
Syndromy predisponující k nádorům v dětském věku – zkušenosti Kliniky dětské onkologie FN Brno
Syndromes Predisposing to Cancer in Children – the Experience of Pediatric Oncology Departmentof University Hospital Brno
There is a broad spectrum of hereditary cancer predisposition syndromes affecting pediatric population. Early genetic testing establishing the diagnosis may assist in patient dispensarisation and management. Secondary prevention and prompt diagnosis of cancer improves the prognosis and overall survival. A centralised dispensarisation of pediatric patients with hereditary cancer predisposition syndromes on pediatric oncology department offers complex multidisciplinary care to the patient and his family.
Key words:
hereditary cancer predisposition syndromes – dysmorphology – pediatric cancer – genetic testing
The author declares she has no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
14. 7. 2015
Accepted:
18. 8. 2015
Authors:
V. Bajčiová
Authors‘ workplace:
Klinika dětské onkologie LF MU a FN Brno
Published in:
Klin Onkol 2016; 29(Supplementum 1): 62-70
Category:
Review
doi:
https://doi.org/10.14735/amko2016S62
Overview
V dětské populaci se vyskytuje široké spektrum hereditárních nádorových predispozičních syndromů. Časné genetické testování a diagnóza syndromu může pomoci v dispenzarizaci pacienta a zdravotní péči. Sekundární prevence a časná diagnostika nádoru zlepšuje prognózu pacienta a jeho celkové přežití. Centralizované sledování dětských pacientů s hereditárním predispozičním syndromem na pracovišti dětské onkologie nabízí pacientovi a jeho rodině komplexní multidisciplinární péči.
Klíčová slova:
hereditární nádorové predispoziční syndromy – dysmorfie – dětské typy nádorů – genetické testování
Úvod
V posledních letech prudce stoupají informace o příčinách a mechanizmu vzniku nádorových onemocnění. Byly definovány molekulární základy mnoha hereditárních nemocí, které vedly k pochopení mechanizmu hereditární predispozice ke vzniku nádoru.
Spektrum a typy nádorů u dětí a mladégenerace dospívajících do 20 let věku se zásadně liší od nádorů dospělé a starší populace. Vyvolávající příčiny, stavy a změny vedoucí ke vzniku nádoru mohou být rozdílné od příčin známých z dospělé onkologie, ale molekulárně genetické mechanizmy postupné proměny zdravé buňky v nádorovou jsou stejné v každém věku. Dětské typy nádorů mají jiné predispoziční syndromy a stavy asociované se vznikem nádoru ve srovnání s dospělou onkologií.
Správná a včasná diagnóza a poznání hereditární zátěže má význam nejen pro samotné dítě, ale i jeho rodinu. Genetické vyšetření a potvrzení hereditární zátěže u dítěte nese s sebou často celou řadu psychosociálních problémů v rodině – hněv, strach, karcinofobii, deprese, pocit hanby a méněcennosti, pocit ztráty kontroly, izolace a nezřídka pocit viny a obviňování rodičů vzájemně. Severoamerická Společnost klinické onkologie (ASCO) doporučuje genetické testování a vyšetření u dětí pod 15 let pouze v případě, kdy součástí hereditární zátěže jsou nádory vyskytující se v dětském věku a jsou doloženy strategie a postupy snižující riziko rozvoje tohoto nádoru při včasném odhalení genetického rizika. Pokud je syndrom spojen s nádorovým onemocněním projevujícím se až v dospělém a starším věku, je doporučeno genetické testování po 15. roku dítěte [1,2].
Příčiny vzniku nádorů u dětí
Nádory u dětí patří mezi vzácné nemoci, z celkového počtu zhoubných onemocnění v populaci tvoří pouze 1 %. V ČR je ročně diagnostikováno téměř 85 000 zhoubných onemocnění, ale z nich je pouze 350 – 380 dětí pod 15 let věku a přibližně stejný počet dospívajících do 20 let věku. Moderní dětská onkologie dnes dokáže vyléčit více než 85 % onkologicky nemocných dětí, přesto jsou zhoubná onemocnění 2. nejčastější příčinou úmrtí u mladé generace do 20 let věku.
Nádorové onemocnění je komplexní, multifaktoriální a vícestupňový proces. V dětské onkologii nemají faktory zevního prostředí zásadní význam. Na vzniku nádoru, především u malého dítěte do pěti let věku, se podílejí pouze nepřímo (životní styl rodičů, kouření v rodině, stravovací návyky rodičů, hygienický standard rodiny, stres, pracovní zátěž).
Výskyt dětských typů nádorů je většinou sporadický (80 %), nádory se vyskytnou náhodně bez známé příčiny či identifikace rizikového faktoru. Častější výskyt nádorů u dětí je pozorován u různých vrozených vývojových vad (kardiovaskulárních, urogenitálních, skeletálních). Kumulativní riziko vzniku nádoru roste s věkem. Děti s vrozenou non‑chromozomální vývojovou vadou mají až dvojnásobně vyšší riziko vzniku nádoru před dosažením 15 let věku. Největší riziko vzniku nádoru je u vrozených vad urogenitálního systému, mikrocefalií a parciálního, tzv. overgrowth syndromu (hemihypertrofie). U těchto dětí je vyšší riziko tzv. embryonálních typů nádorů (hepatoblastom, nefroblastom, neuroblastom). Biologicky tyto nádory mohou představovat konečnou změnu v poruše naprogramovaného procesu organogeneze v průběhu embryonálního života [3,4].
Hereditárně podmíněné nádory tvoří asi 5 – 10 % nádorů dospělého věku. U dětí lze předpokládat, že hereditární zátěž je vyšší a může být příčinou 15 – 29 % zhoubných nádorů dětského věku [5].
Podezření na hereditárně podmíněný nádor lze vyslovit na základě vyšetření:
- a) individuálního pacienta (vícečetné primární nádory, bilaterální nádory v párových orgánech, výskyt vzácného (dospělého) typu nádoru v dětském věku, výskyt velmi vzácného typu nádoru, výskyt nádoru s projevy typickými pro určitý syndrom, výskyt nádoru asociovaného s jinou vzácnou nemocí, nádor asociovaný s vrozenou vývojovou abnormalitou nebo kongenitálním defektem nebo přítomnost dysmorfie);
- b) rodiny (stejný typ nádoru u příbuzného 1. linie, dva a více příbuzných 1. linie s nádorem stejné lokalizace, známý familiární nádorový syndrom, dva a více příbuzných 2. linie ve stejné lokalizaci, dva a více příbuzných 1. linie se vzácným typem nádoru).
