelinLK (2010) Heterologous immunity between viruses. Immunol Rev 235: 244–266.
59. TuuminenT, KekalainenE, MakelaS, Ala-HouhalaI, EnnisFA, et al. (2007) Human CD8+ T cell memory generation in Puumala hantavirus infection occurs after the acute phase and is associated with boosting of EBV-specific CD8+ memory T cells. J Immunol 179: 1988–1995.
60. BjorkstromNK, RieseP, HeutsF, AnderssonS, FauriatC, et al. (2010) Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116: 3853–3864.
61. FosterSL, HargreavesDC, MedzhitovR (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447: 972–978.
62. PathakSK, BasuS, BhattacharyyaA, PathakS, BanerjeeA, et al. (2006) TLR4-dependent NF-kappaB activation and mitogen- and stress-activated protein kinase 1-triggered phosphorylation events are central to Helicobacter pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated induction of IL-6 release from macrophages. J Immunol 177: 7950–7958.
63. LoetscherP, UguccioniM, BordoliL, BaggioliniM, MoserB, et al. (1998) CCR5 is characteristic of Th1 lymphocytes. Nature 391: 344–345.
64. SattlerA, WagnerU, RossolM, SieperJ, WuP, et al. (2009) Cytokine-induced human IFN-gamma-secreting effector-memory Th cells in chronic autoimmune inflammation. Blood 113: 1948–1956.
65. BoyleP, ClementK, GuH, SmithZD, ZillerM, et al. (2012) Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol 13: R92.
66. MartinM (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12.
67. WuTD, NacuS (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881.
68. LiuY, SiegmundKD, LairdPW, BermanBP (2012) Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol 13: R61.
69. AssenovY, MüllerF, LutsikP, WalterJ, LengauerT, et al. (2014) Comprehensive Analysis of DNA Methylation Data with RnBeads. Nature Methods [in press].
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Novel Cyclic di-GMP Effectors of the YajQ Protein Family Control Bacterial Virulence
- MicroRNAs Suppress NB Domain Genes in Tomato That Confer Resistance to
- The ESAT-6 Protein of Interacts with Beta-2-Microglobulin (β2M) Affecting Antigen Presentation Function of Macrophage
- Characterization of Uncultivable Bat Influenza Virus Using a Replicative Synthetic Virus