
PET-CT dokumentovaná kompletní 4letá remise Erdheimovy-Chesterovy nemoci po léčbě kladribinem
PET-CT documented complete remission of Erdheim-Chester disease, lasting more than 4 years from treatment initiation with cladribine
Erdheim-Chester disease is a very rare histiocytic disease. It represents one form of juvenile xanthogranuloma in WHO classification of blood diseases. The disease often causes B symptoms, skeletal pain and also may cause diabetes insipidus and retroperitoneal fibrosis. Selection of therapy depends on published case reports and small clinical trials. There are no recommendations for treatment based on randomized studies. Interferon α is probably the most commonly used drug for this disease. Some remissions have been described after treatment. However, long-term interferon α application is needed which is associated with numerous side effects. There are limited experiences with clabridine in this indication. In Pubmed Medline database, we have found 3 publications dedicated to description of treatment response after cladribine in Erdheim-Chester disease and other 7 papers evaluating effect of cladribine on juvenile xanthogranuloma forms, mostly with positive outcome. Based on these 10 publications we choose cladribine as first-line treatment in our patient. The treatment started in October 2009 with combination of 2-chlorodeoxyadenosine (Litak) 5 mg/m2 sc. + cyclophosphamide 150 mg/m2 iv. + dexamethasone 24 mg iv., five days consecutively. These cycles were repeated monthly. Mentioned formula was submitted 4 times and 3 times in limited application on day 1 – 3. The reason of that was neutropenia grade 3. All symptoms disappeared after treatment. Only diabetes insipidus persisted because damage of pituitary stalk is irreversible. Therapeutic effect was monitored by PET-CT imaging, initially every 6 months, later in 12-month intervals. PET-CT imaging showed complete remission of disease and 4.5 years duration of remission after treatment. The treatment was well tolerated with no complications implying hospitalization. Only mild thrombocytopenia and neutropenia remains after 4.5 years. Based on case report and publications we consider cladribine as appropriate firs-line drug for Erdheim-Chester disease. Therapeutic failure after 3–4 cycles may suggest other options (interferon α, anakinra, vemurafenib), but only in the case if healthcare provider is willing to cover this new and more expansive treatment than therapy with cladribine.
Key words:
anakinra – cladribine – Erdheim-Chester disease –interferon α – juvenile xanthogranuloma – PET-CT in diagnosis of fever of unknown origin – vemurafenib – 2-chlorodeoxydenosin
Autori:
Zdeněk Adam 1; Zdeněk Řehák 2; Renata Koukalová 2; Zbyněk Bortlíček 5; Marta Krejčí 1; Luděk Pour 1; Petr Szturz 1; Jiří Prášek 3; Tomáš Nebeský 1; Zdenka Adamová 6; Zdeněk Král 1; Jiří Mayer 1
Pôsobisko autorov:
Interní hematologická a onkologická klinika LF MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Oddělení nukleární medicíny, PET centrum Masarykova onkologického ústavu Brno, přednosta prim. MUDr. Zdeněk Řehák, Ph. D.
2; Klinika nukleární medicíny LF MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Jiří Prášek, CSc.
3; Radiologická klinika LF MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil A. Válek, CSc., MBA
4; Institut biostatisky a analýz LF MU Brno, ředitel doc. RNDr. Ladislav Dušek, Ph. D.
5; Soukromá ordinace praktického lékaře pro děti a dorost, Obilní trh 9, Brno
6
Vyšlo v časopise:
Vnitř Lék 2014; 60(5-6): 499-511
Kategória:
Kazuistika
Súhrn
Erdheimova-Chesterova nemoc je onemocnění patřící mezi velmi vzácné histiocytární choroby. Představuje jednu z forem chorob, řazených poslední WHO klasifikací krevních chorob do skupiny juvenilního xantogranulomu. Nemoc často působí B symptomy, dále bolesti skeletu a může způsobit i diabetes insipidus i fibrózu retroperitonea. Výběr léčebných postupů se odvíjí od publikovaných popisů případů či malých skupin pacientů. Pro tuto nemoc neexistuje léčebné doporučení založené na randomizovaných studiích. Interferon α je zřejmě nejčastěji používaný lék pro tuto nemoc, byly po něm popsány remise. Je však nutná dlouhodobá aplikace interferonu α a ta je spojena s četnými nežádoucími účinky. Zkušeností s kladribinem v této indikaci je o něco méně. V databázi Medline Pubmed jsme nalezli 3 publikace popisující léčebnou odezvu po kladribinu u Erdheimovy-Chesterovy nemoci a 7 dalších publikací, hodnotících účinek kladribinu u některé z forem juvenilního xantogranulomu, převážně s pozitivním výsledkem. Na základě těchto 10 publikací jsme zvolili pro popisovaného pacienta kladribin jako léčbu první linie. Léčba byla zahájena v říjnu roku 2009 kombinací 2-chlorodeoxyadenozin (Litac) 5 mg/m2 s. c. + cyklofosfamid 150 mg/m2 i. v. + dexametazon 24 mg i.v., vše 5 dní po sobě. Opakování těchto cyklů bylo plánováno v měsíčních intervalech. Chemoterapii jsme podali 4krát ve výše uvedeném složení a 3krát s aplikací omezenou pouze na 1.–3. den. Důvodem pro redukci byla neutropenie grade III. Po ukončení léčby vymizely všechny symptomy nemoci, přetrvává pouze diabetes insipidus, protože poškození stopky hypofýzy touto chorobou je ireverzibilní. Účinek léčby byl sledován PET-CT zobrazením, které bylo prováděno zpočátku v 6měsíčních intervalech, později v 12měsíčních intervalech. PET-CT prokázalo dosažení kompletní remise nemoci a trvání této remise po 4,5 letech od zahájení léčby. Léčba byla dobře tolerována, v průběhu léčby se nevyskytly žádné komplikace, vynucující si hospitalizaci. Po 4,5 letech od ukončení léčby přetrvává mírná trombocytopenie a neutropenie. Na základě popsaného případu a citované literatury považujeme kladribin za vhodný lék první linie pro léčbu Erdheimovy-Chesterovy choroby. Teprve při nedosažení léčebné odezvy po 3–4 cyklech kladribinu je možné otestovat další léčebné postupy (interferon α, anakinra, vemurafenib), pokud ale plátce zdravotní péče bude ochoten hradit tuto novou, ale podstatně dražší léčbu, než je kladribin.
Klíčová slova:
anakinra – Erdheimova-Chesterova choroba – interferon α – juvenilní xantogranulom – kladribin – PET-CT diagnostika při horečce nejasného původu – vemurafenib – 2-chlorodeoxydenosin
Úvod
Krev a krvetvorná tkáň je tvořena buňkami myeloidní, lymfoidní a histiocytární řady. Transformací buněk uvedených linií vznikají maligní onemocnění. Z nejasného důvodu je podstatně nižší výskyt (incidence) maligních chorob odvozených z buněk histiocytární linie než výskyt maligních nemocí odvozených z buněk myeloidní či lymfoidní řady. Také počet histopatologických jednotek je podstatně nižší ve skupině maligních histiocytárních chorob než ve skupině myeloidních či lymfoidních neoplazií. Přehled všech maligních chorob odvozených od histiocytárních buněk ukazuje tab. 1.

Teprve v roce 2008 byla do kategorie maligních histiocytárních nemocí jednoznačně přiřazena skupina chorob, pro něž je typická proliferace histiocytů podobných těm, které jsou obsaženy v kožních xantogranulomech, obvykle mají pěnitou (xantomatózní) komponentu a v jejich okolí jsou obrovské buňky Teutonova typu.
Jednoznačně nejčastějším typem je solitární kožní xantogranulom, který nikdy neprogreduje a nepřechází do diseminované formy. Ložiska tvořená proliferujícími pěnitými histiocyty se však mohou vytvořit v kterémkoliv orgánu a tkáni a poškodit tak svého nositele. Tyto tzv. hluboké (viscerální), solitární či diseminované formy, se objevují ve věku do 10 let a z toho snad polovina v prvním půl roce života. Výjimkou je pouze generalizovaná forma také patřící do skupiny juvenilního xantogranulomu, která se objevuje pouze v dospělosti – Erdheimova-Chesterova choroba [1].
V dospělosti je možné výjimečně se setkat s plošným xantomem (xanthoma planum), s xantogranulomem či nekrobiotickým xantogranulomem, který bývá často v oblasti víček a očnice, ale může být přítomen i kdekoliv jinde. Z nejasného důvodu u dospělých osob s xanthoma planum či s nekrobiotickým xantogranulomem bývá přítomný monoklonální imunoglobulin. Zatím nebyla prokázána příčinná souvislost mezi monoklonálním imunoglobulinem a plošným xantomem či xantogranulomem dospělých [1], přesto jsou tyto nozologické jednotky popisovány v některých monografiích o monoklonálních gamapatiích. Domníváme se, že však na rozdíl od IgA pemfigu či skléredému, kde je poměrně jasně prokázána kauzální souvislost s monoklonálním imunoglobulinem, v případě plošného xantomu či xantogranulomu tomu tak není. Stejně tak není ale souvislost mezi metabolizmem lipidů a plošnými xantomy či xantogranulomy, i když v proliferujících histiocytárních buňkách se akumulují tukové inkluze, tak nejde o důsledek vyšší hladiny tuků a tedy střádací chorobu, ale jde o proliferaci fagocytujících histiocytárních buněk, z nejasných důvodů provázenou někdy přítomností monoklonálního imunoglobulinu.
Erdheimova-Chesterova nemoc je velmi vzácná. První popis zveřejnili roku 1930 vídeňský patolog Jakob Erdheim a jeho americký kolega patolog William Chester a chorobu nazvali lipid granulomatosis [2]. V následujících letech bylo v odborné literatuře popsáno jen několik stovek případů, přičemž většina popisů je z posledních 10 let.
