
5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury
5 Years of Activities of the National Reference Laboratory for Human Prion Diseases Attached to the Department of Pathology and Molecular Medicine of FTNsP: Our Experience and an Overview of Literature
During the existence of the Czech National Reference Laboratory for Transmissive Spongiform Encephalopathies at the Department of Pathology and Molecular Medicine of Thomayer Teaching Hospital in Prague (the laboratory was established in July 2001) as many as 90 brains of patients with suspicion of Creutzfeldt-Jakob disease (CJD) have been examined. The histopathologic criteria for definite CJD were fulfilled in 50 cases. No case of the new variant of CJD has been detected in the Czech Republic. Recently, obligatory testing of brain tissue from corneal graft donors was established in the laboratory.
Key words:
prion diseases – Creutzfeldt-Jakob disease – neuropathology – diagnostics
Authors:
R. Matěj 1; R. Rusina 2; F. Koukolík 1
Authors‘ workplace:
Národní referenční laboratoř lidských TSE/CJN při Oddělení patologie a molekulární medicíny FTNsP, Praha
1; Neurologická klinika IPVZ a FTNsP, Praha
2
Published in:
Cesk Slov Neurol N 2007; 70/103(6): 637-642
Category:
Review Article
Poděkování: Autoři tímto děkují všem, kteří se na činnosti NRL v jejím průběhu podíleli či podílejí.
V abecedním pořadí pak zejména MUDr. Pavlu Bočanovi, CSc., RNDr. Miladě Matějčkové, ing. Janě Novákové, CSc., MUDr. Jakubu Sikorovi, ing. Aleně Srbové.
Overview
Prionová onemocnění tvoří skupinu vzácných neurodegenerací. V roce 2001 byla ustanovena Národní referenční laboratoř lidských TSE/CJN při Oddělení patologie a molekulární medicíny, která se zabývá diagnostikou těchto onemocnění v České republice. Za 5 let činnosti bylo v laboratoři vyšetřeno více než 90 zemřelých s podezřením na prionové onemocnění, u 50 z nich byla definitivní diagnóza neuropatologicky potvrzena. Případ nové varianty Creutzfeldtovy-Jakobovy nemoci nebyl v České Republice doposud zaznamenán. Od 1. 1. 2007 probíhá v laboratoři i povinné testování CNS dárců rohovek.
Klíčová slova:
prionová onemocnění – Creutzfeldtova - Jakobova nemoc – neuropatologie – diagnostika
Úvod
Prionová onemocnění neboli transmisivní spongiformní encefalopatie jsou skupina neurodegenerativních chorob postihujících zvířata a lidi [1]. Lidská prionová onemocnění jsou:
- sporadická, familiární a iatrogenní Creutzfeldtova-Jakobova nemoc (sCJN, fCJN, iCJN)
- Gerstmannova-Sträusslerova-Scheinkerova nemoc, též Gerstmannův-Sträusslerův syndrom (GSS)
- kuru etnika Fore žijícího na Nové Guinei
- fatální familiární i sporadická insomnie (FFI, sFI)
- nová varianta CJN (vCJN)
Poznávání prionových chorob má dlouhou a složitou historii, která není ukončena (tab. 1.)

Prionová hypotéza
Prionová hypotéza, za jejíž formulaci byl v roce 1986 Stanley B. Prusiner oceněn Nobelovou cenou za lékařství a fyziologii, dokazuje, že patogenem je protein PrPSc . Index Sc je odvozen od slova scrapie, onemocnění známého od r. 1738, postihujícího ovce a kozy, česky klusavky. Protein PrPSc svým chemickým složením odpovídá fyziologickému proteinu PrPC exprimovanému na povrchu mnoha buněčných typů, nejen neuronů. Funkce tohoto proteinu není doposud jednoznačně objasněna. Index C je odvozen od slova celulární. PrPC je u lidí kódován prionovým genem PRNP na 20. chromozomu.
Patologický prionový protein PrPSc se od fyziologického celulárního PrPC odlišuje prostorovým uspořádáním. PrPSc je odolný vůči natrávení proteinázou K, takže se někdy označuje jako PrPres. Molekulární struktura PrPSc a PrPC je známa u myší lépe než u lidí [2].
