
Trombocytopenie u pacientů s průkazem antifosfolipidových protilátek nebo s antifosfolipidovým syndromem
Thrombocytopenia in patients with proved antiphospholipid antibodies or with antiphospholipid syndrome
Thrombocytopenia in patients with antiphospholipid antibodies (APA) is relative common finding which is found in approximately 20–40% of cases. There are likely different mechanisms resulting in thrombocytopenia in these situations. Authors summarise current knowledge of pathophysiology, clinical manifestations and management of APA-associated thrombocytopenia and other causes of decreased value of peripheral blood platelets in APA positive persons.
Key words:
thrombocytopenia – antiphospholipid antibodies – pathophysiology – treatment
Autori:
A. Buliková 1; D. Haruštiaková 2; J. Zavřelová 1; L. Dušek 2; M. Penka 1
Pôsobisko autorov:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
1; Institut biostatistiky a analýz Lékařské a Přírodovědecké fakulty MU Brno, přednosta doc. RNDr. Ladislav Dušek, CSc.
2
Vyšlo v časopise:
Vnitř Lék 2010; 56(Supplementum 1): 81-90
Kategória:
XVI. Pařízkovy dny, Ostrava-Poruba, 25.–26. březen 2010
Súhrn
Trombocytopenie u nemocných s antifosfolipidovými protilátkami (APA) je relativně častým nálezem, který je prokazován u přibližně 20–40 % případů. Existuje pravděpodobně více mechanizmů, které vedou v těchto případech k trombocytopenii. Autoři shrnují současné znalosti patofyziologie, klinické manifestace a řešení APA-asociované trombocytopenie a jiných příčin snížené hodnoty trombocytů v periferní krvi u osob s pozitivními APA.
Klíčová slova:
trombocytopenie – antifosfolipidové protilátky – patofyziologie – léčba
Úvod
Trombocyty a jejich zapojení do mechanizmů krevního srážení byly vždy považovány za důležitou součást procesů pravděpodobné patogeneze, diagnostiky a případně i léčby nemocných, u nichž byly prokázány antifosfolipidové protilátky (APA) a/nebo stanovena diagnóza antifosfopidového syndromu (APS). Již od počátečního poznání souvislostí mezi výskytem trombózy, rekurentních samovolných reprodukčních ztrát a výskytem antifosfolipidových protilátek bylo u těchto stavů poukazováno na častý výskyt trombocytopenie [1,2]. Snížený počet trombocytů byl dříve také součástí nezbytných diagnostických kritérií antifosfolipidového syndromu, ať již primárního, nebo sekundárního, v souvislosti s jinými systémovými onemocněními pojiva [3,4]. I v současné době, kdy se poznatky o možném působení antifosfolipidových autoimunitních protilátek významným způsobem obohatily a rozšířily [5], se zdá, že ovlivnění trombocytů, resp. jejich funkčních míst, působením APA může mít vliv na klinickou manifestaci APA [6]. V současné době není trombocytopenie nezbytnou součástí klinické manifestace ani součástí diagnostických kritérií APS (tab. 1) [7]. Nově je však definována jednotka tzv. trombocytopenie související s výskytem antifosfolipidových protilátek (APA asociovaná trombocytopenie), která se u prokázaných antifosfolipidových protilátek vyskytuje relativně často a je přímo vázána na jejich přítomnost, resp. klinické působení. Nicméně ovlivnění počtu anebo funkce trombocytů v systémové cirkulaci spolu s výskytem APA může nastat i z jiných důvodů, a tato skutečnost může mít různé diagnostické či terapeutické konsekvence. Jejich stručný přehled, případně i rámcové diferenciálně diagnostické možnosti, se pokusíme přiblížit v našem sdělení.
![Revidovaná kritéria antifosfolipidového syndromu (Sydney 2004) [7].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/be3f29d2b229979bb7578e0012376c60.jpeg)
Trombocyty a jejich antigeny ve vztahu k působení antifosfolipidových protilátek
Pojem antifosfolipidové protilátky (APA) je tradičně používán pro ty z autoprotilátek, které mají klinický význam u nemocných s antifosfolipidovým syndromem (APS). Nicméně správnější pojem je spíše protilátky závislé na fosfolipidech, resp. fosfolipid-dependentní, neboť již v počátku 90. let minulého století nezávisle na sobě prokázaly tři pracovní skupiny, že klinicky významné protilátky jsou namířené proti makromolekulárním strukturám vázaným na negativně nabité, nejčastěji fosfolipidové povrchy [8–10]. Za nejdůležitější z těchto struktur je považován β2-glykoprotein I (β2-GP I). Z obecně patofyziologického pohledu je možné rozlišit dva hlavní procesy působení APA – reakce protilátky s antigeny navázanými na membránu může ovlivňovat kinetiku reakcí závislých na fosfolipidech, nebo reakce protilátky s antigeny vázanými na buněčné povrchové receptory může měnit přenos signálu a aktivaci buňky [11]. V současné době je s narůstajícími poznatky o úloze buněk v procesech krevního srážení stále častěji zdůrazňována a detailněji popisována úloha buněčných membránových receptorů při působení APA na organizmus. Byly popsány receptory na nejrůznějších buněčných strukturách – endoteliích, trombocytech, na endometriálních deciduálních buňkách, na buňkách sincitium-trofoblastu, na neuronech, které pravděpodobně za účasti APA vážou β2-glykoprotein I [12–14]. Aktivací dalších signálních drah [15,16] dochází ke změně nastavení buňky, většinou je indukován nejen protrombotický, ale i proinflamatorní fenotyp [12]. V posledních letech je opakovaně zdůrazňována role komplementu v případě reprodukčních ztrát zapříčiněných antifosfolipidovými protilátkami [17,18], včetně všech důsledků takto navozeného oxidativního vzplanutí polymorfonukleárů, včetně ovlivnění metabolizmu kysličníku dusného v endotelu.
