
Genetické syndromy predisponující k dětským nádorům centrálního nervového systému
Genetic Syndromes Predisposing to Tumors of Central Nervous System in Children
Background:
The overall incidence of childhood malignancies is rather low. Central nervous system tumours constitute the largest group of solid tumours among children. In contrast to adult population, a genetic predisposition is frequently associated with these malignancies (it is assumed to occur in approximately 15–25% of all childhood tumours) and there is also a number of monogenic hereditary syndromes known to be associated with brain tumours.
Aim:
The purpose of this article is to present an overview of genetic syndromes reported to increase the risk of childhood central nervous system tumours. The outlined tumour predispositions are divided into two groups. Firstly, syndromes with multisystem manifestation, where neoplasia is one of the components, whereas the distinguishing symptom is usually non-oncological. Secondly, there are syndromes that are diagnosed by the associated neoplasm withou any other noticeable phenotypic manifestation. A brief description of particular diseases is provided with a focus on associated central nervous system tumours. Detection of a tumour predisposition in a child is important not only for the child itself, but also for its family relatives. Often, a modification of treatment is necessary in regards to a genetic diagnosis. With the evolution of personalised medicine the possibility of “tailored” therapy will probably be a demanded solution. Last but not least, it is crucial to provide the child with a specialised preventive care owing to the risk of another potential malignancy. The diagnosis of hereditary cancer predisposition has also a big impact on the relatives of the patient. It enables to specify their oncological risk and arrange a specialised preventive care program, if needed. For high-risk parents planning another pregnancy there is a possibility to prevent the transfer of a certain disposition with the aid of preimplantation and prenatal genetic testing.
Key words:
brain tumours – child – hereditary cancer syndromes
The authors declare she has no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
15. 7. 2015
Accepted:
26. 8. 2015
Authors:
V. Krutílková
Authors‘ workplace:
Gennet, Centrum lékařské genetiky a reprodukční medicíny, Praha
Published in:
Klin Onkol 2016; 29(Supplementum 1): 71-77
Category:
Review
doi:
https://doi.org/10.14735/amko2016S71
Overview
Východiska:
Nádorová onemocnění u dětí jsou obecně vzácná. Ze solidních nádorů jsou nejčastější právě nádory centrálního nervového systému. Ve srovnání s dospělými je v těchto případech genetická predispozice relativně častá (obecně se předpokládá asi u 15 – 25 % dětských malignit) a je popsána řada monogenně dědičných syndromů, u kterých je mozkový nádor jedním z hlavních symptomů.
Cíl:
Článek uvádí přehled genetických syndromů asociovaných s vysokým rizikem nádorů centrálního nervového systému manifestujících se v dětském věku. Uvedené nádorové predispozice jsou rozděleny do dvou skupin. V první části jsou uvedeny syndromy s multisystémovou manifestací, u kterých jsou nádorová onemocnění jedním ze symptomů a které jsou většinou diagnostikovány na základě jiných než nádorových projevů. Důležité je u těchto pacientů zajistit preventivní sledování k včasné diagnostice možných tumorů. Ve druhé části pak jsou uvedeny jednotky charakterizované právě nádorovými onemocněními, která vedou k jejich diagnostice. Stručná charakteristika jednotlivých afekcí je zaměřena na asociované tumory centrálního nervového systému. Odhalení nádorové predispozice u dítěte je důležité nejen pro dítě samotné, ale i pro jeho příbuzné. U nemocného dítěte je často nutná modifikace léčby s ohledem na genetickou diagnózu, s rozvojem personalizované medicíny jistě dojde i k možnosti tzv. léčby na míru. V neposlední řadě je pak třeba pro dítě zajistit cílenou preventivní péči s ohledem na riziko dalších malignit. Diagnóza hereditární nádorové predispozice má velký dopad též na příbuzné pacienta. Umožňuje specifikovat jejich onkologické riziko a v případě potřeby zajistit speciální prevenci. Pokud rodiče plánují další těhotenství, je možno předejít přenosu dispozice použitím preimplantační, event. prenatální diagnostiky.
Klíčová slova:
nádory mozku – dítě – dědičné nádorové syndromy
Úvod
Nádory centrální nervové soustavy (CNS) jsou u dětí nejčastějšími solidními nádory a představují asi 25 % všech nádorů dětského věku. Na rozdíl od dospělých je genetická predispozice u dětí s nádory CNS relativně častá. Mutace ve stejných genech se mohou objevovat jak v nádorech vzniklých na základě dědičné predispozice (germinální mutace), tak ve sporadických tumorech (somatické mutace). Dobře známým příkladem je meningiom a neurofibromatóza typu 2. Některé typy nádorů CNS u dětí (např. meduloblastom, ependymom, pilocytický astrocytom) jsou vázány k faktorům podílejícím se na růstu mozku v časném postnatálním životě, se stoupajícím věkem výskyt těchto tumorů klesá. Děti s malformacemi CNS mají zvýšené riziko nádorů mozku. Příčinou je aktivace týchž drah v různých patologických procesech. Například dráha SHH ‑ PTCH ‑ GLI se podílí na rozvoji holoprosencephalie, Smith ‑ Lemli ‑ Opitzova syndromu, meduloblastomu a Gorlinova syndromu.
