
Gender Differences in the CAG Repeats and Clinical Picture Correlations in Huntington’s Disease
Rozdíly v počtu CAG opakování mezi pohlavími a jejich korelace s klinickým obrazem u Huntingtonovy choroby
Cíle:
Huntingtonova choroba (HD) je dědičná neurodegenerativní porucha. Vztah mezi progresí příznaků a počtem CAG opakování u zmutovaného genu IT15 nebyl prozatím z hlediska pohlaví probádán. Cílem bylo zjistit všechny korelace mezi počtem trinukleotidových repeticí CAG u genu IT15 a věkem nástupu HD, symptomy a progresí nemoci, jakož i případné rozdíly mezi výsledky v závislosti na pohlaví.
Materiál a metodika:
41 pacientů (23 žen a 18 mužů) s mutací způsobující HD se podrobilo neurologickému hodnocení podle Jednotné hodnotící škály Huntingtonovy choroby (Unified Huntington’s Disease Rating Scale, UHDRS). Počet CAG repetic na exonu 1 genu IT15 byl stanoven metodou amplifikace DNA pomocí polymerázové řetězové reakce (PCR) a porovnáním výsledného produktu se standardní DNA.
Výsledky:
Signifikantní korelace mezi výsledky na škále UHDRS, počtem CAG opakování a časem propuknutí choroby byly objeveny pouze u žen. Korelace mezi počtem CAG opakování a věkem nástupu nemoci byla objevena jak u žen, tak u mužů.
Závěr:
Výsledky naznačují korelaci mezi klinickým stavem pacientek s HD a délkou CAG repetice. To by mohlo souviset s přítomností dalšího faktoru u žen.
Klíčová slova:
CAG trinukleotidová opakování – Huntingtonova nemoc – UHDRS – pohlaví
Authors:
D. Zielonka 1; A. Niezgoda 2; M. Olejniczak 3; W. Krzyzosiak 3; J. Marcinkowski 1; W. Kozubski 2
Authors‘ workplace:
Department of Social Medicine, Poznan University of Medical Sciences, Poland
1; Department of Neurology, Poznan University of Medical Sciences, Poland
2; Laboratory of Cancer Genetics, Institute of Bioorganic Chemistry, Polish Academy of Sciences in Poznan, Poland
3
Published in:
Cesk Slov Neurol N 2008; 71/104(6): 688-694
Category:
Original Paper
Overview
Aims:
Huntington’s disease (HD) is a hereditary neurodegenerative disorder. The relationship between symptom progression and the number of CAG repeats in the mutated IT15 gene has not been investigated in relation to gender. The aims were to investigate any correlation between the number of CAG trinucleotide repeats in the IT15 gene and the age of onset of HD, its symptoms and progress and whether the findings differed according to gender.
Materials and methodology:
41 patients (23 women, 18 men) with the mutation causing HD were assessed neurologically on the Unified Huntington’s Disease Rating Scale (UHDRS). The number of CAG repeats on exon 1 of the IT15 gene was determined by polymerase chain reaction (PCR) amplification and by comparison of its product with the DNA size standard.
Results:
Significant correlations between all UHDRS subscale scores, the number of CAG repeats and time from onset were found in women only. A correlation between the number of CAG repeats and age of onset was found in both men and women.
Conclusions:
The results indicate a correlation between the clinical status of HD female patients and their CAG repeat lengths. This could be a result of an additional factor present in females.
Key words:
CAG triplet repeats – Huntington’s disease – UHDRS – gender
Introduction
Huntington’s disease (HD) is a hereditary neurodegenerative, progressive disorder in which gender impact on disease progression has not been investigated [1].
The aim of the current study was to analyze the relationship between the clinical progression of the disease, as measured on the Unified Huntington Disease Rating Scale (UHDRS) [2], and the number of CAG repeats, the time from onset and the age of onset in men and women.
In our previous study [3] we found that the clinical picture of HD, as evaluated by UHDRS, correlates with both the number of CAG repeats and the time from the onset of the disease in both genders. However that earlier study considered 11 patients only.
In the current study we decided to check men and women separately in order to determine whether there were any sexual differences in the clinical picture of the disease.