Na základě objevení predispozičních genů pro různé dědičné nádorové syndromy je možné předcházet vzniku nádorů mnohem účinněji než dříve a začít se specializovanou prevencí již v mladém věku, u mnohých syndromů již od dětství. Identifikace dědičné predispozice k nádorům je důležitou součástí cílené onkologické prevence v populaci [6,7].
Familiární výskyt nádorů je spojen se zvýšeným výskytem maligních onemocnění v rodině, kde genetickým vyšetřením není zjištěna žádná kauzální germinální mutace. Familiární výskyt může být způsoben blíže neurčenou interakcí gen ‑ prostředí, nízce penetrujícími geny nebo kombinací více mechanizmů. Je popsán u 10 – 15 % onkologických pacientů dospělého věku a u méně než 10 % dětských typů nádorů [8,9].
Hereditárních nádorových predispozičních syndromů je dosud popsáno kolem 200. Většinou jsou vzácné, s autozomálně dominantním přenosem. Spektrum dědičných nádorových syndromů vedoucích ke vzniku nádoru v dětském věku je poněkud odlišné než v onkologii dospělého věku. Například syndrom familiární adenomatózní polypózy, který u dospělých vede k časnému rozvoji kolorektálního karcinomu, může být příčinou rozvoje hepatoblastomu již v časném věku do tří let. Riziko karcinomu střeva se zvyšuje v období dospívání [10,11].
Kdy myslet na možnost nádorového predispozičního syndromu
Na možnou účast genetické příčiny onemocnění je nutné myslet prakticky u každého dítěte s maligním nádorem. V běžné praxi mohou lékaři pomoci tzv. varovná znamení, zjištěná rodinnou anamnézou, klinickým vyšetřením a vzezřením dítěte, hodnocením jeho psychomotorického vývoje, případně laboratorním vyšetřením [12].
První, kdo detekují příznaky a možné znaky dysmorfizmu, které mohou být projevy a součástí různých genetických syndromů, jsou obvykle pediatři primárního kontaktu. Hereditárních genetických syndromů je celá řada, ale ne všechny jsou asociovány se zvýšeným výskytem onkologických onemocnění v dětském věku. Ty, které jsou asociovány se zvýšeným výskytem jistých typů nádorů, splňují kritéria genetických nádorových predispozičních syndromů. Jejich rozpoznání může být někdy problematické, protože projevy nemusí být vždy plně vyjádřeny a mohou se v různých obměnách objevit i u jiných členů rodiny [13,14].
Různé projevy dysmorfizmu typické pro danou genetickou změnu a specifický predispoziční syndrom jsou u dítěte často jeho jediným klinickým projevem. Při hodnocení dysmorfických znaků (tj. abnormálních tělesných známek) u dětí je nutno hodnotit růst (gigantizmus nebo naopak snížený růst, proporcionalitu těla), obvod hlavy (mikro ‑ nebo makrocefalie), čelo (prominence, šikmost), oči (vzdálenost a tvar očních štěrbin, epikantus, aniridie), tvar nosu, uši (tvar, pozici, velikost), ústa, skus, patro, krk a trup (tvar, symetrie, zadní vlasová hranice), genitál dle věku, končetiny (kostní abnormality, proporce velikosti vzhledem k trupu), prsty (polydaktylie, syndaktylie), kůži (café ‑ au ‑ lait, pigmentová znaménka, fibromy, změny pigmentace) [12].
Při nálezu dysmorfie a vyjádření podezření na genetický predispoziční syndrom je odeslán pacient s rodiči na genetické vyšetření, kde proběhne anamnestické pátrání v rodinné anamnéze (nevyhnutelné jsou údaje o nádorových onemocněních v linii obou rodičů nejméně tří generací). Důležitým údajem je rovněž věk při výskytu nádoru a typy nádorů. Na základě všech údajů lze potvrdit pravděpodobnost určitého dědičného nádorového syndromu a genetik indikuje genetické testování predispozičních genů [15,16]. Při potvrzení predispozičního nádorového syndromu by měla být u dětského pacienta zajištěna dispenzarizace na pracovišti dětské onkologie.
Přehled nejčastějších nádorových predispozičních syndromů u dětí
Potvrzení přítomnosti specifické germinální mutace u pacienta, případně rodinných příslušníků je důkazem genetického nádorového predispozičního syndromu. Mutace může být zděděná od jednoho z rodičů, nebo může vzniknout u pacienta de novo. Rodina je informována o rizicích rozvoje nádorů vázaných na příslušný syndrom, je navržen plán dispenzárních vyšetření dle míry rizika konkrétních typů nádorů v dětském věku [15].
Seznam nejčastějších genetických predispozičních syndromů s rizikem vzniku nádoru v dětském věku je uveden v tab. 1.
Beckwith‑ Wiedemannův syndrom
Incidence 1 : 13 700, až 85 % případů je sporadických, pouze 15 % je familiárních. Příčinou jsou různé genetické alterace v oblasti chromozomu 11p15.5. Typickými klinickými projevy je nadměrný vzrůst, makroglosie, organomegalie, umbilikální hernie, novorozenecká hypoglykemie. Vázán je s rizikem vzniku nádoru ledviny (Wilmsův nádor), hepatoblastomu, neuroblastomu, ale i adrenokortikálního karcinomu, rhabdoidního nádoru či rhabdomyosarkomu. Jedná se především o embryonální typy nádorů s vrcholem výskytu do 3 – 5 let. V sekundární prevenci nádoru je doporučena ultrasonografie bříška každé tři měsíce do šesti let věku [17].