Genetické syndromy asociované s ná-dory CNS u dětí lze rozdělit na dvě skupiny:
- Multisystémové syndromy, u kterých je nádor pouze jednou z manifestací (tab. 1), tyto syndromy jsou často diagnostikovány před rozvojem nádorového onemocnění.
- Čistě nádorové predispozice, u kterých se v průběhu života objevují různé typy tumorů (tab. 2) a které jsou diagnostikovány právě až na základě manifestace tumoru [1 – 4].

Multisystémové syndromy asociované s nádory CNS u dětí
Neurofibromatóza typu 1
Neurofibromatóza typu 1 (NF1) je autozomálně dominantně dědičné onemocnění s výskytem v populaci 1 : 2 500 – 3 000, až v 50 % jde o de novo mutace. Kauzálním genem je gen NF1, jehož produktem je neurofibromin, který aktivuje GTPázu a inhibuje proteiny signální dráhy Ras, plní tedy funkci negativního růstového regulátoru [2].
Onemocnění se projevuje kombinací kožních, skeletálních, oftalmologických a neurologických příznaků. Pro diagnózu NF1 je nutná přítomnost dvou nebo více příznaků [5]:
- skvrny barvy café-au-lait (u dětí pět a více o průměru 0,5 cm nebo větším, u dospělých šest a více o průměru 1,5 cm nebo větších),
- dva a více neurofibromů jakéhokoli typu nebo jeden plexiformní,
- mnohočetné axilární nebo inguinální pihy,
- dysplazie sfenoidálního křídla nebo kongenitální ohnutí nebo ztenčení kortikální části dlouhých kostí,
- bilaterální gliom optického nervu,
- dva a nebo více Lischových nodulů (hamartomů duhovky),
- příbuzný 1. stupně s NF1 dle těchto kritérií.
Z nádorových projevů u dětí jsou nejčastější gliomy optiku (u 15 – 40 % pacientů), které se manifestují do 6., resp. 10. roku věku, později již vzácně. U dětí s gliomy chiasmatu se může objevit předčasná puberta, častější jsou též gliomy mozkového kmene. I u dětí a dospívajících se může objevit maligní tumor z pochvy periferního nervu. Častěji se vyskytují feochromocytomy, leukemie, Wilmsův tumor, vzácně rhabdomyosarkom, nediferencovaný sarkom a neuroblastom. Z nenádorových projevů se již v novorozeneckém věku mohou objevit skvrny café ‑ au ‑ lait, neurofibromy se většinou objevují kolem puberty, Lischovy noduly v předškolním věku. Častá je makrocefalie, skolióza, hydrocefalus, migrény a hypertenze. Lehká mentální retardace postihuje cca 10 % jedinců s NF1, u 60 % se objevují poruchy učení a chování, u 7 % dětí s NF1 se manifestuje epilepsie, převážně sekundární při tumorech CNS [5].
Pro léčbu asymptomatických gliomů optiku u pacientů s NF1 se doporučuje spíše konzervativní postup, u symptomatických optochiasmatických gliomů rostoucích v čase nebo vedoucích ke zhoršování zrakových funkcí či endokrinologickému deficitu se užívá konvenční chemoterapie a potenciálně i biologická léčba [2].
Gorlinův syndrom
Gorlinův syndrom (GS) (Nevoid Basal Cell Carcinoma Syndrom – NBCCS) je autozomálně dominantně dědičná afekce s incidencí 1 : 50 000 – 100 000, příčinou jsou mutace v tumor supresorovém genu PTCH (ve 20 – 30 % případů jde o de novo mutace), který kóduje transmembránový glykoprotein fungující jako antagonista signální dráhy Hedgehog. Popsán byl i další predisponující gen SUFU. Pro GS je typický vysoký vzrůst, makrocefalie, prominující čelo, hypertelorizmus, mohou se vyskytovat vývojové vady (orofaciální rozštěp, malformace žeber a páteře, kongenitální katarakta, mikroftalmie, kolobom), srdeční fibromy (již i prenatálně), přemostění sella turcica, mentální subnorma. Objevují se čelistní cysty, bazaliomy, kožní keratocysty a milia, ovariální fibromy, popsány jsou i hamartomatózní polypy žaludku [6]. Z nádorů CNS je nejčastější meduloblastom, popisován je u 1 – 5 % pacientů, střední věk manifestace jsou dva roky věku (na rozdíl od sporadického, kdy je to šest let) a jde o desmoplastický typ. Léčba je identická se sporadickým meduloblastomem, měla by být však pokud možno eliminována radioterapie. V radiačním poli se rozvíjí velké množství invazivních bazocelulárních karcinomů, popsán je i vývoj dalších tumorů CNS (meningiomu, anaplastického astrocytomu) [7,8]. Bazocelulární karcinomy se u 75 % pacientů objevují do 20. roku věku.
