
Autoimunitní encefalitidy
Autoimmune Encephalitis
Autoimmune encephalitis is a term used to describe a group of acute or subacute monophasic or progressive inflammatory CNS disorders with autoimmune pathogenesis (i.e. induced by pathogenic autoantibodies or autoreactive effector cells). This group includes such diverse conditions as paraneoplastic CNS syndromes, limbic encephalitis, anti-NMDAR encephalitis, Morvan’s syndrome, Rasmussen encephalitis, stiff-person syndrome spectrum disorders including progressive encephalomyelitis with rigidity and myoclonus, autoimmune movement disorders, encephalitis lethargica, neurological syndromes with increased sensitivity to gluten, etc. This area of neuroimmunology has evolved dynamically during the past 10 years. Many new syndromes have been described, understanding of pathogenesis of the already known syndromes has improved and new antibodies were identified. This review aims to provide an overview of current knowledge in the field of autoimmune encephalitis in order to help clinicians navigate the rough waters of this exciting area of neurology.
Key words:
autoimmune diseases – autoantibodies – encephalitis – paraneoplastic syndromes –limbic encephalitis – anti-N-methyl-D-aspartate receptor encephalitis – epilepsy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
D. Krýsl 1; M. Elišák 2
Authors‘ workplace:
Klinisk Neurofysiologi, Sahlgrenska Universitetssjukhuset, Göteborg
1; Neurologická klinika 2. LF UK a FN v Motole, Praha
2
Published in:
Cesk Slov Neurol N 2015; 78/111(1): 7-23
Category:
Minimonography
doi:
https://doi.org/10.14735/amcsnn20151
Overview
Autoimunitní encefalitidy jsou akutní či subakutní monofázická nebo progresivní zánětlivá onemocnění CNS podmíněná autoimunitními mechanizmy (působením patogenních autoprotilátek nebo autoagresivních efektorových buněk). Mezi autoimunitní encefalitidy lze řadit klasické paraneoplastické syndromy s postižením CNS, limbické encefalitidy, anti‑NMDAR encefalitidu, Morvanův syndrom, Rasmussenovu encefalitidu, onemocnění z okruhu Stiff ‑ Person Syndromu včetně progresivní encefalomyelitidy s rigiditou a myoklonem, akutní autoimunitně podmíněné extrapyramidové poruchy hybnosti, encephalitis lethargica, neurologické syndromy se zvýšenou senzitivitou vůči glutenu a další. V posledních letech prodělala tato oblast neuroimunologie prudký rozvoj. Byly popsány nové syndromy, upřesněny klinické projevy a etiopatogenetické mechanizmy některých již známých chorob a identifikována řada nových typů protilátek. Tato minimonografie si klade za cíl podat ucelený přehled současných znalostí v oblasti autoimunitních encefalitid a umožnit tak klinikům snazší orientaci v této oblasti neurologie.
Klíčová slova:
autoimunita – autoprotilátky – encefalitida –paraneoplastické syndromy – limbická encefalitida – NMDAR encefalitida – epilepsie
Úvod
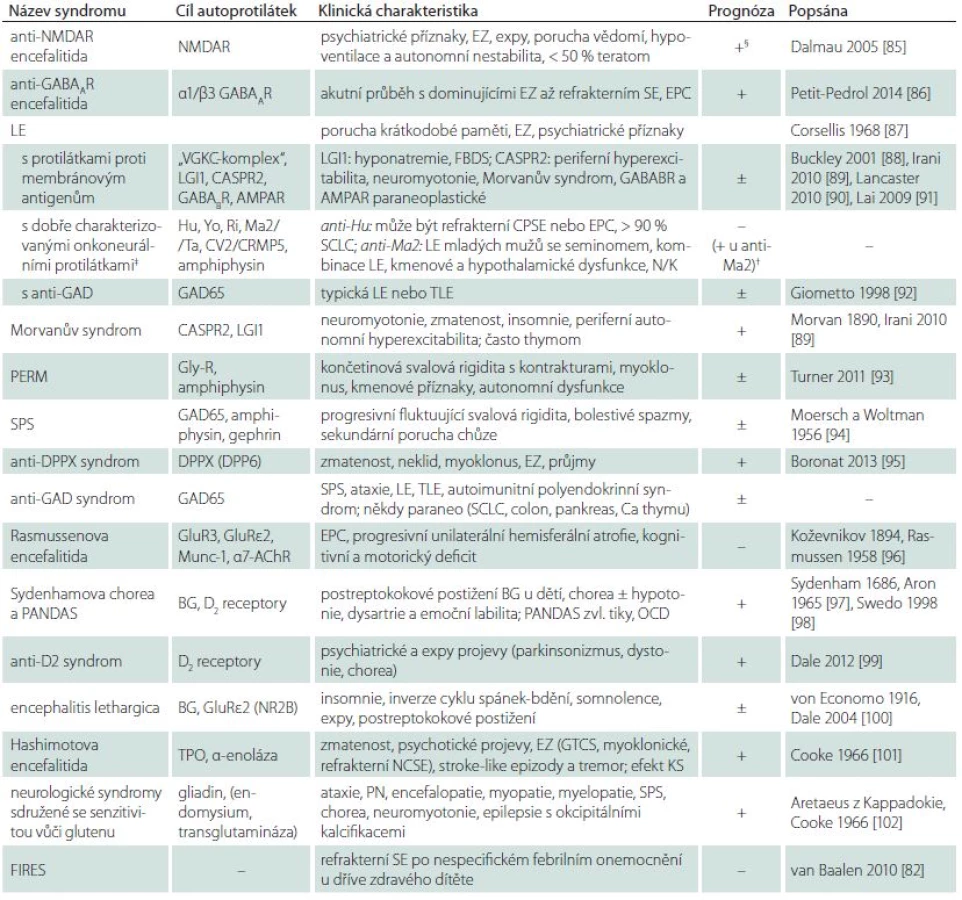
Autoimunitní encefalitidy (AIE) lze charakterizovat jako pestrou skupinu většinou monofázických nebo progresivních chorob, pro které je společné postižení mozku na autoimunitním podkladě. Na rozdíl od demyelinizačních onemocnění dominuje u AIE postižení korové. Klinický obraz a prognóza se u jednotlivých syndromů velmi liší. Na následujících stránkách jsme se pokusili předložit klinicky relevantní přehled patofyziologie, diagnostiky a terapie nejvýznamnějších zástupců této skupiny onemocnění (tab. 1). Důraz je kladen na limbické encefalitidy a encefalitidu s protilátkami proti glutamátovým N ‑ metyl ‑ D ‑ aspartátovým receptorům (anti‑NMDAR encefalitidu) vzhledem k jejich relativně vyšší prevalenci a dobré prognóze při včasné a správně vedené terapii.
Obecné poznámky k patofyziologii autoimunitních encefalitid
Poškození mozku u AIE je per definitionem zprostředkováno autoimunitními mechanizmy. U jednotlivých syndromů může dominovat působení buď patogenních protilátek (angl. directly pathogenic antibodies; tj. protilátek, jejichž vazba na cílový antigen způsobuje sama o sobě poškození anebo funkční ovlivnění cílové tkáně a podílí se tak přímo na vzniku příznaků), nebo autoreaktivních efektorových buněk. Příkladem onemocnění s dominantní úlohou patogenních protilátek jsou AIE sdružené s protilátkami proti membránovým antigenům. Tyto protilátky se váží na extracelulární epitop cílového proteinu lokalizovaného na cytoplazmatické membráně, obvykle v oblasti synapse, a jejich vazba může:
- ovlivňovat funkci dané cílové molekuly (např. protilátky proti receptoru typu B pro kyselinu γ ‑ aminomáselnou – anti‑GABABR inaktivují tento receptor),
- způsobovat internalizaci cílového proteinu (receptoru) (např. protilátky proti glutamátovým N ‑ metyl ‑ D ‑ aspartátovým receptorům – anti‑NMDAR a protilátky proti glutamátovým α ‑ amino ‑ 3 - hydroxy ‑ 5 - metyl ‑ 4 - isoxazol propionátovým receptorům – anti‑AMPAR),
- indukovat fixaci komplementu a následnou komplementem zprostředkovanou buněčnou smrt (např. protilátky proti Leucin‑rich Glioma Inactivated proteinu 1 – anti‑LGI1).
Funkční ovlivnění cílového membránového receptoru protilátkami je reverzibilní děj: v nepřítomnosti protilátek dochází k přirozené obnově funkčních receptorů a k ústupu příznaků onemocnění.
Předpokládá se, že AIE sdružené s protilátkami proti intracelulárním antigenům jsou primárně T buněčně zprostředkovány a protilátky s nimi spojené jsou pravděpodobně nepatogenní. Prokázat roli T buněčných mechanizmů v patogenezi autoimunitního postižení je náročné. Argumenty se opírají o:
- nedostatečnou účinnost léčby cílené na eliminaci a snížení produkce protilátek,
- neuropatologické nálezy (např. infiltráty granzym ‑ B pozitivních cytotoxických T lymfocytů v blízkosti umírajících neuronů u pacientů s paraneoplastickou limbickou encefalitidou anti‑Hu a anti‑Ma2) [1],
- průkaz autoreaktivních T lymfocytů v séru či likvoru (např. nález specifických anti‑Hu a anti‑Yo reaktivních T lymfocytů v séru a likvoru u pacientů s paraneoplastickými syndromy) [2].