Vazba antifosfolipidových protilátek na krevní destičky a jejich následná progresivní aktivace patří k velmi důležitým a velmi častým mechanizmům působení antifosfolipidových protilátek [5,12,13]. Již od poloviny 90. let 20. století se předpokládalo, že depozice imunitních komplexů tvořených antifosfolipidovými protilátkami a β2-GP I na mírně aktivované buňky, resp. trombocyty, které již exprimují negativně nabité fosfolipidy, může vést k jejich další aktivaci, tvorbě mikropartikulí s následným rozšířením fosfolipidového povrchu a se zvýšením generace trombinu [19,20] (obr. 1). V dalším období bylo prokázáno, že vedle úlohy trombocytárního receptoru pro Fc fragment imunoglobulinu, tzv. FcγRIIa, jehož zapojení do patofyziologie působení antifosfolipidových protilátek je předpokládáno nejdéle [22], hrají roli i další struktury. K nim patří zejména receptor, který patří do skupiny receptorů pro lipoproteiny s nízkou denzitou (LDL), tzv. receptor 2‘ pro apolipoprotein E (apoER2‘), resp. i destičkový receptor Ibα [23,24]. Pro působení komplexů β2-glykoproteinu Ia proti němu namířených protilátek je nutná dimerická struktura vazby β2-glykoproteinu I [25] (obr. 1) a předchozí mírná aktivace destiček [25]. Další studie prokázaly přesné určení vazby těchto komplexů na destičkové glykoproteiny či jiné receptory, které mají za následek další aktivaci trombocytů [26,27]. Důležité je zapojení destičkového glykoproteinu Ibα (GP), což je podjednotka receptoru GPIb/IX/V, který je vazebným místem pro řadu ligandů, mezi nimi i von Willebrandova faktoru (vWF), slouží taktéž jako receptor pro přiblížení F XI a trombinu na povrchu destiček. β2-GP I se na podjednotku tohoto receptoru Ibα váže prostřednictvím svých domén II až V [27]. Jsou-li destičkové receptory – prokázáno u apoER2‘ a GPIbα – aktivovány vazbou komplexu β2-GP I a anti-β2-GP I protilátka, pak dochází k aktivaci dalších signálních drah. Tak dochází zejména k aktivaci p38 mitogen-aktivované proteinové kinázy (p38 MAPK) [28]. Tato aktivace má za následek řadu dalších dějů, zejména zvýšení produkce tromboxanu A2, nicméně jeho tvorba může být navýšena i prostřednictvím aktivace cesty fosfoinositidové-3-kinázy/Akt [13]. Tyto procesy mají navíc za následek zvýšení aktivity receptoru GPIIb/IIIa a další aktivaci destiček (obr. 2) [5].
![Působení antifosfolipidových protilátek na trombocyty.
Upraveno podle [21].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/9915aee85fac5acd54cd36b2349a8213.jpeg)
![Vliv aktivace destičkových glykoproteinů antifosfolipidovými protilátkami na další procesy. Upraveno podle [5].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0f5e940fc380a41a44c172fcb0f54bb1.jpeg)
Patofyziologie trombocytopenie v přítomnosti antifosfolipidových protilátek
I když je trombocytopenie v přítomnosti antifosfolipidových protilátek popisována řadu let [1–4], zdají se i dnes příčiny tohoto nálezu co do patofyziologického působení heterogenní. Nejpravděpodobnější příčinou trombocytopenie je různým způsobem navozená zvýšená spotřeba trombocytů. Na destičku navázané komplexy β2-GP I a anti-β2-GP I protilátka mohou být i s vlastním trombocytem odstraňovány v monocyto-makrofágovém systému, zejména ve slezině, cestou vazby Fc receptorů imunoglobulinů. Ke spotřebě trombocytů může docházet v důsledku jejich aktivace a přeměny na mikropartikule s obsahem fosfolipidů, které zvyšují protrombotickou tendenci [13]. Při vzniku trombocytopenie a současné zvýšené tendenci k trombózám mohou však být zapojeny i další mechanizmy. Bylo např. prokázáno, že za fyziologických okolností β2-GP I inhibuje agregaci trombocytů navozenou vWF. V přítomnosti anti-β2-GP I protilátek je toto inhibiční působení blokováno a u pacientů s APA bylo zjišťováno nejméně 1,5násobné navýšení aktivního vWF v cirkulaci [29]. Zvýšená hladina aktivního vWF silně koreluje s počtem trombocytů u deficitů ADAMST13, tedy u trombotické trombocytopenické purpury, resp. u von Willebrandovy choroby typu 2B [30]. Analogie se dá předpokládat i u trombocytopenie v přítomnosti antifosfolipidových protilátek.
Řadu let je známa asociace antifosfolipidových protilátek s případy, které jsou zahrnuty do diagnózy imunitní trombocytopenické purpury (ITP) [31–33]. Průkaz APA je udáván od 37,8 % [32] do 46,3 % [31] u nově diagnostikovaných případů ITP, vyšší je jejich průkaz u případů exacerbace po léčbě, kdy je nález APA popisován až 86 %, případně i u stabilizované choroby až 57 % [33]. Přestože po imunosupresivní léčbě kortikoidy počet trombocytů v těchto případech narůstá a případně dochází i k poklesu specifických antitrombocytárních protilátek, hladina APA se obvykle nemění [31,32]. Navíc po normalizaci počtu destiček může docházet v případě nálezu APA k trombotickým komplikacím [32]. Tato tendence byla prokázána i u jiných studií [34], a z tohoto důvodu je propagován termín s antifosfolipidovými protilátkami asociovaná trombocytopenie [6,34], jejíž diagnostická kritéria jsou dána mezinárodním konsenzem a jsou shrnuta v tab. 2. Trombocytopenie sama o sobě není klinickým kritériem antifosfolipidového syndromu [6,35].