Subjects and methods
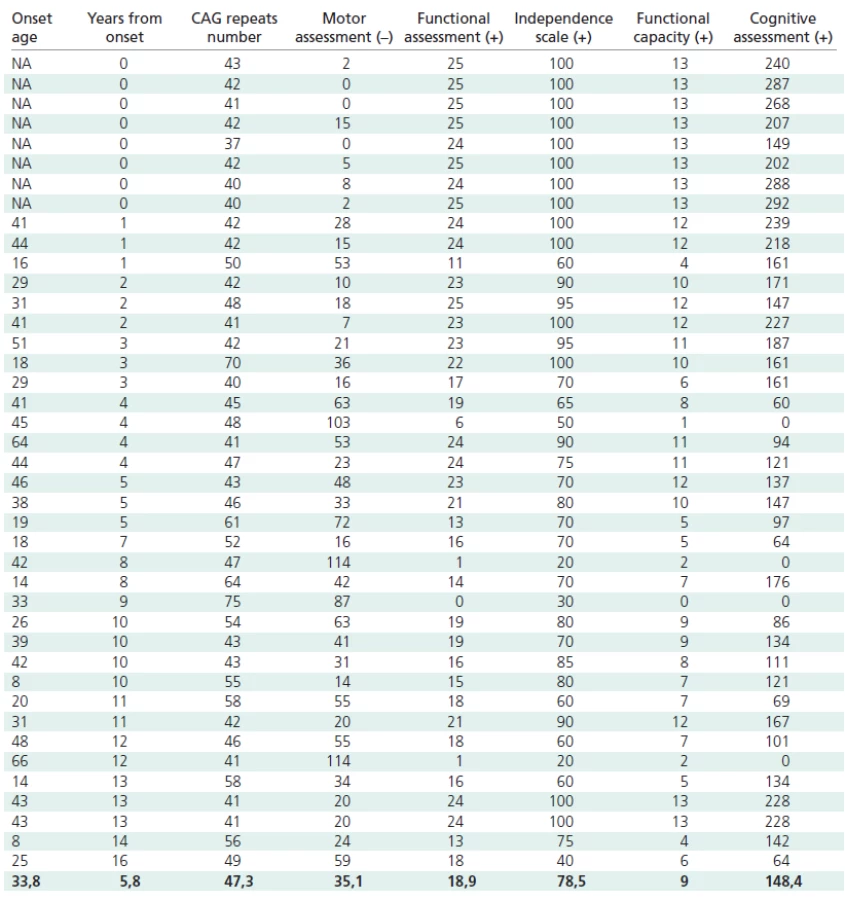
Genetic examinations to confirm HD were carried out on 41 Polish patients and gene carriers (23 women and 18 men) aged 16–78, suspected of HD on the basis of a genetic examination and on their symptoms, including choreic movements, dementia, reduced muscle tone and family history. Thirtythree persons showed full symptoms, and eight were symptomless (one with incomplete gene penetration, Table 1). Nine of our symptomatic probands were affected with the juvenile form of the disease. All the patients agreed to undergo a clinical assessment, including the application of a specific HD clinimetric scale, and all gave their written informed consent [2,4].
The examination procedure was accepted by the Bioethics Committee of Poznań University of Medical Sciences and carried out according to the principles established in Helsinki.
A genetic examination of the number of CAG repeats coding glutamin within exon 1 of IT15 gene (locus 4p 16.3) was carried out. The material examined was DNA isolated from leucocytes of the patients’ peripheral blood. The methods of examination used were PCR and the separation of a radioisotope labeled PCR product against a DNA size marker (pGEM®-3Zf(+) sequencing ladder) in polyacrylamide gel [5].
To evaluate the clinical status of the patient an UHDRS, composed of neurological and psychological parts, was used.
Within the neurological part there are four subscales:
- a) a motor assessment – in which motor disturbances are evaluated,
- b) a functional assessment – which evaluates disturbances in dealing with everyday activities (washing, cleaning, shopping, professional work, etc.),
- c) an independence scale – which evaluates the patient’s ability to function independently when performing everyday activities (eating, washing, dressing etc.),
- d) functional capacity – which evaluates the patient’s potential to function in society and to perform everyday activities. This assessment is based on conversations with the patient’s carers or relatives.
The psychological part contains a cognitive scale, which consists of a verbal fluency test, symbol digit modality test and Stroop test [2,6].
To measure the degree of correspondence between two ordinal-level variables (i.e. CAG repeats and years from onset and scale assessments) and to assess its significance, the Kendall’s Tau test was applied in the study [7].
Moreover, a Weibull regression was adopted using the Bayesian approach [8]. In the model the hazard of onset has been modeled depending on time itself (age of patients at onset) and covariates (number of CAG repeats and gender). Censored observations (gene carriers) have also been taken into account. Two regression analyses have been performed, one for all patients and one for males and females separately.
The computation was performed using WinBUGS version 1.4 [9] based on the simulation technique known as Gibbs sampling. To achieve convergence, two parallel chains were run and the first 1,000 samples of each were discarded (a burn-in), while the following 10,000 cycles (production run) of the Gibbs sampler were used to estimate each quantity of interest. An equilibrium state of streams of values was established via an examination of within chain autocorrelation and a comparison of results of the chains started with over dispersed initial values, including the use of the Gelman-Rubin statistic available within the software.