Předpokládá se, že konverzi fyziologického na patologický prionový protein, která je klíčem onemocnění, spouští malý počet molekul PrPSc (tzv. seeding).
U genetických podob choroby mutovaná forma fyziologického prionového prionu (někdy označovaná PrP M ) nabude odpovídající prostorové uspořádání a agreguje. Získává tím vlastnosti PrPSc a je schopna infikovat pokusná zvířata.
Patologické priony se vyskytují v kmenech, které se projevují fenotypickými rozdíly u inokulovaných hostitelů: odlišnou délkou inkubace, různými druhy neuropatologických změn, stupněm glykosylace PrPSc i stupněm rezistence vůči natrávení proteinázou K. Předpokládá se, že rozdíly mezi kmeny patologických prionů jsou podmíněny rozdíly v jejich konformaci.
Sporadická CJN
Onemocnění se vyskytuje s prevalencí 1-2 případy/1000000 lidí/rok ve věkové skupině nad 40 let, nejčastěji v rozmezí 55–65 let. V městské populaci a populaci lidí starších než 65 let, jakož i v populaci psychiatrických léčeben, může být její výskyt až 5 případů/1 000 000 lidí a rok. Příčina konverze fyziologického na patologický prionový protein není u sCJN známa. Odchylky genu PRNP zjištěny nebyly. Mezi sCJN a scrapie (klusavkou) - prionovým onemocněním ovcí a koz, nebyl zjištěn vztah. Současná data nedokazují, že by mezi sCJN a epidemií bovinní spongiformní encefalopatie byl kauzální vztah [3,4].
Klinický obraz je charakterizován rychle progredující demencí, kterou doprovází myoklonie, extrapyramidové a pyramidové příznaky a/nebo mozečková ataxie [5]. Asi u třetiny nemocných mohou rozvoji demence předcházet nespecifické prodromální příznaky (únava, nespavost, deprese, ztráta na hmotnosti, bolesti hlavy).
Vzácnější je tzv. Heidenhainova varianta, kde převažují projevy postižení zrakově-prostorových funkcí včetně vizuálních halucinací a korové slepoty v důsledku časného postižení primární a asociační zrakové kůry. Demence zpravidla rychle progreduje, onemocnění probíhá nejčastěji několik měsíců, ve většině případů trvání nemoci nepřekračuje jeden rok.
Klinickou diagnózu podporuje typický EEG nález a/nebo nález ß-podjednotky proteinu 14-3-3 v mozkomíšním moku (tab. 2). U rozvinuté sCJN zobrazí EEG generalizované trifazické nebo polyfazické vlny o délce 100–300 ms, které se periodicky opakují v intervalech 0,5–2 s [6].

Protein 14-3-3 je nespecifickým markerem neuronálního rozpadu, který bývá prokazatelný u řady neurologických onemocnění spojených s rozsáhlejší parenchymovou lézí (od hemoragií přes tumory po encefalitidy nebo i rozsáhlejší ischemie). Jeho stanovení má praktický význam pouze v kontextu rychle progredující demence s neurologickými příznaky k potvrzení klinického podezření na sCJN. Předpokládá se, že u neurodegenerativních demencí od Alzheimerovy nemoci přes frontotemporální demenci po demence s Lewyho tělísky dochází k neuronálnímu rozpadu příliš pozvolně na to, aby bylo možno detekovat přítomnost ß-podjednotky proteinu 14-3-3 v likvoru. Naproti tomu v případě sCJN je rychlost progrese onemocnění a s tím spojený zánik neuronů natolik intenzivní, že zvýšené množství uvolněného proteinu 14-3-3 lze detekovat v likvoru [7].
Dalším vyšetřením, které může být přínosné pro stanovení diagnózy sCJN, je MRI. Typickým nálezem bývaji hyperintenzity v oblasti bazálních ganglií (typicky v putamen) a korově, s převahou v inzule a frontálně, v T2 vážených obrazech - ale především ve FLAIR sekvencích. MRI nález nicméně nebyl zahrnut do diagnostických kritérií WHO (tab. 2) [5].
Sporadická CJN se tedy diagnostikuje na základě klinického obrazu (možná sCJN) s přihlédnutím k výsledkům pomocných vyšetření – EEG a likvoru (pravděpodobná sCJN). Definitivní diagnóza CJN (stejně jako dalších prionových onemocnění) je neurohistologická a imunohistochemická [8].