Tuberózní skleróza
Tuberózní skleróza (TSC) je autozomálně dominantně dědičné onemocnění s incidencí 1 : 6 000. Příčinou jsou mutace v genu TSC1 a TSC2, v 60 – 80 % jde o mutace de novo. Gen TSC1 kóduje hamartin, gen TSC2 tuberin. Hamartin s tuberinem vytvářejí heterodimery, které společně regulují buněčnou proliferaci v signální dráze PI3K/ Akt (mTORC1) [9].
Diagnóza TSC je stanovena na základě diagnostických kritérií [10], penetrance je téměř 100 % (většinou do 15. roku věku), exprese klinických příznaků je extrémně variabilní i v rámci rodin. Typické jsou mnohočetné hamartomy mozku, srdce, očí a ledvin a kožní abnormality. Epilepsie se objevuje až u 80 % pacientů, mentální retardace u 40 %, časté jsou i poruchy chování. V CNS se objevují kortikální tubera, subependymální hamartomatózní noduly a intrakraniální kalcifikace. Těžší postižení je u pacientů s mutací v genu TSC1 než TSC2, ženy mívají lehčí formu než muži. U 10 – 15 % pacientů se do 20. roku věku manifestuje subependymální obrovskobuněčný astrocytom, který je benigní, pomalu rostoucí, typicky se vyskytující ve stěně postranní komory. K léčbě rostoucího tumoru je užívána chirurgická léčba a nově mTOR inhibitory [1].
Fanconiho anémie
Fanconiho anémie (FA) je autozomálně recesivně nebo X ‑ recesivně dědičné onemocnění s incidencí 1 : 200 000– – 400 000 charakterizované vrozenými vadami, chromozomální nestabilitou a selháváním kostní dřeně. FA může být způsobena mutacemi nejméně v 15 genech. Tyto geny jsou důležité pro reparaci zkřížených vazeb v DNA.
Bialelickou mutací v genu BRCA2 (FANCD1) je způsobeno 3 – 5 % případů FA. Tento typ FA má často těžký fenotyp s více vrozenými vadami a silnější nádorovou predispozicí. Nádory se často vyvíjejí již v první dekádě života, nejčastěji se jedná o akutní myeloidní leukemii a meduloblastom a na rozdíl od jiných typů FA je spektrum nádorů širší, zahrnující další embryonální tumory jako neuroblastom, hepatoblastom, Wilmsův tumor a také rhabdomyosarkom. Opakovaně byl popsán i vývoj nádorových multiplicit již v první dekádě života [11]. Podobný fenotyp vykazuje i FA způsobena bialelickými mutacemi v genu PALB2 [12].
U dětí s těmito typy FA je v 25 % případů první manifestací tumor CNS. Přičemž diagnóza FA je u nich zcela zásadní, protože vzhledem k extrémní citlivosti k ionizujícímu záření a chemoterapii musí být léčba vedena ve speciálních režimech.
Vyvstává tedy otázka vyšetření genů BRCA2, event. PALB2 u druhého rodiče v případě, že jeden je heterozygot mutace v tomto genu [11].
Cowdenův syndrom
Cowdenův syndrom (CS) je autozomálně dominantně dědičné onemocnění s incidencí 1 : 200 000 – 250 000 způsobené mutacemi v genu PTEN (ve 35 – 60 % jde o mutace de novo). Syndrom je charakterizován mnohočetnými hamartomy a zvýšeným rizikem benigních i maligních tumorů. CS se většinou manifestuje do 20. roku věku mukokutánními lézemi, makrocefalií, časté jsou poruchy psychomotorického vývoje, mohou se objevit křeče. Z nádorů CNS je patognomická Lhermitte ‑ Duclosova choroba, která se obvykle objevuje v dospělém věku, ale popsána je i u dětí. Ostatní popsané nádory jak CNS (meningiom, meduloblastom, gangliocytom, glioblastom), tak i v ostatních lokalizacích (štítnice, prsu, endometria) se objevují v dospělém věku [1].
