
Retrospektivní NGS studie u vysoce rizikových pacientů s hereditární predispozicí k nádorovému onemocnění v Masarykově onkologickém ústavu
Retrospective NGS Study in High-risk Hereditary Cancer Patients at Masaryk Memorial Cancer Institute
Background:
Currently, more than 200 hereditary cancer syndromes have been described, yet, in most countries genetic testing is restricted to a narrow spectrum of genes within a limited group of people tested.
Methods:
For this retrospective study we used the TruSight cancer panel (Illumina) – NGS panel targeting 94 cancer predisposition genes in order to analyze 50 high-risk cancer patients with significant personal and family history of cancer who did not carry mutations in BRCA1, BRCA2, MLH1, MSH2, MSH6, TP53 or APC genes. All pathogenic and potentially pathogenic mutations detected by NGS technology have been confirmed by Sanger sequencing.
Results:
There were several deleterious (frame-shift/nonsense) mutations detected in ATM, BAP1, FANCC, FANCI, PMS2, SBDS, ERCC2, RECQL4 genes. Various pathogenic or potentially pathogenic (missense, predicted splice site, in-frame insertion/deletion) mutations were detected in ATM, BRIP1, CDH1, CHEK2, ERCC2, ERCC3, ERCC4, FANCA, MC1R, MEN1, MRE11A, MUTYH, PALB2, RAD51C, RET, SDHB, STK11. These mutations affect highly conserved protein domains and affect their function as proved by the available functional assays. They were confirmed to be pathogenic as an „Parent No2 “ in serious recessive diseases such as Ataxia telangiectasia or Fanconi anemia. The clinical significance of the majority of detected missense variants still remains to be identified.
Conclusion:
Moderate or low penetrance variants are of limited clinical importance. Panel genetic testing in high-risk individuals with cancer provides important information concerning the cause of the investigated cancer, and may assist in the risk assesment and optimal management of the cancer, as well as in further preventive care.
Key words:
hereditary cancer syndromes – hereditary breast and ovarian cancer syndrome – hereditary nonpolyposis colorectal cancer – high-throughput DNA sequencing – TruSight cancer panel – MiSeq
This work was supported by MH CZ – DRO (MMCI, 00209805) and by the State budget project of CR (OP VaVpI – RECAMO CZ.1.05/2.1.00/03.0101).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
20. 8. 2015
Accepted:
22. 9. 2015
Authors:
E. Macháčková; J. Hazova; E. Sťahlová Hrabincová; P. Vašíčková; M. Navratilova; M. Svoboda; L. Foretová
Authors‘ workplace:
Oddělení epidemiologie a genetiky nádorů, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2016; 29(Supplementum 1): 35-45
Category:
Original Articles
doi:
https://doi.org/10.14735/amko2016S35
Overview
Východiska:
V současné době bylo popsáno již více než 200 nádorových syndromů. Ve většině populací jsou však dostupné pouze informace o mutačním spektru ve vysoce rizikových genech limitované počtem vyšetřených jedinců.
Metody:
V rámci retrospektivní NGS studie v Masarykově onkologickém ústavu bylo provedeno vyšetření TruSight Cancer panelem zahrnujícím 94 genů u 50 vysoce rizikových jedinců se závažnou osobní i rodinnou anamnézou onkologického onemocnění bez detekované kauzální mutace v genech BRCA1, BRCA2, MLH1, MSH2, MSH6, TP53 nebo APC dle indikace. Všechny patogenní nebo pravděpodobně patogenní mutace detekované pomocí NGS technologie byly potvrzeny Sangerovým sekvenováním.
Výsledky:
Ztrátové (frame-shift, nonsense) mutace byly detekovány v genech ATM, BAP1, FANCC, FANCI, PMS2, SBDS, ERCC2, RECQL4. Několik patogenních nebo pravděpodobně patogenních mutací (missense, predikované sestřihové mutace, in-frame delece/ inzerce) bylo zachyceno v genech ATM, BRIP1, CDH1, CHEK2, ERCC2, ERCC3, ERCC4, FANCA, MC1R, MEN1, MRE11A, MUTYH, PALB2, RAD51C, RET, SDHB, STK11. Nacházejí se ve vysoce konzervovaných funkčních doménách proteinů a některé z nich již byly prokázány jako patogenní mutace pomocí funkčních testů nebo u závažných autozomálně recesivních syndromů (Ataxia telangiectasia, Fanconiho anémie). Většina z detekovaných missense variant v řadě dalších genů je nejasného klinického významu a determinace jejich významnosti zůstává otevřená do budoucna.
Závěr:
Detekce variant se střední nebo nízkou penetrancí má pouze limitovanou klinickou využitelnost. Panelové testování u vysoce rizikových osob s nádorovým onemocněním může poskytnout důležitou informaci o příčině nádorové predispozice a může pomoci s výběrem optimální léčby a v preventivní personalizované onkologii.
Klíčová slova:
hereditární nádorové syndromy – hereditární nádor prsu a ovaria – hereditární nepolypózní kolorektální karcinom – sekvenování nové generace – TruSight cancer panel – MiSeq
Úvod
Během posledních 20 let došlo k výraznému rozvoji v oblasti vyšetření genetické predispozice k nádorovým onemocněním. Na základě genetických vyšetření byla do klinické praxe zavedena léčebná a preventivní doporučení (guidelines) vedoucí ke snížení rizika a časnému záchytu onkologického onemocnění u osob s výraznou genetickou predispozicí s detekovanou mutací ve vysoce penetrantním genu: např. Li-Fraumeni syndrom (mutace v genu TP53), Lynchův syndrom (mutace v některém z „mismatch“ reparačních genů –MLH1, MSH2, MSH6, PMS2 – u dědičného nepolypózního kolorektálního karcinomu), dědičná forma nádoru prsu a ovaria (mutace v genech BRCA1, BRCA2).