![Diagnostická kritéria trombocytopenie asociované s antifosfolipidovými protilátkami. Podle [7,34].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/e74e36838b20ec9303aee88985597a0a.jpeg)
Na menší studii bylo prokázáno, že trombocytopenie spojená s primárním antifosfolipidovým syndromem (PAPS), ale i se systémovým lupus erythematodes, je provázena výskytem protilátek namířených proti destičkovým antigenům (GP IIb/IIIa, GP Ib-IX, GP Ia-IIa, GP IV) při vyšetření MAIPA (monoclonal antibody immobilization of platelet antigen) [36]. Bylo zjištěno, že protilátky proti destičkovým glykoproteinům jsou přítomny u sedmi z deseti pacientů s PAPS a s trombocytopenií, zatímco jen u jednoho nemocného s APS a normálním počtem trombocytů [36]. U případů ITP byla MAIPA pozitivní u 46 % (12/26 nemocných).
Situace může být ještě komplikovanější u případů ITP provázených ischemickou chorobou malých cév mozku (small-vessel ischemic brain disease – SVD-ITP) [37], která podobně jako případy APS může být kromě trombocytopenie provázena i recidivujícími epizodami připomínajícími tranzitorní ischemické ataky, tranzitorními poruchami kongnitivních funkcí, zvýšenou hladinou trombocytárních mikropartikulí a nálezem na magnetické rezonanci s průkazem ischemických mikrovaskulárních cévních lézí v podkorových oblastech až k obrazu sclerosis multiplex-like syndromu. Tyto stavy se z klinického pohledu zdají být neodlišitelné od klinické manifestace APS v oblasti CNS provázeného trombocytopenií, i když v etiologii SVD-ITP se předpokládá zejména aktivace trombocytů některými protidestičkovými protilátkami s následnou generací prokoagulační aktivity destičkových mikropartikulí. Tyto vlivy mohou vést k endoteliálnímu poškození a vaskulární dysfunkci cestou potencované trans-endoteliální migrace leukocytů v cévním řečišti mozku [37]. Jiné nálezy však prokazují, že k aktivaci endotelových buněk dochází jen za přítomnosti APA, a ne v případech, které jsou diagnostikovány jako izolovaná ITP [38].
Kromě případů trombocytopenie, které mohou být některými autory hodnoceny jako APA asociovaná trombocytopenie [7], zatímco jinými spíše jako ITP s výskytem antifosfolipidových protilátek [31–34], existuje řada dalších klinických situací, kdy je trombocytopenie provázena nálezem antifosfolipidových protilátek, přesto je nutno zvažovat jiné patofyziologické působení. Rámcový přehled je uveden v tab. 3.
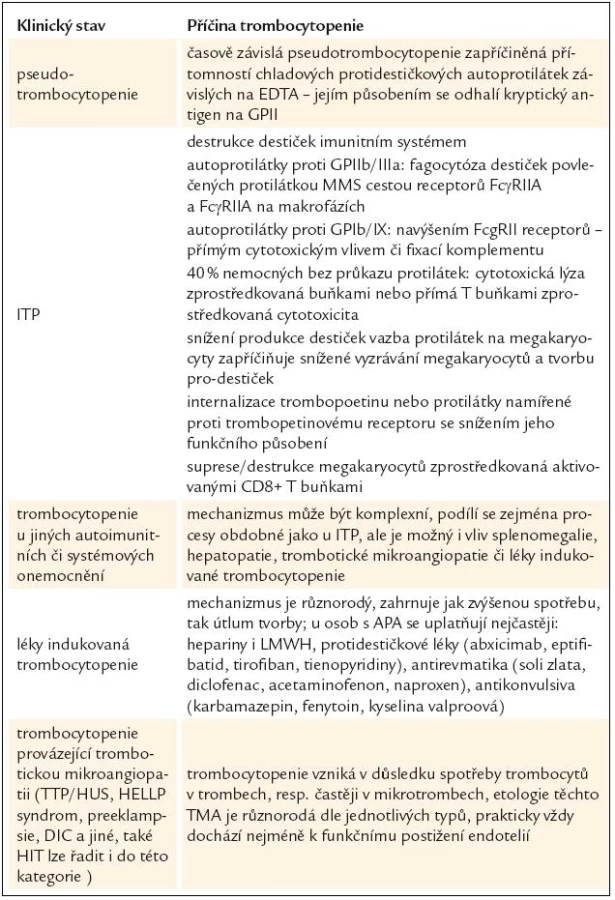
Pseudotrombocytopenie
U většiny případů pseudotrombocytopenie (mimo destičkového satelitizmu a některých makrotrombocytových trombocytopenií) se přepokládá současný vliv přítomnosti etylen-diamino-tetraoctové kyseliny (EDTA), která je ve formě nejčastěji draselné soli součástí odběrových systémů pro analýzu krevního obrazu, a dále protidestičkových protilátek působících za chladu. Působením EDTA se v membráně krevních destiček odhalí kryptogenní antigeny, na které se pak mohou vázat protilátky přítomné v plazmě vyšetřovaného jedince. To vede k aglutinaci trombocytů. Fenomén je laboratorní, při náležitém systému laboratorního provozu rozeznatelný, a zejména bez vlivu na klinický obraz nemocného. Vzhledem k tomu, že je tento jev popisován častěji u nemocných s antifosfolipidovými protilátkami [39], je nutné na něj myslet v diferenciální diagnostice APA-asociované trombocytopenie.