Příkladem AIE s protilátkami proti intracelulárním antigenům jsou syndromy sdružené s protilátkami proti glutamát dekarboxyláze (anti‑GAD), nebo paraneoplastické encefalitidy sdružené s tzv. dobře charakterizovanými onkoneurálními protilátkami (v dalším textu jsou souhrnně označovány jako PNP ‑ Ab – konkrétně jde o anti‑Hu, anti‑Yo, anti‑Ri, anti‑Ma2, anti‑amphiphysin a protilátky proti collapsin response mediator proteinu 5 – anti‑CRMP5/ CV2).
Příčiny vzniku autoagresivní reakce u AIE nejsou objasněny, zvláště v neparaneoplastických případech. U disponovaných pacientů může hrát roli virový či bakteriální spouštěč nebo vakcinace. V paraneoplastických případech se předpokládá následující sekvence dějů [3]: 1. Periferně uložený solidní tumor vystavuje na svém povrchu proteiny, které imunitní systém rozpoznává jako cizí. 2. Části nádoru jsou fagocytovány antigen prezentujícími buňkami, zejm. dendritickými buňkami, které následně migrují do regionálních uzlin, kde indukují klonální expanzi CD4+ a CD8+ T lymfocytů a tumor specifických B lymfocytů. 3. Tumor ‑ specifické T lymfocyty a protilátky zpomalují růst tumoru, současně však reagují zkříženě s tkáněmi vlastního těla (nervového systému). 4. Za určitých podmínek (např. v přítomnosti některých interleukinů) mohou autoreaktivní T a B lymfocyty migrovat přes hematoencefalickou bariéru a způsobovat autoimunitní zánět v CNS. V paraneoplastických případech tedy dochází k primární produkci protilátek v séru a vyšetření PNP ‑ Ab ze séra považujeme za dostačující.
Obecné poznámky k diagnostice autoimunitních encefalitid
Diagnostika autoimunitních encefalitid vyžaduje podrobné zhodnocení všech klinických příznaků a jejich vývoje v čase, posouzení nálezů na MR mozku, vyhodnocení vyšetření likvoru a specifických protilátek. Pokud je klinický obraz charakteristický a výsledky pomocných metod jsou souhlasné, nečiní diagnóza AIE potíže. Častější je bohužel situace, kdy některé střípky mozaiky chybí nebo jsou atypické – není přítomen zánětlivý obraz v likvoru, nález na MR mozku je normální, klinické příznaky nejsou plně vyjádřeny atd. Z pomocných vyšetřovacích metod je třeba vyzdvihnout význam EEG, které může pomoci v diferenciální diagnostice encefalitid a psychogenních poruch, může odhalit subklinické záchvaty a nekonvulzivní status epilepticus (SE) a někdy i navést ke konkrétní diagnóze, viz např. vzorec extreme delta ‑ brush u anti‑NMDAR encefalitidy. Význam pozitronové emisní tomografie mozku (18FDG‑ PET) v diagnostice AIE je prozatím okrajový. Zásadní význam má však celotělový 18FDG ‑ PET/ CT pro diagnostiku okultních nádorů. Biopsie CNS není standardní metoda při podezření na AIE, ale může být indikována z diferenciálně‑diagnostických důvodů.
Zásadní roli v diagnostice AIE má vyšetření specifických protilátek. Přehled hlavních protilátek a s nimi sdružených syndromů je uveden v tab. 2a, b. Mezi protilátky proti intracelulárním antigenům (tab. 2a) patří kromě výše zmíněných ještě protilátky proti BR‑ serin/ threoninkináze, proti Collapsin Response Mediator Proteinu 3– 4 (CRMP3– 4) a proti adenylátkináze 5 – všechny sdružené se syndromem LE. Kromě toho lze do této skupiny zahrnout antimikrozomální protilátky (anti‑TPO). Vysoké sérové titry anti‑TPO patří mezi diagnostická kritéria Hashimotovy encefalitidy (tab. 1). Anti‑TPO jsou ale často přítomny v kombinaci s dalšími protilátkami. Není‑li vyšetřeno celé spektrum protilátek, hrozí nadužívání diagnózy Hashimotovy encefalitidy. Japonští autoři recentně identifikovali protilátky proti α ‑ enoláze, které by měly být specifičtější než anti‑TPO [4] – vyšetření však není v ČR dostupné.


AchR – acetylcholinový receptor, AMPA1,2 – podjednotky glutamátových AMPA receptorů, ANA – antinukleární protilátky, CASPR2 – Contactin Associated Protein 2, CPSE – komplexní parciální status epilepticus, CRMP5 – Collapsin Response Mediator Protein 5, DM1 – diabetes mellitus 1. typu, DPPX (DPP6) – Dipeptidyl-Peptidase like Protein 6, EDS – Excessive Daytime Sleepiness, EPC – Epilepsia Partialis Continua, FBDS – faciobrachiální dystonické záchvaty, GABABR – receptor pro kyselinu γ-aminomáselnou typu B, GAD – glutamát dekarboxyláza, GluRe2 – epsilon-2 podjednotka glutamátových NMDA receptorů, IP – intestinální pseudoobstrukce, KE – kmenová encefalitida, LE – limbická encefalitida, LGI1– Leucinerich Glioma Inactivated protein 1, MA – mozečková ataxie, mf – multifokální, mGluR1 – podjednotka metabotropních glutamátových receptorů, MR – magnetická rezonance, MuSK – Muscle Specifi c Kinase, N/K – narkolepsie/kataplexie, NMDAR – glutamátový NMDA receptor, ON – optická neuritida, PCA-1 – Purkinje cell Cytoplasmic Antibody type 1 (synonymum anti-Yo), PCD – paraneoplastická cerebellární degenerace, PEM – paraneoplastická encefalomyelitida, PERM – progresivní encefalomyelitida s rigiditou a myoklonem, PN – periferní neuropatie, SCLC – malobuněčný karcinom plic, SN – senzorická neuronopatie, SOX1 – Sex determining region Y-box 1, SPS – Stiff -Person Syndrom, TLE – epilepsie temporálního laloku, TPO – thyroidální peroxidáza (anti-TPO – protilátky proti mikrozomům), VGCC – protilátky proti napěťově řízeným vápníkovým kanálům, VGKC – protilátky proti komplexu proteinů sdružených s napěťově řízenými draslíkovými kanály.
Hlavní protilátky proti membránovým antigenům jsou uvedeny v tab. 2b. Jednu z podskupin tvoří protilátky proti proteinům sdruženým s napěťově řízenými draslíkovými kanály (tzv. VGKC complex antibodies). Konkrétně jde o protilátky anti‑VGKC sensu lato, vyšetřované metodou radioimunoeseje (RIA), a jejich později identifikované podtypy: protilátky proti Leucin‑rich Glioma Inactivated Proteinu 1 (anti‑LGI1), Contactin‑Associated Proteinu 2 (anti‑CASPR2), Contactinu‑ 2 a Dipeptidylpeptidase‑Like proteinu 6 (anti‑DPPX). Tyto jednotlivé podtypy jsou vyšetřovány pomocí nepřímé imunofluorescence na tzv. cell‑based esejích. O protilátkách anti‑LGI1 a anti‑CASPR2 bude diskutováno v příslušných oddílech. Ostatní protilátky a syndromy s nimi sdružené jsou uvedeny v tab. 1 a 2b. Pomocí komerčně dostupných kitů využívajících buňky transfektované geny pro LGI1, CASPR2, AMPAR1, AMPAR2, GABABR a NMDAR (Euroimmun AG) lze poměrně spolehlivě identifikovat přítomnost daných protilátek. Pro spolehlivost vyšetření je však nutná pravidelná kontrola kvality u výrobce a dostatečná zkušenost s hodnocením. Vzhledem k tomu, že tyto kity využívají permeabilizované buňky, nelze vyloučit falešné pozitivity při vazbě protilátek intracelulárně. V případě nízkých sérových titrů protilátek existuje také riziko falešně negativních výsledků. Je‑li klinické podezření silné a vyšetření na těchto kitech je opakovaně negativní, je vhodné doplnit vyšetření na živých transfektovaných buňkách – buď u výrobce, nebo v některé z mezinárodně uznávaných laboratoří. Za velmi důležitou pokládáme úzkou spolupráci laboratoře s odesílajícím pracovištěm, jejíž součástí je mj. konzultace klinických příznaků a výsledků pomocných vyšetření.
Vyšetření PNP ‑ Ab se v praxi provádí převážně metodou Western blot – senzitivita a specificita kitů je vesměs vysoká. V případě nejasností lze ověřit přítomnost protilátek pomocí nepřímé imunofluorescence.
Pro stanovení anti‑GAD se nejčastěji využívá RIA či ELISA (Enzyme ‑ Linked Immunosorbent Assay), případně imunohistochemické metody. Imunohistochemické metody detekují vysoké titry anti‑GAD (cca > 2 000 µ/ ml), které jsou pro sdružené neurologické syndromy charakteristické. K odhadu intratékální produkce anti‑GAD je možno využít výpočet anti‑GAD indexu [5]:
QGAD = (Titr GADCSF / Titr GADsérum) / / (albuminCSF (mg/ l) / albuminSérum (mg/ l)).
Hodnoty QGAD vyšší než IgG index, zejména hodnoty > 1, indikují intratékální syntézu specifických protilátek. Výpočet QGAD je ovšem spolehlivý (a smysluplný) pouze u vysokých titrů anti‑GAD. Další z možných doplňujících vyšetření je stanovení anti‑GAD specifických oligoklonálních IgG pásů.