V současné době již bylo popsáno více než 200 různých nádorových syndromů (OMIM databáze), ale molekulárně genetické vyšetření bylo do současné doby dostupné pouze pro nejznámější vysoce rizikové geny.
Pokroky v sekvenační technologii – tzv. sekvenování nové generace (next-generation sequencing – NGS) – umožnily masivní testování sekvence desítek až stovek genů současně, což umožňuje testování známých i potenciálně rizikových genů, které jsou součástí signálních a reparačních drah významných pro nádorovou predispozici. Výrazné rozšíření spektra analyzovaných genů vede nejen k identifikaci kauzálních mutací ve vysoce rizikových genech, ale také k detekci celé řady mutací s dosud nedeterminovanou mírou rizika nebo k detekci středně rizikových variant, které ovšem mohou ve vzájemných kombinacích výrazně navyšovat relativní riziko nádorové predispozice v postižených rodinách.
V současné době jsou nabízeny k vyšetření různé multigenové komerční panely cílené na predikci rizika ke vzniku nádorového onemocnění. K těm rozsáhlejším patří TruSight Cancer Target Genes panel (Illumina), který je jedním z nejčastěji používaných komerčních panelů v laboratořích západní Evropy. TruSight Cancer panel byl vyvinut ve spolupráci Illuminy s týmem profesorky Nazneen Rahman (Institute of Cancer Research ICR, London) v roce 2012 (http://www.illumina.com/products/trusight_cancer.html). Panel je cílen na sekvence 94 genů a 284 SNPs (singe nucleotide polymorphims), které byly v předchozích letech asociovány s hereditární predispozicí k nádorovému onemocnění.
Součástí TruSight Cancer panelu (http:/ / www.illumina.com/ products/ trusight_cancer.html) jsou proby pro analýzu těchto genů: AIP, ALK, APC, ATM, BAP1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, BUB1B, CDC73, CDH1, CDK4, CDKN1C, CDKN2A, CEBPA, CEP57, CHEK2, CYLD, DDB2, DICER1, DIS3L2, EGFR, EPCAM, ERCC2, ERCC3, ERCC4, ERCC5, EXT1, EXT2, EZH2, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FH, FLCN, GATA2, GPC3, HNF1A, HRAS, KIT, MAX, MEN1, MET, MLH1, MSH2, MSH6, MUTYH, NBN, NF1, NF2, NSD1, PALB2, PHOX2B, PMS1, PMS2, PRF1, PRKAR1A, PTCH1, PTEN, RAD51C, RAD51D, RB1, RECQL4, RET, RHBDF2, RUNX1, SBDS, SDHAF2, SDHB, SDHC, SDHD, SLX4, SMAD4, SMARCB1, STK11, SUFU, TMEM127, TP53, TSC1, TSC2, VHL, WRN, WT1, XPA, XPC.
V naší retrospektivní studii jsme se zaměřili na otestování komerčně dostupného TruSight Cancer panelu a možnost jeho využití pro odhalení příčiny nádorové predispozice u dříve neobjasněných retrospektivních případů.
Soubor pacientů
Pilotní retrospektivní studie byla provedena u 50 vzorků genomové DNA izolované z periferní krve. DNA byla získána od pacientů s vysoce rizikovou osobní a rodinnou anamnézou výskytu onkologického onemocnění s různými druhy onkologických diagnóz, u kterých nebyla nalezena riziková mutace v rutinně analyzovaných vysoce rizikových genech. Většina z těchto vybraných pacientů byla již dříve vyšetřena na přítomnost mutací v genech BRCA1, BRCA2 (hereditární syndrom nádoru prsu a ovaria – 28 pacientů), MLH1, MSH2, MSH6 nebo APC (hereditární syndrom nepolypózního nebo polypózního kolorektálního karcinomu – 10 pacientů) nebo TP53 (Li-Fraumeni syndrom), případně na jiné vzácné, s vysokou pravděpodobností hereditární nádorové syndromy bez odhalené příčiny nádorové predispozice (nádory ledvin, mozkové nádory, maligní melanom atd.).
Všichni pacienti zařazení do retrospektivní studie souhlasili s vyšetřením genů, které mohou způsobit predispozici k onkologickému onemocnění, podepsali informovaný souhlas s vyšetřením a souhlasili také s anonymním využitím DNA k lékařskému výzkumu.
Metody
Vyšetření bylo provedeno pomocí Tru-Sight Cancer Target Genes panelu. Principem postupu je enzymatická fragmentace (tagmentace) 50 ng genomové DNA → indexování a pool knihovny → enrichment/ hybridizační (TruSight Rapid Capture protocol) postup s ≈ 4 000 prób cílených na → 1 700 exonů 94 nádorově predispozičních genů (255 Kb cílových sekvencí). Sekvenační běh připravené knihovny (s MiSeq Reagent kit V2) na MiSeq systému (Illumina) → analýza pomocí MiSeq reporter software → anotace VCF souborů pomocí Illumina VariantStudio (www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_trusight_cancer.pdf).
Výběr patogenních a potenciálně patogenních variant (predikce významnosti dle Align-GVGD, PolyPhen-2, SIFT programů u missense variant; predikce ovlivnění sestřihu dle GeneSplicer, NNSPLICE; BDGP Splice Site Prediction) → kontrola správnosti HGVS nomenklatury (www.hgvs.org) u in/del variant → kontrola a případně převedení nálezu na referenční sekvenci doporučenou v příslušných mezinárodních mutačních databázích (HGMD, LOVD, IARC atd.).
Navolení primerů k analýze sledovaných variant pomocí LightScanner Primer Design Software (Idaho Technology Inc.), optimalizace PCR (polymerase chain reaction) podmínek a PCR amplifikace sledované oblasti. Potvrzení přítomnosti významných (patogenních a potenciálně patogenních vhodných k segregační analýze) nálezů pomocí Sangerova sekvenování na 3130 Genetic Analyser (Applied Biosystems) a jeho ověření u nezávisle izolovaného vzorku DNA.