Základním projevem Erdheimovy-Chesterovy nemoci je masivní infiltrace kostní dřeně dlouhých kostí pěnitými histiocyty. Důsledkem je zvýšená tvorba hydroxyapatitu v těchto kostech, vedoucí k zesílení jejich struktury (k osteoskleróze), což může být paradoxně provázeno úbytkem hydroxyapatitové struktury v obratlích nepostižených touto chorobou. Infiltrace dlouhých kostí vyvolává intenzivní bolest. V ostatních projevech nemoci se již jednotliví pacienti liší. Nemoc může postihnout velké cévy a vést k zesílení jejich stěny, může způsobit periaortální fibrózu. Fibróza v okolí ledvin a v retroperitoneu může oblenit tok uretery a připomínat tak idiopatickou retroperitonální fibrózu (Ormondovu chorobu).
Masivní infiltrace kostní dřeně dlouhých kostí nezpůsobuje anémii či cytopenii obecně, protože v dospělém věku je již v těchto dlouhých kostech krvetvorná kostní dřen fyziologicky nahrazena tukovou, krvetvorná kostní dřen zůstává v plochých kostech a obratlích, které nebývají infiltrovány těmito patologickými histiocyty. Nemoc se může přihlásit B symptomy (subfebrilie či febrilie, úbytek hmotnosti, noční pocení). Nemoc lze snadno diagnostikovat, pokud se postupuje dle doporučení pro diagnostiku teploty nejasného původu a provede se celotělové PET-CT s požadavkem, aby byly zobrazeny celé dlouhé končetiny a cílený odběr k histologickému vyšetření z místa nejvyšší akumulace fluorodeoxyglukózy [1–16].
Erdheimova-Chesterova nemoc byla již v českém a slovenském písemnictví popsána [17–25]. Erdheimovu-Chesterovu nemoc je třeba zásadně odlišit od histiocytózy z Langerhansových buněk, neboť průběh Erdheimovy-Chesterovy nemoci je podstatně méně příznivý než je průběh histiocytózy z Langerhansových buněk [26–29]. Podrobněji o patologii non Langerhans cell histiocytóz pojednává článek prof. Planka [30].
Osud nemocných s Erdheimovou-Chesterovou chorobou je velmi individuální a odpovídá stupni poškození organizmu, nezřídka byl popisován fatální průběh. Údajů o prognóze je málo, největší soubor zveřejnil Veyssier Belot, který uvádí, že z 37 nemocných 22 zemřelo v průběhu 2,7letého sledování. Nejčastějšími příčinami úmrtí je plicní fibróza s dušností a srdeční selhání. Neurologické postižení může způsobovat ataxii či parézy, poruchu kognitivních funkcí a vést k postupnému zhoršování funkce CNS [31].
Pro léčbu této nemoci nebyla zatím publikována žádná doporučení, ošetřující lékaři se musí rozhodovat na základě publikací popisujících menší soubory nemocných či jednotlivé případy. Proto chceme upozornit na vynikající efekt 2-chlorodexyadenozinu, potvrzený PET-CT monitorováním.
Popis případu
Anamnéza a výsledky laboratorních vyšetření
Muž, narozený roku 1953, se až do roku 2000 těšil ze svého života při plném zdraví, bez vážnějších chorob. V roce 2001 jej začala trápit polyurie. Endokrinologové v roce 2001 potvrdili diabetes insipidus a podali odpovídající substituční léčbu. V roce 2004 jej začaly trápit undulující horečky s třesavkou, pocit zimnice, trvající vždy po několik dní, pak se teplota spontánně normalizovala a ale jen na několik dní, než opět vzestoupila.
Na odborné pracoviště byl odeslán poprvé v roce 2005 s podezřením na maligní lymfom. Byla provedena následující vyšetření: CT mediastina a plic bez patologie, CT břicha bez lymfadenopatie, trepanobiopsie lopaty kosti kyčelní bez patologie, celotělová PET: difuzně vyšší akumulace fluorodeoxyglukózy v obou femurech. Dále podstoupil pacient endoskopická vyšetření (gastroskopii, kolonoskopii, echokardiografii), vše bez patologického nálezu.
Pacient měl normální hodnoty krevního obrazu a normální biochemické vyšetření, vyjma hodnoty zvýšené hodnoty CRP (63 mg/l) a zvýšené hodnoty LD na 6,9 µkat/l (norma do 3,75 µkat/l). Také hodnota sedimentace byla zvýšená (52/72). Závěr vyšetřujícího lékaře byl: „Maligní lymfom nebyl dostupnými vyšetřeními prokázán, a tedy protinádorová léčba není indikována”, a tímto stručným závěrem byl pacient odeslán zpět do péče endokrinologa.
V roce 2009 byl pacient odeslán na Interní hematologickou a onkologickou kliniku, na ambulanci pro monoklonální gamapatie a vzácné nemoci. Pořád jej trápily intermitentní horečky kolem 38 °C, které trvaly vždy několik dní, a pak spontánně ustaly. Oproti roku 2005 však přibyly v roce 2009 neurčité bolesti v bércích, kolenou, kotnících a kyčlích. Bolesti postihovaly obě končetiny stejně, na vizuální analogové škále intenzity bolesti byly pacientem hodnoty jako 4/10. Bolesti byly lokalizované, tupého charakteru, nejevily žádnou návaznost na tělesnou námahu a objevovaly se v záchvatech 1–2krát denně. Objektivní nález na pacientovi byl zcela normální. V roce 2009 však již měl podstatně vyšší hodnotu CRP než v roce 2005, opakovaně se pohybovala kolem 80–16 mg/l. Jinak v laboratorních výsledcích nebylo nic nápadného, monoklonální imunoglobulin nebyl prokazatelný.
V roce 2005 ponechal vyšetřující lékař bez interpretace a dalšího pátrání vyšší akumulaci fluorodeoxyglukózy v oblasti femorů na PET vyšetření. To bylo totiž jasným signálem přítomnosti Erdheimovy-Chesterovy nemoci. Pro vysvětlení připomeneme, že při klasickém vyšetření PET se snímá člověk od lební baze do půli stehen. Pouze pokud lékař, odesílající nemocného k vyšetření, požádá o vyšetření celé postavy, tak se snímají i bérce. První PET díky tomu, že nebyly snímány celé dolní končetiny, tak nemohla přinést typický obraz zvýšení aktivity v dolních končetinách.
Zobrazení skeletu pomocí PET-CT, scintigrafie skeletu a celotělového MRI skeletu
Vzhledem k nejasným potížím a neobjasněné zvýšené aktivitě v oblasti femorů dle PET-CT vyšetření z roku 2005 jsme v srpnu roku 2009 provedli další zobrazení celého skeletu, již včetně bérců a chodidel (PET-CT vyšetření, scintigrafii skeletu Tc pyrofosfátem, celotělové MR zobrazení skeletu). Stručně shrneme výsledky těchto 3 zobrazení skeletu.
Scintigrafie skeletu ze 7. 7. 2009
Radioaktivní techneciumpyrofosfát byl zvýšeně vychytáván v oblasti pravého zygomatického oblouku, v pravé frontální oblasti, v mandibule mediálně, ve střední části levého humeru, v obou radiích proximálně, v obou femorech v proximální třetině a v distální polovině (vynechána prostřední třetina femorů),v obou tibiích a dále v talu a kalkaneu vlevo. Místa zvýšeného vychytávání techneciumpyrofosfátu se shodují s místy maximální akumulace fluorodeoxyglukózy.
Výsledky vstupního vyšetření dokumentuje obr. 1 a 2.