Obecné poznámky k terapii autoimunitních encefalitid
Léčbu AIE lze rozdělit na symptomatickou terapii, imunoterapii a onkologickou léčbu (u paraneoplastických syndromů).
Symptomatická léčba ovlivňuje příznaky onemocnění (epileptické záchvaty a SE, psychiatrické příznaky, extrapyramidové projevy, poruchy spánku, vegetativní příznaky, bolest, spasticitu aj.), z nichž některé mohou být pro pacienta život ohrožující. Pozornost je třeba věnovat nežádoucím účinkům této léčby, které mohou klinický obraz samotné AIE ovlivnit.
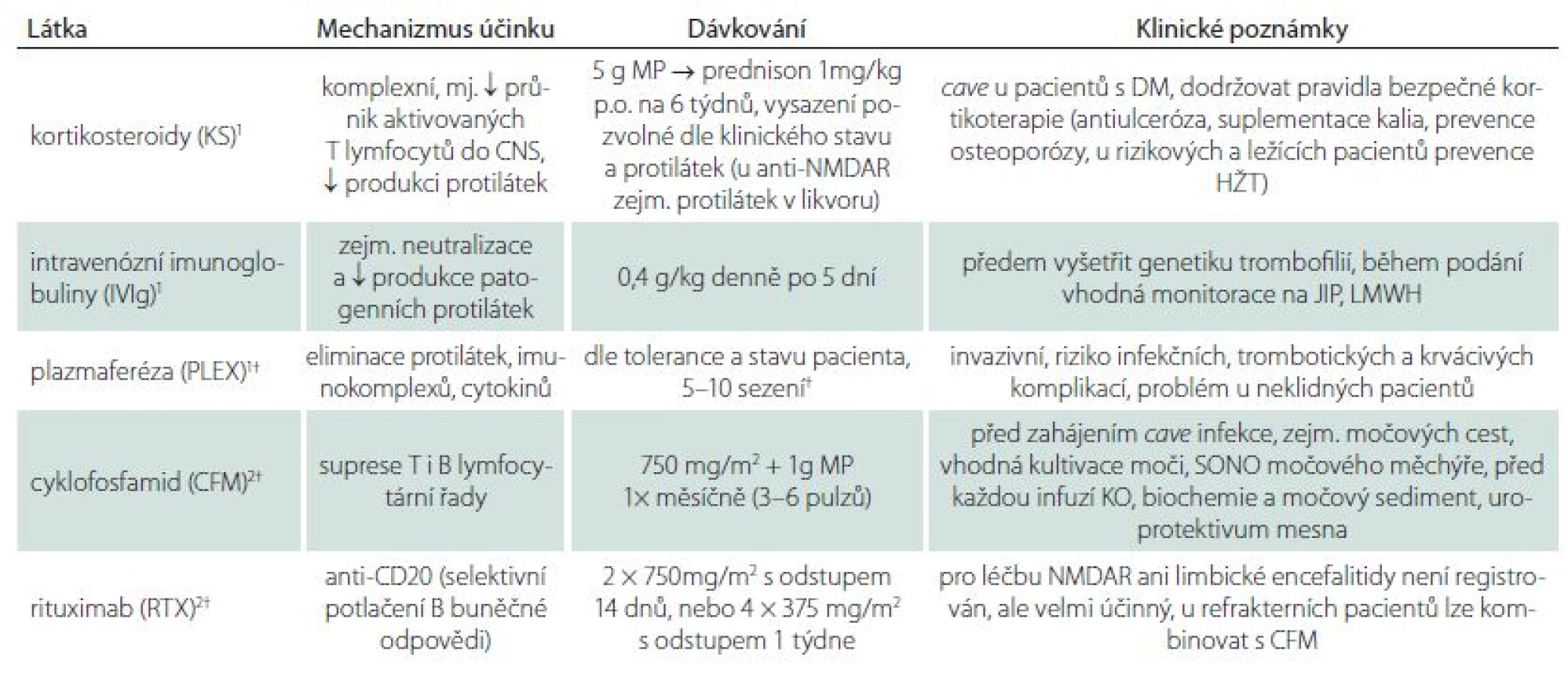
Imunoterapii lze rozdělit na léčebné postupy 1. a 2. linie (tab. 3). Léčba první linie je zaměřena na eliminaci a snížení produkce protilátek a zahrnuje kortikosteroidy (KS), intravenózní imunoglobuliny (IVIg) a eliminační metody. Kromě plazmaferézy (PLEX) se z eliminačních metod v současné době prosazuje selektivní IgG imunoadsorpce, která je stejně účinná, ale pro pacienta komfortnější a bezpečnější [6 – 8]. Problémem je její nízká dostupnost a vysoká cena. V současné době neexistují randomizované studie prokazující vyšší účinnost IVIg nebo eliminačních metod. Racionálním přístupem je volba takové léčby, s níž má dané pracoviště dostatečné zkušenosti, aby bylo minimalizováno riziko komplikací. Léčba 2. linie zahrnuje cytostatika, zejm. cyklofosfamid (CFM) a rituximab (RTX), který způsobuje selektivní depleci CD20+ B lymfocytů [9]. Rozhodneme‑li se zahájit imunoterapii, je třeba tak učinit co nejdříve a léčit agresivně. Nedostatečná léčba či její oddálené zahájení zhoršuje u řady syndromů prognózu a zvyšuje riziko relapsů. Zároveň je ale nutné respektovat rizika léčby (zvláště v podobě infekčních komplikací) a přístup diferencovat podle jednotlivých typů AIE a dle konkrétního pacienta. Anti‑NMDAR encefalitida je příkladem onemocnění, u něhož je oprávněn agresivní přístup. Naproti tomu u klasických paraneoplastických syndromů zpravidla dostačuje léčba KS, přičemž hlavní důraz je kladen na brzké odhalení okultního tumoru a zahájení onkologické léčby. Větší problém než indikace imunoterapie činí zpravidla rozhodnutí o jejím vysazení. Vysazení udržovací léčby u LE nebo anti‑NMDAR encefalitidy v remisi se dá zvažovat přibližně za rok od navození remise, za předpokladu, že u pacienta nejsou přítomny nové nebo fluktuující neurologické příznaky. I po vysazení je však nutné vždy pacienta nadále sledovat. Pro správné vedení léčby je důležité objektivní hodnocení klinického stavu, a to jak na počátku léčby, tak během pravidelných kontrol. Kognitivní výkon a psychiatrické příznaky je nutné kvantifikovat pomocí některé ze skríningových škál (Montreal Cognitive Assessment – MoCA, Mini Mental Status Examination – MMSE, Beckova škála deprese aj.), dle možností též neuropsychologickým vyšetřením. Vzhledem k vzácnému výskytu AIE je, zvláště v komplikovaných případech, vhodná konzultace léčby na některém ze specializovaných pracovišť.
Bližší charakteristiky jednotlivých autoimunitních encefalitid
Klasické paraneoplastické syndromy s postižením CNS
Paraneoplastické syndromy (PS) postihují pouze malou část pacientů s nádory. Nejčastěji je s PS sdružen malobuněčný karcinom plic (SCLC) (3 – 5 % pacientů), thymom (15 – 20 %) a nádory z B buněk a plazmocytů (3 – 10 %). U ostatních nádorů se PS vyskytují u méně než 1 % nemocných. Diagnóza PS předchází v 70 % diagnóze tumoru a doba od počátku neurologických příznaků k odhalení nádoru se pohybuje zpravidla v řádu měsíců, výjimečně několika let. Příznaky PS se rozvíjejí obvykle subakutně a bývají doprovázeny zánětlivým obrazem v likvoru – lymfo ‑ monocytární pleocytózou a zvýšenou celkovou bílkovinou, později přítomností oligoklonálních IgG pásů. Pro diagnózu PS má velkou váhu přítomnost tzv. dobře charakterizovaných onkoneurálních protilátek (PNP ‑ Ab) v séru. U séronegativních pacientů je možné diagnózu PS považovat za jistou, jsou‑li splněna následující kritéria:
- charakteristický klinický obraz,
- přítomnost okultního tumoru,
- zánětlivý nález v likvoru nebo bioptickém materiálu a
- vyloučení jiných komplikací onkologického onemocnění.