Segregační analýza u rodin zatížených patogenní nebo potenciálně patogenní variantou.
V případě potvrzování nálezů u genů s přítomností pseudogenů byla provedena dvoukolová „nested“ PCR vycházející z publikovaných primerů, kde autoři deklarovali amplifikaci specifických produktů bez přítomnosti pseudogenů. Pro analýzu genu PMS2 byly použity primery pro Long-Range PCR (s použitím Expand Long Template PCR system, Roche) a vnitřní primery dle Vaughna et al [1]. Pro analýzu exonu 2. genu SBDS byly použity vnější primery dle Kawakamiho et al [2] a vnitřní primery byly navoleny softwarem LightScanner Primer Design (Idaho Technology Inc.).
Výsledky a vybrané kazuistiky
Téměř u poloviny z vyšetřených vzorků DNA byly detekovány patogenní nebo možné/ pravděpodobně patogenní (IARC klasifikace UV-4: „likely pathogenic“) mutace v některém z 94 analyzovaných genů.
Ve skupině 28 pacientů s podezřením na dědičnou predispozici nádoru prsu a/nebo ovaria (dříve indikováni k analýze v genech BRCA1, BRCA2 nebo v některých případech také k analýze genu TP53 bez detekované kauzální mutace) byly nejčastěji detekovány mutace v dráhách reparace dvouřetězcových zlomů (geny ATM, CHEK2) a ICL (interstrand cross-links) reparace křížových vazeb v DNA (Fanconi anemia geny – FANC geny) spřažené s homologní rekombinací (geny BRIP1, RAD51C, PALB2). Další dráhou s opakovanými záchyty pravděpodobně významných nálezů byla dráha nukleotidové excizní reparace (NER – geny ERCC2, ERCC3) a geny z rodiny RecQ helikáz (RECQL4, WRN), které se účastní DNA reparačních procesů. Souhrnné výsledky nálezů u skupiny s podezřením na dědičnou predispozici nádoru prsu a/ nebo ovaria jsou uvedeny v tab. 1. Ztrátové (nonsense, frame-shift) mutace byly u této skupiny vyšetřených detekovány v genech: ATM, FANCI, FANCC, RECQL4 a SBDS.
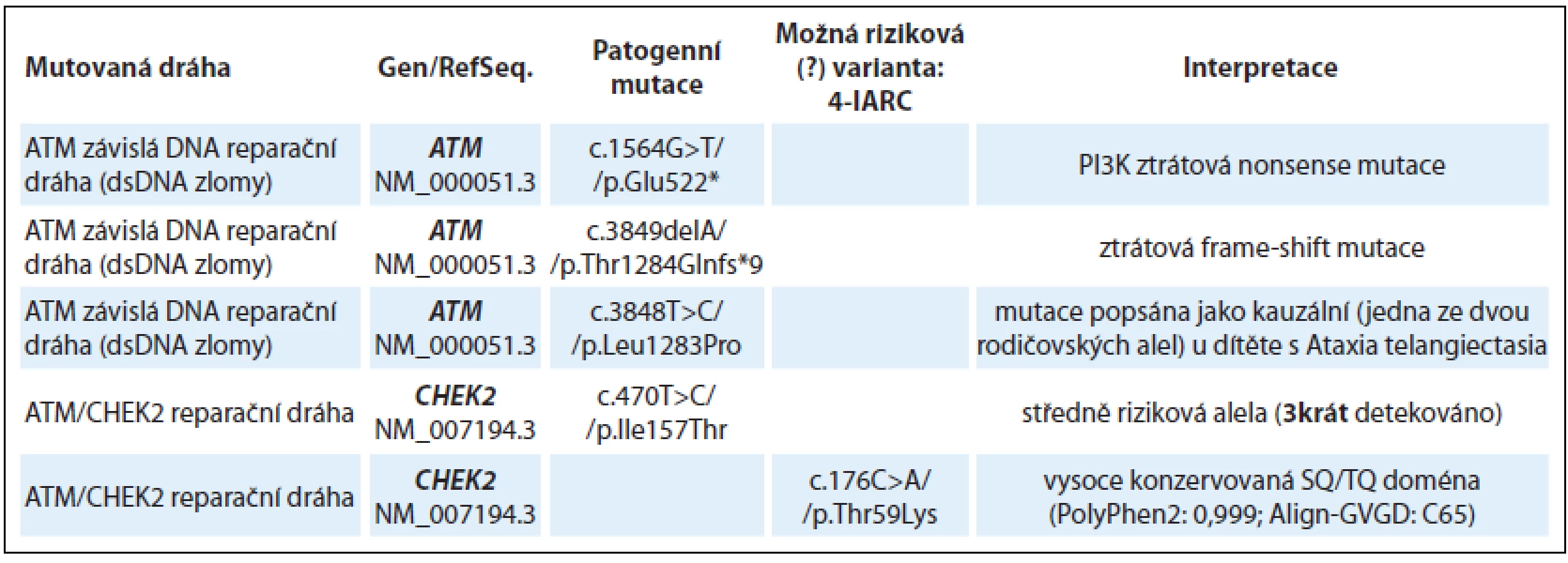

Patogenní missense mutace byly detekovány v genech FANCA, BRIP1 a dále bylo zachyceno několik středně rizikových (≈ 2násobné zvýšení rizika) missense mutací v CHEK2 genu a MC1R receptoru.
Nově detekovaným nálezem je missense mutace v genu BRIP1 (synonymum FANCJ, BACH1) p.Cys350Tyr u ženy s karcinomem prsu (kazuistika 1). BRIP1 missense mutace p.Cys350Tyr je lokalizována v jednom ze čtyř vazebných cysteinů Fe-S domény a je naprosto ztrátová pro helikázovou funkci BRIP1 [3]. BRIP1 helikáza interaguje s komplexem BRCA1/ BRCA2/ RAD51/ PALB2 a je nezbytná pro reparaci dsDNA zlomů a homologní rekombinaci. BRIP1 mutace p.Cys350Tyr zatím nebyla popsána germinálně, ale ztrátové mutace v genu BRIP1 jsou asociovány s predispozicí k nádorům prsu [4].