Rubinstein‑Taybi syndrom
Rubinstein‑Taybi syndrom (RTS) je autozomálně dominantně dědičné onemocnění s incidencí 1 : 100 000 – 125 000, příčinou jsou mutace v genu CREBBP a EP300C, event. mikrodelece v oblasti 16p13.3. Téměř vždy je důsledkem mutace de novo. Pacienti s RTS mají typickou faciální stigmatizaci, široké palce na rukou i nohou, malý vzrůst a středně těžkou až těžkou mentální retardaci. RTS je spojen se zvýšeným rizikem malignit, v dětství především meduloblastomu a leukemie [13,14].
Ataxia teleangiektasia
Ataxia teleangiektasia (AT) je autozomálně recesivně dědičné onemocnění s incidencí 1 : 40 000 – 100 000, příčinou jsou mutace v genu ATM. Gen ATM kóduje proteinkinázu, která se účastní buněčných reparačních procesů. Hlavním příznakem je růstová retardace, okulokutánní teleangiektázie, imunodeficience a ataxie. Jedinci s AT mají vysoké riziko nádorových onemocnění, v dětství jsou nejčastější leukemie a lymfomy, z nádorů CNS je popisován meduloblastom, později je vysoké riziko dalších malignit (prsu, vaječníku, žaludku, melanomu, leiomyomu, popsány jsou i sarkomy). Typická je hypersenzitivita vůči radiačnímu záření, proto je důležitá znalost diagnózy AT před zahájením léčby (musí být použito modifikované léčebné schéma) [15].
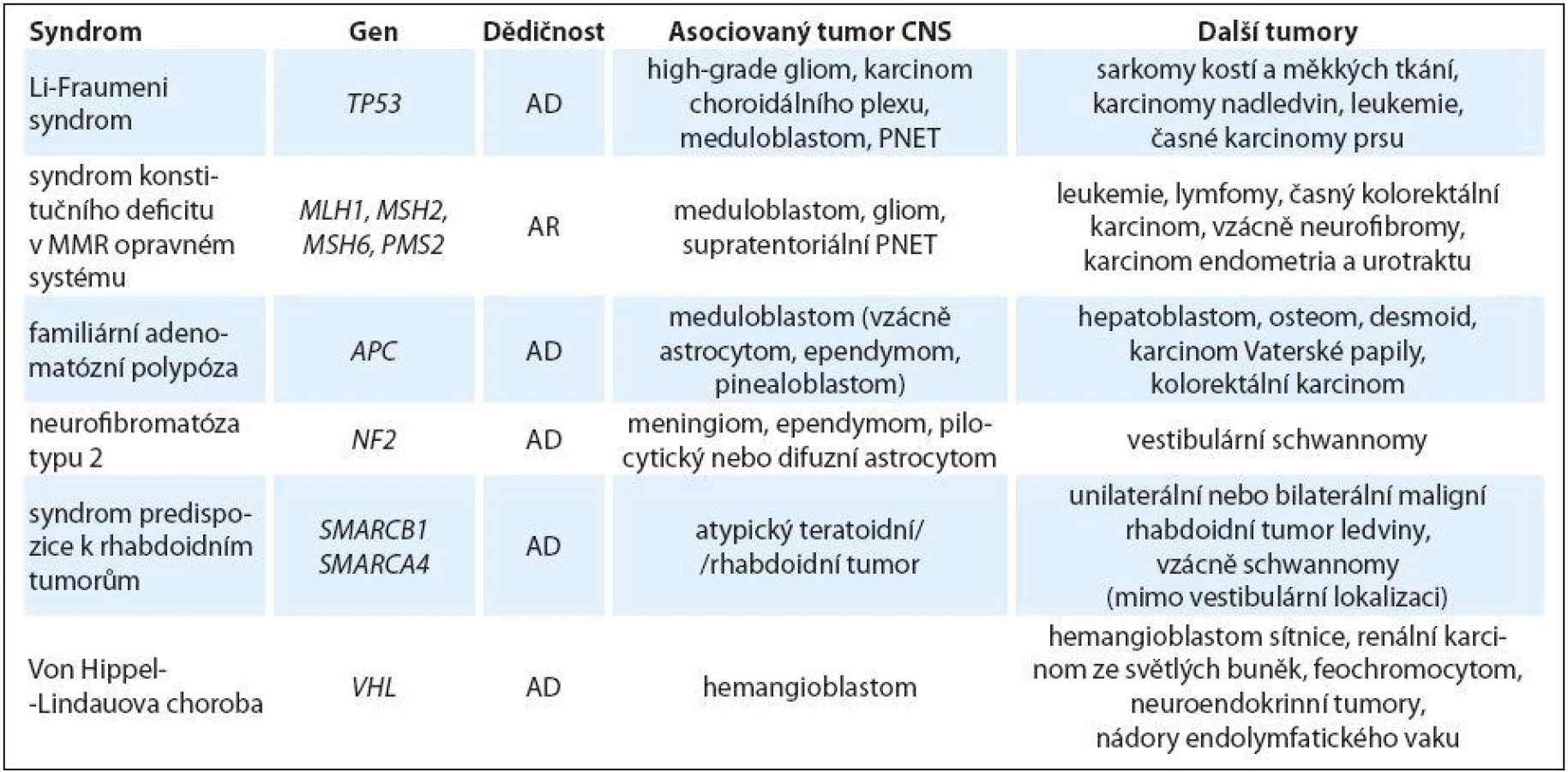
Hereditární nádorové syndromy asociované s tumory CNS u dětí bez další primárně nápadné systémové manifestace
Li-Fraumeni syndrom
Li ‑ Fraumeni syndrom je autozomálně dominantně dědičná afekce charakterizovaná mnohočetnými časnými nádory, s incidencí 1 : 5 000 – 20 000, v 7 – 20 % jde o mutace de novo [16]. Typické jsou tumory CNS, sarkomy kostí a měkkých tkání, adrenokortikální karcinomy a časné karcinomy prsu. Vyskytnout se ale mohou prakticky jakékoliv typy malignit. Příčinou Li ‑ Fraumeni syndromu jsou mutace v genu TP53, který má klíčovou úlohu v regulaci buněčného cyklu a apoptózy. Vyšetření genu TP53 je indikováno nejčastěji na základě modifikovaných kritérií dle Chompretové [17]. Nádorové riziko do 18. roku věku je 30 – 40 % [14]. Riziko nádorů CNS je u jedinců s Li ‑ Fraumeni syndromem velmi vysoké – nejčastější je astrocy-tom, glioblastom, meduloblastom (často sonic ‑ Hedgehog subtyp) a karcinom choroidálního plexu (CPC). U pacientů s