PS lze dělit na klasické a tzv. neklasické [10]. Přehled hlavních PS s postižením CNS je uveden v tab. 4. Odkazujeme též na přehledové články [11,12]. Přítomnost některého z klasických PS anebo přítomnost PNP ‑ Ab by vždy měla vést k podrobnému vyšetření za účelem vyloučení okultního tumoru. Při plánování onkoskríningu je vhodné postupovat dle platných doporučení European Federation of Neurological Societies (EFNS) [13]. Při podezření na karcinom plic a thymom je indikováno CT hrudníku; při negativním nálezu pokračujeme vyšetřením 18FDG ‑ PET/ CT. Při suspektním nálezu karcinomu plic následuje obvykle bronchoskopie s využitím endobronchiální ultrasonografie a odběrem bioptického materiálu. U PS sdružených s nádory prsu odhalí mamografie nádor asi v 80 %. Vyšší citlivost má MR prsů a zejm. 18FDG ‑ PET. U ovariálních a testikulárních nádorů je první volbou ultrasonografie, v případě podezření na ovariální teratom u žen MR pánve. Zralé teratomy nevykazují hypermetabolizmus na 18FDG ‑ PET, a toto vyšetření proto není při podezření na teratom přínosné. U PS sdružených s teratomy (např. anti‑NMDAR encefalitida) je při negativitě ultrazvukového/ MR vyšetření pánve vhodné provést též vyšetření CT hrudníku k vyloučení mediastinální lokalizace teratomu. Je‑li podezření na ovariální tumor vysoké, a všechny nálezy na zobrazovacích metodách negativní, lze v odůvodněných případech indikovat probatorní laparoskopii. Podobně doporučují někteří autoři orchiektomii při nálezu mikrokalcifikace testes u pacientů s LE a protilátkami anti‑Ma2. Tento názor je podpořen vysokým záchytem mikroskopických tumorů [14]. Význam onkomarkerů při diagnostice okultních tumorů je jen okrajový, jistý přínos může mít vyšetření SOX1 a neuron ‑ specifické enolázy pro SCLC, protilátek proti titinu u thymomu, CA ‑ 125 pro karcinom ovaria, β ‑ hCG a α ‑ fetoproteinu pro nádory varlat. Při negativitě vyšetření se doporučuje onkoskríning opakovat každých šest měsíců do celkové doby pěti let.
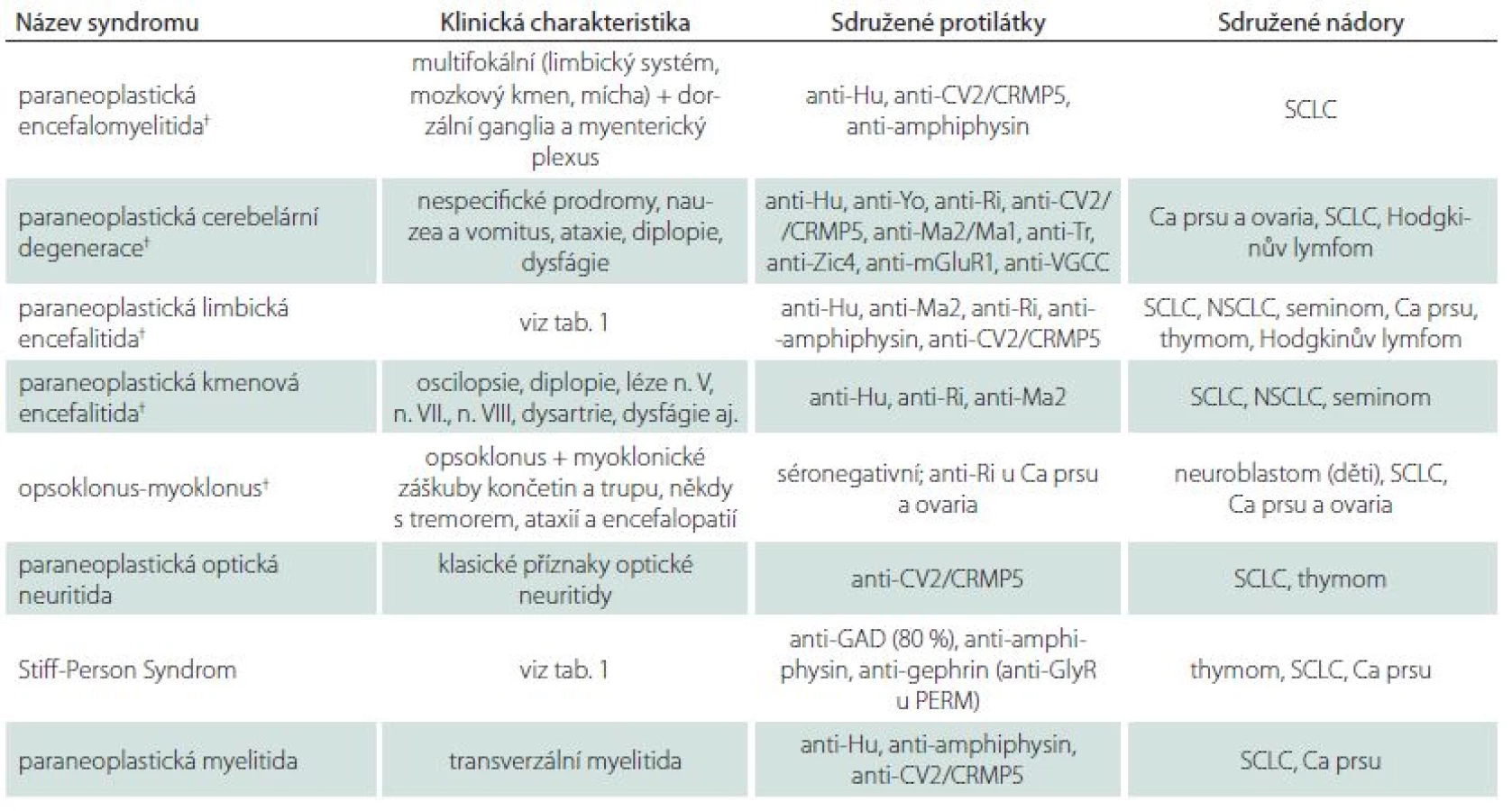
Léčba klasických PS spočívá především v léčbě přidruženého nádoru. Imunoterapie je u většiny klasických PS málo účinná, přesto se zpravidla zkouší KS, IVIg či eliminační metody. Výjimku tvoří relativně dobrá odpověď na imunoterapii u paraneoplastického opsoklonu ‑ myoklonu sdruženého s neuroblastomem u dětí a částečný efekt imunoterapie u mladých mužů s LE anti‑Ma2.
Limbické encefalitidy
Limbickou encefalitidu (LE) popsal jako první v roce 1968 britský neuropatolog Corsellis u pacienta s bronchogenním karcinomem. První série neparaneoplastických LE byla publikována v roce 2001. Současně byl identifikován nový typ protilátek s tímto typem LE spojený – protilátky anti‑VGKC (viz oddíl Obecné poznámky k diagnostice autoimunitních encefalitid) [15]. Později se ukázalo, že u většiny pacientů s touto LE jsou přítomny protilátky proti Leucin‑rich Glioma Inactivated proteinu 1 (LGI1) a Contactin‑Associated Proteinu 2 (CASPR2) [16]. Kromě autoimunitní etiologie (která je nejčastější) může mít LE též infekční příčinu (např. v rámci Whippleovy choroby nebo způsobenou virem HHV6 u pacientů po transplantacích). LE nepatří mezi běžná onemocnění, i když přesné incidenční a prevalenční populační údaje nejsou k dispozici. Během dvou let sledování této problematiky jsme ve FN v Motole zaznamenali 12 případů (4× anti‑LGI1, 1× anti‑GAD, 1× anti‑AMPAR1, 2× anti‑Ma2, 1× anti‑Hu,1× anti‑Ri/ anti‑Yo, 2× séronegativní). Za stejné období jsme pro srovnání zaznamenali 15 případů anti‑NMDAR encefalitidy.
Charakteristickou trias klinických příznaků LE tvoří:
- epileptické záchvaty;
- subakutně rozvinutá porucha krátkodobé paměti, která může postupně progredovat do obrazu demence;
- psychiatrické příznaky, zahrnující poruchy chování s agresivitou nebo apatií, afektivní poruchy, či psychotické projevy.
Dále se mohou se objevit hypothalamické příznaky (poruchy spánku, termoregulace, autonomní a endokrinní dysfunkce, minerálové dysbalance – zvl. SIADH charakteristický pro LE s protilátkami anti‑LGI1) a extrapyramidové příznaky. Poruchy spánku mohou např. u LE anti‑VGKC zahrnovat insomnii, hypersomnii, absenci REM spánku, ztrátu atonie nebo poruchu chování v REM spánku. U LE anti‑Ma2 se specificky vyskytuje syndrom narkolepsie ‑ kataplexie doprovázený snížením hladin hypokretinu v likvoru [17,18]. Příznaky LE mohou být kombinovány s příznaky kmenové encefalitidy, cerebelitidy nebo s příznaky spinálními a periferními.