U některých jedinců bylo nalezeno několik patogenních a potenciálně patogenních variant, zatímco u jiných byly přítomny pouze běžné (nízkorizikové) nálezy.
U pacientky (ročník 1977) s nádorem ovaria ve 29 letech a pozdější diagnózou nádoru štítné žlázy byla detekována ztrátová mutace v genu ATM c.3849delA, přičemž nádory vaječníků nepatří do typického spektra nádorů u ztrátových ATM mutací. U této pacientky však byla zároveň zachycena missense mutace v genu RAD51C p.Thr287Ala, která byla popsána jako riziková mutace na základě funkčního testu [5,6]. RAD51C je protein funkční v procesech homologní rekombinace a DNA reparace spřažené s homologní rekombinací. Patogenní mutace v genu RAD51C/ FANCO jsou popisovány především v souvislosti s HBOC fenotypem v postižených rodinách a v případě homozygotní nebo bialelické mutace s projevy recesivního syndromu Fanconiho anémie (FA) [7]. A právě mutace v genu RAD51C jsou častěji přítomny u ovariálních nádorů a mohlo by se v tomto případě jednat o významný modifikační faktor. Analýza probandek s karcinomem ovaria z HOC a HBOC rodin v ČR prokázala přítomnost patogenních mutací v genech RAD51C a RAD51D u 3 % vyšetřovaných [8].
Jiným příkladem vícečetných nálezů je žena (ročník 1970) s nádorem prsu v 32 letech s missense mutací ve vysoce konzervované doméně genu ATM p.Leu1283Pro, která byla již dříve zachycena jako jedna z rodičovských alel u dítěte s recesivním syndromem Ataxia Telangiectasia (Ataxia-Telangiectasia databáze). Jako další riziková mutace byla u této ženy missense mutace v genu FANCA p.Arg517Trp, která se opět nachází ve vysoce konzervované doméně a byla zachycena germinálně jako druhá alelická mutace v německé populaci u ženy s relativně pozdními projevy FA (AML po 40. roce věku) a s nádorem prsu ve 37 letech [9].
U pacientky s nádorem slinivky (ročník 1992) s rodinnou anamnézou karcinomu prsu a karcinomu slinivky byla zachycena nonsense mutace v genu FANCI p.Arg1285* (kazuistika 2). Autozomálně recesivní syndrom FA je způsoben bi-alelickými mutacemi v FA genech. Relativní riziko u heterozygotních nosičů mutace v některém z FANC genů je značně variabilní [10]. Mutace v genu FANCI jsou celosvětově velmi vzácné, riziko solidních nádorů u heterozygotních nosičů ve FANCI genu není známo (LOVD Fanconi anemia database [11]).

Pacienti s podezřením na HNPCC nebo FAP
Ve skupině 10 pacientů s podezřením na dědičnou predispozici nádoru k nádoru kolorekta bez detekované kauzální mutace v genech MLH1, MSH2, MSH6 (nepolypózní kolorektální karcinom) nebo APC (familiární polypózní syndrom) byly detekovány patogenní mutace v reparační dráze bázové excizní reparace BER (gen MUTYH způsobující autozomálně recesivní formu polypózy), středně rizikové mutace v genu CHEK2 a „in-frame“ delece v genu MEN1 c.809_817del, která již byla popsána u jedinců s MEN1 syndromem [12] a v naší studii byla zachycena u mladého muže (ročník 1986) s rodinnou anamnézou nádoru kolorekta, gastrinomu a nádoru žaludku. Souhrnné výsledky nálezů u skupiny s podezřením na dědičnou predispozici k nádorům GIT jsou uvedeny v tab. 2. Je zde zařazena také žena (ročník 1946) s triplicitou: duplicitní nádor prsu (52, 62 let) s adenokarcinomem tlustého střeva a maligním melanomem, která byla jako jediná zařazena v obou výše zmíněných skupinách. U pacientky byla detekována frame-shift mutace v genu FANCC c.455dupA (tab. 1) spolu s pravděpodobně patogenní in/ del mutací v genu STK11 (synonymum LKB1) c.740_743delinsTCAC/p.Asn247_Ile248delinsIleThr nacházející se ve vysoce konzervované katalytické doméně STK11 kinázy (tab. 2). V tomto případě můžeme spekulovat, že FANCC mutace je pravděpodobným rizikovým faktorem predispozice k nádoru prsu a STK11 mutace ke vzniku adenokarcinomu.