Iatrogenní prionové nemoci
Iatrogenní CJN (v současnosti se užívá pojem náhodně přenesené, accidentally transmitted CJN) byly způsobeny transplantací rohovky, užitím nedostatečně sterilizovaných intracerebrálních elektrod, transplantací dura mater, užitím růstového hormonu získaného z kadaverózních hypofýz. Případy, které byly důsledkem intracerebrální nebo intraokulární inokulace, jsou klinicky blízké sCJN s rychle progredující demencí. Případy dané periferní inokulací se projevují častěji jako cerebelární ataxie připomínající kuru. Inkubace onemocnění po transplatanci dura mater se pohybovala mezi 19-46 měsíci, inkubace po periferní inokulaci byla v průměru 15letá, nejdelší známá inkubace přesáhla 23 let [9].
Hrubý statistický odhad potenciálního rizika přenosu CJN při transplantacích je v České republice 3 promile (2 případy prionových chorob /100 000 lidí/rok při 1 500 transplantacích).
Dalším problémem je možnost přenosu patogenního prionového proteinu prostřednictvím (neuro)chirurgických výkonů. Naše laboratoř zastihla 2 případy potenciálního přenosu CJN po biopsii mozku provedené na neurochirurgickém pracovišti při nejasném neurologickém onemocnění, které jsme po úmrtí pacientky diagnostikovali jako sCJN. Retrospektivně jsme prokázali priony i v zapůjčeném bioptickém vzorku. Nástroje byly sterilizovány standardním způsobem, což priony neovlivňuje. 1 ze 2 následně operovaných pacientů zemřel z jiného důvodu. Druhý je, pokud je nám známo, živ a v odstupu 2 let bez příznaků prionového onemocnění.
Samostatnou otázkou je problematika přenosu patologické formy prionového proteinu krevní transfuzí, která je však v současnosti omezena na popis 4 případů u nové varianty CJN ve Velké Británii [10].
Nová varianta CJN
Nová varianta CJN (vCJN, Willova nemoc) byla popsána roku 1996. Její vztah k epidemii BSE je podrobně popsán [3,11]. Předpokládá se, že epidemie BSE mohla být důsledkem zamoření krmných směsí pro hovězí dobytek priony z ovcí stižených klusavkou, nebo že je výsledkem „spontánní“ konverze prionů u hovězího dobytku a jejich následného šíření rovněž nejspíše krmnou směsí. V prvním případě by priony překonaly mezidruhovou bariéru ovce – hovězí dobytek.
Nová varianta CJN u lidí se považuje za důsledek další překonané mezidruhové bariéry a to hovězí dobytek - lidé. Předpokládá se přenos potravou. Na vCJN do současnosti zemřelo téměř 200 lidí. Až na 22 případů ve Francii a jednotlivá úmrtí v různých zemích po celém světě jsou všichni zemřelí Britové. Klinické příznaky těchto pacientů se zřetelně liší od klasické sporadické CJN, v popředí je ataxie, poruchy čití (včetně bolestivých parestezií a dysestezií na končetinách) a časné psychiatrické projevy. Postupně se rozvíjí demence, myoklonie však nemusí být pravidlem. Onemocnění začíná často v mladším věku, už od druhé dekády. U žádného z pacientů nebyly zjištěny změny EEG charakteristické pro sCJN, vyšetřování proteinu 14-3-3 v likvoru je také vysoce nespecifické a jen málo senzitivní. Paraklinickým vyšetřením, které může být přínosné pro stanovení diagnózy vCJN, je MRI. Typickým nálezem bývají hyperintenzity v oblasti pulvinaru talamu v T2 vážených obrazech a především ve FLAIR sekvencích s charakteristickým obrazem tzv. znamení L nebo hokejky (L, hockey-stick pulvinar sign) [5].
Všechny ověřené případy vCJN jsou M/M (methionin-methionin) homozygoti kodonu 129 prionového genu. Jedinou výjimkou je asymptomatický heterozygotní pacient 129 methionin (M)/valin (V), jenž dostal transfuzi krve od dárce, u něhož později propukla vCJN. Tento asymptomatický pacient zemřel z jiného důvodu ještě před propuknutím příznaků. Z toho plyne, že vývoj vCJN je u heterozygotů M/V a homozygotů V/V patrně pomalejší, než je odhadovaných 10 let inkubace vCJN u homozygotů M/M, a tito lidé mohou být nositeli subklinického onemocnění.