Neurofibromatóza typ I
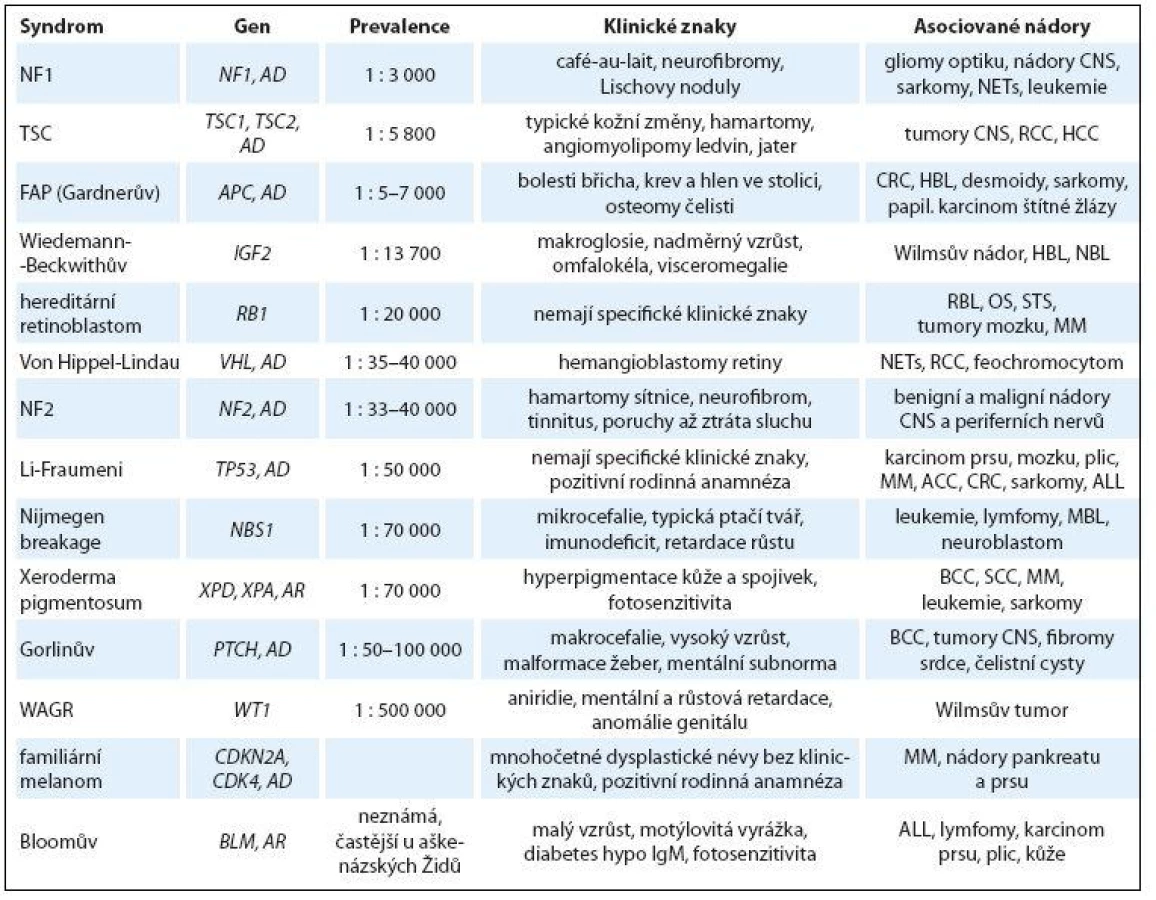
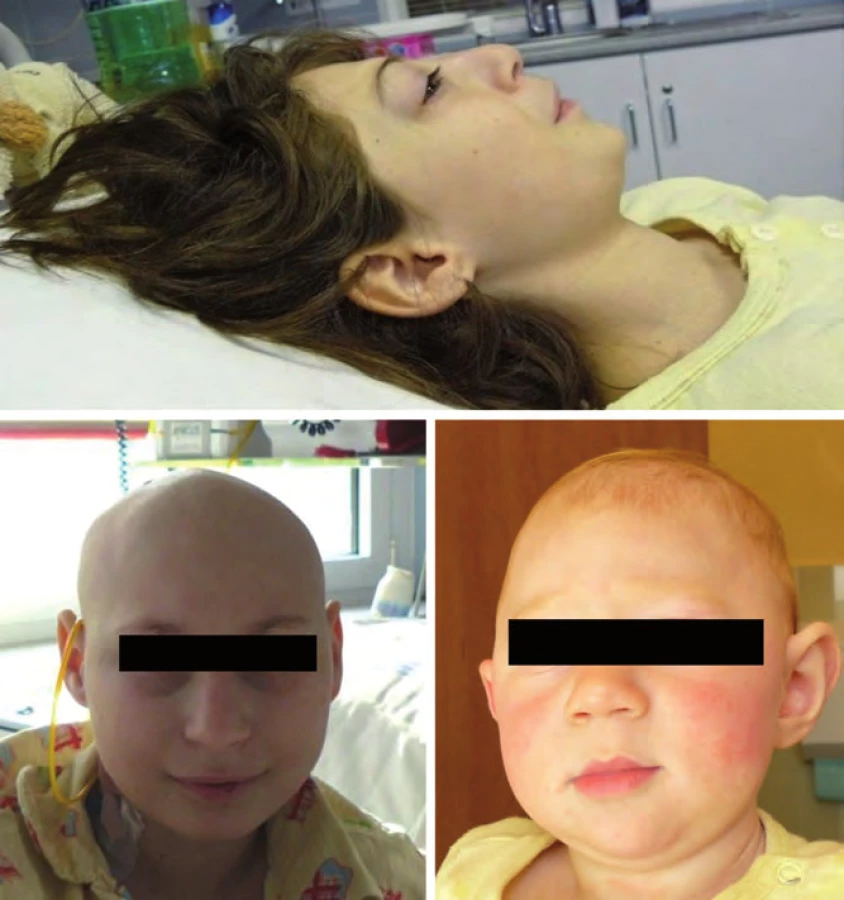
Incidence 1 : 3 500, jedná se autozomálně dominantní dědičnost mutace tumor supresorového genu NF1 (17q11.2). Typickým klinickým projevem jsou café ‑ au ‑ lait skvrny na kůži (počet > 6 s rozměrem > 5 mm), axilární pihy, vícečetné kožní neurofibromy, hamartomy duhovky, kostní anomálie, někdy lehká mentální retardace. Neurofibromatóza typu I (NF1) je asociována se zvýšeným rizikem vzniku především nádorů CNS (gliomy optiku, hamartomy mozku, astrocytomy, plexiformní neurofibromy, ependymomy) ve věku pod 10 let, ve věku dospívání je riziko rozvoje fibrosarkomů, endokrinních nádorů a leukemií. Pacienti vyžadují komplexní multidisciplinární sledování (oční, neurologické, evokované potenciály zrakové i sluchové, MRI mozku) (obr. 1) [18,19].