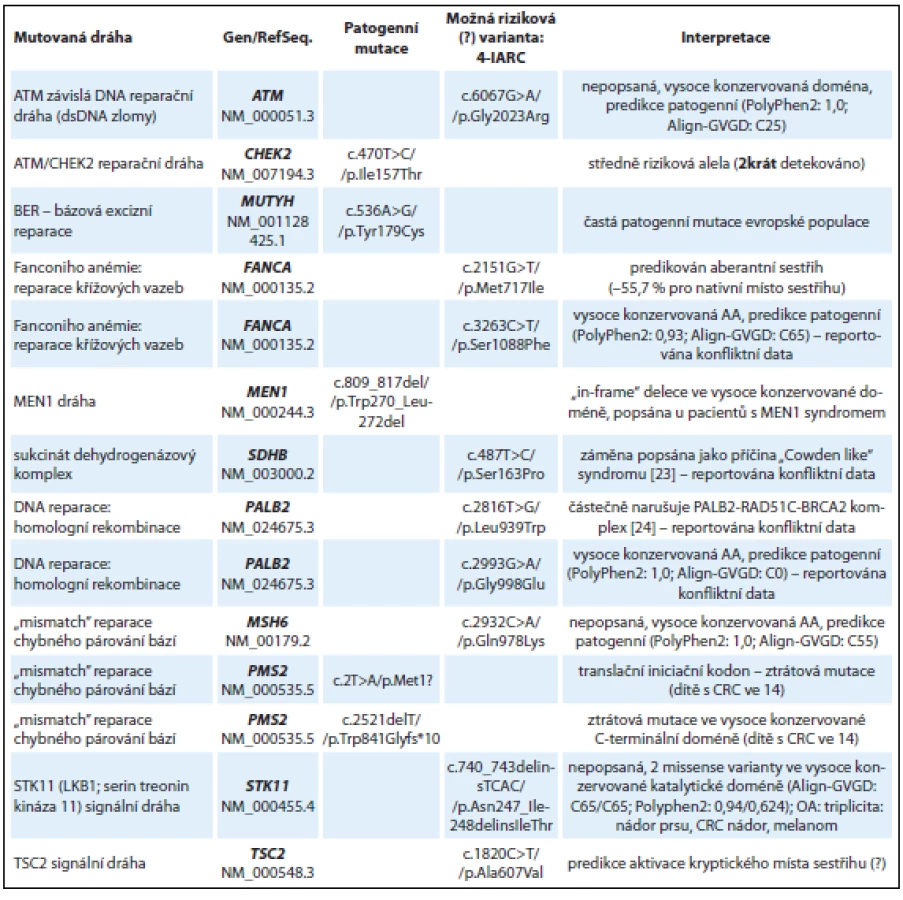
U dítěte (chlapec, ročník 1999) diagnostikovaného s kolorektálním karcinomem ve 14 letech byly detekovány dvě patogenní mutace v genu PMS2 (kazuistika 3). Inaktivující PMS2 mutace c.2T>A v iniciačním kodonu pro metionin p.Met1? je patogenní mutací znemožňující syntézu proteinu. Druhou detekovanou PMS2 mutací je c.2521delT, která způsobuje posun čtecího rámce (frame-shift) a předčasnou terminaci translace v C-terminální dimerizační doméně PMS2 proteinu, která je klíčová pro vazbu s MLH1 proteinem [13]. Bialelické mutace v PMS2 genu způsobují závažnou MMR deficienci s manifestací intestinálních nádorů v dětském věku [14].

Pacienti se sarkomy nebo nádory mozku
V tab. 3 je uveden souhrn nálezů u skupiny pěti pacientů s diagnózami sarkomu nebo nádory mozku bez detekované mutace v genu TP53. Bezpochyby významným nálezem je missense mutace v genu ATM p.Asn2875Ser, kde Asn2875 je katalytickou aminokyselinou v aktivním místě PI3K domény, a bylo experimentálně prokázáno, že substituce za jinou aminokyselinu (Asn > Lys, Asn > Ser) inaktivuje kinázovou aktivitu ATM [15,16]. ATM mutace p.Asn2875Ser zatím nebyla germinálně popsána (kazuistika 4).


Pacienti s různými typy nádorů
V tab. 4 jsou uvedeny souhrnné výsledky u osmi pacientů s různými druhy nádorů s pravděpodobnou genetickou predispozicí, kteří již byli dříve vyšetřeni na mutace v různých genech (dle diagnózy TP53, MLH1, MSH2, MSH6, FLCN, VHL, RB1) bez nálezu kauzální patogenní mutace.
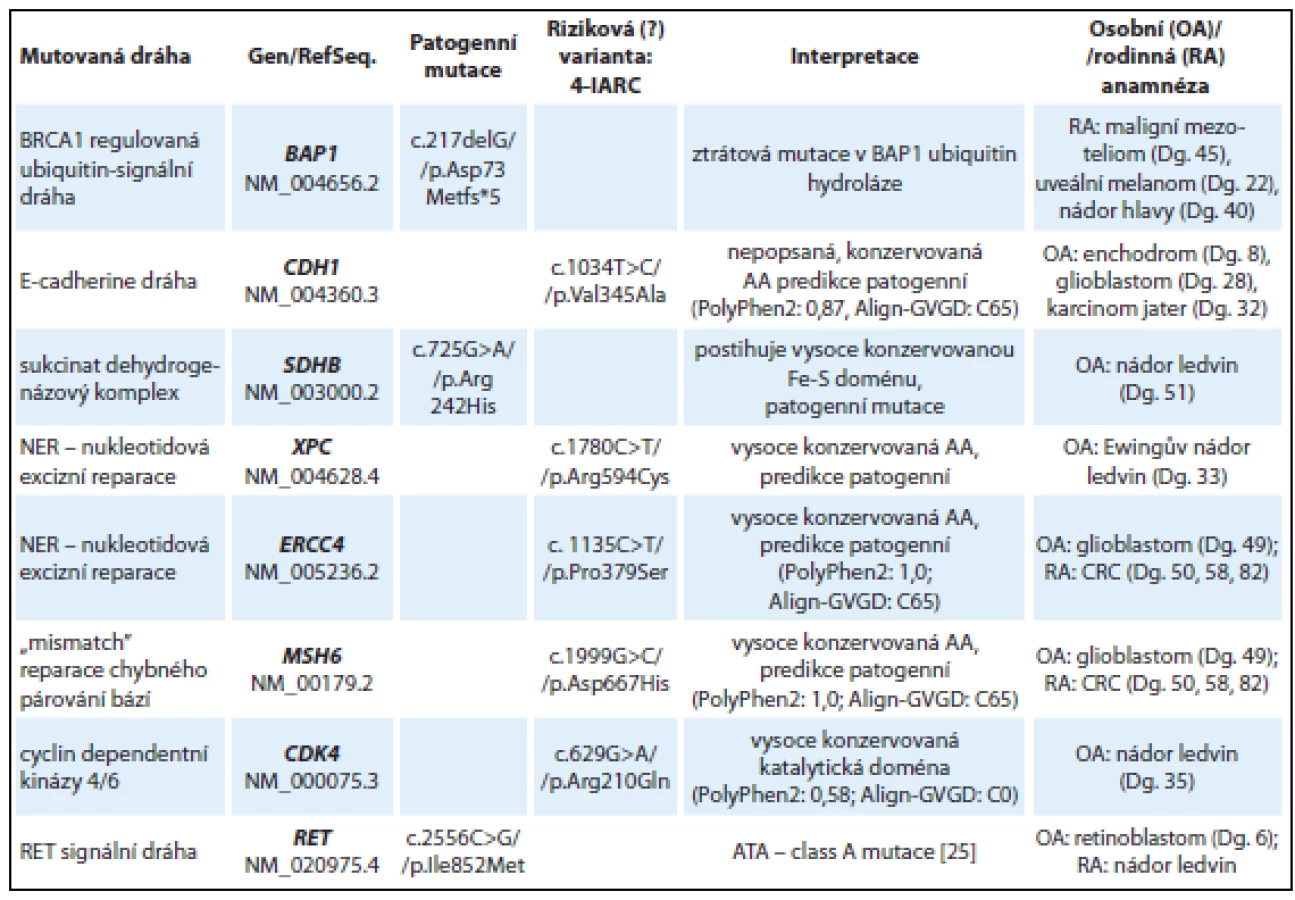
U mladé ženy s rodinnou anamnézou maligního mezoteliomu a uveálního melanomu byla detekována frame-shift mutace v genu BAP1: c.217delG (kazuistika 5). Gen BAP1 kóduje ubikvitin-karboxyterminální hydrolázu, která se nachází v komplexu s BRCA1 proteinem a účastní se DNA reparačních procesů, regulace transkripce a buněčné proliferace. Mutace v genu BAP1 byly popsány u autozomálně dominantního nádorového syndromu, který je asociován především s výskytem maligního mezoteliomu a uveálního melanomu [17].

U pacienta s karcinomem ledviny v 51 letech byla detekována patogenní missense mutace v genu SDHB p.Arg242His, která se nachází ve vysoce konzervované Fe-S doméně SDHB proteinu. Jedná se o již dříve popsanou mutaci u pacientů s diagnózou feochromocytomu, paragangliomu a GIT stromálních tumorů (LOVD databáze, [18]).
Při analýze 94 genů je u každého jedince detekováno také velké množství variant nejasného významu (IARC klasifikace UV-3, nejasného významu – neklasifikované [19]) a frekvenčně častých nízko rizikových polymorfních variant. Tyto varianty však mají minimální klinickou využitelnost [20] a v tomto stručném sdělení nebudou rozebírány.