Podle očekávání je transgenní myš exprimující lidskou podobu 129 V PrPC vůči infekci priony BSE a vCJN značně odolná. Pokud u těchto myší onemocnění propuklo, odlišovalo se od vCJN neurohistologicky, kromě toho byl zjištěn jiný typ PrPSc než typ odpovídající vCJN. Naproti tomu a rovněž podle očekávání transgenní myš exprimující lidskou podobu 129 M PrPC má po inokulaci priony BSE a vCJN znaky vCJN. Zatím u jediné myši inokulované priony BSE se objevily známky neodlišitelné od lidské sCJN [2]. Usuzovat z toho, že možný nárůst případů sCJN prokazovaný v roce 2001 ve Švýcarsku je důsledkem expozice prionům BSE, se však zdá předčasné a je nejspíše důsledkem dokonalejších a přesnějších metod surveillance prionových onemocnění v této zemi [13]. Dlouhodobá rozsáhlá studie sCJN ve Spojeném království totiž žádné změny v jejím profilu nezjistila. Kromě toho první diagnostikované případy sCJN předcházely prvním diagnostikovaným případům BSE o více než 60 let [2].
Familiární podoba CJN
Dědičné formy Creutzfeldtovy-Jakobovy nemoci jsou způsobeny mutacemi v PRNP, jichž je v současnosti známo více než 20. Jde o autosomálně dominantní přenos s různou mírou penetrance pro různé mutace. Rodokmeny postižené těmito onemocněními se vyskytují mimo jiné v sociogeografických clusterech na severozápadním Slovensku v oblasti Kysuce a na Oravě s přesahem do severovýchodních částí Moravy a na jižním Slovensku s přesahem do přilehlé oblasti Maďarska [14].
Gerstmannova-Sträusslerova-Scheinkerova nemoc
GSS je krajně vzácné autosomálně dominantní onemocnění podmíněné několika druhy mutací PRNP.
Klinicky se projevuje mozečkovou ataxií s pyramidovými příznaky, později nastupuje demence. Kromě těchto příznaků se objevuje dysfagie, dysartrie, hyporeflexie. Průběh onemocnění je obvykle 5letý, tedy podstatně delší než u CJN.
Má poměrně charakteristický neurohistologický obraz s tvarově i tinkčně typickými velkými plakami, které se objevují i v bílé hmotě. U GSS se prokazuje několik typů mutací PRNP, například P102L, nebo G131V [5]. V České republice byla koncem 80. let minulého století zachycena rodina stižená tímto onemocněním. Molekulárně genetické vyšetření zatím nebylo provedeno.
Fatální familiární a sporadická insomnie
Příčinou fatální familiární insomnie je D178N mutace prionového genu (PRNP) při kodonu 129 M/M [15]. Existuje i sporadická forma tohoto onemocnění, do současnosti jí bylo celosvětově popsáno 8 případů.
V klinickém obrazu převažuje nespavost a známky dysautonomie, později se objevuje ataxie, dysartrie a myoklonie; časté bývají pyramidové příznaky. V závěru onemocnění je insomnie úplná, doprovází ji demence, rigidita, dystonie a mutizmus [15].
V České republice nebyla dosud zastižena žádná forma tohoto onemocnění.
Laboratorní diagnostika prionových chorob
Postmortální, případně bioptická diagnostika prionových chorob se opírá o vyšetření neurohistologické, imunohistochemické prováděné několika typy protilátek, imunologické vyšetření metodou westernblot a určení polymorfizmu kodonu 129 genu PRNP pomocí molekulárně biologických metodik [16]. V případě familiárních podob se sekvenuje gen PRNP. Vztah polymorfizmu kodonu 129 k sCJN bude nutné revidovat [17]. Priony je možné detekovat dalšími metodami užívanými ve výzkumu.