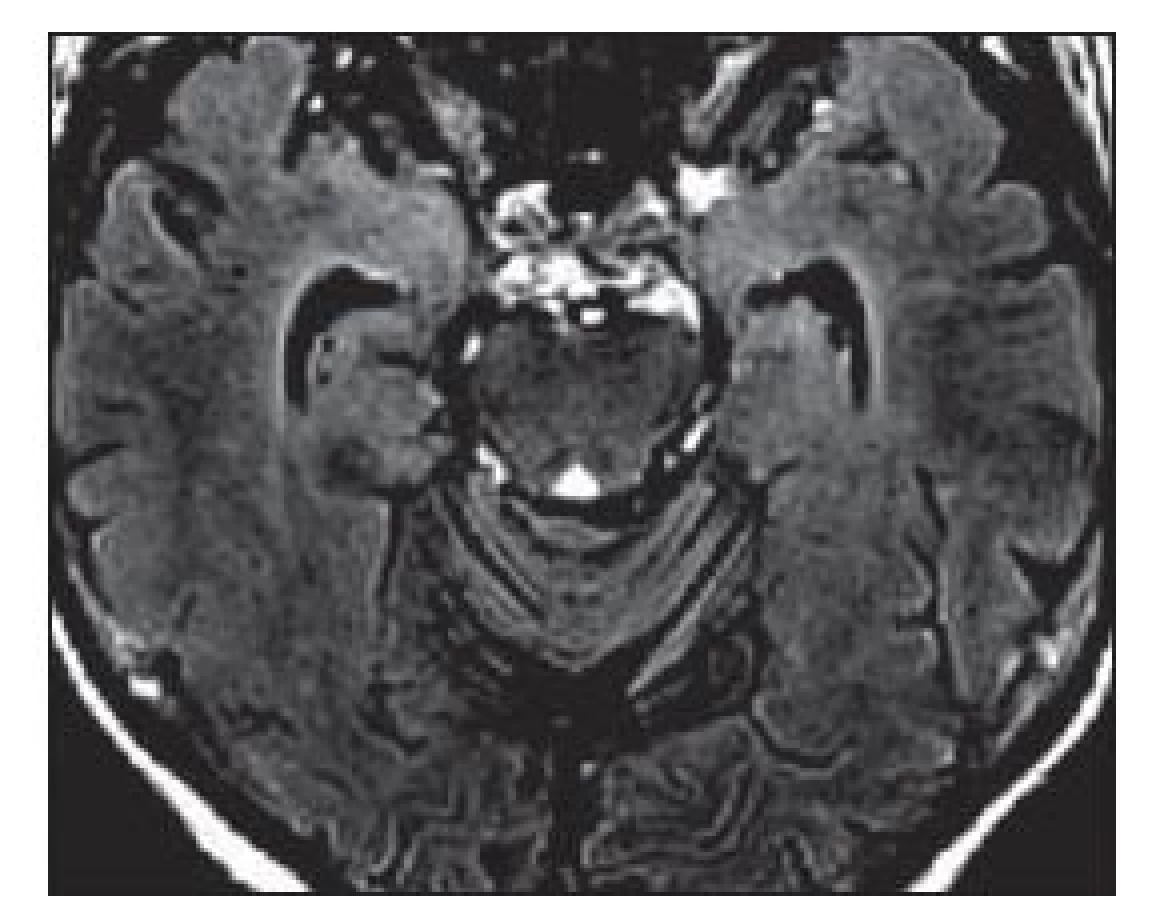
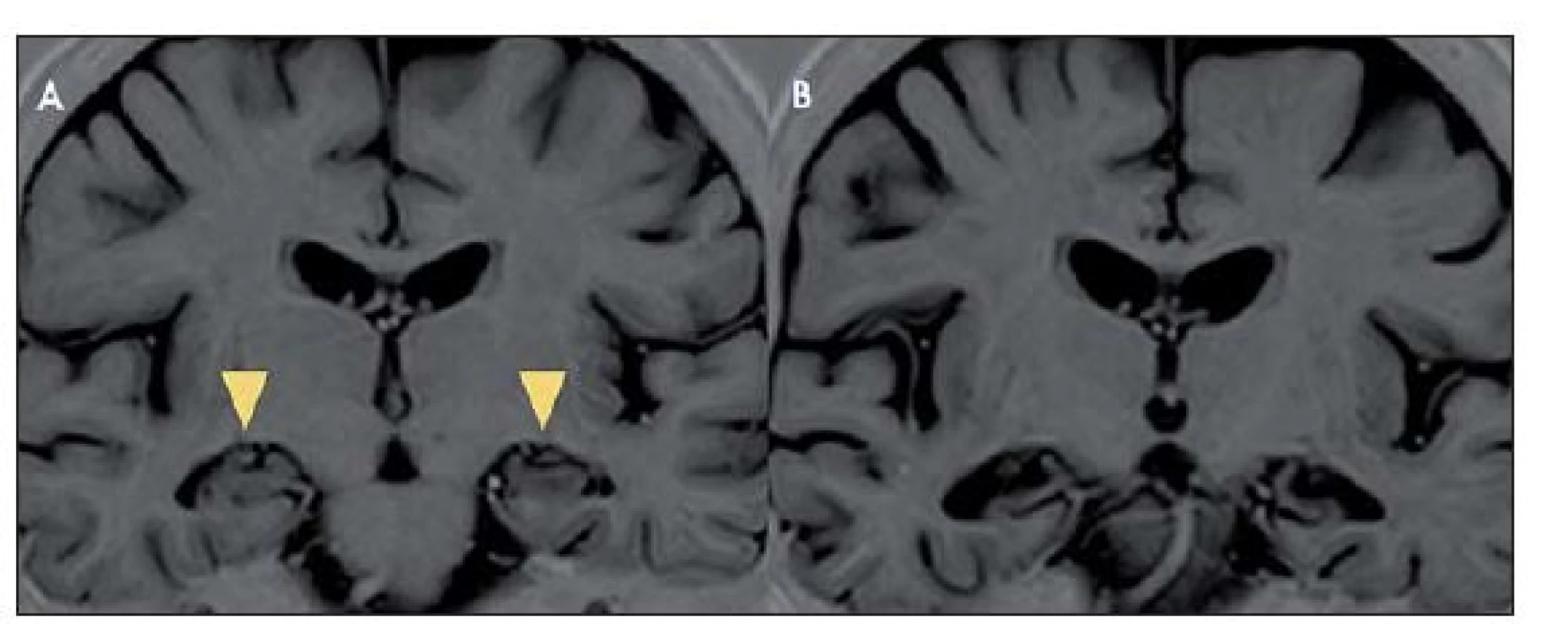
Na MR mozku se často vyskytuje charakteristické zvýšení signálu mediotemporálních struktur (amygdaly, hipokampu a temporálních pólů) na T2 vážených obrazech (T2 - v.o.) a FLAIR (Fluid ‑ Attenuated Inversion Recovery), mnohdy asymetrické, vzácněji unilaterální (obr. 1). Tyto změny mohou být i relativně nenápadné. Vzácněji může být přítomno postkontrastní sycení mediotemporálně. V průběhu LE dochází k postupné atrofizaci mediotemporálních struktur (obr. 2), někdy až s rozvojem sekundární hipokampální sklerózy [19]. U pacientů s probíhajícím či recentně proběhlým SE může být těžké rozhodnout, zda jsou signálové změny na MR způsobeny zánětlivým postižením či tímto statem. Normální MR nález se vyskytuje asi u 10 – 20 % LE. Nález v likvoru je často abnormní, ale nespecifický. Lymfocytární pleocytózu nacházíme u většiny LE spíše na počátku onemocnění, později je v likvoru patrná intratékální syntéza protilátek a oligoklonální IgG pásy. Proto je vhodné v nejistých případech vyšetření s odstupem několika týdnů opakovat. Diferenciální diagnostika LE je pestrá (tab. 5).

Paraneoplastické limbické encefalitidy
Klasické paraneoplastické limbické encefalitidy (PLE) jsou nejčastěji sdruženy s protilátkami anti‑Hu a anti‑Ma2, vzácně s protilátkami anti‑Ri, anti‑CRMP5/ CV2 a anti‑amfifyzin. Řada PLE je séronegativních. PLE se mohou vzácně vyskytovat i u dětí (zejm. ve spojení s neuroblastomem) [20]. PLE s protilátkami anti‑Hu je nejčastěji prototypická LE s obvyklou triádou příznaků a abnormním nálezem na MR mozku. Častý je výskyt komplexního parciálního SE. PLE s protilátkami anti‑Hu se může vyskytovat též v kontextu paraneoplastické encefalomyelitidy (PEM), v takovém případě ji může doprovázet syndrom epilepsia partialis continua. Nejčastějším nádorem sdruženým s PLE s protilátkami anti‑Hu je SCLC. Efekt imunoterapie je bohužel nevýrazný a prognóza i s ohledem na základní onemocnění nepříznivá. PLE s protilátkami anti‑Ma2 se často vyskytuje v kombinaci s postižením hypothalamu a mozkového kmene a příznaky LE mohou být kombinovány s poruchou spánku včetně sekundární narkolepsie ‑ kataplexie, hyperfagií, extrapyramidovými příznaky (hypokineze, atypický parkinsonizmus, orofaciální dystonie) a kmenovými symptomy (obrna vertikálního pohledu, dysfagie, dysartrie). Postihuje typicky mladé muže, kdy bývá sdružena s testikulárním seminomem, často mikroskopickým. Druhý vrchol výskytu je u žen v šesté dekádě, kdy bývá nalézán nemalobuněčný Ca plic (NSCLC) a Ca prsu. V diferenciální diagnóze může být zvažována Whippleova choroba s postižením CNS. PLE s protilátkami anti‑Ma2 má ze všech nejlepší prognózu, a je proto mandatorní aktivní přístup k terapii a onkoskríningu.