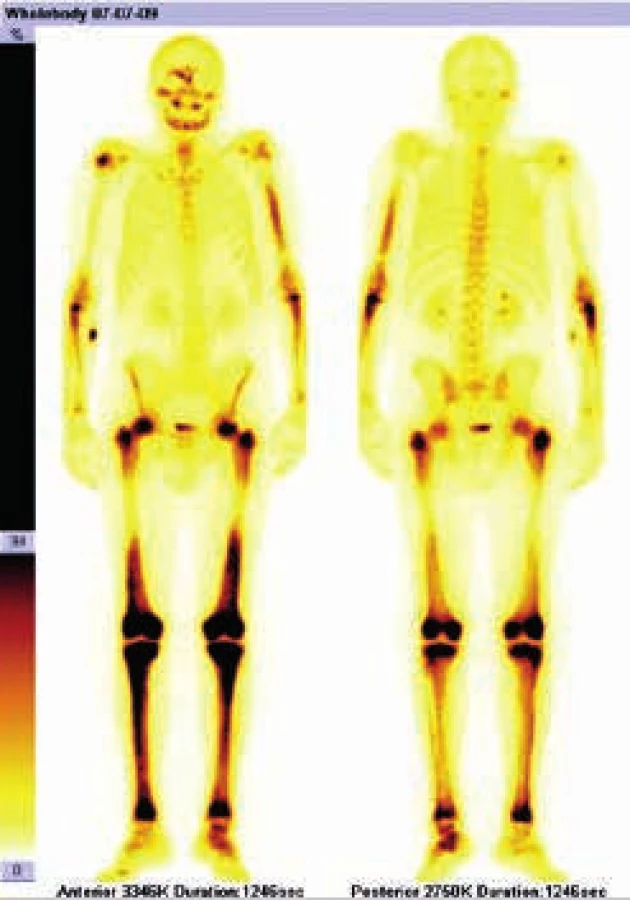
PET-CT vyšetření z 11. 8. 2009
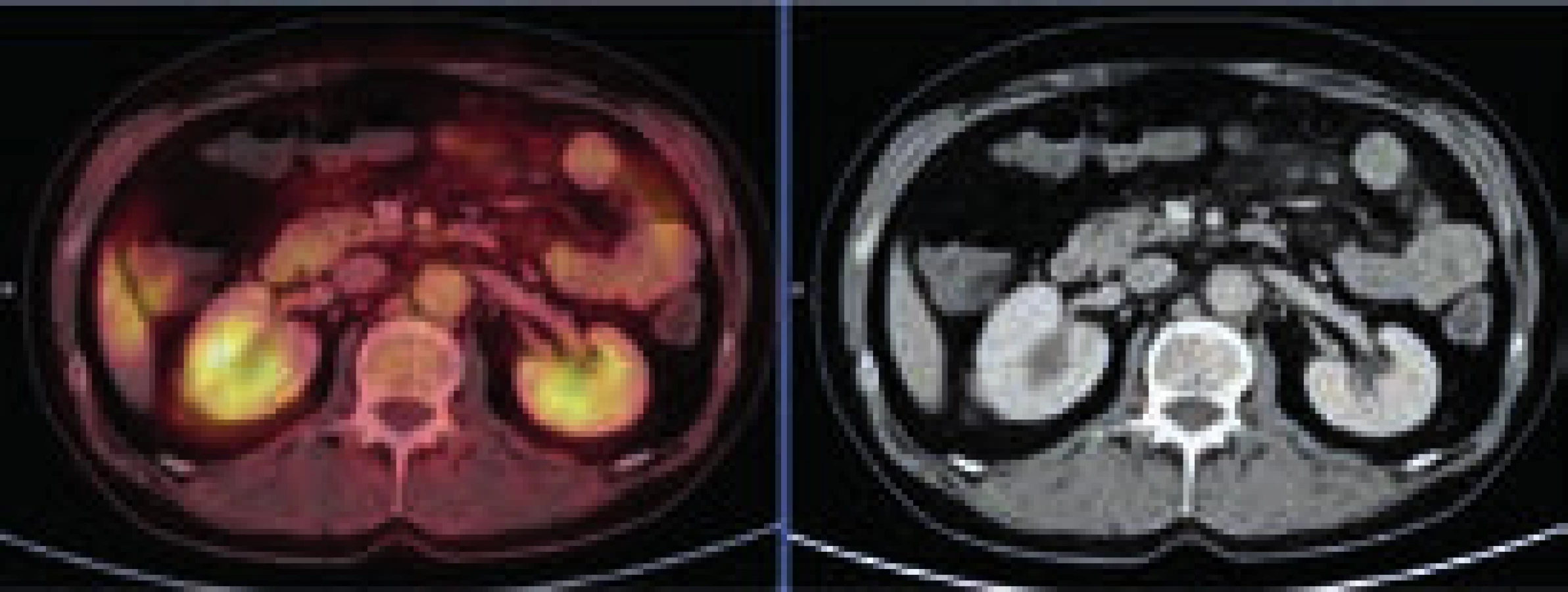
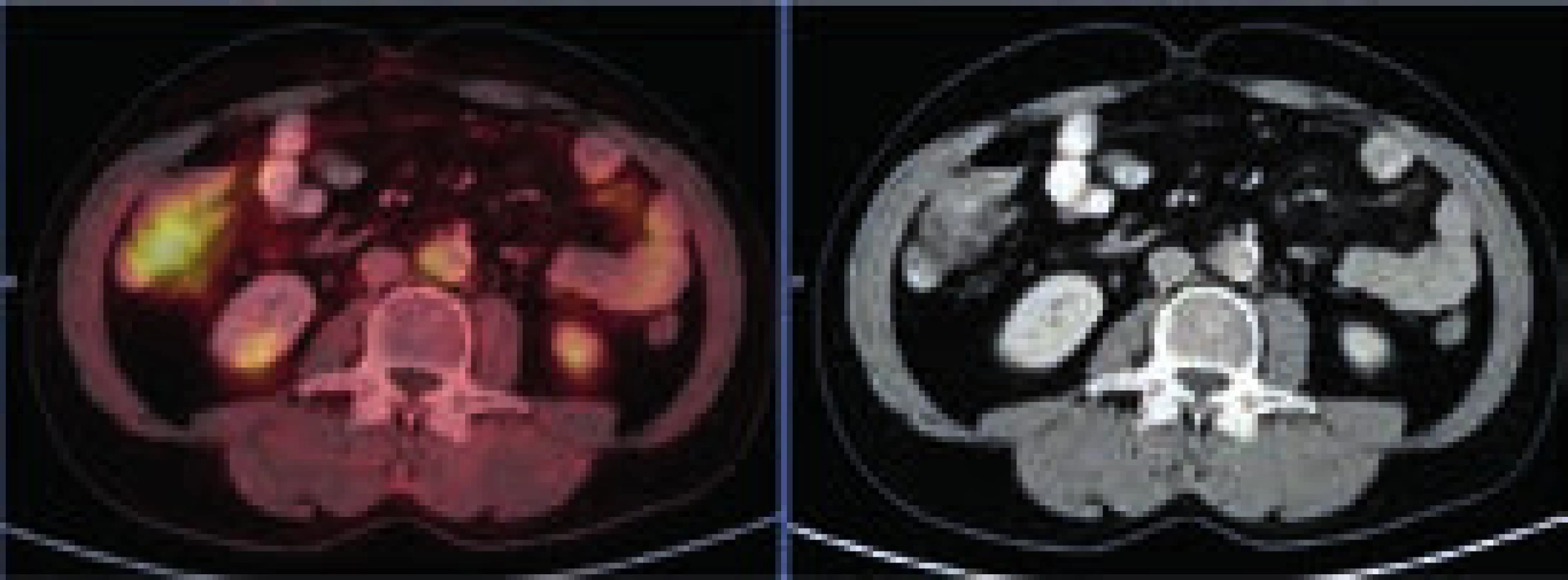
Při CT zobrazení byl v oblasti hrudníku normální nález vyjma zesílené stěny aorty s maximem v oblouku aorty a odstupu velkých cév, kde šíře stěny aorty dosahovala 5 mm. Zesílená stěna hrudní aorty nevykazovala hypermetabolizmus. V břišní dutině nebyla popsána žádná patologická struktura, pouze byly neostré kontury ledviny a zvýšená denzita mezenteriálního tuku. V břišní dutině bylo patrné také zesílení stěny aorty, místy s hypermetabolizmem v cévní stěně. Zevní kontury aorty byly neostré.
Změněná byla struktura dlouhých kostí dolních, ale i horních končetin. Výrazně sklerotická struktura těchto kostí byla prostoupena osteolytickými okrsky. V úseku diafýz femorů bylo patrné zesílení kompakty a byla patologická struktura dřeňové dutiny se zvýšenou denzitou. Patologické změny byly patrné v obou femorech s vynecháním střední třetiny diafýz. Stejné změny byly patrné v celých tibiích, inkompletně v obou fibulách. Vlevo postihly změny i kost patní. Stejné patologické změny byly zřetelné v oblasti humerů.
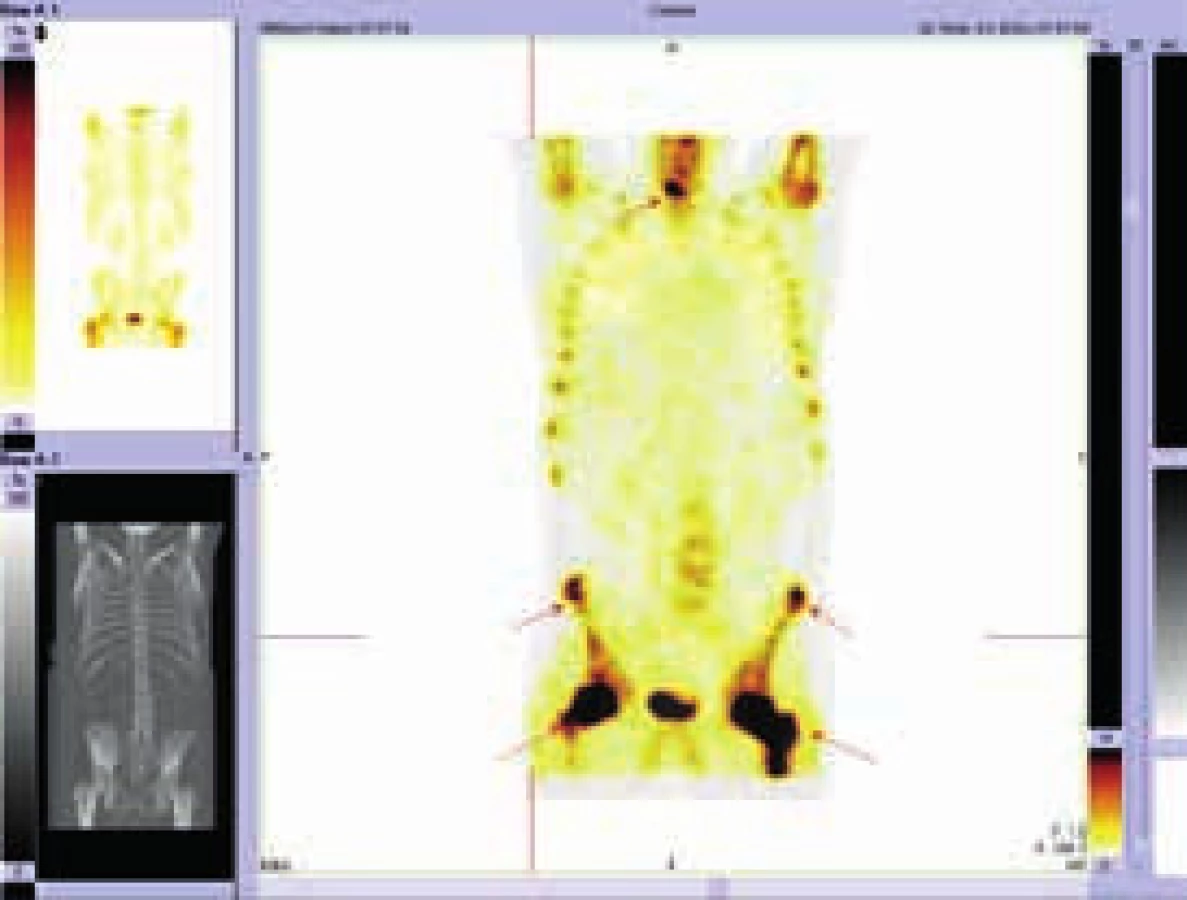
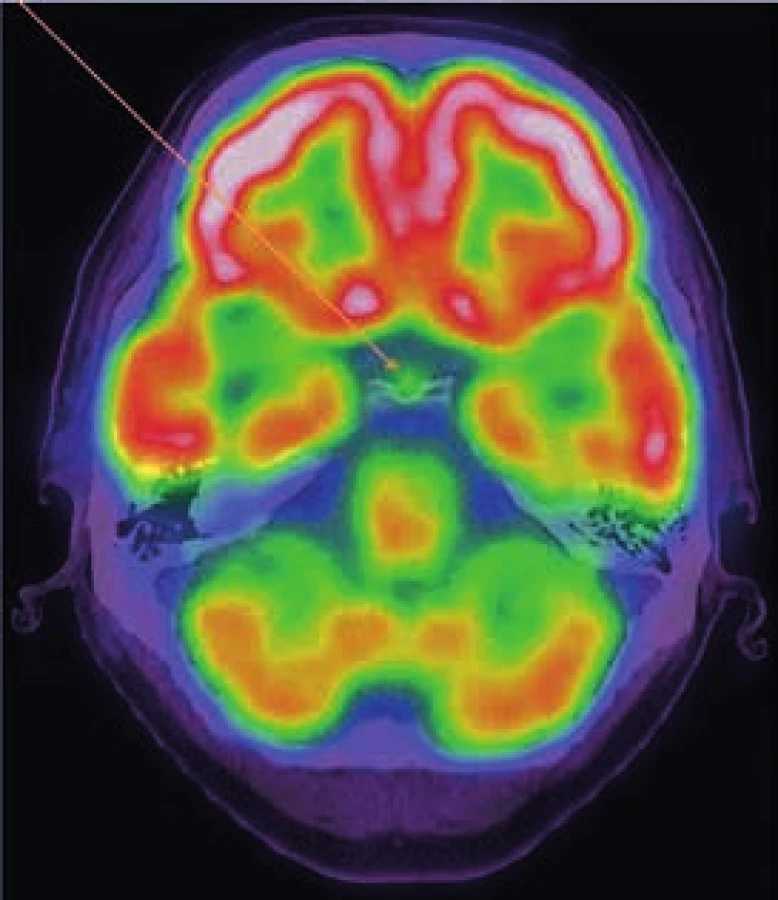
Při hodnocení akumulace fluorodeoxyglukózy bylo konstatováno, že nejvyšší akumulace fluorodeoxyglukózy je v oblasti obou femorů proximálně, SUVmax 9,5 a 9,8 a také v distálních částech femorů s SUVmax 9,7 a 11,52. Střední části diafýz femorů nevykazovaly zvýšenou akumulaci. Zvýšená akumulace byla také v kostech bérců (SUVmax 5,4) a také v obou humerech distálně (SUVmax 6,46 a 7,58). Některá zobrazení ze vstupního vyšetření v roce 2009 ukazují obr. 3–5.