Imunitní trombocytopenická purpura
Celá problematika ITP a APA-asociované trombocytopenie, jejich odlišností, podobností, ale také rozmanitého pohledu různých autorů, byla nastíněna v předchozích odstavcích [31–33,36–38]. V diferenciální diagnóze nepomůže pozitivní či negativní nález protilátek včetně MAIPA. Patofyziologie ITP je však mnohem komplexnější než ta, která se přepokládá u trombocytopenie provázející antifosfolipidové protilátky. Nejenže je předpokládáno několik způsobů zvýšené degradace trombocytů, často v závislosti na cílovém antigenu, ale současně dochází ke snížené produkci z megakaryocytů [40–44]. Přehled možného působení přináší tab. 3. Praktickým řešením v diferenciální diagnostice obou stavů, a tím i předpokládaného převládajícího patogenetického mechanizmu, je dle našeho názoru rozlišení klinické; nemocní, kteří budou s větší pravděpodobností krvácet nebo již krvácejí, by měli být považováni a řešeni jako ITP.
Trombocytopenie navozená systémovým onemocněním
Ať už je trombocytopenie provázející systémové onemocnění jakékoli geneze a mohou se na tom podílet jak mechanizmy zmíněné výše, či vlivy, které budou zmíněny následovně, je trombocytopenie < 100 × 109/l s výjimkou polékových stavů považována za nezávislé diagnostické kritérium systémového lupus erythematodes (SLE). Studie zahrnující poměrně velký počet nemocných nezjistily rozdíl ve významnosti trombocytopenie mezi nemocnými se SLE, ať již nemocní měli, či neměli antifosfolipidové protilátky [45]. Podobné závěry přinesla i jiná pozorování [46]. Nicméně současná doporučení kladou důraz na to, zda je APA-asociovaná trombocytopenie provázena systémovým procesem, či nikoli [7]. Každopádně je nutno nahlížet na mechanizmus vzniku první situace jako na více komplexní proces.
Léky indukovaná trombocytopenie
Léků, které mohou vést k poklesu krevních destiček, jsou tucty a možná i stovky [47]. V tab. 3 jsme se pokusili uvést některé, které mohou být v přítomnosti antifosfolipidových protilátek či antifosfolipidového syndromu užity častěji než v jiných případech. Taktéž mechanizmy vzniku takto indukované trombocytopenie jsou poměrně heterogenní a přesahují rámec tohoto sdělení. Z toho se vymyká patofyziologie, diagnostika a diferenciální diagnostika APA-asociované trombocytopenie a heparinem indukované trombocytopenie (HIT). Oba tyto stavy mají řadu podobností, odlišností a dle současných nálezů i diagnostických a diferenciálně diagnostických problémů. O jisté asociaci nálezu obou typů protilátek i patofyziologického mechanizmu se publikuje již řadu let [19,48]. Předpokládalo se, že jistá aktivace trombocytů navozená antifosfolipidovými protilátkami může být příčinou častější indukce protilátek proti komplexu PF4-heparin, tak jak je to známo z jiných situací, zejména v případech extrakorporálního oběhu či ortopedických výkonů [49]. V současné době se zdá situace o to komplikovanější, že se ukazuje, že velká část pacientů s APS může mít falešně pozitivní testy na přítomnost HIT antigenu, resp. protilátek namířených proti komplexu PF4-heparin, které však nejsou potvrzeny ani při použití EIA (enzyme immunoassay) s nadbytkem heparinu, ani funkčními studiemi [50,51]. Z tohoto pohledu se zdá, že diagnóza HIT u nemocných s APA či APS může být „přediagnostikována“ a jistotu přináší velmi podrobná diferenciální diagnostika stavu, která může být z laboratorního pohledu poměrně komplikovaná [51–53], leč z důvodů klinických nezbytně nutná.
Trombocytopenie vznikající při trombotické mikroangiopatii (TMA)
Situace provázené trombotickou mikroangiopatií, která může být v zásadě velmi heterogenní patofyziologie (tab. 3), jsou zřetelně častěji popisovány u nemocných s antifosfolipidovými protilátkami [54]. Případy recidivující trombocytopenické mikroangiopatie jsou i v naší literatuře popisovány v souvislostí s diagnózu systémového onemocnění [55]. TMA může být vážnou, dokonce i primární klinickou manifestací antifosfolipidového syndromu a podtypy této klinické manifestace si mohou vyžádat odlišnou léčebnou strategii. Každopádně by měli být nemocní, zejména ti, u nichž nelze vysvětlit tento klinicko-laboratorní syndrom jinými důvody, vyšetřeni na přítomnost APA s ohledem zvážení protitrombotické terapie a taktéž by měl být vyloučen katastrofický antifosfolipidový syndrom [56]. Asherson et al navrhuje odlišovat pojmy „mikroangiopatický antifosfolipidový syndrom“ pro subtypy katastrofického antifosfolipidového syndromu, který se manifestuje postižením mikrocirkulace zejména s postižením střeva a u něhož se předpokládá patogenetická role APA [57], a dále „mikroangiopatický syndrom asociovaný s antifosfolipidovými protilátkami“, v jehož patogenezi hraje dominantní úlohu základní onemocnění, zatímco výskyt APA je přídatným fenoménem [58].