Diskuze
Technologie NGS v současné době přechází z výzkumu do diagnostické praxe. Rutinní klinické využití vyžaduje vysokou senzitivitu, vysokou specificitu, pokud možno jednoduchost přípravy sekvenačních knihoven, cenovou přijatelnost a uživatelsky zvládnutelnou analýzu dat.
Genetické testy by měly být hodnoceny na základě analytické validity, klinické validity a klinické využitelnosti [20].
Analytická validita vyjadřuje stupeň citlivosti a specificity testu, zda správně vyhodnotí přítomnost nebo nepřítomnost mutace (varianty) ve vyšetřovaném genu. Nezbytné je dostatečné pokrytí (min. 50krát v případě germinálních mutací) kódujících oblastí analyzovaných genů včetně sestřihových míst v intronech (min. ± 20 bp). S klesajícím pokrytím narůstá množství falešně pozitivních záchytů.
Při detekci patogenní germinální mutace v případě, že tyto výsledky mají být použity ke klinickým účelům, je nezbytný průkaz její přítomnosti Sangerovým sekvenováním a potvrzení z nezávisle izolovaného vzorku DNA (nejlépe z nového odběru). To zahrnuje navolení primerů specifických pro analyzovanou oblast, optimalizaci podmínek amplifikace a sekvenování „klasickou“ Sangerovou metodou. Tyto primery jsou pak dále využity pro segregační analýzu v postižené rodině.
V případě detekce missense variant s „pouze“ predikovaným patogenním efektem jsou zapotřebí další analýzy, jako je segregační analýza, funkční testy zaměřené na funkční domény s přítomností detekované varianty, analýza sestřihu na mRNA úrovni u predikovaných sestřihových mutací.
U GC bohatých oblastí často dochází k chybnému alignmentu (mapování na referenční sekvenci) až k úplnému výpadku analyzovaných sekvencí (V TruSight Cancer panelu je to markantní např. u genů PHOX2B nebo CDKN1C). Je proto nezbytné kontrolovat nejen detekované varianty, ale je třeba brát v úvahu také nepokryté úseky analyzovaných genů a v případě indikace vyšetření těchto genů, doplnit vyšetření chybějících oblastí alternativní metodou.
Z našich zkušeností jsou NGS technologie méně citlivé na rozsáhlejší delece/ duplikace přesahující cca 10 párů bází. Obecně ale nelze definovat pouze rozsah, protože záleží na sekvenci analyzovaného úseků a lokalizaci delece/inzerce vzhledem k pozici oligonukleotidů hybridizujících na cílené sekvence (u enrichment/ hybridizačního postupu). Se zvyšujícím se rozsahem delece/ duplikace klesá podíl zastoupení alely s delecí v celkovém pokrytí. Enrichment/hybridizační postup je výrazně specifičtější než amplifikační příprava knihovny a omezuje zanášení chyb v průběhu amplifikace [21].