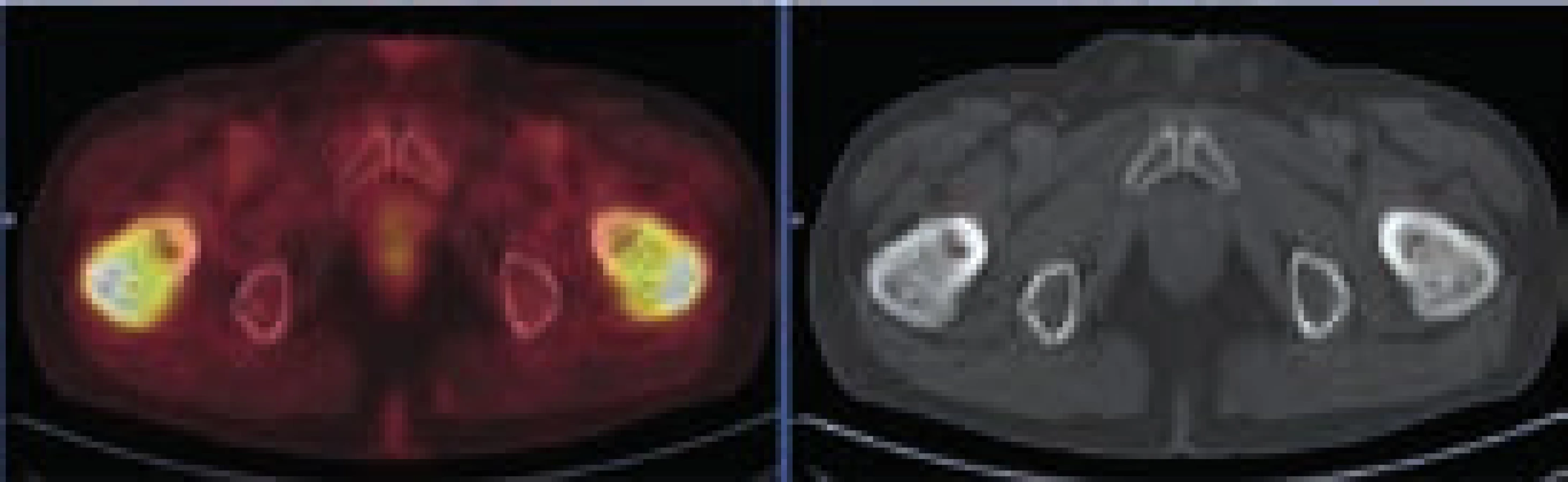
Vyšetření pomocí MRI
Při celotělovém MR zobrazení skeletu se prokázala rozsáhlá infiltrace kostní dřeně s převahou v oblasti dolních končetin. MRI a PET-CT prokázaly také infiltraci stopky hypofýzy (obr. 6 a 7).
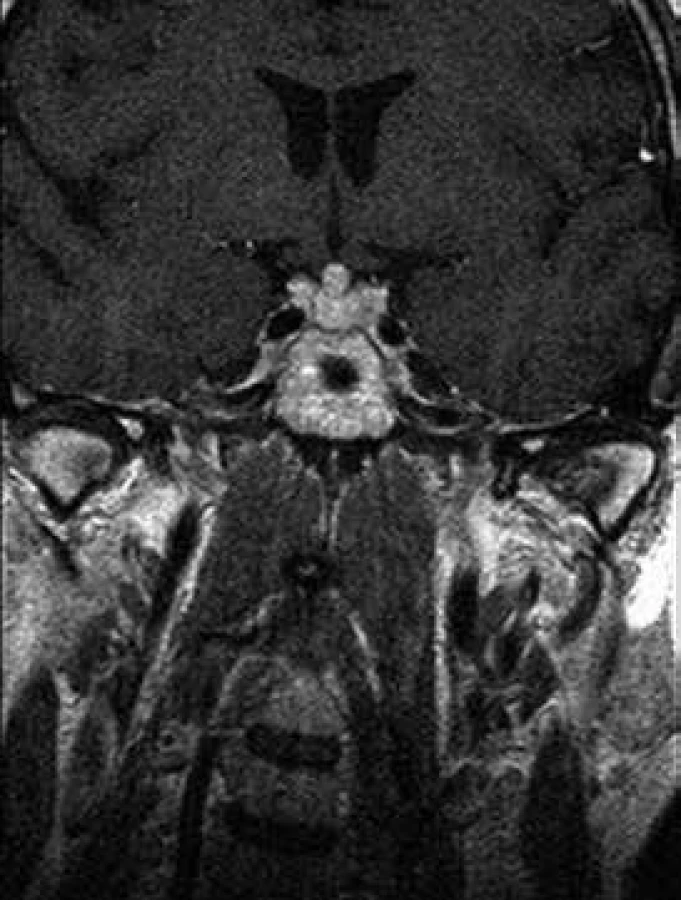
Vstupní denzitometrické vyšetření skeletu
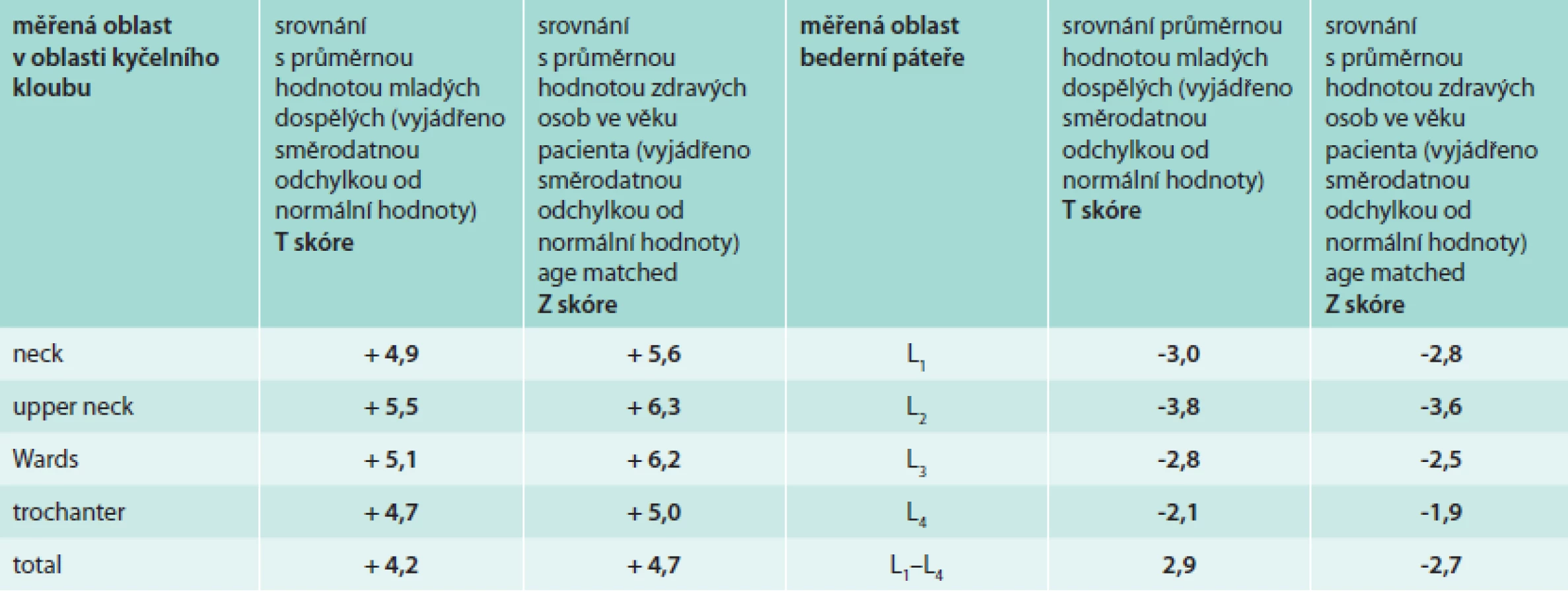
Vstupní denzitometrické vyšetření (DEXA) proběhlo v klasicky měřených oblastech, bederní páteři a krčku femoru. Hustota bederních obratlů byla v pásmu osteoporózy, tedy Z skóre výrazně pod hodnotou -2,0, zatímco hodnota Z skóre v oblasti krčku femoru byla v pozitivních hodnotách. Výsledky kostní denzitometrie přináší tab. 2. Naměřená vysoká denzita krčku femoru odpovídá zesílené kostní struktuře zřetelné při RTG a CT zobrazení.