Klinická manifestace APA-asociované trombocytopenie
APA-asociovaná trombocytopenie je dána opakovaným průkazem poklesu trombocytů pod hladinu 100 × 109/l v časovém odstupu 12 a více týdnů, diagnostická kritéria shrnuje tab. 2 [7]. Případy takto definovaného snížení počtu destiček budou méně časté než obecně trombocytopenie provázející výskyt antifosfolipidových protilátek. Ta je přítomna zhruba u 20–40 % nemocných [34,46,59]. Trombocytopenie < 100 × 109/l se v italském registru antifosfolipidových protilátek (IR-APA; 319 osob) nacházela u 26 % nemocných, významnější – tedy < 50 × 109/l u 11 % [60]. Atsumi et al [34] vyšetřili 342 pacientů se systémovým onemocněním, u 175 z nich našli APA; trombocytopenii < 100 × 109/l pak zjistili u 22,3 % nemocných s průkazem APA, zatímco jen u 5,4 % nemocných, u nichž antifosfolipidové protilátky prokázány nebyly. V této studii byla vyloučena jiná příčina trombocytopenie (DIC, TTP, ITP či léky indukovaná trombocytopenie). Cuadradová et al [46] zjišťovala trombocytopenii u 23,4 % ze 171 nemocných, závažnou (< 50 × 109/l) jen u šesti z nich. Vyšší výskyt trombocytopenie u nemocných s APA je popisován v evropské multicentrické studii – 34 % [61]. Sami jsme trombocytopenii < 150 × 109/l zjišťovali u 29 ze 124 nemocných s nálezem APA, tj. 23,4 %, současně jsme prokázali statisticky významně nižší počet trombocytů u nemocných s průkazem některého typu antitrombocytárních protilátek, resp. s nálezem antikardiolipinů ve třídě IgG (obr. 3 a 4) [62].
![Naše výsledky vlivu antitrombocytárních protilátek na počet trombocytů u osob s APA [62].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/7dc3d14b2900abed672dd47fd83add99.jpeg)
![Počet trombocytů u nemocných s APA je ovlivněn nálezem ACLA IgG [62].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/6c940a79a7f8ec0581b036f66f646595.jpeg)
Krvácivé projevy jsou u trombocytopenie provázející výskyt APA vzácné, ve výše zmíněném IR-APA měli vážnější krvácivé projevy jen čtyři nemocní. Ve sdělení Cuadradové et al [46] nejsou žádné krvácivé projevy popisovány.
V souladu s koncepcí nálezu antifosfolipidových protilátek jako stavutrombofilního není naopak výskyt trombocytopenie „na překážku“ trombotickým či jiným projevům působení těchto autoprotilátek. Cuadradová [46] nenalezla statisticky významný rozdíl mezi výskytem arteriálních, venózních či rekurentních trombóz u nemocných s trombocytopenií či bez ní (42,5 vs 53 %, 45 vs 54 %, 62,5 vs 61 %) a rozdíl nebyl statisticky významný ani v ztrátách plodu, i když byla jistá tendence vyššího zastoupení komplikací i nemocných s trombocytopenií (57 vs 44 %). V IR-APA [60] taktéž zjistili podobný výskyt trombotických komplikací u nemocných se sníženým počtem destiček či bez něj (32 vs 40 %), i když u těžké trombocytopenie byl tento výskyt vzácný (9 %, p < 0,01). Nicméně i u významně sníženého počtu trombocytů se trombotické komplikace mohou objevit [63]. Význam nálezu antifosfolipidových protilátek ve vztahu ke klinické manifestaci APS a trombocytopenii v dlouhodobém sledování narůstá (32). U nemocných původně diagnostikovaných jako ITP se klinická manifestace APS objevila u 45,1 % nemocných trvale pozitivních na APA a trombotické komplikace nastaly u 97,7 % z nich do pěti let sledování, zatímco u nemocných s ITP bez APA „jen“ u 39,9 %. Také v našem souboru za použití analýzy hlavních komponent jsme zjistili, že snížení počtu trombocytů současně s nálezem lupus antikoagulans silně ovlivňuje výskyt žilního tromboembolizmu (obr. 5) [62].
![Vztah sníženého počtu trombocytů a klinické manifestace žilního tromboembolizmu [62].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/01c7ba966c6323a0b5d8618309636c91.jpeg)
Léčebná strategie trombocytopenie provázené výskytem antifosfolipidových protilátek
Pokud je APA-asociovaná trombocytopenie jedinou klinickou manifestací působení antifosfolipidových protilátek, pak sama o sobě v naprosté většině případů léčbu nevyžaduje [64]. Potřeba navýšení počtu trombocytů je u trombocytopenie spojená s výskytem APA zvažována podobně, jako je tomu u nemocných s ITP; tj. při počtu trombocytů mezi 20–30 × 109/l či v případě krvácivých projevů nebo v případě potřeby provedení invazivních výkonů i při vyšším počtu trombocytů. U APA-asociované trombocytopenie navíc může vyvstat potřeba zavedení antitrombotické léčby – v tomto případě by bylo zapotřebí, aby se počet destiček pohyboval mezi 30 a 50 × 109/l [65]. Spektrum požívaných léků je prakticky totožné, jaké je zavedeno pro ITP, tj. kortikosteroidy, intravenózní imunoglobulin, imunosupresiva [65]. U refrakterních případů, zejména těch s dalšími klinickými projevy autoimunitního onemocnění, lze využít i náročnějších postupů, jako je splenektomie [66], ovlivnění imunity za pomocí anti-CD20 působících monoklonálních protilátek [67,68], resp. vzácně může být přistoupeno i k léčbě využívající aplikace kmenových hemopoetických buněk [69]. V tuto chvíli nelze s jistotou vyhodnotit event. efekt agens napodobujících efekt trombopoietinu, resp. agonistů jeho receptoru, které se prokázaly být účinné jak u ITP, tak i u trombocytopenie provázející myelodysplastický syndrom či jiné případy periferních trombocytopenií [70]. Z některých studií hodnotících účinnost těchto preparátů u ITP byli totiž vyloučeni nemocní s APA protilátkami. V individuálních případech byl použit danazol [71], případně dapson [72].