Komplex tuberózní sklerózy
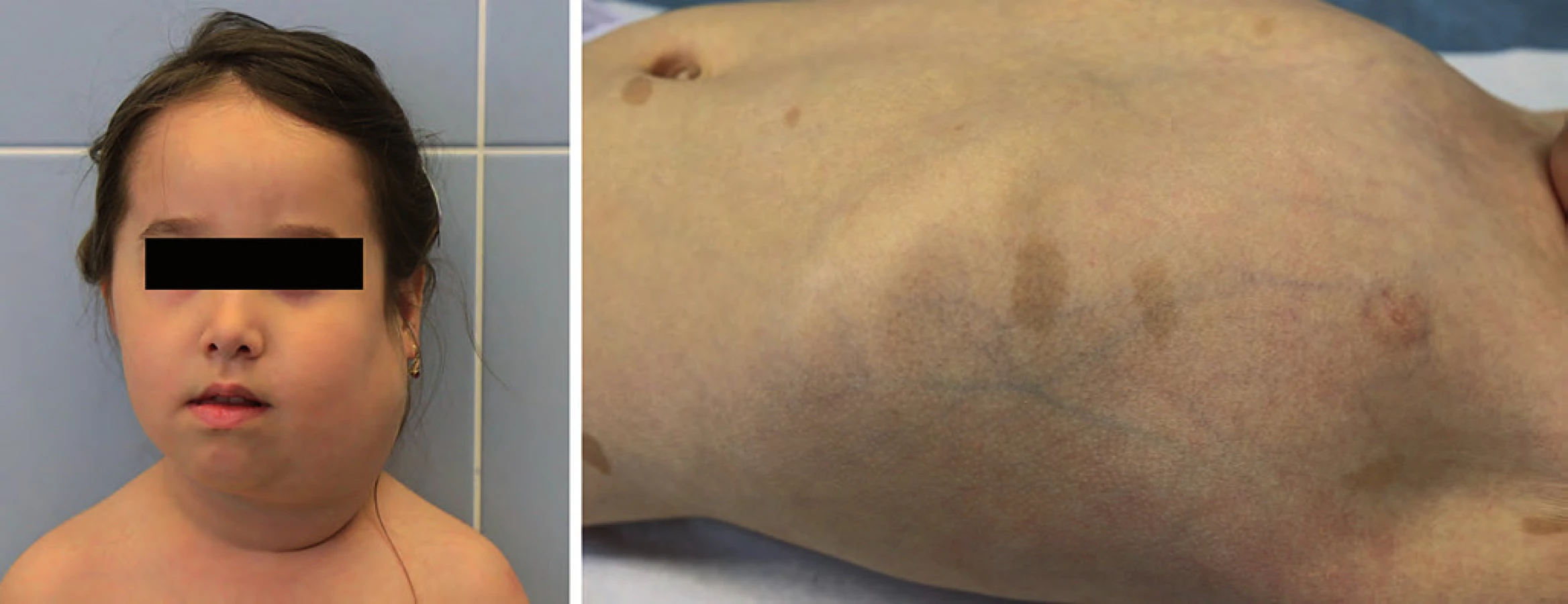
Komplex tuberózní sklerózy (TSC) je autozomálně dominantně dědičný syndrom s incidencí 1 : 5 800. Většinou se jedná o de novo mutaci genu TSC1 (lokus 9q34) nebo TSC2 (lokus 16p13). Projevuje se tvorbou hamartomů prakticky v kterémkoli orgánu (kůže, mozek, ledviny, srdce). Typické jsou faciální angiofibromy, depigmentace kůže, fibrózní plaky a sítnicové hamartomy (obr. 2). U novorozenců jsou prvním příznakem tuberózní sklerózy vícečetné rhabdomyomy srdce, které většinou spontánně regredují. Indikací k operaci je pouze symptomatický rhabdomyom srdce (arytmie, obstrukční příznaky, poruchy kontraktility). Mezi další projevy patří tvorba častých angiomyolipomů jater a ledvin nebo drobné cysty ledvin. U dospívajících se může projevit velkobuněčný subependymální astrocytom. Je zvýšené riziko vzniku karcinomu ledviny [18].