Histologické stanovení diagnózy
Uvedená scintigrafická zobrazení vysvětlují, proč trepanobiopsie lopaty kosti pánevní neprokázala v roce 2005 žádnou patologii. Lopaty kosti pánevní nebyly u tohoto člověka postiženy. Proto jsme v roce 2009 již neopakovali necílenou biopsii lopaty kosti pánevní a místo toho jsme požádali ortopedy o odběr materiálu z oblasti femoru, v němž byla dle celotělového MRI skeletu i PET vyšetření značná patologická infiltrace. V celkové narkóze byl na operačním sále odebrán ortopedy vzorek kostní dřeně z té části femoru, kde bylo dle zobrazovacích vyšetření maximum patologického nálezu.
Částečka spongiózní kosti byla prostoupena agregáty pěnitých makrofágů s fibrózou. Místy byly přimíšeny nehojné lymfoidní a plazmatické buňky. Imunohistochemie: CD68 pozitivní, CD1a negativní, S100 fokálně pozitivní, α1-antitrypsin pozitivní, lyzozym pozitivní. Histologický obraz a imunofenotyp korelují s Erdheimovou-Chesterovou nemocí.
Léčebné schéma
Léčba byla zahájena 13. 10. 2009 kombinací 2-chlorodeoxyadenozin (Litac) 5 mg/m2 s. c. + cyklofosfamid 150 mg/m2 i. v. + dexametazon 24 mg i.v. vše 1.–5. den. Opakování těchto cyklů bylo plánováno v měsíčních intervalech.
Chemoterapii jsme podali 4krát ve výše uvedeném složení a 3krát s redukovanou aplikací pouze 1.–3. den. Důvodem pro redukci byla cytopenie, neutropenie grade III.
Chemoterapie byla ukončena v červnu roku 2010. Vzhledem k redukci dávek přípravku Litac v průběhu chemoterapie jsme místo původních 6 cyklů podali celkem 7 cyklů ve snaze aplikovat celkovou plánovanou dávku.
Pro kombinaci osteolytických a osteosklerotických změn skeletu byl podáván klondronát (Bonefos).
Vyhodnocení účinku léčby první linie
Pacient již v průběhu léčby udával úplné vymizení bolestí v dolních končetinách a vymizely také teploty. Při opakovaném laboratorním vyšetření bylo zřetelné klesání hodnot markerů zánětu (CRP). Zároveň však postupně docházelo ke snižování hodnot neutrofilů a lymfocytů jako nežádoucí důsledek této chemoterapie (tab. 3).
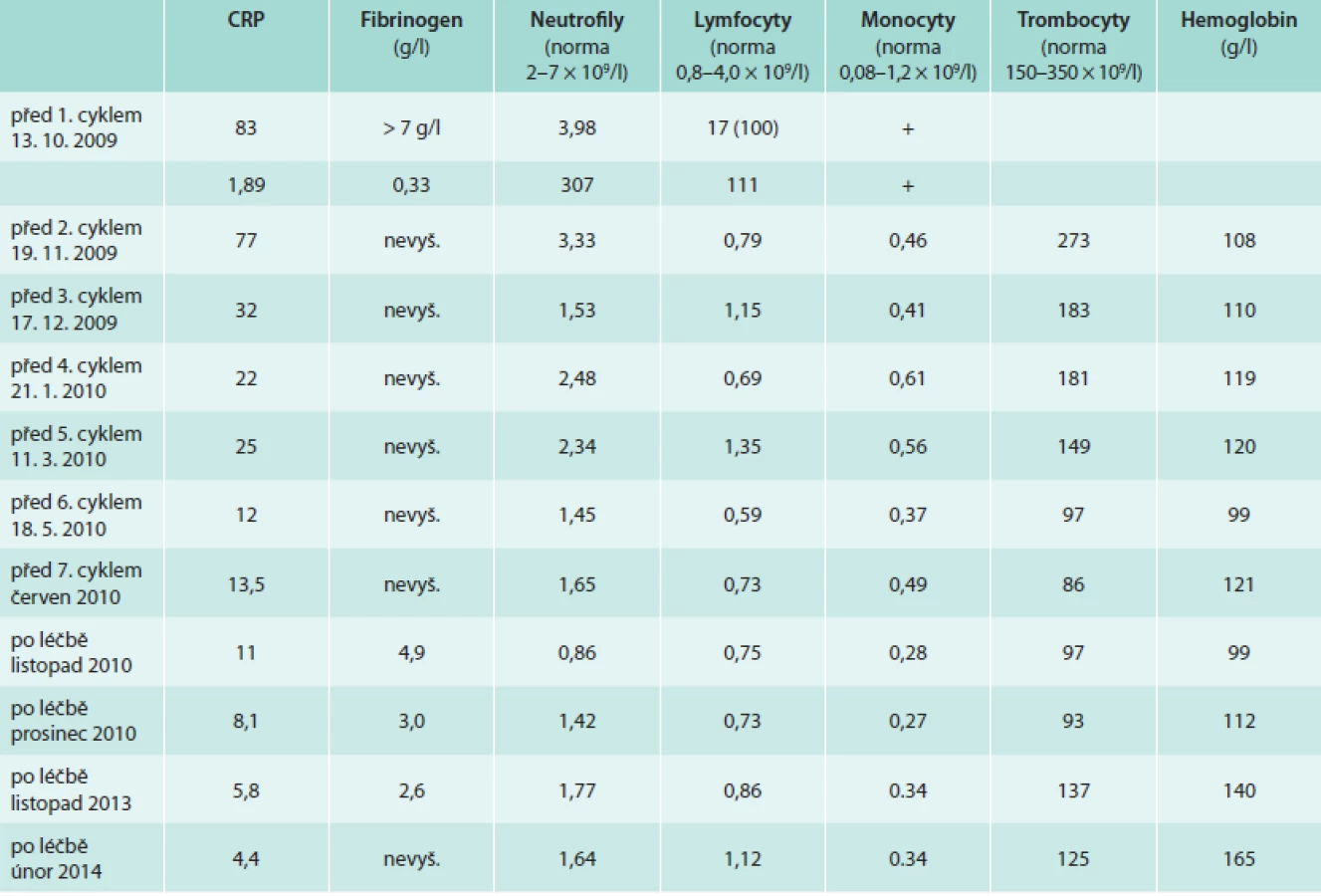
Základem pro hodnocení léčebné odezvy jsou však změny na opakovaně prováděných zobrazovacích vyšetřeních.
PET-CT zobrazení
U pacienta bylo provedeno PET-CT vyšetření před léčbou, v průběhu léčby a po jejím ukončení a následně byl pravidelně sledován zprvu v půlročních, později v ročních intervalech. Při každém vyšetření byla měřena hodnota SUV vždy ve stejných bodech se zvýšenou akumulací fluorodeoxyglukózy. Aby bylo možné srovnání, byla míra aktivity v měřených místech vyjádřena jak hodnotou SUVmax, tak poměrnou hodnotou SUVmax v ložisku ku SUVmax v játrech. Tento poměr je vhodnější pro srovnávání jednotlivých vyšetření mezi sebou, neboť eliminuje vlivy odchylek množství podané radioaktivity při jednotlivých vyšetřeních.
Při kontrolním PET-CT vyšetření bylo použito tzv. low dose CT zobrazení (neboli zobrazení s menší dávkou záření) a tím se mírně zmenšila rozlišitelnost. Ale i tak bylo zřejmé, že v dutině břišní došlo k postupné regresi fibrózních změn paraaortálně, maximální postižení bylo původně při mediální kontuře aorty přibližně v úrovni pupku. Při vstupním vyšetření byla šíře aorty včetně fibrózních změn 26 mm v průměru. Ke zlepšení došlo nenápadně, postupně od 1. kontroly. Podobný vývoj byl patrný i vpravo perirenálně: při CT z ledna roku 2011 byla patrná jen diskrétní, nespecifická rezidua původně velmi jasně zřetelných fibrotických změn a při posledním PET-CT vyšetření již byl nález bez patologických odchylek.
Doba sledování od stanovení diagnózy je 48 měsíců. Na obr. 8 jsou pod sebou vyšetření z roku 2009, 2011 a 2013.

Hodnoty poměru SUVmax v ložisku ku SUVmax v játrech, které jsou optimální pro dlouhodobé sledování ze všech provedených vyšetření, ukazuje graf 1 a 2. Graf dokumentuje 4 roky trvající kompletní remisi nemoci.

Nežádoucí účinky léčby
Subjektivní tolerance léčby byla po celou dobu dobrá, žádná alopecie, žádné gastrointestinální potíže. V průběhu léčby klesal absolutní počet lymfocytů i neutrofilů a závažný imunodeficit, navozený touto léčbou, se projevil až po ukončení léčby pásovým oparem, který pro přerušení léčby recidivoval. To vedlo k dlouhodobému podávání profylaktické dávky acykloviru (400 mg denně) po dobu 6 měsíců. V průběhu léčby nebyla však nutná ani jedna hospitalizace pro infekční komplikace, přijetí si vynutil až recidivující herpes zoster, který vzniknul až 2. měsíc po ukončení léčby. Léčba měla však dlouhodobé myelosupresivní důsledky.
Trombocyty byly v září roku 2009 před zahájením léčby 308 × 109/1, v průběhu léčby klesaly, nejnižší dosažená hodnota v průběhu léčby byla 32 × 109/1 a nad 100 × 109/1 vystoupily až v prosinci roku 2011.