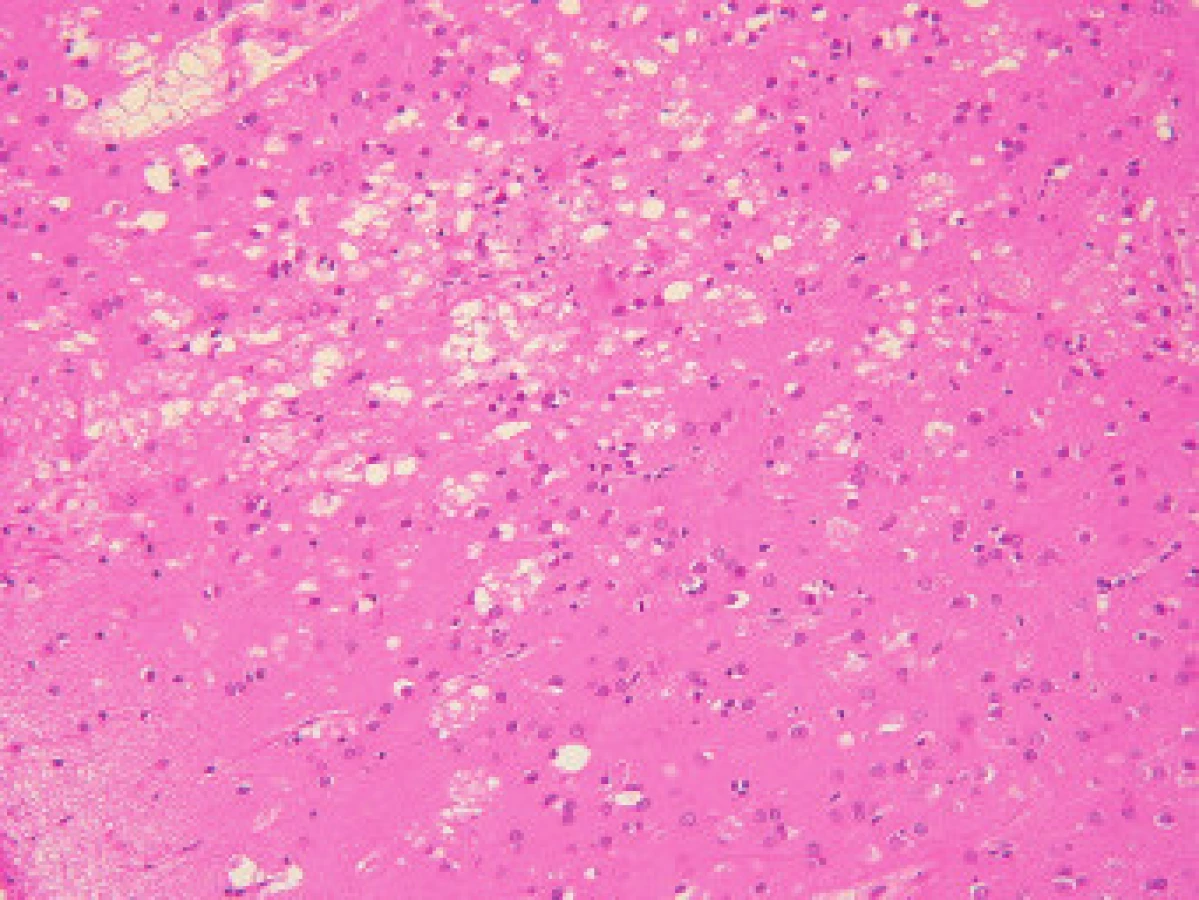
Klasickou neurohistologickou trojicí změn při sCJN (a dalších prionových nemocích) je spongiformní dystrofie, numerická atrofie neuronů a glióza (obr. 1). Spongiformní dystrofii charakterizují vakuoly v neuropilu, jejichž průměr je 2-20 µm. Objevují se ložiskově, je nutné odlišit je od artefaktů a od vakuol, které se objevují v horních korových vrstvách při různých příčinách korové atrofie.
Imunohistochemické vyšetření užívá různé typy protilátek ozřejmujících výskyt prionů ve tkáni (obr. 2).