Results
The number of CAG repeats in mutant allels ranged from 37 to 75, with a mean number of 47.3 ± 8.7. All the patients were heterozygous for the mutant allele. The time from the onset of symptoms ranged from 0 to 16 years, with a mean of 5.6 ± 4.9 years. The age of onset of the disease varied from 8 to 66 years, with a mean of 33.8 ± 14.7 years.
The following results were obtained in the particular subscales: motor scale from 0 to 114 (mean 35.1 ± 30.6); functional scale from 0 to 25 (mean 18.8 ± 6.8); independence scale from 20 to 100 (mean 78.5 ± 23.0); functional efficiency from 0 to 13 (mean 8.9 ± 3.9). The total score in the psychological examination varied from 0 to 292 (mean 148.4 ± 79.9).
Statistically significant negative correlations were found between the number of CAG repeats and the age at onset (t = –0.48; p = 0.000072) in both male and female patients.
In addition, we found many significant correlations between the CAG repeat number and the UHDRS score in female patients with onset in adulthood only, in female HD patients only (with onset in adulthood and juvenile onset), and in all female mutation carriers and female patients (Table 2).
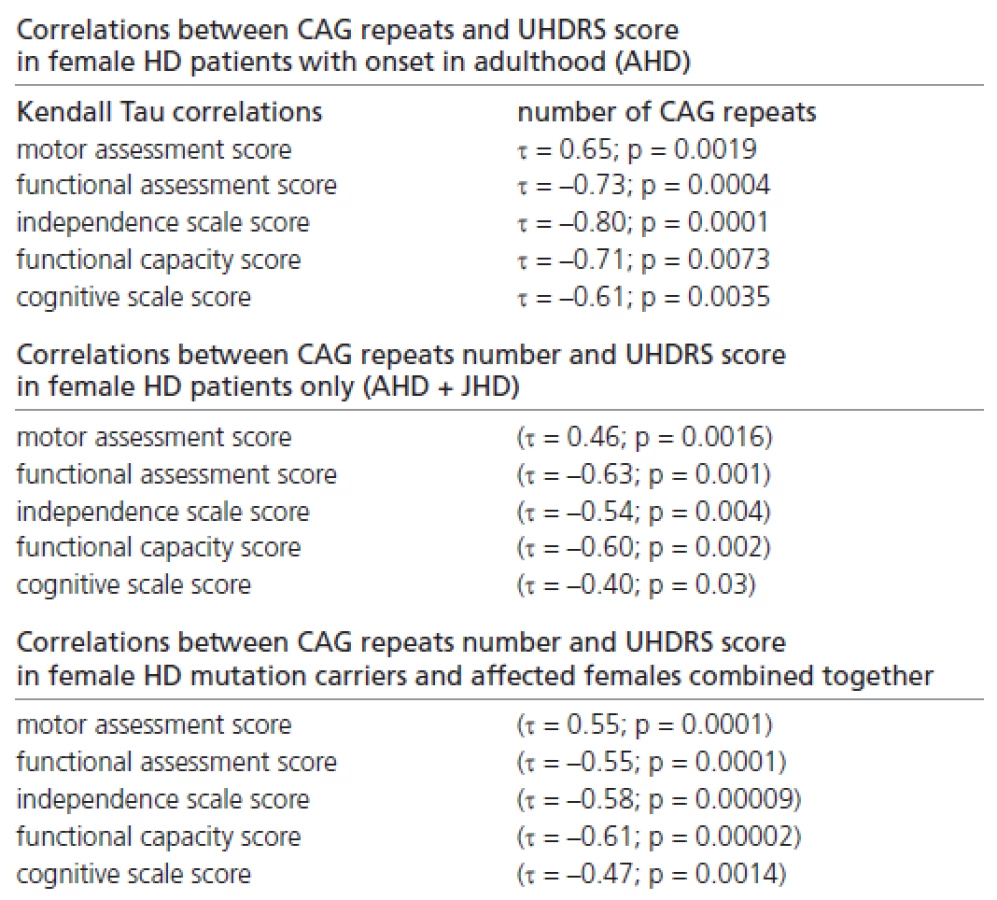
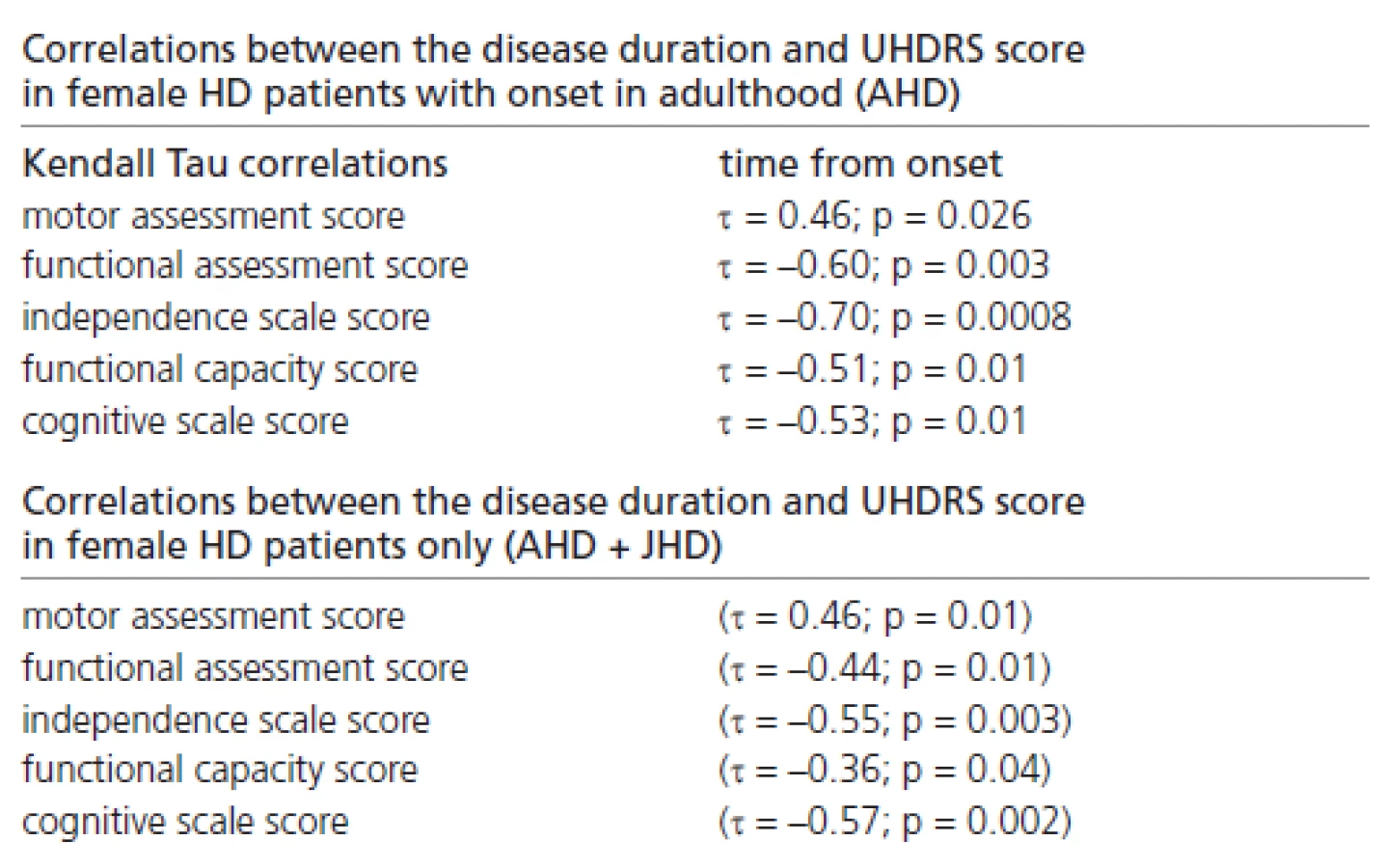
Significant correlations were also found between the disease duration and the UHDRS score, reflecting the probands’ clinical status in females with onset in adulthood only, and in those females (with onset in adulthood and juvenile onset) combined together (Table 3).
Interrelations, on the borderline of significance between CAG number and cognitive assessment, were found in the group of men with HD onset in adulthood (Table 4) and between CAG number and motor assessment in seven male JHD patients (Table 5).