Svůj význam mohou u APA-asociované trombocytopenie hrát paradoxně i léky s antitrombotickým účinkem. Mezi nimi je častější použití kyseliny acetylosalicylové, jejíž podání může snižovat protrombotické a prozánětlivé nastavení endotelu navozené APA [73], i když její role v primární profylaxi APS není s jistotou prokázána [74]. Aspirin však může cestou inhibice agregačních vlastností trombocytů vést ke snížení exprese destičkových fosfolipidů, a tím i vazby antifosfolipidových protilátek na své povrchy. Takto může zamezit mechanizmu působení antifosfolipidových protilátek [75], a tím i trombocytopenii. Sami jsme tento efekt opakovaně pozorovali při běžném podávání kyseliny acetylosalicylové u lehké či středně významné trombocytopenie. Jiným lékem s potenciálně antitrombotickým působením jsou antimalarika, která ruší expresi membránových glykoproteinů trombocytů (GP IIb/IIIa) navozenou antifosfolipidovými protilátkami [5,76]. I jejich podání vedlo v ojedinělých případech i k úpravě trombocytopenie [77]. V případech, kde je trombocytopenie u APA spojená s možným výskytem HIT, jsou obvykle užívány léky s antitrombotickým působením, avšak bez obsahu i nízkomolekulárních heparinů [78,79]. U APS spojených s trombotickou mikroangiopatií, u nichž vzniká trombocytopenie komplexním působením, se v léčbě využívají obvykle steroidy, výměnná plazmaferéza a intravenózní imunoglobuliny [54,56].
Závěr
Trombocytopenie spojená s výskytem antifosfolipidových protilátek je relativně častým nálezem. Většina těchto případů neovlivňuje ani klinický obraz, ani základní léčebný přístup u těchto nemocných. Průkaz sníženého počtu krevních destiček by však měl vést ošetřujícího lékaře k úvaze nad možným patofyziologickým mechanizmem, neboť tento může být více komplexní než jen vliv přítomnosti APA. To pak umožňuje cílenou terapii v situacích, kdy je léčebné ovlivnění nezbytné.
MUDr. Alena Buliková, Ph.D.
www.fnbrno.cz
e-mail: abulik @fnbrno.cz
Doručeno do redakce: 24. 3. 2010
Zdroje
1. Harris EN, Asherson RA, Gharavi AE et al. Thrombocytopenia in SLE and related autoimmune disorders: Association with anticardiolipin antibodies. Br J Haematol 1985; 59: 227–230.
2. Harris EN, Cahn JKH, Asherson RA et al. Thrombosis, recurrent fetal loss, and thrombocytopenia. Arch Intern Med 1986; 146: 2153–2156.
3. Asherson RA, Khamashta M, Ordi-Ros J. The “primary” antiphospholipid syndrome. Major clinical and serological features. Medicine 1989; 68: 366–375.
4. Alacrón-Segovia D, Pérez-Vézquez ME, Villa AR et al. Preliminary classification criteria for the antiphospholipid syndrome within systemic lupus erythematosus. Sem Arthr Rheumat 1992; 21: 275–285.
5. Pierangeli SS, Chen PP, Raschi E et al. Antiphospholipid antibodies and the antiphospholipid syndrome: pathogenetic mechanisms. Semin Throm Hemost 2008; 34: 236–250.
6. Pennings MTT, Derksen RHWM, van Lummel M et al. Platelet adhesion to dimeric β2-glycoprotein I under conditions of flow is mediated by at least two receptors: glycoprotein Ibα and apoliporotein E receptor 2’. J Thromb Haemost 2007; 5: 369–377.
7. Miyakis S, Lockshin MD, Atsumi T et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4: 295–306.
8. McNeil HP, Simpson RJ, Chesterman CN et al. Anti-phospholipid antibodies are directed a complex antigen that includes a lipid-binding inhibitor of coagulation. Proc Natl Acad Sci 1990; 87: 4120–4124.
9. Galli M, Comfurius P, Maassen C et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990: 335: 1544–1547.
10. Matsuura E, Igarashi Y, Fujimoto M et al. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet 1990; 336: 177–178.
11. Roubey RAS. Mechanisms of autoantibody-mediated thrombosis. Lupus 1998; 7: 114–119.
12. Pierangeli SS, Chen PP, Gonzalez EB. Antiphospholipid antibodies and the antiphospholipid syndrome: an update on treatment and pathogenic mechanisms. Curr Opin in Hematology 2006; 13: 366–375.
13. Giannakopoulos B, Passam F, Rahgozar S et al. Current concepts on the pathogenesis of the antiphospholipd syndrome. Blood 2007, 109; 422–430.
14. Meroni PL. Pathogenesis of the antiphospholipid syndrome: An additional example of the mosaic of autoimmunity. Blod 2008; 30: 99–103.
15. Raschi E, Testoni C, Bosisio D et al. Role of the MyD66 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 2003; 101: 3495–3500.
16. Vega-Ostertag M, Casper K, Swerlick Ret al. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheumat 2005; 52: 1545–1554.
17. Holers VM, Girardi G, Mo L et al. C3 activation is required for anti-phospholipid antibody-induced fetal loss. J Ex Med 2002; 1995: 211–220.
18. Redecha P, Tilley R, Tencati M et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blod 2007; 110: 2423–2431.
19. Arnout J. The pathogenesis of the antiphospholipid syndrome: a hypothesis based on parallelisms with heparin-induces thrombocytopenia. Thromb Haemost 1996; 75: 536–541.
20. Vermylen J, Hoylaerts MF, Arnout J. Antibody-mediated thrombosis. Thromb Haemost 1997; 78: 420–426.
21. Arnout J, Vermylen J. Current status and implication of autoimmune antiphospholipid antibodies in relation to thrombotic disease. J Throm Haemost 2003; 1: 931–942.
22. Arnout J. The pathogenesis of the antiphospholipid syndrome: a hypothesis based on parallelisms with heparin-induced thrombocytopenia. Thromb Haemost 1996; 78: 420–426.