Li‑ Fraumeni syndrom
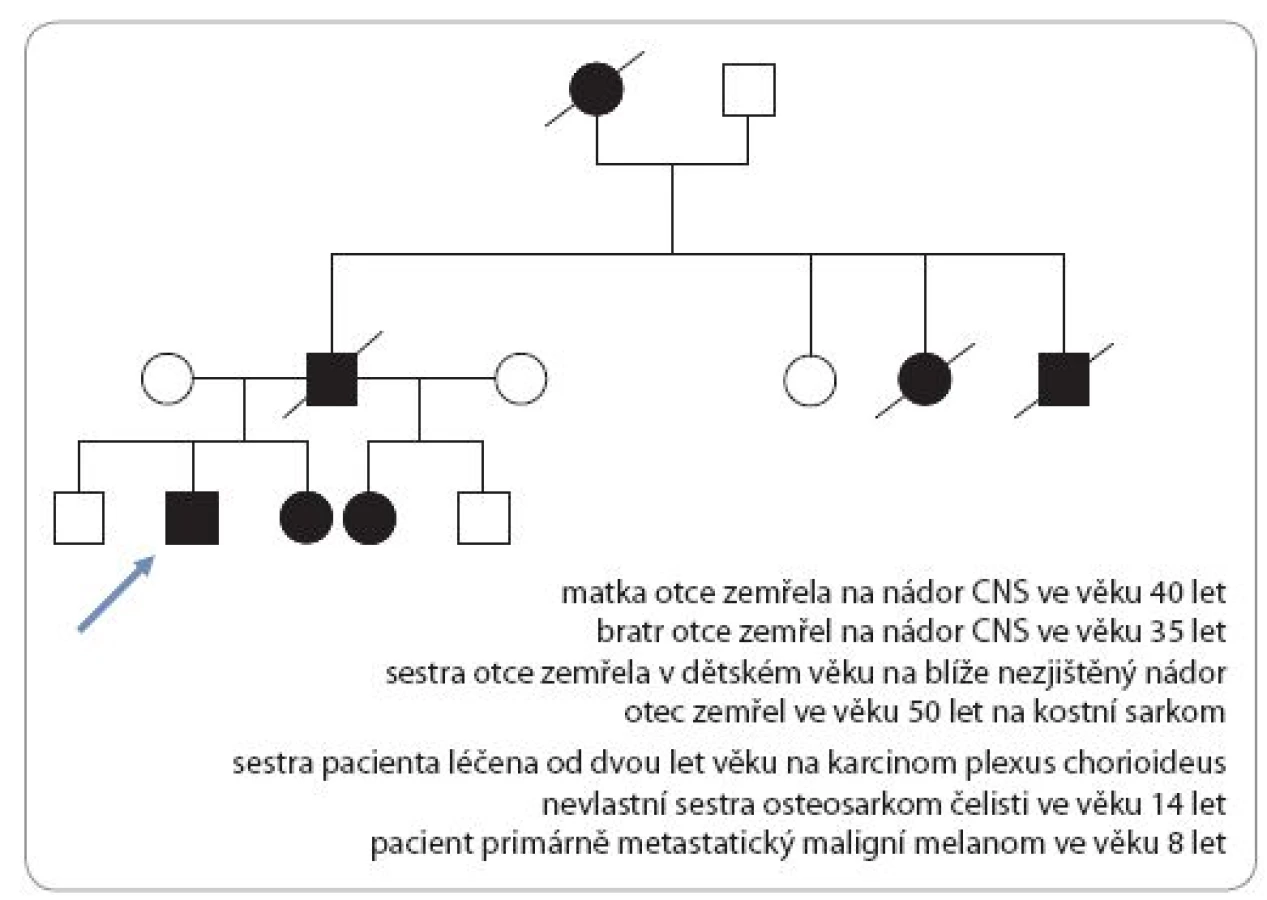
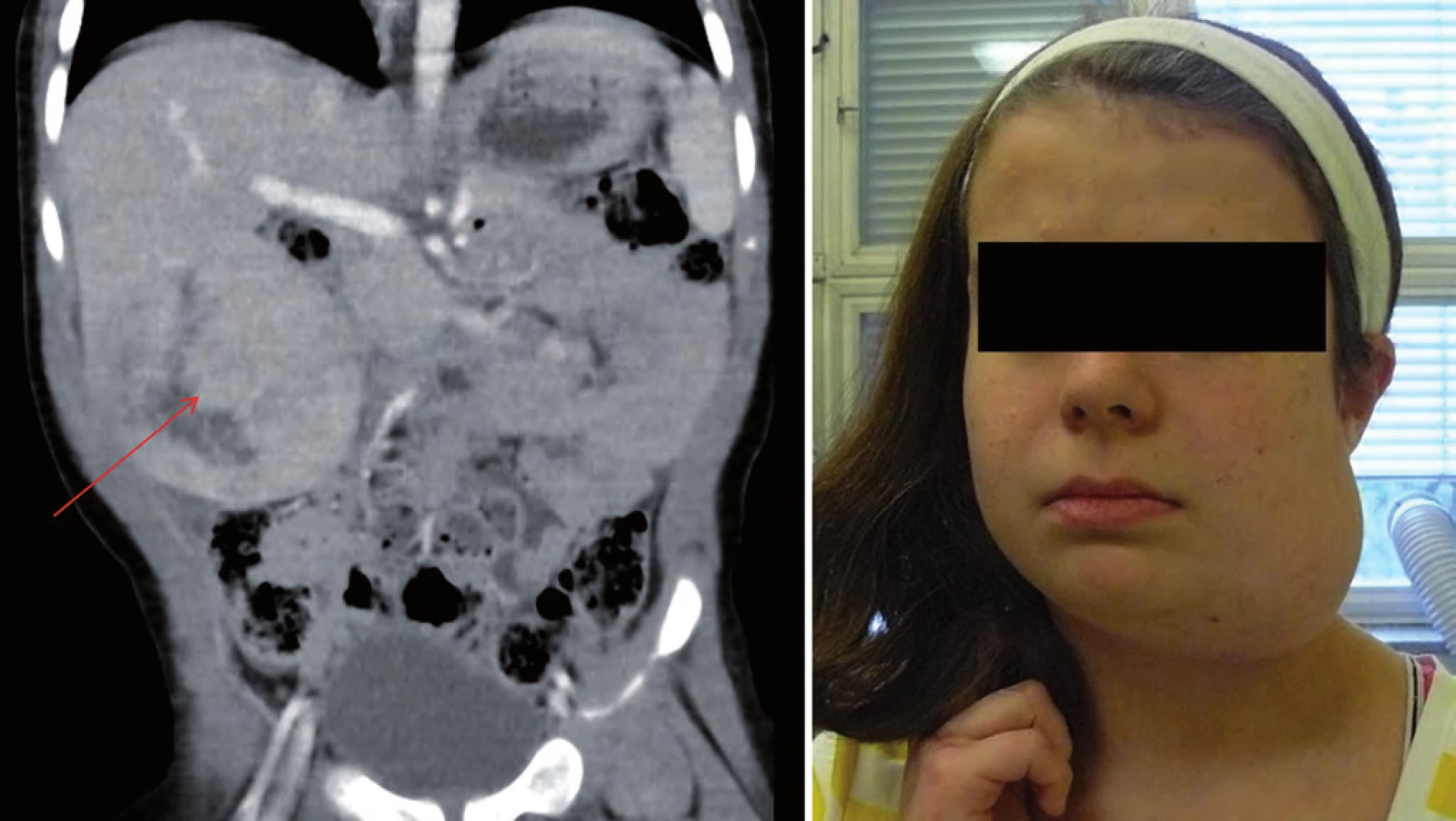
Incidence je 1 : 50 000 s autozomálně dominantním typem dědičnosti. Dispozice se dědí cestou alely tumor supresorového genu TP53 s bodovou mutací (17p13). Nositelé mutace TP53 nemají specifické klinické změny vizáže, typická je rodinná anamnéza. U Li ‑ Fraumeni syndromu je vysoké riziko vzniku širokého spektra nádorů (v dětském věku především sarkomů kostí a měkkých tkání, nádorů mozku – vzácného karcinomu chorioidálního plexu, kožního maligního melanomu). V dospívajícím a mladém dospělém věku hrozí rozvoj karcinomu prsu, leukemie a různých typů karcinomů (obr. 3) [17,19].