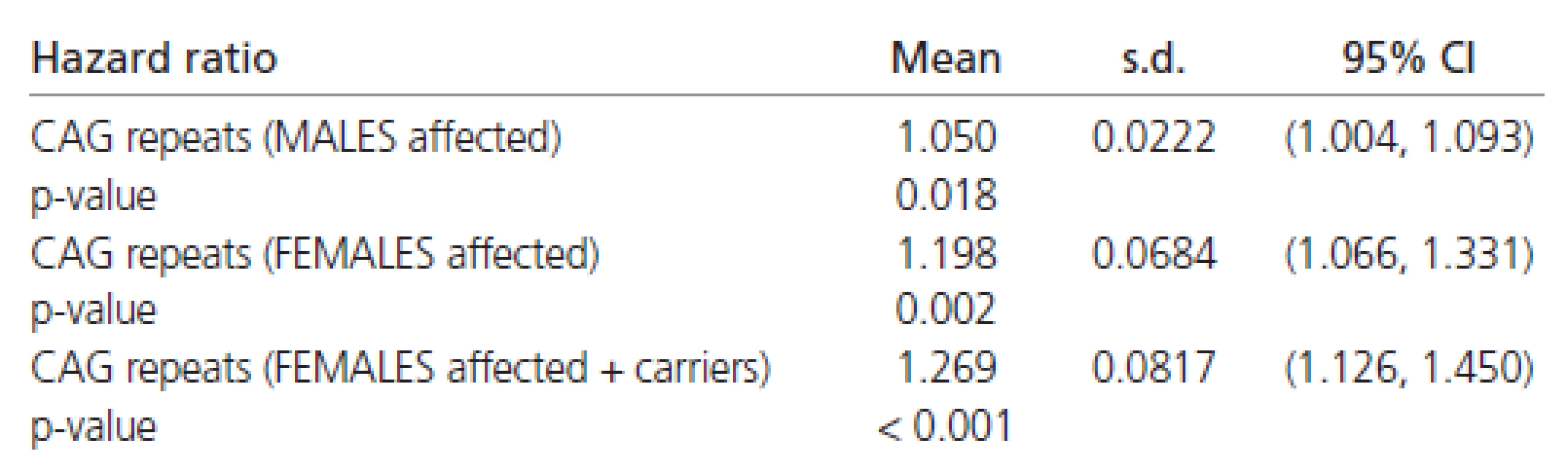
The estimated hazard ratios derived from the Weibull’s regression in all the patients are shown in Table 6. These values indicate that females have about a 50 % chance of early disease affection in comparison to males (Table 6). Moreover, the affection risk increases with the number of CAG repeats in all patients (approximately 8 % for each additional CAG repeat).
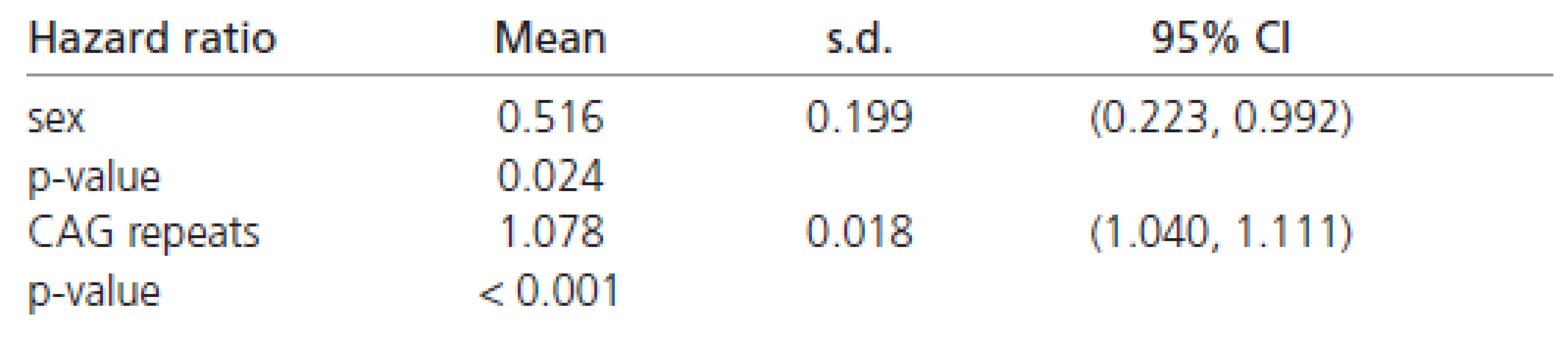
The estimated Weibull’s regression hazard ratios in males and females separately are reported in Table 7. The estimates shown there indicate a four to five times stronger impact of CAG repeats on the rate of disease progression in females than in males. Combining all the results (Tables 6 and 7) confirming the finding that females have about half the chance of early affection by the disease in comparison to males. However, the influence of CAG repeats on the rate of disease progression is at least four times greater in females than in males.
Discussion
Huntington’s disease (HD) is recognized as a disorder in which symptom progression is not entirely dependent on the number of CAG repeats [10–15]. However, in homozygous cases, in respect of a mutant allele, progress of the disease is quicker than in heterozygous cases [16,17].
It is believed that the number of CAG repeats in the IT15 gene correlates with the age of disease onset [18,19]. However, it has been observed that other factors determining significant variations in onset age exist. Such factors as the GluR6 kainate glutamate receptor (GRIK2), apolipoprotein E (APOE), the transcriptional coactivator CA150 (TCERG1), the ubiquitin carboxy‑terminal hydrolase L1 (UCHL1), p53 (TP53), caspase‑activated DNase (DFFB), and the NR2A and NR2B glutamate receptor subunits (GRIN2A, GRIN2B) have all been considered as possible factors influencing variance of onset [18,20]. It has recently been confirmed that GRIN2A and TCERG1 show an association with the residual age of HD onset [20,21]. It has also been reported that GRIN2A influences the age of onset in relation to the gender of the patient [21]. A number of research reports concerning GRIK2 and APOE give conflicting results. While a correlation between the number of CAG repeats and age of onset has been considered as stronger if the number of TAA triplet repeats within the gene for GRIK2 is taken into account [18], other studies have not confirmed this relationship [20]. In 1999 a claim was made that the apolipoprotein’s E genotype influences age of onset in an age specific manner [22]. However, Saft et al later denied this allegation [23].