Limbická encefalitida s protilátkami anti‑LGI1
LE s protilátkami anti‑LGI1 postihuje nejčastěji muže v šesté dekádě. Kromě klasické triády příznaků LE se u více než poloviny případů vyskytuje hyponatremie, podmíněná syndromem inadekvátní sekrece ADH (SIADH). U 40 % pacientů se na samém počátku onemocnění vyskytují faciobrachiální dystonické záchvaty (FBDS). Ty jsou charakterizovány mimovolními krátkými spazmy v oblasti mimického svalstva a svalstva končetin – typická je krátká unilaterální tonická grimasa doprovázená abdukcí a flexí ipsilaterální horní končetiny. FBDS se mohou dostavovat v sériích, často mnohokrát za den. Vzhledem k tomu, že jsou FBDS v některých případech doprovázeny iktálním vzorcem v EEG, jedná se pravděpodobně o epileptické záchvaty a nikoli dystonické projevy. FBDS velmi dobře reagují na léčbu KS, zatímco antiepileptická léčba bývá neúčinná. Na MR mozku jsou u LE s protilátkami anti‑LGI1 přítomny typické signálové změny, zánětlivý obraz v likvoru naopak často chybí. I když se jedná o LE většinou neparaneoplastickou, u menšiny pacientů se nádory vyskytují. Nejčastěji se jedná o NSCLC a thymom. Z tohoto důvodu je i u LE anti‑LGI1 indikován podrobný onkoskríning. I když je efekt imunoterapie u LE anti‑LGI1 poměrně dobrý, encefalitida může relabovat, zvláště je‑li iniciální léčba nedostatečně agresivní. Imunoterapie 1. linie zahrnuje KS (i.v. metylprednisolon v dávce 4 – 5 g s následným převedením na p.o. prednison 1mg/ kg denně) a návazně IVIg v dávce 0,4g/ kg/ den po dobu pěti dní. Nedostaví‑li se cca během dvou týdnů zlepšení, je možno pokračovat léčbou 2. linie, která zahrnuje CFM nebo RTX (viz též oddíl Obecné poznámky k terapii autoimunitních encefalitid a tab. 3). Prednison p.o. se doporučuje ponechat cca šest týdnů s následným pomalým snižováním a převedením na udržovací dávku.
Limbické encefalitidy s protilátkami anti‑GABABR a anti‑AMPAR
LE s protilátkami proti GABAB receptoru je vzácná. Postihuje nejčastěji muže a ženy v šesté dekádě. Klinický obraz je velmi podobný jako u LE anti‑LGI1, nebývá však přítomna hyponatremie a FBDS. V likvoru bývá zánětlivý obraz včetně lymfo ‑ monocytární pleocytózy. MR mozku bývá abnormní s charakteristickým obrazem LE. Ve většině případů je LE anti‑GABABR sdružena s nádory, zejm. SCLC [21]. Anti‑GABABR a anti‑AMPAR protilátky se mohou vyskytovat společně s dalšími typy protilátek (zvláště anti‑GAD, anti‑VGCC, anti‑TPO a ANA). Protilátky anti‑GABABR byly v jedné ze studií přítomny u tří z pěti paraneoplastických syndromů sdružených s protilátkami anti‑GAD, z toho u dvou pacientů šlo o LE [22]. Není proto vyloučeno, že u části pacientů s paraneoplastickou LE anti‑GAD může symptomy podmiňovat autoimunitní reakce vůči GABAB receptoru. Anti‑GABABR protilátky zřejmě inaktivují GABAB receptory a nevedou tak per se k ireverzibilnímu neuronálnímu poškození.
LE s protilátkami proti AMPA receptoru postihuje častěji ženy. Podobně jako LE anti‑GABABR je velmi vzácná a bývá paraneoplastická. Nejčastěji sdruženým nádorem bývá SCLC, Ca prsu nebo thymom. Podobně jako u anti‑NMDAR encefalitidy dochází v důsledku anti‑AMPAR protilátek k internalizaci cílových receptorů [23]. Efekt imunoterapie je proto u tohoto syndromu, stejně jako u LE anti‑GABABR poměrně dobrý.
Limbická encefalitida s protilátkami anti‑GAD
Protilátky anti‑GAD jsou nepoměrně častěji než s LE sdruženy s mozečkovou ataxií s pozdním nástupem a se Stiff ‑ Person Syndromem (SPS) [5]. Kromě toho bývají v nízkých titrech pozitivní u pacientů s DM 1. typu. U LE anti‑GAD jsou sérové titry zvýšené v řádu tisíců U/ ml. LE anti‑GAD postihuje nejčastěji ženy mladého věku (medián 23 let) a relativně častěji se vyskytuje u dětí. Může probíhat pod typickým obrazem LE, nebo jako epilepsie temporálního laloku (TLE). Nově byla popsána anti‑GAD séropozitivní extralimbická AIE s MR nálezem postkontrastně sytících ložisek imitujících tumor [24]. I když je LE anti‑GAD častěji neparaneoplastická, může být sdružena s tumory (SCLC, NSCLC, Ca pankreatu, Ca colon, Ca thymu). Diagnostika LE ‑ anti GAD se neliší od jiných typů LE. Uvážlivý postup je však nutný při hodnocení protilátek. Sérový titr anti‑GAD by u neurologických syndromů měl být vyšší než 2 000 U/ ml (v některých laboratořích nejsou rutinně vyšetřovány titry vyšší než 120 U/ ml a je nutné o doplnění titrace cíleně požádat). Při nálezu vysoké hodnoty anti‑GAD v séru je vhodné provést kontrolní test s paralelním vyšetřením likvoru. U titrů anti‑GAD v séru < 2 000 U/ ml je diagnosticky významná zejm. přítomnost zánětlivého obrazu v likvoru.
Efekt imunoterapie nebývá u LE anti‑GAD a zvláště u TLE anti‑GAD výrazný, byť existují kazuistiky léčené s úspěchem [25]. Jedinou randomizovanou studií hodnotící efekt imunologické léčby u anti‑GAD syndromů je studie prokazující příznivý efekt IVIg u anti‑GAD asociovaného SPS [26]. Imunoterapii u LE anti‑GAD lze zahájit standardní léčbou 1. linie, tj. i.v. KS a IVIg (viz oddíl Obecné poznámky k terapii autoimunitních encefalitid). U pacientů s přidruženým DM je během podávání KS nutné monitorovat glykemii nebo léčbu zahájit samotnými IVIg. Zahájení terapie 2. linie (RTX, CFM) mohou podporovat např. následující faktory:
nedostatečný efekt terapie 1. linie a progresivní (objektivně dokumentované) horšení kognitivního výkonu,
- progrese zánětlivého obrazu v likvoru,
- progrese titru protilátek, zejm. v likvoru,
- aktivita procesu na MR mozku, např. nově vzniklé postkontrastní sycení nebo progrese signálových změn na T2 - v.o. a FLAIR.
U pacientů s TLE anti‑GAD se lze řídit podobnými kritérii – v případě, že přetrvávají pouze epileptické záchvaty, není přítomna objektivní kognitivní deteriorace, neprogreduje nález v likvoru a nezvyšují se titry protilátek, je po vyzkoušení imunoterapie 1. linie indikováno pokračovat již pouze antiepileptickou léčbou.
Séronegativní limbické encefalitidy
Přibližně u 10 – 15 % nemocných se syndromem LE nejsou přítomny specifické protilátky. V těchto případech je třeba spolehnout se na přesvědčivý klinický obraz, souhlasné nálezy na MR mozku a v likvoru (případně na EEG). Zároveň je třeba vyloučit alternativní diagnózy. Jedním z diagnostických kritérií je i dobrá odezva na imunomodulační léčbu, ve které jsou využívány většinou léky 1. linie.
Morvanův syndrom
Fibrilující choreu, dnes označovanou jako Morvanův syndrom, popsal v roce 1890 bretaňský lékař Augustin Morvan. Jde o autoimunitní onemocnění projevující se periferní autonomní hyperexcitabilitou, neuromyotonií a neuropsychiatrickými příznaky, zejm. zmateností a insomnií. Neuromyotonie je charakterizována přítomností difuzních svalových kontrakcí s křečemi, fascikulacemi a myokymiemi [27]. Zobrazovací vyšetření včetně MR mozku bývají normální [28]. Morvanův syndrom je sdružen nejčastěji s protilátkami anti‑CASPR2, vzácněji s protilátkami anti‑LGI1. V sérii 29 pacientů z roku 2012 byli postiženi převážně muži (93 %) a nádorové onemocnění bylo zjištěno téměř u poloviny nemocných (41 %). Nejčastějším přidruženým nádorem byl thymom (zvláště u pacientů s pozitivitou anti‑CASPR2). V těchto paraneoplastických případech byla prognóza onemocnění i přes léčbu nepříznivá [29].
Anti‑NMDAR encefalitida
V roce 2007 předložili Dalmau et al soubor 12 pacientek s akutně rozvinutými psychiatrickými příznaky, epileptickými záchvaty, poruchou paměti, dyskinézami, autonomní dysfunkcí a progredující poruchou vědomí vyžadující UPV. U všech pacientek zjistili autoři přítomnost protilátek proti heterodimerům podjednotek NR1/ NR2B (či méně často NR1/ NR2A) glutamátových NMDA receptorů v séru a v likvoru. U 11 pacientek byla zjištěna přítomnost ovariálního teratomu a u jedné pacientky zralého mediastinálního teratomu. Odstranění nádoru a agresivní imunoterapie vedla u osmi z devíti takto léčených pacientek k poklesu titrů protilátek a k úzdravě. Dvě ze tří pacientek, u kterých nebyl nádor odstraněn, zemřely v důsledku neurologických komplikací. Autoptický nález zahrnoval rozsáhlou mikrogliózu, ojedinělé T lymfocytární infiltráty a neuronální ztrátu vyjádřenou nejvýrazněji v oblasti hipokampu [30].
Anti‑NMDAR encefalitida je dnes považována za druhou nejčastější autoimunitní encefalitidu (po akutní demyelinizující encefalomyelitidě) [31]. Vícečetné studie prokázaly, že vazba anti‑NMDAR protilátek vede k dislokaci NMDA receptorů ze synaptické štěrbiny a jejich internalizaci. Snížení počtu NMDA receptorů na postsynaptické membráně způsobuje utlumení glutamátergní transmise a sekundární postižení dalších neurotransmiterových systémů (dopaminergního, noradrenergního a cholinergního).