Neutrofilní leukocyty byly před léčbou 4,07 × 109/1, v průběhu léčby byla nejnižší hodnota 0,45 × 109/1 a na hodnotu nad 1,0 × 109/1se dostaly v prosinci roku 2010. Poslední hodnota z roku 2013 byla 1,7 × 109/1.
Lymfocyty byly před léčbou 2,05 × 109/1, v průběhu léčby výrazně klesal jejich počet, nejnižší hodnota byla 0,37 × 109/1. Takže po 4 letech je stále počet lymfocytů, neutrofilů a trombocytů nižší než byl před zahájením léčby.
Diskuse
Pro léčbu Erdheimovy-Chesterovy nemoci nebyla publikována žádná doporučení, ověřená randomizovanými studiemi, protože těchto nemocných je velmi málo. K dispozici jsou pouze popisy případů či menších skupin pacientů a několik přehledových publikací.
Interferon α
Poměrně často byl pro léčbu používán interferon α, který vedl v mnoha případech ke stabilizaci nemoci a v mnoha publikacích je stále uváděn jako lék první volby.
Interferon α byl podáván od dávky 3 miliony jednotek 3krát týdně až po dávku 9 milionů jednotek 3krát týdně. Pegylovaný interferon α-2a je ekvivalentem. Byl podáván v dávce 135–200 µg týdně [32–35]. V jedné z posledních publikací popisujících 24 nemocných léčených vysokými dávkami interferonu α (> 18 milionů jednotek týdně) nebo vysokou dávkou pegylovaného interferonu α-2a (>185 µg týdně) uvádějí ústup kožních ložisek a také ložisek v mozku, v hypofýze a v její stopce, v plicích a v srdci, což jsou lokalizace této nemoci velmi těžce léčebně ovlivnitelné [36].
Další autoři popisují regresi jen v určitých oblastech. Braiteh popisuje dlouhodobou regresi v oblasti retroorbitálních ložisek, zlepšení v oblasti skeletu a také zmírnění diabetes insipidus u 3 léčených pacientů [34]. Arnaud popisuje jen malý pozitivní vliv interferonové léčby na průběh plicní formy této nemoci [37].
Prediktivní faktory předpovídající léčebnou odpověď na interferon α se nepodařilo najít. Nicméně autoři největšího souboru 53 nemocných uvádějí, že léčba interferonem α byla nezávislým prognostickým faktorem pro prodloužení přežití [37].
Léčba interferonem je však léčba dlouhodobá a při dlouhodobé léčbě se dostávají do popředí nežádoucí účinky léčby, celková slabost, únava, deprese, hypotenze, takže pacienti, kteří tuto léčbu zpočátku tolerovali, ji po více týdnech či měsících přestávají tolerovat [38].
Glukokortikoidy
Kortikoidy nemají na průběh tohoto onemocnění žádný zásadní vliv, jak vyplynulo z jednotlivých pozorování [32,34,38].
Chemoterapie
Klasická cytostatika byla v různých chemoterapeutických režimech také testována. Jejich výsledkem však byl maximálně jen dočasný ústup aktivity nemoci [39–41].
2-chlorodeoxyadenozin
U našeho pacienta jsme se rozhodli pro léčbu kladribinem. Připomeneme proto základní farmakodynamické údaje, neboť tato fakta a nečetné publikované popisy případů, kdy pacienti byli léčeni dávkou 0,07–0,14 mg/kg/den 5 dní po sobě, nás vedly k upřednostnění kladribinu před interferonem α. Kladribin (2-chlorodeoxyadenozin) je derivát adenozinu, u něhož na 2. uhlíku purinového kruhu byl vodík substituován chlórem. Tato změna je dostatečná, aby zabránila deaminaci 2-chlorodeoxyadenozinu. Kladribin je v buňce metabolizován a fosforylován až na 2-chloroadenozintrifosfát, který je vlastní účinnou formou léku. Akumulace deoxyadenozintrifosfátu je největší v buňkách, jejichž aktivační (fosforylační) enzym, deoxycytidinkináza, má největší aktivitu, zatímco inaktivační (defosforylační) enzym, cytoplazmatická 5-nukleotidáza, má nejmenší aktivitu. Velmi příznivý poměr uvedených 2 enzymů pro účinnost kladribinu je v klidových a proliferujících lymfocytech, v monocytech, v histiocytech a v Langerhansových buňkách. V nich dosahují nitrobuněčné koncentrace 2-chloroadenozintrifosfátů několiksetkrát vyšší hladiny, než je jeho plazmatická koncentrace. Díky této vlastnosti je kladribin vysoce selektivním cytostatikem, má intenzivní cytotoxický účinek na pomalu progredující maligní lymfatické a některé histiocytární buňky. V buňkách ostatních tkání je akumulace 2-deoxyadenozintrifosfátu malá, takže na ně cytotoxicky nepůsobí. Kladribin proniká v účinné dávce do CNS, intratekální koncentrace kladribinu dosahuje 25 % plazmatické koncentrace, což je dostačující hladina pro tumoricidní efekt na granulomy z lymfoidních či histiocytárních buněk. Biologická dostupnost podkožní aplikace je 100%, takže je možné jak nitrožilní, tak podkožní podání. Farmakologické vlastnosti kladribinu (100% resorpce z podkoží, několikasetnásobná akumulace v cílových senzitivních buňkách a 15–30hodinový intracelulární poločas) umožňují aplikaci formou podkožních injekcí.
V literární databázi Medline jsme našli jen 3 publikace, které popisují pozitivní účinek kladribinu u této nemoci. Jako první popsal podání kladribinu v této indikaci s pozitivním výsledkem Sheidow v roce 2000 [42].
Na tuto zkušenost navázal Myra et al, kteří v rámci iniciální léčby svému pacientovi podali etopozid, při absenci léčebné odezvy pak cyklofosfamid a cyklosporin. Když uvedené 3 testované léčebné alternativy nebyly dostatečně účinné, tak jako 4. léčebnou linii podávali pacientovi kladribin v dávce 0,14 mg/mg/kg/den 5 dní po sobě kontinuální infuzí Hickmanovým katétrem. Po 2 cyklech byla léčebná odezva již zřetelná. Celkem aplikovali 6 cyklů a uvádějí, že při hodnocení po 2 letech léčebná odezva stále trvala [43]. Pozitivní zkušenosti s kladribinem v této indikaci uvádí také Aouba [44].
Náš popis dlouhodobého léčebného efektu u pacienta s Erdheimovou-Chesterovou nemocí je tedy teprve 4. ve světové literatuře. Erdheimova-Chesterova nemoc je však jen jednou z forem tzv. juvenilního xantogranulomu. Je pravděpodobné, že všechny formy této nemoci budou na léčebné postupy podobně citlivé.
Blouin et al popisují podání 2-chlorodeoxyadenozinu a cytozin-arabinozidu po selhání předchozí léčby kombinací etopozidu, kortikoidu a vinblastinu. Tato léčba vedla u pacienta s juvenilním xantogranulomem ke kompletní remisi [45].
Podobně u dalšího dítěte s postižením CNS juvenilním xantogranulomem byla léčba zahájená prednizonem a vinblastinem bez efektu a teprve až aplikace kladribinu v monoterapii navodila léčebnou odezvu [46].
A z roku 2011 je zpráva o úspěšné léčbě xanthoma planum pomocí 2-chlorodeoxyadenozinu [47]. V roce 2013 se objevily další popisy úspěšné léčby nekrobiotického granulomu a juvenilního xantogranulomu kladribinem [48,49].
Popsán je ale i případ, v němž byly buňky juvenilního xantogranulomu na tuto léčbu zcela rezistentní [50] anebo případ dlouhodobé myelosuprese [51].
Literatura tedy obsahuje dosti důkazů účinnosti 2-chlorodeoxyadenozinu čili kladribinu u různých klinických forem juvenilního xantogranulomu, a proto jsme jej zvolili jako lék první linie. Zkušenost vyplývající z námi popsaného případu potvrzuje citované údaje o účinnosti kladribinu u Erdheimovy-Chesterovy nemoci a je dalším střípkem do mozaiky poznání nemocí ze skupiny juvenilního xantogranulomu.
Radioterapie a operační léčba
Radioterapie v této indikaci také selhala, v ozářené oblasti nedošlo k žádné klinicky zásadní regresi [34]. Operační odstranění největších mas představuje dočasné řešení, protože velmi rychle dochází k recidivě [52].
Bisfosfonáty
Léčba bisfosfonáty vede jen k částečnému zpomalení progrese kostního postižení [53–55].
Anakinra
V poslední době se objevily četné zprávy o značném přínosu dlouhodobé léčby preparátem blokujícím receptor pro interleukin 1, označovaným jako anakinra. Tato léčba však potřebuje pravidelnou aplikaci. Po zahájení léčby se rychle odstraní patologická únava související s vysokými hladinami některých interleukinů a pomalu regredují i orgánové projevy nemoci. Příznivých účinek blokády receptoru interleukinu 1 u pacientů s Erdheimovou- Chesterovou chorobou byl ověřen na více pacientech [56–64].
Vemurafenib – inhibitor BRAF (V600E)
Vemurafenib je inhibitor mutovaného peptidu B-RAF (V600E). Tato mutace je častá u melanomu a proto vemurafenib prodlužuje přežití pacientů s melanomem a dle současného poznání bude mít širší použití. Vemurafenib (Zelboraf) je v ČR hrazen u některých pacientů s neresekabilním či metastazujícím BRAF V600E pozitivním melanomem, počet léčených pacientů v ČR tímto lékem je asi v řádu desítek.
Zkratkou RAF se označují serin/treoninové kinázy. Protein B-RAF má klíčovou důležitost v tzv. MAP kinázové mitogenní signální dráze RAS/RAF/MEK/ERK. Proteiny A-RAF, B-RAF a C-RAF jsou schopné aktivovat kinázy MEK. Nejčastější mutací B-RAF je V600E, která má za následek asi 138násobné zvýšení aktivity kinázy oproti nemutované variantě a vede ke konstitutivní aktivaci MAPK dráhy a tedy ke zvýšení mitogenní aktivity buněk postižených uvedenou mutací.