CPC je pravděpodobnost nálezu mutace v genu TP53 36 – 44 % [18,19], u tohoto typu nádoru by mělo být vyšetření genu TP53 indikováno vždy. Průměrný věk manifestace tumoru CNS je 16 let, více než polovina s Li ‑ Fraumeni syndromem asociovaných tumorů CNS se vyvine u dětí do 18 let. U pacientů s Li ‑ Fraumeni syndromem by měla být minimalizována expozice jak diagnostické, tak terapeutické radiaci vzhledem k riziku indukce dalších malignit [1].
Syndrom konstitučního deficitu v MMR (mismatch repair) opravném systému
Syndrom konstitučního deficitu v MMR (mismatch repair) opravném systému (constitutional mismatch repair ‑ deficiency syndrome – CMMR ‑ D) je vzácná autozomálně recesivně dědičná afekce způsobená bialelickými mutacemi v genech MMR. Geny MMR systému (MLH1, MSH2, MSH6, PMS2) hrají zásadní roli v udržení integrity genomu korekcí chyb vzniklých při replikaci DNA. Klinicky je jednotka naprosto odlišná od Lynchova syndromu s výrazně horší prognózou.
U poloviny pacientů s CMMR ‑ D se rozvine nádor CNS (nejčastěji glioblastom a další high‑grade gliomy, PNET, meduloblastom), u poloviny nádor zažívacího traktu (přítomny jsou mnohočetné polypy) a cca u třetiny se objeví hematologická malignita (lymfom, akutní lymfoblastická i myeloidní leukemie). Mohou se objevit další nádory ze spektra Lynchova syndromu. U 40 % pacientů se objevuje metachronní tumor. Nádory mozku a hematologické malignity jsou nejčastěji diagnostikovány v první dekádě života, kolorektální karcinom a karcinom tenkého střeva ve druhé a třetí. Karcinom endometria a urotraktu se objevuje u mladých dospělých.
Krom nádorových onemocnění jsou pro jednotku typické kožní projevy typu skvrn café ‑ au ‑ lait, hypopigmentace, může být lehká imunodeficience i vrozené vývojové vady.
Rodinná anamnéza často ani u jednoho z rodičů Lynchovu syndromu neodpovídá (nejčastější příčinou jsou mutace v genu PMS2).
Vyšetření genů MMR by mělo být indikováno u dětí s hematologickou malignitou nebo nádorem CNS a anamnézou podezřelou z Lynchova syndromu u jednoho z rodičů a/ nebo při současném výskytu skvrn café ‑ au ‑ lait a/ nebo sourozence s dětským nádorem. Vždy je též indikováno při výskytu nádoru GIT v období adolescence nebo při nádorové duplicitě odpovídající spektru CMMR ‑ D.