Další problematickou oblastí jsou geny s pseudogeny. V analyzovaném souboru jsme řešili průkaz detekované přítomnosti mutace v genech PMS2 a SBDS. V těchto případech není možná přímá amplifikace analyzovaných úseků z důvodů vysoké homologie mezi genem a jeho pseudogeny. V případě genu PMS2 jsme při potvrzení obou mutací mohli vyjít z publikovaného zdroje a optimalizovat „nested“ PCR s publikovanými primery [1]. V případě nonsense mutace v genu SBDS c.184A>T/p.Lys62* byla situace složitější. Homologie mezi SBDS genem a pseudogenem SBDSP1 je 97 % a navíc v pozici 184 pseudogenu je T jako standardní nukleotid (SBDSP1: NR_024110.1: n.647T; g.72301327T). Již při prvotní přímé amplifikaci exonu 2 bylo ze srovnání vzorku pacientky a několika kontrolních vzorků patrné, že zastoupení c.184A>T odpovídá 50 % u kontrolních vzorků a cca 75 % u vzorku pacientky a zároveň byly ve všech vzorcích přítomny čtyři další varianty odpovídající sekvenci SBDSP1 pseudogenu. Ke specifické amplifikaci exonu 2 jsme použili publikované vnější primery [2] a vnitřní primery pro „nested“ PCR již byly navoleny softwarem LightScanner Primer Design (Idaho Technology Inc.). Tímto způsobem bylo možné získat produkt specifický pouze pro exon 2 SBDS genu bez sekvence pseudogenu.
Klinická validita zahrnuje hodnocení významnosti detekované varianty a odhad míry rizika pro nosiče této varianty. Odhad míry rizika genetických nálezů je klíčový pro klinickou využitelnost panelového testování, které by mělo vést k individuálnímu doporučení pro plán preventivního sledování, cílené medikaci nebo chirurgickým profylaktickým zákrokům, které mohou riziko nádorového onemocnění snížit.
Abychom mohli stanovit klinickou validitu, musíme znát odpověď na tyto základní otázky [20]:
- Jsou mutace v konkrétním genu asociovány s nádorovou predispozicí?
- Které varianty nebo skupiny variant jsou rizikové mutace a pro které typy nádorů?
- Jaká je výše nádorové predispozice vzhledem k incidenci onemocnění?
V oblasti klinického hodnocení je nutné vycházet z výsledků biologického hodnocení nalezené varianty, ze segregačních studií a z dosavadních klinických znalostí o uvedeném genu. U nosičů vysoce rizikových patogenních mutací v genech velkého účinku (vyšší než čtyřnásobné riziko vzhledem k běžné populaci: např. mutace v genech TP53, BRCA1, BRCA2, CDH1, MLH1, PTEN, APC atd.) lze navrhnout dispenzarizaci nosičů dle platných doporučení. V případě neznámé klinické validity není vhodné nabízet prediktivní testování zdravých příbuzných, ale odhadnout riziko a navrhnout prevenci dle rodinné anamnézy.
Pro řadu genů z TruSight panelu ale zatím nebyla doporučení stanovena a míra relativního rizika pro heterozygotní nosiče mutací je nejasná.
Dispenzarizace u středně rizikových genů většinou zatím nebyla navržena ani publikována. Incidence středního rizika je uváděna jako cca 2–4násobná v porovnání s rizikem průměrné populace [22]. Kumulací získaných dat (v rámci multicentrických studií) o středně rizikových genech je možné postupně odhalit předpokládanou výši a spektrum nádorových rizik a navrhnout rozsah preventivní péče. Zatím je možné se opřít o publikované informace jednotlivých studií, a především o empirická rizika v testované rodině.
Odhad rizika založený na analýzách v přísně selektované vysoce rizikové skupině rodinných případů nemusí vždy odrážet správnou hodnotu průměrného rizika u nosičů v běžné populaci [20]. V současné době chybí pro naši populaci srovnávací výsledky analýz v dostatečně velké kontrolní skupině – zdravé věkově starší populaci bez nádorové predispozice v osobní nebo rodinné anamnéze.
Detekce nízkorizikových mutací a častých polymorfních variant, u kterých je uváděna spíše modifikace rizika, nemají z klinického hlediska uplatnění. Na velkých genových panelech jsou jich detekovány desítky a jsou s podobnou frekvencí detekovány také u kontrolních vzorků.
U nosičů patogenních mutací v genech pro známé recesivní syndromy (FA, AT, XP, aj.) je vhodné zvážit podání informace o mutaci z důvodů možného výskytu tohoto syndromu v dalších generacích. Frekvence nosičství mutací v těchto genech je však velmi vzácná a pravděpodobnost autozomálně recesiního syndromu v dalších generacích je nízká. Proto dosud není jasný přístup k této problematice a je vhodné postupovat individuálně.
Závěr
Téměř u poloviny z vyšetřených jedinců byly detekovány patogenní nebo pravděpodobně patogenní (IARC-4) mutace v některém z analyzovaných genů. Ztrátové (nonsense, frame-shift) mutace byly detekovány v genech: ATM, BAP1, FANCI, FANCC, PMS2, RECQL4, SBDS. Dále bylo nalezeno několik s vysokou pravděpodobností patogenních missense variant nebo „in-frame inzercí/delecí v genech: ATM, BRIP1, CDH1, CHEK2, ERCC2, ERCC3, ERCC4, FANCA, MC1R, MEN1, MRE11A, MUTYH, PALB2, RAD51C, RET, SDHB, STK11, které se nacházejí ve vysoce konzervovaných funkčních doménách proteinu a některé z nich již byly prokázány jako patogenní mutace pomocí funkčních testů nebo u závažných autozomálně recesivních syndromů (Ataxia telangiectasia, FA).
Pro klinické využití je nezbytné stanovení relativního rizika, což je v řadě případů ve fázi výzkumných studií. Detekce variant s nízkou penetrancí má pouze limitovanou klinickou využitelnost. Rozšíření spektra analyzovaných genů představuje výzvu pro zajištění odpovídajícího preventivního sledování a léčebných postupů u osob v riziku. Je zapotřebí úzká spolupráce onkologů, klinických genetiků, molekulárních biologů a bioinformatiků, aby bylo možné plně využít potenciál NGS technologií v běžné diagnostické praxi a preventivní personalizované onkologii.