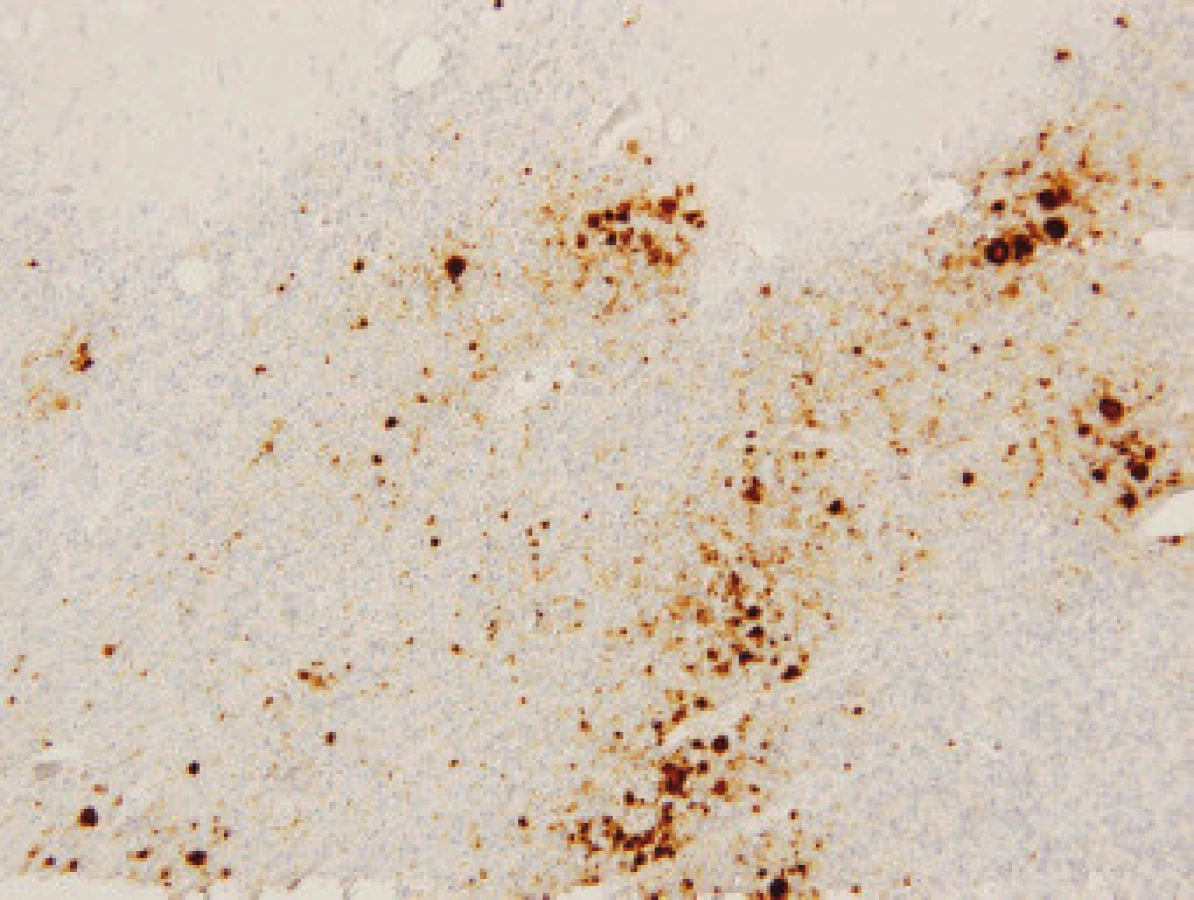
Nález při vCJN se od změn při sCJN odlišuje histologicky, imunohistochemicky a imunologicky [6]. Novou variantu CJN charakterizuje velký počet „floridních” plak v kůře mozku i mozečku, shluky malých plak v imunohistochemickém obraze, amorfní pericelulární a perivaskulární kupení PrP, těžké spongiformní změny a perineuronální a axonální kupení PrP v neostriatu, výrazná astrocytóza a numerická atrofie neuronů v zadních talamických jádrech a mezencefalu, perineurální a retikulární kupení PrP v šedé hmotě kmene a prodloužené míchy.