Anti‑NMDAR encefalitida postihuje především mladé ženy (v sérii 100 pacientů medián 23 let s rozmezím 5 – 76 let) [32]. Vzácně se toto onemocnění může vyskytnout v těhotenství [33]. Literární údaje [34,35], ale též naše vlastní zkušenosti ukazují relativně vysoký počet postižených dětí – ve FN v Motole bylo např. od roku 2012 do roku 2014 diagnostikováno 15 pacientů, z toho šest dětí. Nejmladšímu úspěšně léčenému pacientovi popsanému v literatuře byly necelé dva roky [36]. U mužů se onemocnění vyskytuje méně často, ale u chlapců není vzácností [9,37].
Klinický obraz anti‑NMDAR encefalitidy je charakteristický. Přibližně u čtvrtiny pacientů předchází rozvoji neurologických příznaků nespecifické virové onemocnění, febrilie, či vakcinace [38]. Po několika dnech až týdnech se dostavují závažné psychiatrické příznaky. Encefalitida může probíhat pod obrazem akutní mánie či deprese, často s psychotickými rysy [39]. Psychotická produkce může zahrnovat paranoidní bludy a halucinace. Tyto projevy jsou doprovázeny buď výraznou iritabilitou a agitací, nebo naopak behaviorálním útlumem, který může přejít až do obrazu maligní katatonie [40]. Mohou se střídat epizody akineze a agitace a byla popsána přítomnost paradoxních reakcí na zevní stimuly (např. nepřítomnost reakce na bolest a zároveň odpor při pokusu o pasivní otevření očí) [32]. U dětí je na počátku onemocnění v popředí většinou porucha chování s agresí a agitací, úzkostné stavy, zmatenost, regres řeči a dalších dříve nabytých schopností [37,41,42]. V úvodní fázi anti‑NMDAR encefalitidy jsou proto pacienti často hospitalizováni na psychiatrických odděleních. Význam diferenciálně diagnostické úvahy směrem k anti‑NMDAR encefalitidě u akutních psychotických stavů u mladých žen byl v literatuře mnohokrát zdůrazněn.
K psychiatrickým příznakům se u 76 – 82 % pacientů (zejm. dospělých) v dalším průběhu přidávají epileptické záchvaty [32,38]. Převažují záchvaty generalizované tonicko‑klonické (53 % pacientů) a komplexní parciální (10 %), relativně častý je též výskyt SE (cca 6 % pacientů) [32]. Vzhledem k přítomnosti autonomní dysfunkce není překvapující výskyt poruch srdečního rytmu až iktální asystolie během záchvatu.
V chronologickém rozvoji příznaků anti‑NMDAR encefalitidy následují extrapyramidové příznaky (až 89 % pacientů). U dětí mohou být extrapyramidové poruchy hybnosti prvním příznakem onemocnění. Nejčastěji se vyskytují orofaciální dyskineze s grimasováním, pohyby připomínajícími přežvykování, či prudkým svíráním a otevíráním úst vedoucím často k poranění rtů, jazyka anebo zubů [32]. Z dalších příznaků byly popsány mimovolní choreoatetoidní pohyby končetin a pánve, choreatické pohyby prstů připomínající hru na klavír [43], dystonické postury končetin, nucené stáčení očí či tzv. ocular dipping, myoklonus, tremor a balizmus. Diferenciálně diagnosticky je třeba brát v úvahu možnou polékovou etiologii dyskinéz.
Porucha vědomí a centrální hypoventilace se následně dostavuje u 45 – 69 % pacientů. Medián doby od vzniku příznaků k UPV je 8 týdnů, s rozmezím 2 – 40 týdnů [32]. Známky autonomní nestability se vyskytují u 69 % pacientů a zahrnují: tachykardii, bradykardii, asystolii, hypotenzi, hypertenzi, hypotermii, hypertermii, hyperhidrózu, sialoreu a u 4 % pacientů adynamický ileus. Relativně častá je dysfunkce sinoatriálního uzlu se sinusovou tachykardií a epizodami bradykardie navozenými vagovými stimuly [44]. Někteří pacienti mohou vyžadovat dočasnou kardiostimulaci [45]. V této fázi onemocnění bývají pacienti hospitalizováni na neurologických JIP či ARO. Průběh anti‑NMDAR encefalitidy bývá protrahovaný – jedna z dětských pacientek diagnostikovaných ve FN v Motole absolvovala více než roční hospitalizaci [46]. Dlouhodobá intenzivní péče s sebou přináší řadu rizik.
Diagnostika anti‑NMDAR encefalitidy spočívá ve stanovení specifických protilátek v séru a likvoru. Relativně vysoká senzitivita a specificita komerčně dostupných kitů umožňuje poměrně rychlou a spolehlivou diagnostiku anti‑NMDAR encefalitidy [47]. Při použití komerčně dostupných kitů existuje (byť velmi nízké) riziko falešných pozitivit při vazbě protilátek nikoli na membráně, ale intracelulárně (viz oddíl Obecné poznámky k diagnostice autoimunitních encefalitid). Při vyšetřování protilátek je vhodné párové vyšetření séra a likvoru – zaznamenali jsme opakovaně pacienty s negativitou anti‑NMDAR protilátek v séru a současnou pozitivitou v likvoru. Klinické zlepšení koreluje s poklesem titru protilátek, zvláště v likvoru. Podobně dochází k opětovnému vzestupu titrů protilátek při relapsu onemocnění. Je třeba též upozornit na možnost pozitivity anti‑NMDAR protilátek u jiných diagnóz – konkrétně Creutzfeldt ‑ Jakobovy choroby [48] a Herpes ‑ Simplex Encefalitidy (HSE) [49].
Z pomocných metod je pro diagnostiku anti‑NMDAR encefalitidy přínosné především vyšetření likvoru. Zánětlivý obraz v likvoru je přítomen až u 95 % nemocných. Lymfo ‑ monocytární pleocytóza se vyskytuje u 68 – 91 % pacientů (v rozmezí 5 – 380 buněk v µl, medián 32 buněk v µl) [32]. Pravděpodobnost zachycení pleocytózy je nejvyšší na počátku onemocnění. Později je častější přítomnost intratékální produkce IgG a oligoklonálních IgG pásů (až u 43 % nemocných po 35. dnu onemocnění) [38]. Lymfo ‑ monocytární pleocytóza se může znovu objevit v případě relapsu encefalitidy. Nález na MR mozku je u 45 – 89 % pacientů normální. U zbývajících pacientů jsou přítomny diskrétní, nespecifické abnormity, zahrnující zvýšení signálu na T2 - v.o. a FLAIR mediotemporálně, extratemporálně kortikálně, v oblasti mozečku, bazálních ganglií, mozkového kmene, hypothalamu, corpus callosum či multifokálně [32]. Hlavní přínos MR mozku u pacientů s podezřením na anti‑NMDAR encefalitidu spočívá ve vyloučení jiné etiologie (tab. 5). Specifickou situací je rozvoj sekundární anti‑NMDAR encefalitidy po proběhlé HSE [50]. Narozdíl od pravého relapsu HSE nebývají v tomto případě na MR přítomny nové nekrotické léze a v mozkomíšním moku nedetekujeme DNA herpes simplex viru, ale naopak přítomnost anti‑NMDAR protilátek. EEG nálezy u anti‑NMDAR encefalitidy jsou ovlivněny výskytem epileptických záchvatů a tíží poruchy vědomí. Může být přítomno zpomalení základní aktivity, intermitentní či perzistentní pomalá generalizovaná či lokalizovaná aktivita, fokální a multifokální epileptické grafoelementy, iktální vzorce. Záchyt epileptiformní abnormity je pravděpodobnější v časných fázích onemocnění, později dominuje generalizované zpomalení záznamu v pásmu theta či delta. Epizody nekonvulzivního SE mohou být provázeny generalizovanou rytmickou delta aktivitou s vývojem co do morfologie, frekvence a pole [51], anebo střídáním generalizované delta aktivity s epizodami ostré theta 5 – 6 Hz s maximem nad předními kvadranty [52]. U přibližně 30 % pacientů s anti‑NMDAR encefalitidou může být přítomen charakteristický EEG vzorec, tzv. extreme delta brush (EDB) [53]. Jedná se o pomalou aktivitu v pásmu delta s převahou nad předními kvadranty se superponovanou rychlou frekvencí kolem 20 Hz. Význam tohoto vzorce je dvojí:
- při kongruentním klinickém obrazu by přítomnost EDB měla vést k podezření na anti‑NMDAR encefalitidu a vyšetření protilátek,
- EDB je negativním prognostickým markerem z pohledu výsledného funkčního stavu.
18FDG ‑ PET nemá v současné podobě pro diagnostiku anti‑NMDAR encefalitidy zásadní přínos – nálezy zobrazují zejm. iktální hypermetabolizmus u pacientů s opakovanými epileptickými záchvaty či SE [54].
Anti‑NMDAR encefalitida je ve 26 – 58 % sdružena s ovariálním teratomem [9,32,38]. Výskyt teratomu je častější u Afroameričanek, Asiatek a mladších žen – vrchol výskytu se nachází mezi 13. a 30. rokem. U dětí mladších 12 let jsou nádory přítomny méně často [55]. Teratomy se kromě ovaria mohou vzácněji vyskytovat i v jiných lokalizacích a mohou být mikroskopické (viz Klasické paraneoplastické syndromy s postižením CNS). U mužů byly v jednotlivých případech anti‑NMDAR encefalitidy nalezeny SCLC a teratom varlat [56].