Nijmegen‑ breakage syndrom
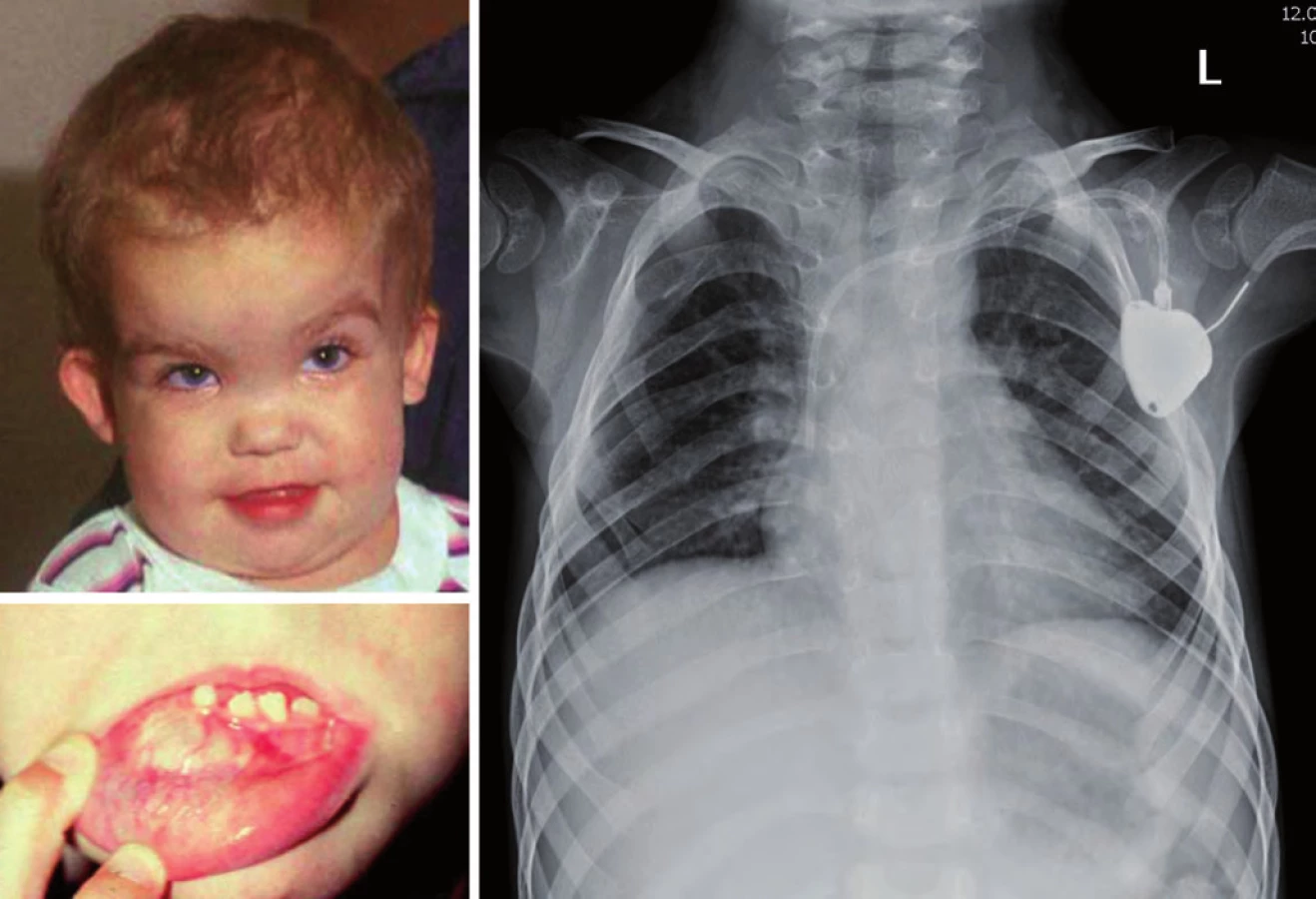
Nijmegen ‑ breakage syndrom je způsoben mutací NBS1 genu s incidencí 1 : 70 000, častější je ve střední a východní Evropě u slovanské populace (Polsko, Čechy, Slovensko, Ukrajina, Rusko). Jedná se o autozomálně recesivní syndrom poruchy opravních (repair) genů. V klinice dominuje především mikrocefalie zvýrazňující se s věkem, faciální dysmorfizmus (typická tzv. ptačí tvář „tukana“ s ustupujícím čelem, prominujícím nosem a mikrognacií), růstová retardace, ovariální selhání u dívek, radiosenzitivita a poruchy imunity. Intelekt je obvykle normální nebo lehká subnorma. Hrozí riziko vzniku především hematologických malignit (Hodgkinův lymfom, non‑Hodgkinovy lymfomy, leukemie), obvykle po 10. roku věku (obr. 4) [18].
Syndrom familiární adenomatózní polypózy
Syndrom familiární adenomatózní polypózy (FAP) je poměrně častý s incidencí 1 : 5 000 – 7 500. Dědičnost je autozomálně dominantní, příčinou je mutace genu APC (lokus 5q21 – 22) s tvorbou množství (stovek až tisíců) adenomatózních polypů tlustého střeva a rekta již od věku kolem 10 let. Hrozí vysoké riziko maligního zvratu a karcinom střeva již v dětském a adolescentním věku. Endoskopické vyšetření střeva a kolonoskopie se provádějí 1krát ročně od věku 10 – 12 let, často je v prevenci rozvoje karcinomu nutná totální kolektomie před 20. rokem věku. Genetické vyšetření dětí v rodině s potvrzenou mutací APC genu se v současnosti posouvá do nejnižší věkové skupiny pro zvýšené riziko vzniku embryonálního nádoru jater – hepatoblastomu (10 – 15 % dětí s hepatoblastomem má potvrzenou mutaci APC genu) nebo dědičného hepatocelulárního karcinomu ve věku do 15 let. Součástí syndromu jsou často osteomy čelisti, zubní abnormality nebo desmoidy (obr. 5) [7,18,19]. V současnosti u dětí s potvrzenou mutací APC genu a nálezem mnohočetných polypů a tubulárních střevních adenomů lze použít chemoprevenci vzniku karcinomu střeva, oddálit maligní transformaci, snížit novotvorbu dalších polypů a oddálit tak provedení totální kolektomie do staršího věku podáváním cox ‑ 2 inhibitorů (celecoxib) [20,21].