Národní referenční laboratoř transmisivních spongiformních encefalopatií a Creuzfeldtovy-Jakobovy nemoci České republiky
Laboratoř byla vybudována z rozhodnutí ministra zdravotnictví ČR při oddělení patologie Fakultní Thomayerovy nemocnice. Současně byla pro klinickou diagnostiku prionových onemocnění vyčleněna lůžka na Neurologické klinice IPVZ a FTNsP. Činnost zahájila laboratoř 1. 7. 2001. Je plně vybavena přístrojově a provádí standardní diagnostické vyšetření zemřelých s podezřením na prionové onemocnění, kdy se soustředí na diferenciální diagnostiku neurodegenerativních onemocnění s cílem vyhledávat případy prionových onemocnění. Základním smyslem činnosti je dohled nad možným výskytem vCJN. Zavedeny jsou neurohistologické a imunohistochemické metody nejen pro diagnostiku prionových chorob, ale i pro široké diferenciálně diagnostické pole neurodegenerativních onemocnění na současné evropské úrovni.
V letech 2002-2006 bylo v NRL vyšetřeno přes 90 autoptických vzorků tkáně CNS zemřelých, u nichž bylo vysloveno podezření z prionového onemocnění na úrovni pravděpodobné či možné CJN, nebo byla CJN zmíněna v diferenciálně diagnostické rozvaze. V 50 případech byla neuropatologicky potvrzena definitivní diagnóza prionového onemocnění typu CJN s roční distribucí odpovídající přibližně odhadované incidenci CJN v populaci (tab. 3). Většina případů byla klinicky alespoň v úrovni možné CJN rozpoznána, v některých případech však šlo o náhodný záchyt; je tedy zřejmé, že skutečný počet CJN bude v ČR vyšší. V ostatních případech se jednalo o široké spektrum jiných neurodegenerativních, zánětlivých, nádorových a cévních onemocnění CNS, přehled nejčastějších uvádí tab. 4. Z tabulky je zřejmé, že se jednalo o široké spektrum klasických diferenciálně diagnostických entit. Velmi zajímavé byly případy kombinací několika různých neuropatologických jednotek, kdy jsme v 1 případě diagnostikovali kombinované postižení Alzheimerovou nemocí spolu s difuzními vaskulárními změnami a vyvinutou difuzní korovou chorobou s Lewyho tělísky a ve 2 případech se jednalo o kombinované postižení Alzheimerovou nemocí a vaskulární subkortikální demencí Binswangerova typu. Ve 2 případech nebylo možné současnými diagnostickými metodikami dospět k jednoznačné definitivní diagnóze, a to i přes konzultace na špičkových referenčních neuropatologických pracovištích v Rakousku a ve Velké Británii.
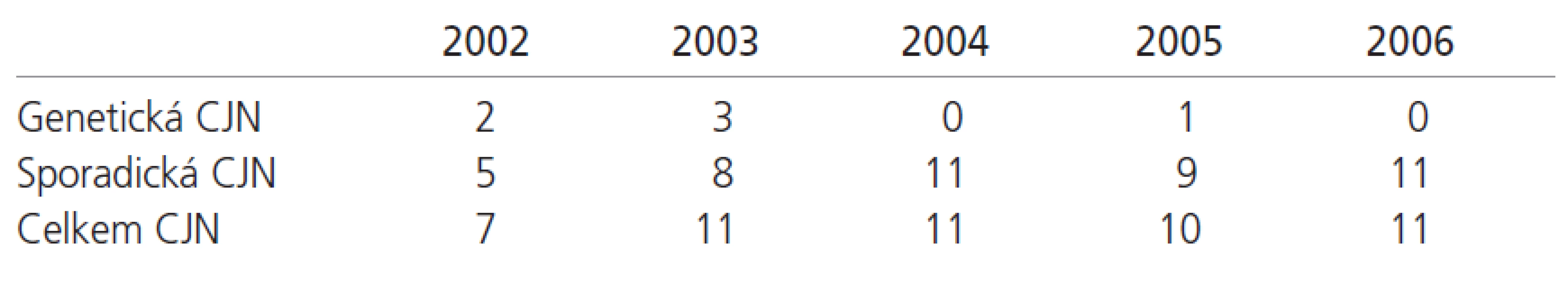

Kromě metodik autoptického vyšetřování tkání CNS laboratoř rutinně provádí intravitální testování ß - podjednotky proteinu 14-3-3 v mozkomíšním moku metodou western blot. Přítomnost tohoto nespecifického markeru s relativně vysokou mírou senzitivity a specificity svědčí u pacientů s rychle progredující demencí pro postižení prionovým onemocněním. Senzitivita i specificita tohoto vyšetření je poměrně široce diskutována v literatuře, je významně ovlivněna indikačními kritérii, přesto patří v současnosti do standardního vyšetřovacího postupu dle doporučení WHO (tab. 2).