Some special forms of HD have been described. The most different form of HD, in terms of both symptoms and progression, is juvenile HD (JHD) [24,25]. A correlation between the number of CAG repeats and age of onset is different in this group of patients, in which each CAG repeat has a weaker impact on age of onset than in an adult HD population [26]. It appears to be related to differences in the pathogenetic model of JHD [27].
A relationship between gender and progression of the disease has not been investigated. However it has been found, in the mouse HD model, that gender appears to play a role in the neuropathological changes and phenotype differences [28]. In the mouse model it has been observed that female offspring show a tendency to a reduction in the number of trinucleotide repeats when they inherit the disease from their male parents [29]. This suggests the existence of a repair mechanism present in female fetuses, probably connected with chromosome X. On the other hand, it has been reported that the disease develops earlier in those sons who inherit the mutation from their father. [29–32].
The Weibull’s analysis in this study indicates that the gradient of risk of being affected increases more steeply in females than in males. This means that there is a relatively larger number of women with the same number of CAG repeats who will start symptoms. In the male the opposite applies, with the risk of being affected earlier being higher, but the number of men with the same CAG number who will start to exhibit symptoms in the same time is smaller. The analysis thus indicates populational differences between genders in the area of early HD affectation. However the size of population in this study is not large enough to draw a general conclusion.
In carrying out a Kendall correlation, the following factors were taken into consideration: evaluation of the clinical findings in UHDRS and the overall picture, the number of CAG repeats and the time from onset. A significant reverse correlation between the age at onset and the number of CAG repeats was observed in the patients in both the male and female groups. The lower UHDRS scores in the functional assessment, independence scale, functional capacity and cognitive assessments, and the higher motor assessment score obtained in the group of women, was connected with a larger number of CAG repeats and a longer time from onset.
Our proband population has been divided into subgroups according to the type of HD (juvenile or adult) and also according to the presence or absence of symptoms. We first tried to find any relations in a subpopulation of patients with HD onset in adulthood. Statistically significant correlations were found in the female cohort only. We then decided to add juvenile HD females to the cohort in order to find out what influence a juvenile pathogenetic model had upon the earlier results. In many previously published reports it has been suggested that JHD patients should be considered separately, because of differences in the pathogenesis and symptomatology of the disease, although both types are related to a mutation in the same gene [24,27]. The effect of another gene, or of a non‑genetic factor being the cause of the different clinical course of JHD have also been suggested [24,33,34]. In the light of these latter suggestions we decided to check what effect the addition of our JHD female patient population would have on the total cohort’s results. Reconsideration of the correlations we found in the larger group of female patients (those with adult and juvenile onset combined) did not alter the tendency previously observed. Finally, we decided to add a group of eight pre‑symptomatic females to this analysis and, again, the tendency previously observed did not change. In addition, a similar result has been confirmed in a group of pre‑symptomatic women considered separately.
A statistically significant correlation has only been reported previously between the age of onset and the number of CAG repeats [26,35–40]. One report, concerning a Russian population, is an exception [41] but this was conducted with the use of different clinical quantification tools. These authors reported finding a statistically significant correlation between the number of CAG repeats and the patients’ psychological and neurological condition, but they did not divide their subjects by gender.
The correlations in the female patients reported here were very strong in contrast to the male group. If similar results are found in a sufficiently large group of patients this would confirm that there is a factor in women which is a condition for compatibility between clinical findings and the genetic picture. The genetic aspects of chromosome X and the hormonal differences between women and men should be taken into consideration. This research is the first of its type conducted on a Polish population and therefore the cause of the differences mentioned above requires further investigation in the field of HD pathogenesis and with particular reference to phenotype conditioning cofactors.
It is worth emphasizing the fact that dating disease onset in HD patients is difficult especially in retrospective studies. According to recent studies some HD symptoms may even start as much as 10 years before the estimated disease onset [42].
A recent report of research conducted on a Slavic population reported longer survival rates in female HD patients than in males and suggests the existence of sexual differences in disease progression [43].
In future in order to achieve a more complete picture of the pathological changes which occur in the course of HD and to make the diagnostics more appropriate to the stage of the disease found when the patient first attends, the number of probands should be increased and the diagnostics should be extended to include imaging techniques e. g. – woxel‑based magnetic resonance imaging (MRI) volumometry, MRI spectroscopy and three-dimensional emission tomography of the single photon, if available. Quantification of the patients’ clinical status after consecutive time intervals should also be carried out.
Dr. Daniel Zielonka M.D., Ph.D.