Otázka odlišné senzitivity těchto pacientů na onkologickou léčbu je předmětem probíhajících studií [20].
Familiární adenomatózní polypóza
Familiární adenomatózní polypóza (FAP) je autozomálně dominantně dědičná afekce charakterizovaná těžkou polypózou s incidencí 1 : 10 000. Příčinou jsou mutace v genu APC, ve 20 – 25 % jde o mutace de novo.
Gen APC ovlivňuje proliferaci a diferenciaci buněk.
Vzácně může být první manifestací meduloblastom v dětství, popsány jsou i další typy nádorů CNS (astrocytom, ependymom, pinealoblastom). Asociace FAP a extraintestinálních projevů (kožní léze typu fibromů, lipomů, sebaceózních a epidermoidních cyst, dále nazopharyngeální aniofibromy, osteomy, desmoidní tumory, kongenitální hypertrofie retinálního pigmentového epitelu) je označována jako Gardnerův syndrom. Kombinace meduloblastomu (vzácně jiného tumoru CNS) a FAP je známa jako Turcotův syndrom typu 2. Z dalších dětských malignit je popisován hepatoblastom [21].
Neurofibromatóza typu 2
Neurofibromatóza typu 2 (NF2) je autozomálně dominantně dědičná afekce s incidencí 1 : 33 000 – 40 000. Příčinou jsou mutace v tumor supresorovém genu NF2, až v 50 % jde o de novo mutace.
Pro onemocnění je typický rozvoj bilaterálních vestibulárních schwannomů, které se klinicky začínají projevovat většinou v pubertě. Mohou se objevovat i další benigní nádory jednak intrakraniální (meningiomy a ependymomy), jednak schwannomy centrálních a periferních nervů, vzácně astrocytomy a neurofibromy. Skvrny café ‑ au ‑ lait jsou popisovány asi u 40 % pacientů, ale jen asi 1 % jich má více než šest. Již v dětském věku může být přítomna kata-rakta, retinální hamartomy a epiretinální membrány.
Na rozdíl od NF1 jsou klinické projevy u NF2 v rámci rodiny podobné.
Časná diagnostika je zásadní pro správné načasování chirurgického řešení neurinomů akustiku a pro zachování sluchu [5]. Ve vybraných případech je přínosné radiochirurgické ozáření na gama noži či X ‑ noži [22].
Syndrom predispozice k rhabdoidním tumorům
Syndrom predispozice k rhabdoidním tumorům (RTPS) je vzácná autozomálně dominantně dědičná afekce. Příčinou jsou mutace v genu SMARCB1, cca v 75 % jde o mutace de novo, penetrance je neúplná (v řadě rodin je rodič asymptomatický nositel mutace), opakovaně je popsán i germinální mozaicizmus u jednoho z rodičů. Ztráta funkce genu SMARCB1 vede k vysoké chromozomální nestabilitě. Dalším predisponujícím genem je gen SMARCA4.
Pro jednotku je typický časný výskyt intrakraniálních rhabdoidních tumorů (atypický teratoidní rhabdoidní tumor) a renálních či extrarenálních (v dutině břišní lokalizovaných) maligních rhabdoidních tumorů. Jde o vysoce maligní onemocnění se špatnou prognózou, u dětí s diagnostikovanou germinální mutací v genu SMARCB1 je popisován časnější věk manifestace a horší prognóza přežití. V rodinách s RTPS byly dále popsány karcinom choroidálního plexu, meduloblastom a supratentoriální PNET [1,23].
Germinální mutace v genu SMARCB1 jsou popisovány i v rodinách se schwannomatózou, která se manifestuje v dospělosti (schwannomy nepostihují vestibulární nerv), výjimečné jsou rodiny s kombinací fenotypu RTPS a schwannomů [1].
Na riziko RTPS je nutno pomýšlet u dětí s germinální delecí 22q11, pokud zahrnuje i oblast genu SMARCB1 (syndrom je charakterizován vývojovými vadami obličeje a srdce, imunodeficiencí, poruchami psychomotorického vývoje – klinicky je znám jako DiGeorgův syndrom).
Von Hippel‑ Lindauova choroba
Von Hippel ‑ Lindauova choroba (VHL) je autozomálně dominantně dědičná afekce s incidencí 1 : 35 000 – 40 000, příčinou jsou mutace v genu VHL, cca v 20 % jde o mutace de novo.