Mutace BRAF (V600E) byla prokázána při hodnocení vzorků v tkáňových bankách u 38–69 % pacientů s histiocytózou z Langerhansových buněk. Sahm prokázal tuto mutaci ve 34 z 89 (38 %), Badalian v 35 z 61 (57 %) archivovaných vzorků. Satoh studoval mutace BRAF ve vzorcích od 16 pacientů, mutaci BRAF identifikoval celkem u 11 (69 %) pacientů, z toho se v 9 (56 %) případech jednalo o mutaci B-RAF V600E [65–67]. Toto poznání potvrdili i další autoři [68–70].
Haroche udělal analýzu přítomnosti této mutace ve tkáňových blocích od 127 pacientů s různými histiocytárními chorobami. Mutace BRAF (V600E) byla zjištěna u 13 z 24 (54 %) pacientů s Erdheimovou-Chesterovou chorobou a u 11 z 29 (38 %) pacientů s histiocytózou z Langerhansových buněk. U dalších typů histiocytárních chorob ji nenašli [71].
Toto teoretické poznání se již dostalo do klinické praxe. Haroche popsal 3 pacienty s Erdheimovou-Chesterovou nemocí, z nichž 2 měli současně kožní nebo uzlinovou formu histiocytózy z Langerhansových buněk. Všichni 3 pacienti měli onemocnění, které nereagovalo na léčbu interferonem α. Proto u nich použili vemurafenib v počáteční dávce 1 920 mg/den. Výchozí dávka byla po čase snížena u všech 3 nemocných na 960 mg/den. Důvodem byly kožní nežádoucí účinky léčby grade II, jiné nežádoucí účinky nebyly pozorovány. Překvapující byla rychlost nástupu léčebné odezvy, u všech nemocných bylo zásadní zlepšení pozorováno již v průběhu prvních dnů léčby. Léčebná odpověď byla dokumentována PET-CT vyšetřením. U pacientů, kteří měli souběh histiocytózy z Langerhansových buněk a Erdheimovy-Chesterovy nemoci, došlo k regresi obou histiocytárních nemocí. Vemurafenib je novým velmi účinným lékem pro pacienty s Erdheimovou-Chesterovou nemocí a s histiocytózou z Langerhansových buněk obsahující mutaci BRAF (V600E) [72].
Vemurafenib je tedy slibným lékem alespoň pro ty nemocné s Erdheimovou-Chesterovou chorobou, kteří mají uvedenou mutaci.
Inhibitory tyrozinkináz
Léčba novými léky (imatinib a sunitinib), které inhibují mimo jiné i PDGF signální cestu, dosáhla jen mírného zlepšení u pacientů s Erdheimovou-Chesterovou nemocí [72,73]. Jde zatím o ojedinělé zprávy.
Tocilizuman a infliximab
V klinické praxi byly dále testovány monoklonální protilátky zaměřené proti interleukinu 6 (tocilizumab) a proti TNFα (infliximab). Tyto postupy navodily léčebnou odezvu. Jde však zatím o první zkušenost, kterou bude nutno ověřit na dalších nemocných. A není zatím jasné, jak dlouho léčebná odezva bez udržovací léčby vydrží [74–82].
Závěr
Interferon α je zřejmě nejčastěji používaný lék pro léčbu Erdheimovy-Chesterovy nemoci, po němž byly popsány remise. Nespornou nevýhodou této léčby je nutnost trvalé aplikace, tedy udržovací léčby, až do progrese. Dle našich zkušeností s aplikací interferonu v jiných indikacích je dlouhodobá aplikace interferonu obvykle velmi špatně tolerovaná a výrazně zhoršuje kvalitu života.
Kladribin je oproti interferonu α excelentně tolerován a má potenciál u této nemoci dosáhnout dlouhodobé remise, jak dokládá popsaný případ. Léčba kladribinem aplikovaným současně s alkylačními cytostatiky, v našem případě s cyklofosfamidem, měla však za následek dlouhodobější myelosupresi.
Současný farmaceutický průmysl nabízí pro tyto nemocné další velmi úspěšné léky (anakinra, vemurafenib), náklady na ně jsou však mnohonásobně vyšší než na léčbu kladribinem. Na základě literárních dat a našich zkušeností doporučujeme pro pacienty s uvedenou chorobou použít v rámci léčby první linie kladribin v monoterapii a při neúspěchu po 3–4 měsících léčby testovat jiné, v diskuzi popsané léčebné postupy.
Text byl vytvořen na podporu následujících grantových aktivit: NT 12215–4/2011, MUNI/A/0723/2012, MZ ČR-RVO (MOÚ, 00209805), NT12215, MZ ČR-RVO (FNBr, 65269705) a dále OP VaVpl – RECAMO, CZ.1.05/2.1.00/03.0101, MZ ČR-RVO (MOÚ 00209805). A také MH CZ-DRO (MMCI, 00209805) RECAMO, CZ.1.05/2.1.00/03.0101.
prof. MUDr. Zdeněk Adam, CSc.
z.adam@fnbrno.cz
Interní hematologická a onkologická klinika LF MU a FN Brno
www.fnbrno.cz
Doručeno do redakce: 2. 12. 2013
Přijato po recenzi: 17. 2. 2014
Zdroje
1. Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol.2. 4th edition. WHO Press: Lyon 2008. ISBN 9789283224310.
2. Chester W. Über lipoidgranulomatose. Virchows Arch Pathol Anat 1930; 279: 561–602.
3. Křivanová A, Adam Z, Mayer J et al. Horečka nejasného původu – etiologie a diagnostický algoritmus. Vnitř Lék 2007; 53(2): 169–178.
4. Alušík S. Horečka nejasného původu. Vnitř Lék 1995; 41(12): 827–831.
5. Saudek F. Horečka nejasného původu. Čas Lék Česk 1983; 122(15): 452–458.
6. Salzberger B, Müller-Schilling M, Fleck M. Fever of unknown origin. Z Rheumatol 2013; 72(3): 255–266.
7. Horowitz HW. Fever of unknown origin or fever of too many origins? N Engl J 2013; 368(3): 197–199.
8. Koranda P, Mysliveček M. Pozitronová emisní tomografie při stanovení diagnózy vaskulitidy velkých cév – jedné z příčin horeček nejasného původu. Vnitř Lék 2006; 52(11): 1002–1003.
9. Buysschaert I, Vanderschueren S, Blockmans D et al. Contribution of (18)fluoro-deoxyglucose positron emission tomography to the work-up of patients with fever of unknown origin. Eur J Intern Med 2004; 15(3): 151–156.
10. Blockmans D, Knockaert D, Maes A et al. Clinical value of [(18)F]fluoro-deoxyglucose positron emission tomography for patients with fever of unknown origin. Clin Infect Dis 2001; 32(2): 191–196.
11. Fleck M. Fever of unknown origin – differential diagnosis and diagnostic evaluation. Dtsch Med Wochenschr 2013; 138(37): 1828–1832.
12. Kouijzer IJ, Bleeker-Rovers CP, Oyen WJ. FDG-PET in fever of unknown origin. Semin Nucl Med 2013; 43(5): 333–339.
13. Hao R, Yuan L, Kan Y et al. Diagnostic performance of 18F-FDG PET/CT in patients with fever of unknown origin: a meta-analysis. Nucl Med Commun 2013; 34(7): 682–688.
14. Halcin A, Kinova S. Contribution of PET/CT imaging to differential diagnosis of fever of unknown origin. Bratisl Lek Listy 2013; 114(2): 67–70.
15. Kim YJ, Kim SI, Hong KW et al. Diagnostic value of 18F-FDG PET/CT in patients with fever of unknown origin. Intern Med J 2012; 42(7): 834–837.
16. Qiu L, Chen Y. The role of 18F-FDG PET or PET/CT in the detection of fever of unknown origin. Eur J Radiol 2012; 81(11): 3524–3529.
17. Kinkor Z, Koudela K, Koudela K et al. Warfarinem vyvolaná hemorhagická pseudocysta malé pánve u ženy s vrozeným genetickým defektem koagulace komplikovaná usuračním pseudoxanthomem pánevní kosti napodobující Erdheimovu-Chesterovu nemoc. Acta Chir Ortop Traum Čech 2007; 74(2): 114–117.
18. Kinkor Z. Severe pulmonary involvement in Erdheim-Chester disease (case report). Cesk Patol 2001; 37(3): 114–117.
19. Kinkror Z. Závažné plicní postižení u Erdheimovy-Chesterovy nemoci. Čes Slov Patol Soudní Lék 2001; 37(3): 114–117.
20. Kolář J, Kučera V, Povýšil C et al. Erdheim-Chester disease. Rofo 1984; 141(6): 698–701.
21. Mergancová J, Kubes L, Elleder M. Xanthogranulomatous processes in the area of the large vessels. Cesk Patol 1986; 22(3): 145–150.
22. Mergancová J, Kubeš L, Elleder M. A xantogranulomatous process encircling large blood vessels (Erdheim-Chester disease). Czech Med 1988; 11(1): 57–64.
23. Kučera, V, Čáp V, Kužel J et al. Vzácná příčina osteosklerózy: Erdheimův Chesterův syndrome. Cesk Radiol 1984; 38(6): 393–402.
24. Janková H, Říhová E. Juvenilní xantogranulom. Oftalmologie v kasuistikách 2007; 3: 214–218.
25. Vašáková M. Co je to Erdheimova nemoc? Kazuist. Alergol. Pneumol. ORL 2006; 3 (4): 22–28.
26. Mottl H, Starý J, Chánová M et al. Treatment of recurrent Langerhans cell histiocytosis in children with 2-chlorodeoxyadenosine. Leuk Lymphoma 2006; 47(9): 1881–1884.
27. Mottl H, Rob L, Stary J et al. Langerhans cell histiocytosis of vulva in adolescent. Int J Gynecol Cancer 2007; 17(2): 520–524.
28. Mottl H, Ganevová M, Radvanská J et al. Treatment results of Langerhans cell histiocytosis with LSH II protocol. Cas Lek Cesk 2005; 144(11): 753–755.