Syndrom konstitučního deficitu MMR systému
Bialelické mutace v MMR genech (především MLH1, MSH2, MSH6, PMS2) mohou způsobovat syndrom konstitučního deficitu MMR systému (CMMR ‑ D). Je charakterizován velmi časným výskytem nádoru tlustého střeva, již ve druhé dekádě života (hereditary non‑polyposis colorectal cancer – HNPCC), hematologických malignit (lymfomy a leukemie), nádorů mozku (astrocytomy a glioblastomy) a kožními příznaky NF1 (café ‑ au ‑ lait). První nádorová onemocnění, leukemie a nádory mozku se mohou objevit již od dvou let věku. V mladém dospělém věku se rovněž vyskytuje endometriální karcinom a nádory močových cest. Téměř u 40 % pacientů s CMMR ‑ D se vyvine metachronní sekundární malignita [22,23].
Von Hippel‑ Lindau syndrom
Incidence 1 : 35 000 s autozomálně dominantní dědičností mutace genu VHL (lokus 3p25). Typické jsou hemangioblastomy v retině, mozečku, mozkovém kmeni a míše už v dětském věku. Z nádorů jsou časté neuroendokrinní nádory, feochromocytom/ paragangliom, v dospělém věku je zvýšené riziko vzniku karcinomu ledviny. Komplexní preventivní péče musí být zahájena už v dětském věku pro nízký věk výskytu nádorů [17,18].
Gorlinův syndrom
Gorlinův syndrom je podmíněn autozomálně dominantním přenosem mutace PTCH genu (lokus 9q22.3). Je spojen především s rozvojem meduloblastomu u malých dětí do dvou let, basocelulárním karcinomem kůže do 10 let věku. Typické jsou dysmorfické znaky – makrocefalie, prominující čelo, hypertelorizmus, vysoký vzrůst, malformace žeber (vidlicovitá žebra) a páteře, odontogenní cysty, mentální subnorma (obr. 6) [18].
Syndrom familiárního melanomu
Tento syndrom je způsoben autozomálně dominantním přenosem mutace v genu CDKN2A, vzácněji v CDK4 nebo MCIR. Na kůži jsou obvykle desítky dysplastických pigmentových névů. Nosiči mutace CDKN2A mají celoživotní riziko vzniku melanomu v 60 – 92 %, kromě toho mají zvýšené riziko vzniku dalších typů nádorů (astrocytom mozku, nádory slinivky). Mutace CDKN2A by měla být vyšetřena u všech pacientů s maligním melanomem v dětském věku [18].
Noonanové syndrom
S incidencí 1 : 1 000– 2 500 je Noonanové syndrom způsoben autozomálně dominantní mutací PTPN11 genu na chromozomu 12. V posledních letech se u části pacientů s Noonanové syndromem prokázala mutace KRAS genu, zařaďuje se tedy mezi tzv. rasopatie. Pro děti je typický nízký vzrůst, vrozené srdeční vady (pulmonální stenóza) a typické dysmorfické rysy – hypertelorizmus, ptóza víček, nízce posazené uši, deformity hrudníku (pectus carinatum), svalová hypotonie a různé stupně mentálního deficitu. Pacienti mají rovněž poruchy srážlivosti krve a jsou ohroženi vznikem hematologických malignit, u dětí převážně akutní leukemií, myelodysplastickým syndromem, případně rozvojem juvenilní myelomonocytární leukemií.
Soubor pacientů Kliniky dětské onkologie s predispozičními nádorovými syndromy
Na Klinice dětské onkologie FN Brno dispenzarizujeme děti s nádorovými predispozičními syndromy od roku 2009. Za toto relativně krátké časové období sledujeme 33 genetických nádorových predispozičních syndromů u 157 dětí (tab. 2). Zastoupení dle pohlaví bylo bez predominance (počet chlapců 81, počet dívek 76, poměr chlapců a dívek je 1 : 1,06), věk v čase zahájení dispenzarizace byl různý dle typu syndromu a klinických potíží (od novorozeneckého věku po 18 let). V časovém sledu je jednoznačně vidět stoupající počet dispenzarizovaných hereditárních predispozičních syndromů (graf 1).


Z celkového počtu 157 dětí bylo odesláno na ambulanci dětské onkologie primárně s diagnózou nádoru 73 pacientů (46 %), u kterých byl až následně diagnostikován predispoziční nádorový syndrom. V posledních 2 – 3 letech se zvyšuje počet pacientů odeslaných k dispenzarizaci na základě pozitivní rodinné anamnézy, typických známek dysmorfizmu nebo potvrzené germinální mutace ještě před rozvojem zhoubného onemocnění. Ze sledovaného souboru pacientů zemřelo 11 dětí (7 %). Jedna pacientka s neurofibromatózou typu Izemřela na terciární malignitu, čtyři pacienti zemřeli na sekundární malignitu – dvě adolescentní dívky s čistou formou gonadální dysgeneze (Swyerův syndrom) zemřely na sekundární akutní myeloidní leukemii, jeden chlapec s hereditárním retinoblastomem zemřel na sekundární osteosarkom sedm let od diagnózy retinoblastomu a jedna dívka s Nijmegen ‑ breakage syndromem zemřela na sekundární non‑Hodgkinův lymfom. Jedna dívka s Mafucciho syndromem zemřela ve věku 21 let na komplikaci – hemoragický šok při krvácení do břicha při ruptuře jaterního hemangiomu. Ostatních pět pacientů zemřelo na maligní nádor v souvislosti se svou genetickou zátěží (graf 2).

Závěr
Hereditární nádorové predispoziční syndromy se v posledních letech stávají novým problémem na poli dětské onkologie, což prokazuje i vlastní soubor pacientů s těmito syndromy. Význam této problematiky dokumentuje fakt, že v roce 2012 na pravidelném setkání ASPHO (American Society of Pediatric Hematology/ Oncology) byl organizován workshop věnovaný predispozičním nádorovým syndromům v dětském věku [1].
Dle výsledků analýzy našeho souboru pacientů s hereditární zátěží je téměř u poloviny pacientů predispoziční syndrom nepoznán a na onkologii se ocitnou až s diagnózou zhoubného nádoru.
Dalším problémem je fakt, že i v případě již diagnostikovaného predispozičního syndromu je dětský pacient často sledován u jiného specialisty (např. neurolog, oční lékař, kožní lékař, ortoped, kardiolog atd.), který ovšem málokdy sleduje dítě komplexně z hlediska možného rozvoje nádoru a sleduje pacienta pouze z hlediska své odbornosti.
Klíčovou roli v rozpoznání prvních známek možného genetického predispozičního nádorového syndromu hraje pediatr primárního kontaktu, který nejlépe zná dítě i jeho rodinu.