V oblasti klinického hodnocení je nutné vycházet z výsledků biologického hodnocení nalezené varianty, ze segregační studie a z dosavadních klinických znalostí o uvedeném genu. V případě neznámé klinické validity není vhodné nabízet prediktivní testování zdravých příbuzných, ale odhadnout riziko a navrhnout prevenci dle rodinné anamnézy.
Využití NGS technologie a vyšetření panelů mnoha desítek genů pro hereditární nádorová onemocnění je významným posunem nejen v možnostech prevence nádorových onemocnění u vysoce rizikových osob.
Práce byla podpořena MZ ČR – RVO (MOÚ, 00209805) a Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO CZ.1.05/2.1.00/03.0101).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
RNDr. Eva Macháčková, Ph.D.
Oddělení epidemiologie a genetiky nádorů
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: emachack@mou.cz
Obdrženo: 20. 8. 2015
Přijato: 22. 9. 2015
Sources
1. Vaughn CP, Robles J, Swensen JJ et al. Clinical analysis of PMS2: mutation detection and avoidance of pseudogenes. Hum Mutat 2010; 31(5): 588–593. doi: 10.1002/ humu.21230.
2. Kawakami T, Mitsui T, Kanai M et al. Genetic analysis of Shwachman-diamond syndrome: phenotypic heterogeneity in patients carrying identical SBDS mutations. Tohoku J Exp Med 2005; 206(3): 253–259.
3. Wu Y, Brosh RM. DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res 2012; 40(10): 4247–4260. doi: 10.1093/ nar/ gks039.
4. De Nicolo A, Tancredi M, Lombardi G et al. A novel breast cancer – associated BRIP1 (FANCJ/ BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res 2008; 14(14): 4672–4680. doi: 10.1158/ 1078-0432.CCR-08-0087.
5. Vaz F, Hanenberg H, Schuster B et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 2010, 42(5): 406–409.
6. Meindl A, Hellebrand H, Wiek C et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 2010; 42(5): 410–414. doi: 10.1038/ ng.569.
7. Clague J, Wilhoite G, Adamson A et al. RAD51C germ-line mutations in breast and ovarian cancer cases from high-risk families. PLoS One 2011; 6(9): e25632. doi: 10.1371/ journal.pone.0025632.
8. Janatova M, Soukupova J, Stribrna J et al. Mutation analysis of the RAD51C and RAD51D genes in high-risk ovarian cancer patients and families from the Czech Republic. PLoS One 2015; 10(6): e0127711. doi: 10.1371/ journal.pone.0127711.
9. Huck K, Hanenberg H, Gudowius S et al. Delayed diagnosis and complications of Fanconi anaemia at advanced age – a paradigm. Br J Haematol 2006; 133(2): 188–197.
10. Berwick M, Satagopan JM, Ben-Porat L et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res 2007; 67(19): 9591–9596.
11. Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol 2006; 24(23): 3799–3808.
12. Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29(1): 22–32.
13. Wu X, Platt JL, Cascalho M. Dimerization of MLH1 and PMS2 limits nuclear localization of MutLalpha. Mol Cell Biol 2003; 23(9): 3320–3328.
14. Herkert J, Niessen R, Olderode-Berends M et al. Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: case series, review and follow-up guidelines. Eur J Cancer 2011; 47: 965–982. doi: 10.1016/ j.ejca.2011.01.013.
15. Rahal EA, Henricksen LA, Li Y et al. ATM regulates Mre11-dependent DNA end-degradation and microhomology – mediated end joining. Cell Cycle 2010; 9(14): 2866–2877.
16. Edvadsen H, Tefre T, Jansen L et al. Linkage disequilibrium pattern of the ATM gene in breast cancer patients and controls; association of SNPs and haplotypes to radio-sensitivity and post-lumpectomy local recurrence. Radiat Oncol 2007; 2: 25–33.
17. Testa JR, Cheung M, Pei J et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011; 43(10): 1022–1025. doi: 10.1038/ ng.912.
18. Janeway KA, Kim SY, Lodish M et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 2011; 108(1): 314–318. doi: 10.1073/ pnas.1009199108.
19. Plon SE, Eccles D, Easton D et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29(11): 1282–1291. doi: 10.1002/ humu.20880.
20. Easton DF, Pharoah PD, Antoniou AC et al. Gene-Panel sequencing and the prediction of breast cancer risk. N Engl J Med 2015; 372: 2243–2257. doi: 10.1056/ NEJMsr1501341.
21. Desai AN, Jere A. Next-generation sequencing: redy for the clinics? Clin Gene 2012; 81(6): 503–510. doi: 10.1111/ j.1399-0004.2012.01865.x.
22. Hollestelle A, Wasielewski M, Martens JW et al. Discovering moderate-risk breast cancer susceptibility genes. Curr Opin Genet Dev 2010; 20(3): 268–276. doi: 10.1016/ j.gde.2010.02.009.
23. Ni Y, Zbuk KM, Sadler T et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet 2008; 83(2): 261–268. doi: 10.1016/ j.ajhg.2008.07.011.
24. Park JY, Singh TR, Nassar N et al. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014; 33(40): 4803–4812. doi: 10.1038/ onc.2013.421.
25. Machens A, Spitschak A, Lorenz K et al. Germline RET sequence variation I852M and occult medullary thyroid cancer: harmless polymorphism or causative mutation? Clin Endocrinol 2011; 75(6): 801–805. doi: 10.1111/ j.1365-2265.2011.04158.x.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 1
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
- Spasmolytic Effect of Metamizole
- Obstacle Called Vasospasm: Which Solution Is Most Effective in Microsurgery and How to Pharmacologically Assist It?
Most read in this issue
- PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice
- Hepatoblastom, etiologie, kazuistiky
- Genetika tumorigenézy nádorov kolorekta (možnosti testovania a screeningovej predikcie dedičnej formy ochorenia – Lynchovho syndrómu)
- Fanconiho anémie, komplementační skupina D1 v důsledku bialelické mutace genu BRCA2 – kazuistika