Po částečné rekonstrukci a přístrojovém dovybavení v roce 2006 laboratoř provádí i typizaci prionových proteinů metodikou westernblot a molekulárně genetické vyšetřování PRNP genu přímým sekvenováním ke zjištění přítomnosti patogenní variace a polymorfizmu kodonu 129. Do současné doby bylo identifikováno 6 případů genetické formy CJN se 2 známými a nejčastějšími mutacemi v Evropské populaci E200K a D178N.
Laboratoř spolupracuje s referenčními laboratořemi prionových nemocí WHO ve skotském Edinburghu a Vídni v Rakousku. Všechna oddělení patologie ČR byla písemně informována o způsobu spolupráce v případě podezření na lidské prionové onemocnění.
Od 1. 1. 2007 začala NRL na základě doplňku transplantačního zákona povinně testovat mozkovou tkáň všech dárců rohovek na přítomnost patologické formy prionového proteinu metodou westernblot. NRL spolupracuje se všemi očními bankami v České Republice a od doby spuštění testování bylo vyšetřeno již přes 160 vzorků s negativním výsledkem.
Přijato k recenzi: 20. 4. 2007
Přijato do tisku: 15. 5. 2007
MUDr. Radoslav Matěj, Ph.D.
Národní referenční laboratoř TSE-CJN
Oddělení patologie a molekulární medicíny
Fakultní Thomayerova nemocnice s poliklinikou
Vídeňská 800
14059 Praha 4 - Krč
e-mail: radoslav.matej@ftn.cz
Sources
1. Jirák R, Koukolík F. Demence. Neurobiologie, klinický obraz, terapie. Praha: Galén 2004.
2. Watts JC, Balachandran A, Westaway D. The expanding universe of prion diseases. PloS Pathog 2(3) http://pathogens.plosjournals.org/perlserv/?request=get-document&doi=10.1371/journal.ppat.0020026
3. Smith PG, Cousens SN, d' Huillard Aignaux JN, Ward HJ, Will RG. The epidemiology of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol 2004; 284 : 161–191.
4. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005; 64(9): 1586-1591.
5. WHO: WHO Manual for Surveillance of Human Transmissible Spongiform Encephalopathies. (2003).
6. Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129 : 2278-2287.
7. Sanchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Saanchez-Valle R, Mitrova E et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2006; 67 : 637-643.
8. Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003; 126 : 1333-1346.
9. Preusser M, Strobel T, Gelpi E, Eiler M, Broessner G, Schmutzhard E, Budka H. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry 2006; 77 : 413-416.
10. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364 : 527–529.
11. Ironside JW, Head MW. Neuropathology and molecular biology of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol 2004; 284 : 133–159.
12. Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000; 37 : 1-9.
13. Glatzel M, Rogivue C, Ghani A, Streffer JR, Amsler L, Aguzzi A. Incidence of Creutzfeldt-Jakob disease in Switzerland.Lancet. 2002; 360 : 139-141.
14. Kovács GG, Majtényi C. Creutzfeldt-Jakob disease in Hungary. Folia Neuropathol 2005; 43 : 279-285.
15. Tabernero C, Polo JM, Sevillami MD, Muñoz R, Berciano J, Cabello A et al. Fatal familial insomnia: clinical, neuropathological, and genetic description of a Spanish family J Neurol Neurosurg Psychiatry 2000; 68 : 774-777.
16. Cali I, Castellani R, Yuan J, Al-Shekhlee A, Cohen ML, Xiao X et al. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain 2006; 129 : 2266-2277.
17. Mitrová E, Mayer V, Jovankovičová V, Slivarichová, D, Sólová L. Creutzfeldt–Jakob disease risk and PRNP codon 129 polymorphism: necessity to revalue current data. Eur J Neurol 2005; 12 : 998-1001.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2007 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Obrna lícního nervu
- Poruchy polykání ve vztahu k vertebrogenním dysfunkcím
- Protilátky proti glykokonjugátům v diagnostice autoimunitních neuropatií
- Dercumova choroba (lipomatosis dolorosa) – zriedkavo diagnostikované ochorenie: kazuistika