Pro syndrom jsou charakteristické hemangioblastom CNS a retiny, feochromocytom, renální cysty a karcinom ledvin ze světlých buněk. VHL syndrom se většinou manifestuje v dospělosti, jsou ale popsány hemangioblastomy i u dětí a dospívajících, stejně tak i retinální hemangioblastomy. Většina pacientů s VHL má mnohočetné hemangioblastomy v mozkovém kmeni, míše a okolí nervových kořenů ve srovnání se sporadickými hemangioblastomy, které se nejčastěji vyskytují v mozečku [24].
Závěr
U dospělých pacientů je příčinou nádorových onemocnění hereditární predispozice v 5 – 10 %, dle recentních studií se předpokládá, že u dětí je to v 15 – 25 % [25,26].
Pro odhalení genetické dispozice je nezbytná podrobná rodinná anamnéza. Je však třeba si uvědomit, že řada uváděných syndromů vzniká na podkladě mutace de novo, rodinná anamnéza tedy nemusí být nijak nápadná.
U dítěte samotného je pak třeba klinické vyšetření zaměřené na specifické příznaky, např. kožní změny, koincidence s vrozenými vadami a jinými onemocněními (např. imunodeficit, ataxie). S ohledem na vlastní nádorové onemocnění je třeba si všímat neobvyklé histologie, lokalizace tumoru či nezvykle časné manifestace onemocnění. Podezřelé by vždy měly být nádorové multiplicity či výskyt malignity u sourozenců.
Diagnóza genetického onemocnění u dítěte je velmi důležitá jednak s ohledem na případnou modifikaci léčby (speciální léčebné režimy u syndromů chromozomální nestability, v řadě případů je nutné omezit nebo vynechat radioterapii), s rozvojem personalizované medicíny se postupně otevírají možnosti tzv. léčby na míru a v neposlední řadě je nutné pro tyto děti zajistit speciální sledování k časné diagnostice event. dalších malignit. S rozvojem nových diagnostických možností i na tomto poli dochází k významnému zlepšení (příkladem je užití celotělové MRI u pacientů s Li ‑ Fraumeniho syndromem).
Rozvoj metod molekulární diagnostiky v poslední době přinesl významné zlepšení diagnostických možností těchto predispozic. Využívány jsou metody sekvenace příští generace, k dispozici je řada panelů umožňujících vyšetření velkého množství genů v rámci jednoho testu, pro klinické použití se otevírají i možnosti sekvenace exomu, či dokonce genomu. Nutno podotknout, že velkým problémem stále zůstává klinická interpretace laboratorních nálezů.
Diagnóza hereditární nádorové predispozice má velký dopad též na příbuzné pacienta. Umožňuje specifikovat jejich onkologické riziko a v případě potřeby zajistit i pro ně speciální prevenci. Pokud rodiče plánují další těhotenství, je možno předejít přenosu dispozice použitím preimplantační, event. prenatální diagnostiky. Je ale nutné, aby o těchto možnostech byli správně informováni.
Autorka deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Věra Krutílková
Gennet
Centrum lékařské genetiky a reprodukční medicíny
Kostelní 9
170 00 Praha 7
e-mail: vera.krutilkova@gennet.cz
Obdrženo: 15. 7. 2015
Přijato: 26. 8. 2015
Sources
1. Bleeker FE, Hopman SM, Merks JH et al. Brain tumors and syndromes in children. Neuropediatrics 2014; 45(3): 137 – 161. doi: 10.1055/ s ‑ 0034 ‑ 1368116.
2. Villani A, Malkin D, Tabori U. Syndromes predisposing to pediatric central nervous system tumors: lessons learned and new promises. Curr Neurol Neurosci Rep 2012; 12(2): 153 – 164. doi: 10.1007/ s11910 ‑ 011 ‑ 0244 ‑ 5.
3. Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog ‑ patched ‑ gli pathway in human development and disease. Am J Hum Genet 2000; 67(5): 1047 – 1054.
4. Poduri A, Evrony GD, Cai X et al. Somatic mutation, genomic variation, and neurological disease. Science 2013; 341(6141): 1237758. doi: 10.1126/ science.1237758.
5. Petrák B, Plevová P, Novotný J et al. Neurfibromatosis von recklinghausen. Klin Onkol 2009; 22 (Suppl): S38 – S44.
6. Plevová P, Krutílková V, Puchmajerová A, et al. Gorlinův syndrom. Klin Onkol 2009; 22 (Suppl): S34 – S35.
7. Amlashi SF, Riffaud L, Brassier G et al. Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population‑based study and review of the literature. Cancer 2003; 98(3): 618 – 624.
8. Choudry Q, Patel HC, Gurusinghe NT et al. Radiation‑induced brain tumors in nevoid basal cell carcinoma syndrome: implications for treatment and surveillance. Child Nerv Syst 2007; 23(1): 133 – 136.