29. Mottl H, Mrácek J, Kabelka Z et al. Langerhans-cell histiocytosis in children. Česk Pediatr 1992; 47(9): 530–533.
30. Plank L. Diagnostická patológia non-Langerhans cell histiocytóz. Vnitř Lék 2010; 56 (Suppl 2): S27-S38.
31. Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D et al. Erdheim-Chester disease: Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75(3): 157–169.
32. Arnaud L, Hervier B, Neel A et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 2011; 117(10): 2778–2782.
33. Drier A, Haroche J, Savatovsky J et al. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings. Radiology 2010; 255(2): 586–594.
34. Braiteh F, Boxrud C, Esmaeli B et al. Successful treatment of Erdheim-Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood 2005; 106(9): 2992–2994.
35. Suzuki HI, Hosoya N, Miyagawa K et al. Erdheim-Chester disease: multisystem involvement and management with interferon-alpha. Leuk Res 2010, 34(1): e21-e24. Dostupné z DOI: <http://doi: 10.1016/j.leukres.2009.07.026>.
36. Hervier B, Arnaud L, Charlotte F et al. Treatment of Erdheim-Chester disease with long-term high-dose interferon-alpha. Semin Arthritis Rheum 2012; 41(6): 907–913.
37. Arnaud L, Pierre I, Beigelman-Aubry C et al. Pulmonary involvement in Erdheim-Chester disease: a single-center study of thirty-four patients and a review of the literature. Arthritis Rheum 2010; 62(11): 3504–3512.
38. Adam Z, Pour L, Svobodník A et al. Kvalita života a tolerance udržovací léčby u mnohočetného myelomu. Vnitř Lék 2002; 48(3): 216–229.
39. Veyssier-Belot C, Cacoub P, Caparros -Lefebvre D et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine 1996; 75: 157–169.
40. Broccoli A, Stefoni V, Faccioli L et al. Bilateral orbital Erdheim-Chester disease treated with 12 weekly administrations of VNCOP-B chemotherapy: a case report and a review of literature. Rheumatol Int 2012; 32(7): 2209–2213.
41. Jendro MC, Zeidler H, Rosenthal H et al. Improvement of Erdheim-Chester disease in two patients by sequential treatment with vinblastine and mycophenolate mofetil. Clin Rheumatol 2004; 23(1): 52–56.
42. Sheidow TG, Nicolle DA, Heathcote JG. Erdheim-Chester disease: two cases of orbital involvement. Eye (Lond) 2000;14 (Pt 4): 606–612.
43. Myra C, Sloper L, Tighe PJ et al. Treatment of Erdheim-Chester disease with cladribine: a rational approach. Br J Ophthalmol 2004; 88(6): 844–847.
44. Aouba A, Larousserie F, Le Guern V et al. Spumous histiocytic oligoarthritis coexisting with systemic Langerhans’ cell histiocytosis: case report and literature review. Joint Bone Spine 2009; 76(6): 701–704.
45. Blouin P, Yvert M, Arbion F et al. Juvenile xanthogranuloma with hematological dysfunction treated with 2CDA-AraC. Pediatr Blood Cancer 2010; 55(4): 757–760.
46. Rajendra B, Duncan A, Parslew R et al. Successful treatment of central nervous system juvenile xanthogranulomatosis with cladribine. Pediatr Blood Cancer 2009; 52(3): 413–415.
47. Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011; 147(4): 459–464.
48. Sutton L, Sutton S, Sutton M. Treatment of necrobiotic xanthogranuloma with 2-chlorodeoxyadenosine. Skinmed 2013; 11(2): 121–123.
49. Tamir I, Davir R, Fellig Y et al. Solitary juvenile xanthogranuloma mimicking intracranial tumor in children. J Clin Neurosci 2013; 20(1): 183–188.
50. Orsey A, Paessler M, Lange BJ et al. Central nervous system juvenile xanthogranuloma with malignant transformation. Pediatr Blood Cancer 2008; 50(4): 927–930.
51. Yamada K, Yasui M, Sawada A et al. Severe persistent bone marrow failure following therapy with 2-chlorodeoxyadenosine for relapsing juvenile xanthogranuloma of the brain. Pediatr Blood Cancer 2012; 58(2): 300–302.
52. Oweity T, Scheithauer BW, Ching HS et al. Multiple system Erdheim-Chester disease with massive hypothalamic-sellar involvement and hypopituitarism. J Neurosurg 2002; 96(2): 344–351.
53. Mossetti G, Rendina D, Numis FG et al. Biochemical markers of bone turnover, serum levels of interleukin-6/interleukin-6 soluble receptor and bisphosphonate treatment in Erdheim-Chester disease. Clin Exp Rheumatol 2003; 21(2): 232–236.
54. Srikulmontree T, Massey HD, Roberts WN. Treatment of skeletal Erdheim-Chester disease with zoledronic acid: case report and proposed mechanisms of action. Rheumatol Int 2007; 27(3): 303–307.
55. Eyigor S, Kirazli Y, Memis A et al. Erdheim-Chester disease: the effect of bisphosphonate treatment–a case report. Arch Phys Med Rehabil 2005; 86(5): 1053–1057.
56. Clerico A, Ragni G, Cappelli C et al. Erdheim-Chester disease in a child. Med Pediatr Oncol 2003; 41(6): 575–577.
57. Arnaud L, Gorochov G, Charlotte F et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood 2011; 117(10): 2783–2790.
58. Stoppacciaro A, Ferrarini M, Salmaggi C et al. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim-Chester disease: implications for pathogenesis. Arthritis Rheum 2006; 54(12): 4018–4022.
59. Aubert O, Aouba A, Deshayes S et al. Favorable radiological outcome of skeletal Erdheim-Chester disease involvement with anakinra. Joint Bone Spine 2012; 80(2): 206–207.
60. Courcoul A, Vignot E, Chapurlat R. Successful treatment of Erdheim-Chester disease by interleukin-1 receptor antagonist protein. Joint Bone Spine 2014; 81(2):175–177.
61. Killu AM, Liang JJ, Jaffe AS. Erdheim-Chester disease with cardiac involvement successfully treated with anakinra. Int J Cardio 2013; 167(5): e115-e117. Dostupné z DOI: <http://doi: 10.1016/j.ijcard.2013.04.057>.
62. Tran TA, Pariente D, Lecron JC et al. Treatment of pediatric Erdheim-Chester disease with interleukin-1-targeting drugs. Arthritis Rheum 2011; 63(12): 4031–4032.
63. Aouba A, Georgin-Lavialle S, Pagnoux C et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood 2010; 116(20): 4070–4076.
64. Adam Z, Szturz P, Bučková P et al. Blokáda receptoru pro interleukin-1 preparátem anakinra vedla u pacienta s Erdheimovou-Chesterovou nemocí k vymizení patologické únavy, k poklesu markerů zánětu a ústupu fibrózy v retroperitoneu. Vnitř Lék 2012; 58(4): 313–318.
65. Sahm F, Capper D, Preusser M et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 2012; 120(12): e28-e34.
66. Badalian-Very G, Vergilio JA, Degar BA et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010; 116(11): 1919–1923.
67. Satoh T, Smith A, Sarde A et al. B-RAF Mutant Alleles Associated with Langerhans Cell Histiocytosis, a Granulomatous Pediatric Disease. PLoS One 2012; 7(4): e33891. Dostupné z DOI: <http://doi: 10.1371/journal.pone.0033891>.
68. Yousem SA, Dacic S, Nikiforov YE et al. Pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. Chest 2013; 143(6): 1679–1684.
69. Kansal R, Quintanilla-Martinez L, Datta V. Identification of the V600D mutation in Exon 15 of the BRAF oncogene in congenital, benign langerhans cell histiocytosis. Genes Chromosomes Cancer 2013; 52(1): 99–106.
70. Tadmor T, Tiacci E, Falini B et al. The BRAF-V600E mutation in hematological malignancies: a new player in hairy cell leukemia and Langerhans cell histiocytosis. Leuk Lymphoma 2012; 53(12): 2339–2340.
71. Haroche J, Cohen-Aubart F, Emile JF et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013; 121(9): 1495–1500.
72. Haroche J, Amoura Z, Charlotte F et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim-Chester histiocytosis. Blood 2008; 111(11): 5413–5415.
73. Janku F, Amin HM, Yang D et al. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 2010; 28(31): e633-e636. Dostupné z DOI: <http:// doi: 10.1200/JCO.2010.29.9073>.
74. Dagna L, Corti A, Langheim S et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol 2012; 30(28): e286-e290.
75. Ferrero E, Belloni D, Corti A et al. TNF-alpha in Erdheim-Chester disease pericardial effusion promotes endothelial leakage in vitro and is neutralized by infliximab. Rheumatology (Oxford) 2014; 53(1):198–200.
76. Chohan G, Barnett Y, Gibson J et al. Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 2012; 83(5): 573–575.
77. De Knop KJ, Aerts NE, Ebo DG et al. Multicentric reticulohistiocytosis associated arthritis responding to anti-TNF and methotrexate. Acta Clin Belg 2011; 66(1): 66–69.
78. Sakaguchi M, Nagai H, Tsuji G et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol 2011; 147(1): 131–133.
79. Aouba A, De Bandt M, Aslangul E et al. Haemophagocytic syndrome in a rheumatoid arthritis patient treated with infliximab. Rheumatology (Oxford) 2003; 42(6): 800–802.
80. Sellam J, Deslandre CJ, Dubreuil F et al. Refractory multicentric reticulohistiocytosis treated by infliximab: two cases. Clin Exp Rheumatol 2005; 23(1): 97–99.
81. Lee MW, Lee EY, Jeong YI et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol 2004; 84(6): 478–479.
82. Henzan T, Nagafuji K, Tsukamoto H et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol 2006; 81(1): 59–61.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2014 Číslo 5-6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Statiny indukovaná myopatie: Jak na diferenciální diagnostiku?
- Statinová intolerance
- Projekt MedPed
Najčítanejšie v tomto čísle
- Stillova nemoc dospělých – obtížná cesta k diagnóze přes horečku a výpotky nejasné etiologie
- Purple urine bag syndrome – raritní, ale nepřehlédnutelný příznak močové infekce
- Sentinelová uzlina pri malígnom melanóme
- Difuzní idiopatická skeletární hyperostóza