23. Pennings MTT, Derksen RHWM, van Lummer M et al. Platelet adhesion to dimeric β2-glycoprotein I under conditions of flow is mediated by at least two receptors: glycoprotein Ibα and apoliprotein E receptor 2‘. J Throm Haemost 2007; 5: 369–377.
24. Urbanus RT, Pennings MTT, Derksen RHWM et al. Platelet activation by dimeric β2-glycoprotein I requires signaling via both glycoprotein Ibα and apolipoprotein E receptor 2’. J Thromb Haemost 2008; 6: 1405–1412.
25. Lutters BCH, Derksen RHWM, Tekelenburg WL et al. Dimers of β2-Glykoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2’. J Biolog Chem 2003; 278: 33831–33838.
26. van Lummel M, Pennings MTT, Derksen RHWM et al. The binding site in β2-glycoprotein I for ApoER2’ on platelet is located in domain V. J Biological Chem 2005; 280: 36729–36736.
27. Shi T, Giannakopoulos B, Yan X et al. Anti-β2-glykoprotein I antibodies in complex with – β2-glykoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Artritis Rheumat 2006; 54: 2558–2567.
28. Vega-Ostertak ME, Ferrata DE, Romay-Penabad Z et al. Role of p38 mitogen-activated protein kinase in antiphospholipid antibody-mediated thrombosis and endothelial cell activation. J Thromb Haemost 2007; 5: 1828–1834.
29. Hulstein JJJ, Lenting PJ, de Laat B et al. β2-glycoprotein I inhibits von Willebrand factor-dependent platelet adhesion and aggregation. Blood 2007; 110: 1483–1491.
30. Hulstein JJJ, de Groot PG, Silence K at al. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood 2005; 106: 3035–3042.
31. Stasi R, Stipa E, Masi M et al. Prevalence and clinical significance of elevated antiphospholipid antibodies in patients with idiopatic thrombocytopenic purpura. Blood 1994; 84: 4203–4208.
32. Diz-Kücükkaya R, Hacihanefioglu A, Yenerel M et al. Antiphospholipid antibodies and antiphospholipid syndome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood 2001; 98: 1760–1764.
33. Bidot CJ, Jy W, Horstman LJ et al. Antiphosholipid antibodies in immune thrombocytopenic purpura tend to emerge in exacerbation and decline in remission. British J Haematol 2004; 128: 366–372.
34. Atsumi T, Furukawa S, Amengual O et al. Antiphospholipid antipody associated thromocytopenie and the paradoxical risk of thrombosis. Lupus 2006; 14: 499–504.
35. Wilson WA, Gharavi AE, Koike T et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheumat 1999; 42: 1309–1311.
36. Macchi L, Rispal P, Clofent-Sanchez G et al. Anti-platelet antibodies in patients with systemic lupus erythematosus and primary antiphopholipid antibody syndrome: their relationship with observed thromocytopenia. British J Haematol 1997; 98: 336–341.
37. Jimenez JJ, Jy W, Mauro LM et al. Transendothelial migration of leukocytes is promoted by plasma from a subgroup if immune thrombocytopenic purpura patients with small-vessel ischemic brain disease. Am J Hematol 2008; 83: 206–211.
38. Dunoyer-Geindre S, Bochlen F, Favier R et al. Endothelial cell activation by immunuglobulins from patients with immune thromobcytopenic purpura or with antiphopholipid syndrome. Haematologica 2008; 84: 635–636.
39. Bizzato N, Brandaliase M. EDTA-dependent pseudothrombocytopenia. Association with antiplatelet and antiphospholipid antibodies. Am J Clin Patol 1995; 103; 103–107.
40. Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002; 346: 995–1008.
41. Go RS, Johnston KL, Bruden KC. The association between platelet autoantibody specificity and response to intravenous immunoglobulin G in the treatment of patients with immune thromobcytopenie. Haematologica 2007; 92: 283–284.
42. Ollsson B, Andersson PD, Jernas M et al. T-cell mediated lysis of autologous platelets in chronic thromocytopenic purpura. Nat Med 2003; 9: 1123–1124.
43. Zang F, Chu X, Wang L et al. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol 2006; 76: 427–431.
44. McMillian R, Wang L, Tomer A et al. Suppression of in vitro megakaryocyte production by antiplatetet autoantibodies from adult patients with chronic ITP. Blood 2004; 103: 1364–1369.
45. Cervera R, Khamashta MS, Font J et al. Systemic lupus erythematosus: Clinical and immunological patterns of disease expression in a cohort of 1000 patients. Medicine 1993; 72: 113–124.
46. Cuadrado MJ, Mujic F, Muňoz E et al. Thrombocytopenia in the antiphospholipid syndrome. Ann Rheum Dis 1997; 56: 194–196.
47. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl Med 2007; 357: 580–587.
48. Gruel Y, Rupin A, Watier H et al. Anticardiopipin antibodies in heparin-associated thrombocytopenia. Thromb Res 1992; 67: 601–606.
49. Socher I, Kroll H, Jorsk S et al. Heparin-independent activation of platelet by heparin-induced thrombocytopenia antibodies: a common occurrence. J Thromb Haemost 2008; 6: 197–200.
50. Pauzner R, Greinacher A, Selleng K et al. False-positive tests for heparin-induced thrombocytopenia in patients with antiphospholipid syndrome and systemic lupus erythematosus. J Tromb Haemost 2009; 7: 1070–1074.
51. Martion-Toutain I, Piette JC, Diewmert MC et al. High prevalence of antibodies to platelet factor 4 heparin in patients with antiphospholipid antibodies in absence of heparin induced thromocytopenia. Lupus 2007; 16: 79–83.
52. Lo GK, Sigouin ChS, Warkentin TE. What is the potential overdiagnosis of heparin-induced thrombocytopenia. Am J Hematol 2007; 82: 1037–1043.