9. Foretová L, Macháčková E, Gaillyová R et al. Hereditární nádorová onemocnění. In: Foretová L, Svoboda M, Slabý Oet al (eds). Molekulární genetika v onkologii. 1. vyd. Praha: Mladá fronta 2014.
10. Vrtěl R, Filipová H, Vodička R et al. Tuberózní skleróza. Klin Onkol 2009; 22 (Suppl): S50 – S53.
11. Meyer S, Tischkowitz M, Chandler K et al. Fanconi anaemia, BRCA2 mutations and childhood cancer: a developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J Med Genet 2014; 51(2): 71 – 75. doi: 10.1136/ jmedgenet ‑ 2013 ‑ 101642.
12. Tischkowitz M, Xia B. PALB2/ FANCN – recombining cancer and Fanconi anemia. Cancer Res 2010; 70(19): 7353 – 7359. doi: 10.1158/ 0008 – 5472.CAN ‑ 10 - 1012.
13. Roelfsema JH, Peters DJ. Rubinstein‑Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med 2007; 9(23): 1 – 16.
14. Bourdeaut F, Miquel C, Richer W et al. Rubinstein‑Taybi syndrome predisposing to non‑WNT, non‑SHH, group 3medulloblastoma. Pediatr Blood Cancer 2014; 61(2): 383 – 386. doi: 10.1002/ pbc.24765.
15. Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys 2009; 74(5): 1323 – 1331. doi: 10.1016/ j.ijrobp.2009.02.057.
16. Gonzales KD, Buzin CH, Noktner KA et al. High frequency of de novo mutations in Li ‑ Fraumeni syndrome: impact on age at first diagnosis. J Med Genet 2009; 46(10): 686 – 693. doi: 10.1136/ jmg.2008.058958.
17. Tinat J, Bougeard L, Ronsin M et al. 2009 version of the Chompret criteria for Li ‑ Fraumeni syndrome. J Clin Oncol 2009; 27(26): e108 – e109. doi: 10.1200/ JCO.2009.22.7967.
18. Gozali AE, Britt B, Shane L et al. Choroid plexus tumors; management, outcome, and association with the Li ‑ Fraumeni syndrome: the Children’s Hospital Los Angeles (CHLA) experience, 1991 – 2010. Pediatr Blood Cancer 2012; 58(6): 905 – 909. doi: 10.1002/ pbc.23349.
19. Krutílková V, Trková M, Fleitz J et al. Identification of five families strengthens the link between childhood choroid plexus carcinoma and germline TP53 mutations. Eur J Cancer 2005; 41(11): 1597 – 1603.
20. Vasen HF, Ghorbanoghli Z, Bourdeaut F et al. Guidelines for surveillance of individuals with constitutional mismatch repair ‑ deficiency proposed by the European Consortium „Care for CMmR ‑ D“(C4CMmR ‑ D). J Med Genet 2014; 51(5): 283 – 293. doi: 10.1136/ jmedgenet ‑ 2013 ‑ 102238.
21. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009; 4 : 22. doi: 10.1186 - 1750 - 1172 - 4 - 22.
22. Sun S, Liu AJ. Long/ term follow‑up studies of Gamma Knife surgery for patients with neurofibromatosis type 2. Neurosurg 2014; 121 (Suppl): 143 – 149. doi: 10.3171/ 2014.8.GKS141503.
23. Bruggers CS, Bleyl SB, Pysher T et al. Clinicopathologic comparison of familial versus sporadic atypical teratoid/ rhabdoid tumors (AT/ RT) of the central nervous system. Pediatr Blood Cancer 2011; 56(7): 1026 – 1031. doi:10.1002/ ppbc.22757.
24. Kanno H, Kuratsu J, Nishikawa R et al. Clinical features of patients bearing central nervous system hemangioblastoma in von Hippel ‑ Lindau disease. Acta Neurochir (Wien) 2013; 155(1): 1 – 7. doi: 10.1007/ s00701 ‑ 012 ‑ 1514 ‑ y.
25. Scheinemann K, Bouffet E (eds). Pediatric neuro‑on-cology. New York: Springer ‑ Verlag 2015.
26. Schiffman JD, Geller JI, Mundt E et al. Update on pediatric cancer predisposition syndromes. Pediatr Blood Cancer 2013; 60(8): 1247 – 1252. doi: 10.1002/ pbc.24555.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice
- Hepatoblastom, etiologie, kazuistiky
- Genetika tumorigenézy nádorov kolorekta (možnosti testovania a screeningovej predikcie dedičnej formy ochorenia – Lynchovho syndrómu)
- Fanconiho anémie, komplementační skupina D1 v důsledku bialelické mutace genu BRCA2 – kazuistika