Rokietnicka Str., No. 5”C”
60–806 Poznan, Poland
phone: +48 504609951
fax: +48 618547390
e‑mail: daniel.zielonka@wp.pl
Admitted to review: 25. 5. 2008
Accepted to publication: 28. 8. 2008
Sources
1. Martin JB, Gusella JF. Huntington‘s disease. Pathogenesis and management. N Engl J Med 1986; 315(20): 1267–1276.
2. Zielonka D, de Mezer M, Niezgoda A, Reperowicz K, Krzyzosiak W, Kozubski W. Clinical picture of patients with Huntington‘s disease in relation to the number of trinucleotide CAG repeats in IT–15 gene. Neurol Neurochir Pol 2002; 36(5): 903–909.
3. Huntington Study Group. Uniform Huntington’s Disease Rating Scale: reliability and consistency. Mov Dis 1996; 11 : 136–142.
4. Sharma KR, Romano JG, Ayyar DR, Rotta FT, Facca A, Sanchez-Ramos J. Sympathetic skin response and heart rate variability in patients with Huntington’s disease. Arch Neurol 1999; 56(10): 1248–1252.
5. Peel A, Rao R, Cottrell B, Hayden MR, Ellerby LM, Bredesen DE. Double–stranded RNA-depended protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet 2001; 10(15): 1531–1538.
6. Zielonka D, Reperowicz K, Murawa D, Krzyżosiak W, Kozubski W. Obraz kliniczny w kontekście badań genetycznych i neuroobrazowych u pacjentów z pląsawicą Huntingtona. Nowiny Lekarskie 2000; 3 : 307–404.
7. Kendall M. A new measure of rank correlation. Biometrika 1938; 30 : 81–89.
8. Congdon P. Bayesian Statistical Modelling. Chichester (UK): Wiley 2001 : 429–430.
9. Lunn DJ, Thomas A, Best N, Spiegelhalter D. WinBUGS – a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000; 10 : 325–337.
10. Kieburtz K, MacDonald M, Shih C, Feigin A, Steinberg K, Bordwell K et al. Trinucleotide repeat length and progression of illness in Huntington‘s disease. J Med Genet 1994; 31(11): 872–874.
11. Claes S, Van Zand K, Legius E, Dom R, Malfroid M, Baro F et al. Correlations between triplet repeat expansion and clinical features in Huntington‘s disease. Arch Neurol 1995; 52(8): 749–753.
12. Benitez J, Fernandez E, Garcia Ruiz P, Robledo M, Ayuso C, Garcia Yebenes J. The gene responsible for Huntington‘s disease in Spanish families: its diagnostic value and the relation between trinucleotide expansion and the clinical characteristics. Rev Clin Esp 1994; 194(8): 591–593.
13. Feigin A, Kieburtz K, Bordwell K, Como P, Steinberg K, Sotack J et al. Functional decline in Huntington’s disease. Mov Disord 1995; 10(2): 211–214.
14. Siesling S, van Vugt J, Zwinderman KA, Kieburtz K, Roos R. Unified Huntington’s disease rating scale: a follow up. Mov Disord 1998; 13(6): 915–919.
15. Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K et al. Rate of functional decline in Huntington’s disease. Neurology 2000; 54(2): 452–458.
16. Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 2003; 126(4): 946–955.
17. Dürr A, Hahn-Barma V, Brice A, Pêcheux C, Dodé C, Feingold J. Homozygosity in Huntington‘s disease. J Med Genet 1999; 36(2): 172–173.
18. MacDonald M, Vonsattel J, Shrinidhi J, Couropmitree NN, Cupples LA, Bird ED et al. Evidence for the GluR6 gene associated with younger onset age of Huntington‘s disease. Neurology 1999; 53(6): 1330–1332.
19. de Rooij KE, de Koning Gans PA, Losekoot M, Bakker E, den Dunnen JT, Vegter-van der Vlis M et al. Borderline repeat expansion in Huntington‘s disease. Lancet 1993 11; 342(8885): 1491–1492.
20. Andresen JM, Gayán J, Cherny SS, Brocklebank D, Alkorta-Aranburu G, Addis EA. Replication of twelve association studies for Huntington‘s disease residual age of onset in large Venezuelan kindreds. J Med Genet 2007; 44(1): 44–50.
21. Arning L, Saft C, Wieczorek S, Andrich J, Kraus PH, Epplen JT. NR2A and NR2B receptor gene variations modify age at onset in Huntington’s disease in a sex-specific manner. Hum Genet 2007; 122(2): 175–182.
22. Kehoe P, Krawczak M, Harper PS, Owen MJ, Jones AL. Age of onset in Huntington disease: sex specific influence of apolipoprotein E genotype and normal CAG repeat length. J Med Genet 1999; 36(2): 108–111.