53. Warkentin TE. Antiphospholipid and anti-PF4 antibodies: an association affecting anti-PF4/heparin assay analysis. J Thromb Haemost 2009; 7: 1067–1069.
54. Espinosa G, Bucciarelli S, Cervera R et al. Thrombotic microangiopathic haemolytic anaemia and antiphospholipid antibodies. Ann Rheum Dis 2004; 63: 730–736.
55. Fojtík Z, Kořístek Z, Klabussay M et al. Recidivující trombotická trombocytopenická purpura u nemocné se systémovým lupus erythermatodes. Čes Revmatol 2003; 11: 157–160.
56. Cervera R, Font J, Gómez-Puerta JA et al. Validation of the preliminary criteria for the classification of catastrophic antiphosholipid syndrome. Ann Rheum Dis 2005; 64: 1205–1209.
57. Asherson RA, Pierangeli S, Cervera R. Is there a microangiopathic antiphospholipid syndrome? Ann Rheum Dis 2007; 66: 429–432.
58. Asherson RA, Pierangeli S, Cervera R. Microangiopathic antiphospholipid-associated syndromes revisited – new concepts relating to antiphospholipid antibodies and syndromes. J Rheumatol 2007; 34: 1793–1795.
59. Galli M, Finazzi G, Barbui T. Thromocytopenia in the antiphospholipid syndrome. Br J Haematol 1996; 93: 1–5.
60. Finazzi G. The Italian registry od antiphospholipid antibodies. Haematologica 1997; 82: 101–105.
61. Vianna JL, Khamashta MA, Ordi-Ros J et al. Comparison of the primary and secondary antiphospholipid syndrome: European multicentre study of 114 patients. Am J Med 1994; 96: 3–9.
62. Buliková A, Haruštiaková D, Zavřelová J et al. Long-term follow-up in patients with positive antiphospholipid antibodies – single centre experience. J Thromb Haemost 2009; Suppl. 2: PP–MO–259.
63. Leuzzi RA, Davis GH, Cowchock FS et al. Management of immune thromobocytopenic purpura associated with the antiphospholipid antibody syndrome. Clin Exp Rhematol 1997; 15: 197–200.
64. Galli M, Barbui T. Management of thrombocytopenia in Hughes syndrome. In: Khamashta MA (Ed.) Hughes syndrome. London: Springer Verlag 2000; 408–418.
65. Lim W. Antiphospholipid syndrome. Hematology (Am Soc Hematol Edu Program) 2009: 233–239.
66. Galindo M, Khamashta MA, Hughes GRV. Splenectomy for refractory thrombocytopenia in the antiphospholipid syndrome. Rheumatology 1999; 38: 848–853.
67. Garvey B. Rituximab in the treatment of autoimmune haematological disorders. Br J Heaematol 2008; 141: 149–169.
68. Ruckert A, Glimm H, Lubbert M et al. Successful treatment of life-threatening Evans syndrome due to antiphospholipid antibody syndrome by rituximab-based regimen: a case with long-term follow-up. Lupus 2008; 17: 757–760.
69. Snowden JA, Martin-Rendon E, Watt SM. Clinical stem cell therapies for severe autoimmune diseases. Transfusion medicine 2009; 19: 223–234.
70. Orita T, Tsunoda H, Yabuta N et al. A novel therapeutic approach for thrombocytopenia by miniantibody agonist of the thrombopeitein receptor. Blood 2005; 105: 562–566.
71. Kavanaugh A. Danazol therapy in throm-bocytopenia associated with the antiphospholipid antibody syndrome. Ann Int Med 1994; 121: 767–768.
72. Durand JM, Lefevre P, Kaplanski G et al. Correction of thromocytopenia with dapson in the primary antiphospholipid syndrome. J Rheumatol 1993; 20: 1777–1778.
73. Dunoyer-Geindre S, Kruithof EKO, Boehledn F et al. Aspirin inhibits endothelial cell activation induced by antiphospholipid antibodies. J Throm Haemost 2004; 2: 1176–1181.
74. Erkan D, Harrison MJ, Levy R et al. Aspirin for primary thrombosis prevention in the antiphospholipid syndrome. Arthritis and rheumatism 2007; 56: 2382–2391.
75. Alliot C, Messouak D, Albert F. Correction of thromocytopenia with aspirin in the primary antiphospholipid syndrome. Am J Hematol 2001; 68: 215–217.
76. Rand JH, Wu X-X, Quinn AS et al. Hydrochloroquine directly reduces the binding of antiphospholipid antibody-β2-glycoprotein I complex to phospholipid bilayers. Blood 2008; 112: 1687–1695.
77. Suarez IM, Diaz RA, Aguayo CD et al. Correction of severe thrombocytopenia with choroquine in the primary antiphospholipid syndrome. Lupus 1996; 25: 81–83.
78. Hotlan SG, Knox SK, Terreri A. Use of fondaparinux in patient with antiphospholipid syndrome and heparin-associated thrombocytopenia. J Thromb Haemost 2006; 4: 1632–1634.
79. Athar U, Husain J, Hudson J et al. Prolonged half-life od argatroban in patients with renal dysfunction and antiphospholipid antibody syndrome being treated for heparin-induced thrombocytopenia. Am J Hematol 2007; 83: 245–246.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2010 Číslo Supplementum 1
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Intermitentní hladovění v prevenci a léčbě chorob
- Statiny indukovaná myopatie: Jak na diferenciální diagnostiku?
- Nech brouka žít… Ať žije astma!
Najčítanejšie v tomto čísle
- Trombocytopenie a koagulopatie u hepatopatie: úvod do problematiky
- Monitorace parametrů koagulace a možnosti jejich ovlivnění u pacientů s jaterní cirhózou před invazivními výkony
- Diferenciální diagnostika trombocytopenie v těhotenství
- Využití parametru IPF (Immature platelet fraction) v laboratorní diagnostice