Ten by měl při potvrzení hereditárního predispozičního syndromu odeslat pacienta k dispenzarizaci na specializované pracoviště dětské onkologie. Dětský onkolog vypracuje plán a časový rozvrh dispenzárních prohlídek, organizuje nejenom zobrazovací vyšetření v doporučených časových intervalech, ale i konziliární a laboratorní vyšetření dle individuální míry rizika a typu syndromu.
Časná identifikace predispozičního syndromu znamená adekvátní genetické poradenství pro rodinu, účinnou sekundární nádorovou prevenci, časnější záchyt nádoru u pacienta a zahájení efektivní onkologické léčby. Je prokázáno, že potvrzení genetického predispozičního nádorového syndromu u dětí s následnou dispenzarizací na onkologii vede ke snížení morbidity a mortality na zhoubné nádory u dětí a dospívajících [12,19].
Autorka deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Viera Bajčiová, CSc.
Klinika dětské onkologie
LF MU a FN Brno
Černopolní 9
625 00 Brno
e-mail: vbajciova@fnbrno.cz
Obdrženo: 14. 7. 2015
Přijato: 18. 8. 2015
Sources
1. Schiffman JD, Keller JI, Jundy E et al. Update on pediatric cancer predisposition syndromes. Pediatr Blood Cancer 2013; 60(8): 1247 – 1252. doi: 10.1002/ pbc.24555.
2. American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21(12): 2397 – 2406.
3. Bjorge T, Cnattingius S, Lie RT et al. Cancer risk in children with birth defects and their families: a population based cohort study of 5.2 million children from Norway and Sweden. Cancer Epidemiol Biomarkers Prev 2008; 17(3): 500 – 506. doi: 10.1158/ 1055 ‑ 9965.EPI ‑ 07 ‑ 2630.
4. Botto LD, Flood T, Little J et al. Cancer risk in children and adolescents with birth defects: a population‑based kohort study. PLoS One 2013; 8(7): e69077. doi: 10.1371/ journal.pone.0069077.
5. Testa JR, Malkin D, Schiffman JD. Connecting molecular pathways to hereditary cancer risk syndromes. Am Soc Clin Oncol Educ Book 2013; 81 – 90. doi: 10.1200/ EdBook_AM.2013.33.81.
6. Foretová L. Hereditární malignity si zaslouží víc pozornosti než dosud. Kongresová review. XXXIX. brněnské onkologické dny a XXIX. konference pro nelékařské zdravotnické pracovníky, 8. – 10. 4. 2015, Brno.
7. Závodná K, Milly M, Vavrová L et al. Dědičné nádorové syndromy. Onkológia 2015; 10(2): 84 – 89.
8. Frank C, Fallad M, Sundquist J et al. Population landscape of familial cancer. Sci Rep 2015; 5 : 12891. doi: 10.1038/ srep12891.
9. Clarke AJ, Gaff C. Challenges in the genetic testing of children for familial cancer. Arch Dis Child 2008; 93(11): 911 – 914. doi: 10.1136/ adc.2006.113381.
10. Malkin D, Nichols KE, Zelley K et al. Predisposition to pediatric and hematologic cancer: a moving target. Am Soc Clin Oncol Educ Book 2014; e44 – e55. doi: 10.14694/ EdBook_AM.2014.34.e44.
11. Quinn E, McGee R, Nuccie R et al. Genetic predisposition to neonatal tumors. Curr Pediatr Reviews 2015; 11(3): 164 – 178.
12. Škvor J, Průhová Š. Základy klinické genetiky pro klinickou praxi. Genetika v primární péči: 5 – 6. Mladá fronta 2014.
13. Papakasama S, Tomlinson GE. Genetic predisposition and screening in pediatric cancer. Pediatr Clin North Am 2002; 49(6): 1393 – 1413.
14. Rao A, Rothman J, Nichols KE. Genetic testing and tumor surveillance for children with cancer predisposition syndromes. Curr Opin Pediatr 2008; 20(1): 1 – 7. doi: 10.1097/ MOP.0b013e3282f4249a.
15. Foretová L, Petráková K. Dispenzarizace dědičných nádorových syndromů. Klin Onkol 2009; 22 (Suppl): S6 – S77.
16. Ilenčíková D, Amidová O, Konečný M et al. Genetická konzultácia jako súčasť genetického vyšetrenia. Onkológia 2015; 10(2): 74 – 76.
17. Teplick A, Kowalski M, Biegel JA et al. Screening in cancer predisposition syndromes: guidelines for the general pediatricians. Eur J Pediatr 2011; 170(3): 285 – 294. doi: 10.1007/ s00431 ‑ 010 ‑ 1377 ‑ 2.
18. Foretová L, Macháčková E, Gaillyová R et al. Hereditární nádorová onemocnění. In: Foretová L, Svoboda M,Slabý O et al (eds). Molekulární genetika v onkologii. Praha: Mladá fronta 2014 : 116 – 155.
19. Field M, Shanley S, Kirk J. Inhereted cancer susceptibility syndromes in paediatric practice. J Pediatr Child Health 2007; 4(4): 219 – 229.
20. Lang M, Gasche C. Chemoprevention of colorectal cancer. Dig Dis 2015; 33(1): 58 – 67. doi: 10.1159/ 000366037.
21. Thakkar K, Fishman DS, Gilger MA. Colorectal polyps in childhood. Curr Opin Ped 2012; 24(5): 632 – 637. doi: 10.1097/ MOP.0b013e328357419f.
22. Ilenčíková D. Syndrom konstitučního deficitu mismč opravného systému (CMMR ‑ D). Kazuistika rodiny s bialelickou MSH6 mutáciou. Klin Onkol 2012; 25 (Suppl): S34 – S38.
23. Vasen HF, Ghorbanoghli Z, Bourdeaut F et al. Guidelines for surveillance of individuals with constitutional mismatch realit ‑ deficiency proposed by the European Consortium „Care for CMmR ‑ D“ (C4CMmR ‑ D). J Med Genet 2014; 51(5): 283 – 293. doi: 10.1136/ jmedgenet ‑ 2013 ‑ 102238.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice
- Hepatoblastom, etiologie, kazuistiky
- Genetika tumorigenézy nádorov kolorekta (možnosti testovania a screeningovej predikcie dedičnej formy ochorenia – Lynchovho syndrómu)
- Fanconiho anémie, komplementační skupina D1 v důsledku bialelické mutace genu BRCA2 – kazuistika