23. Saft C, Andrich JE, Brune N, Gencik M, Kraus PH, Przuntek H et al. Apolipoprotein E genotypes do not influence the age of onset in Huntington‘s disease. J Neurol Neurosurg Psychiatry 2004; 75(12): 1692–1696.
24. Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O. Juvenile Huntington’s disease: Does a dosage-effect pathogenic mechanism differ from the classical adult disease? Mech of Ageing Dev 2006; 127(2): 208–212.
25. Židovská J, Klempíř J, Kebrdlová V, Uhrová T, Koblihová J, Anders M et al. Huntingtonova nemoc: zkušenosti s genetickým testováním v letech 1994–2005. Cesk Slov Neurol N 2007; 70/103(1): 72–77.
26. Andresen JM, Gayán J, Djoussé L, Roberts S, Brocklebank D, Cherny SS. The relationship between CAG repeat length and age of onset differs for Huntington‘s disease patients with juvenile onset or adult onset. Ann Hum Genet 2007;71(3): 295–301.
27. Seneca S, Fagnart D, Keymolen K, Lissens W, Hasaerts D, Debulpaep S et al. Early onset Huntington disease: a neuronal degeneration syndrome. Eur J Pediatr 2004; 163(12): 717–721.
28. Dorner JL, Miller BR, Barton SJ, Brock TJ, Rebec GV. Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington‘s disease. Behav Brain Res 2007; 178(1): 90–97.
29. Kovtun I, Therneau T, McMurray C. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington‘s disease gene. Hum Mol Genet 2000; 9(18): 2767–2775.
30. MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH et al. Gametic but not somatic instability of CAG repeat length in Huntington‘s disease. J Med Genet 1993; 30(12): 982–986.
31. Zühlke C, Riess O, Bockel B, Lange H, Thies U. Mitotic stability and meiotic variability of the (CAG)n repeat in the Huntington’s disease gene. Hum Mol Genet 1993; 2(12): 2063–2067.
32. Leeflang E, Tavare S, Marjoram P, Neal CO, Srinidhi J, MacFarlane H et al. Analysis of germline mutation spectra at the Huntington‘s disease locus supports a mitotic mutation mechanism. Hum Mol Genet 1999; 8(2): 173–183.
33. Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington‘s disease age of onset. Proc Natl Acad Sci USA 2004; 101(10): 3498–3503.
34. Cannella M, Gellera C, Maglione V, Giallonardo P, Cislaghi G, Muglia M. The gender effect in juvenile Huntington disease patients of Italian origin. Am J Med Genet B Neuropsychiatr Genet 2004; 125B(1): 92–98.
35. Sánchez A, Mìla M, Castellvi-Bel S, Calopa M, Genis D, Jiménez D et al. Molecular analysis of the IT-15 gene in 79 Spanish families with Huntington‘s disease: diagnostic confirmation and presymptomatic diagnosis. Med Clin (Barc) 1997; 108(18): 687–690.
36. Vojvodić N, Culjković B, Romac S, Stojković O, Sternić N, Sokić D et al. Importance of the number of trinucleotide repeat expansions in the clinical manifestations of Huntington‘s chorea. Srp Arh Celok Lek 1998; 126(3–4): 77–82.
37. Snell R, MacMillan J, Cheadle J, Fenton I, Lazarou LP, Davies P et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington‘s disease. Nat Genet 1993; 4(4): 393–397.
38. Ranen NG, Stine OC, Abbott MH, Sherr M, Codori AM, Franz ML et al. Anticipation and instability of IT-15 (CAG)n repeats in parent‑offspring pairs with Huntington disease. Am J Hum Genet 1995; 57(3): 593–602.
39. Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Roos CA. Trinucleotyde repeat length and clinical progression in Huntington’s disease. Neurology 1996; 46(2): 527–531.
40. Squitieri F, Cannella M, Simonelli M. CAG mutation effect on rate of progression in Huntington’s disease. Neurol Sci 2002; 23 (Suppl 2): S107–S108.
41. Illarioshkin SN, Igarashi S, Onodera O, Markova ED, Nikolskaya NN, Tanaka H et al. Trinucleotide repeat length and rate of progression of Huntington‘s disease. Ann Neurol 1994; 36(4): 630–635.
42. Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M et al. Detection of Huntington‘s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008; 79(8): 874–880.
43. Pekmezovic T, Svetel M, Maric J, Dujmovic-Basuroski I, Dragasevic N, Keckarevic M et al. Survival of Huntington‘s disease patients in Serbia: longer survival in female patients. Eur J Epidemiol 2007; 22(8): 523–526.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Multiple Sclerosis and Magnetic Resonance Imaging: Present Status and New Trends
- Adult Age Sleep Apnoea
- Subacute Hypertensive Reversible Leukoencephalopathy – a Case Report
- Protein 14-3-3 Detection in Cerebrospinal Fluid – Clinico-Pathological Correlation