Imunoterapii je nutné zahájit co nejdříve a léčit agresivně. Schémata používaná pro léčbu anti‑NMDAR encefalitidy jsou založena pouze na publikovaných expertních stanoviscích, randomizované studie chybí. Mezi léky používané v 1. linii patří KS (u dospělých i.v. metylprednisolon v dávce 5 × 1g i.v. s následným převedením na p.o. prednison v dávce 1 mg/ kg//den), IVIg (v dávce 5 × 0,4g/ kg/ den) a eliminační metody (PLEX nebo IgG imunoadsorbce, 5 – 10 sezení). V případě neúspěchu léčby 1. linie je vhodné přistoupit k 2. linii – léčbě cytostatiky. Doporučuje se RTX v dávce 2 × 750 mg/ m2 (max. 1 g) v odstupu 14 dnů nebo měsíční pulzy CFM v dávce 750 mg/ m2 (dle literatury obvykle 3 – 6 pulzů, max. 12 pulzů). Jinou citovanou variantou je kombinace RTX a CFM [32,37,55,57]. Na velké kohortě více než 500 pacientů bylo prokázáno, že opakování léčby 1. linie není v případě nedostatečné odpovědi na první cyklus přínosné [9]. Někteří autoři doporučují přistoupit k terapii 2. linie, nedojde‑li ke zlepšení pacientova stavu do 10 dní po ukončení léčby 1. linie. I přes výraznou agresivitu cytostatické terapie byly zaznamenány jen ojedinělé komplikace této léčby, obvykle v podobě infekcí, v izolovaných případech se vyskytla závažná lymfopenie (CFM) a anafylaktická reakce (RTX). Žádná z těchto komplikací nebyla fatální. U menšiny pacientů může být neúspěšná i léčba 2. linie – negativními prognostickými faktory jsou nepřítomnost nádoru, závažný klinický průběh s rychlou progresí poruchy vědomí, přítomnost EDB vzorce v EEG a iniciálně vysoké titry protilátek v séru a likvoru. Pro léčbu pacientů neodpovídajících na léčbu 2. linie neexistují žádná jednotná doporučení. Mezi jinými byl v jednotlivých kazuistikách zkoušen tacrolimus, mykofenolát mofetil, metotrexát či alemtuzumab [46]. V případě úspěšné léčby se doporučuje pokračovat v KS s postupným snižováním dávky z původních 60 – 80 mg prednisonu denně po 10 mg týdně na dávku 10 mg denně. Vzhledem k tomu, že až u 25 % pacientů může dojít k relapsu encefalitidy, doporučuje řada autorů pokračovat v imunoterapii ještě jeden rok po ukončení akutní léčby [55]. Pro tuto udržovací léčbu lze použít prednison, kombinaci prednisonu s azathioprinem, monoterapii azathioprinem či mykofenolát mofetil. V případě relapsu encefalitidy je možné vrátit se k plné dávce kortikoterapie, případně podat novou kúru léčby 1. linie.
Při včasné diagnóze, rychlém zahájení agresivní imunoterapie a případně onkologické léčby je prognóza anti‑NMDAR encefalitidy příznivá, a to jak z hlediska přežití, tak i výsledného funkčního stavu. Téměř polovina pacientů se uzdraví ad integrum [32]. Naopak u neléčených nebo nedostatečně léčených případů byla pozorována značně vysoká mortalita (podle různých studií 7 – 25 %) [32,58]. Mezi vzácněji se vyskytující reziduální deficity patří dysexekutivní syndrom, impulzivita, případně poruchy recentní paměti [59]. Řada pacientů (27 %) trpí poruchami spánku, zejm. inverzí cyklu spánku a bdění a hypersomnií [60]. I tyto potíže jsou méně časté u pacientů léčených včas a agresivně. K relapsu encefalitidy dochází dle různých autorů u 7 – 28 % [9,32,38]. Z 15 pacientů diagnostikovaných ve FN v Motole se relaps objevil u jedné z dospělých pacientek s neparaneoplastickou formou encefalitidy. U této nemocné se půl roku po úspěšném zaléčení tohoto relapsu rozvinula retrobulbární neuritida a v dalším průběhu u ní byla diagnostikována RS.
Stiff‑ Person Syndrom a progresivní encefalomyelitida s rigiditou a myoklonem
Stiff ‑ Person Syndrom (SPS) je vzácné onemocnění s pravděpodobně autoimunitní patogenezí. První popsané případy pocházejí z 50. let minulého století [61]. Klinický obraz SPS zahrnuje progresivní fluktuující svalovou rigiditu v důsledku kontinuální spontánní svalové aktivity, bolestivé svalové spazmy a sekundární poruchu chůze. Objektivní neurologický nález bývá normální. Kontinuální spontánní svalová aktivita u SPS je podmíněna desinhibicí polysynaptických kmenových a spinálních reflexů a má centrální původ. V neuropatologických nálezech byl u SPS pozorován obraz difuzní encefalomyelitidy s převahou postižení šedé hmoty [62]. U většiny pacientů jsou v séru a likvoru přítomny protilátky anti‑GAD (cca 60 % případů) nebo anti‑amphiphysin. Tíže příznaků SPS koreluje s titrem protilátek [63]. Existují tři hlavní klinické formy SPS: „konvenční SPS“ s dominující axiální rigiditou a bolestivými spazmy trupu a končetin, „stiff ‑ leg syndrom“ s dominující rigiditou a spazmy dolních končetin a konečně PERM – syndrom progresivní encefalomyelitidy s rigiditou a myoklonem [61]. U PERM syndromu je vystupňovaná končetinová svalová rigidita s kontrakturami doprovázena projevy encefalitidy – zejm. kmenovými příznaky a závažnou autonomní dysfunkcí [62]. Většina případů PERM syndromu je zprostředkována protilátkami proti glycinovým receptorům (anti‑GlyR). V likvoru bývá u SPS a PERM přítomna buď mírná lymfo ‑ monocytární pleocytóza či známky intratékální produkce protilátek včetně přítomnosti oligoklonálních IgG pásů. SPS se může vyskytovat v paraneoplastické anebo neparaneoplastické podobě. Nejčastějším přidruženým nádorem je SCLC [64]. V léčbě se zkouší imunoterapie 1. linie (KS, IVIg, případně eliminační metody) s poměrně dobrým efektem. Bolestivé spazmy a svalovou rigiditu lze zmírnit podáváním baklofenu, gabapentinu, pregabalinu a benzodiazepinů [63,65,66].
Syndromy sdružené se zvýšenou citlivostí vůči glutenu
Celiakii popsal již přibližně v 1. století n. l. Aretaeus z Kappadokie, spojitost mezi gastrointestinálními příznaky a expozicí glutenu však byla objasněna až v 50. letech minulého století. Následně byly identifikovány protilátky proti gliadinu jakožto nespecifický marker tohoto onemocnění. Zvýšená senzitivita vůči glutenu (GS) se neprojevuje pouze gastrointestinálními příznaky. Kromě kožních symptomů (dermatitis herpetiformis) se vyskytují i pestré neurologické projevy [67], které zahrnují (v pořadí dle četnosti): ataxii s pozdním nástupem, periferní neuropatii (motorická a senzitivní axonální polyneuropatie, mononeuritis multiplex, čistě motorická neuropatie, či small‑fiber neuropatie), encefalopatii, myopatii, myelopatii, SPS, choreu, neuromyotonii a epilepsii s přítomností okcipitálních kalcifikací na CT. Diagnostika GS se opírá o pozitivitu protilátek proti gliadinu. Do vyšetřovacího panelu je však vhodné zahrnout též protilátky proti transglutamináze a endomyziu (které jsou více specifické pro přítomnost enteropatie) a vyšetření HLA‑DQ2 nebo DQ8 (tyto subtypy se vyskytují u 70 % pacientů s celiakií a neurologickými příznaky). U všech pacientů s neurologickými příznaky a pozitivitou antigliadinových protilátek je vhodné doplnění enterální biopsie k upřesnění diagnózy. Nepřítomnost enteropatie nicméně souvislost neurologických příznaků s GS nevylučuje. Diagnostickou past představuje pozitivita antigliadinových protilátek u 5 – 12 % zdravé populace. Léčba GS spočívá v bezlepkové dietě, jejíž efekt však lze očekávat až cca jeden rok po jejím zahájení [68].
Rasmussenova encefalitida
Autoimunitní podklad má pravděpodobně i Rasmussenova encefalitida (RE). Tento názor se opírá především o neuropatologické nálezy, přítomnost různých typů autoprotilátek a efekt imunoterapie u části pacientů s touto diagnózou. RE je charakterizována:
- postižením převážně jedné mozkové hemisféry s progresivní hemisferální atrofií,
- přítomností refrakterních fokálních epileptických záchvatů a EPC,
- narůstajícím neurologickým a kognitivním deficitem.
Podrobnější popis tohoto syndromu lze nalézt v řadě přehledových článků [69 – 77]. V léčbě byly testovány IVIg [78,79], plazmaferéza [80], selektivní IgG imunoadsorpce [7], tacrolimus [79] aj. Jedinou prospektivní randomizovanou studií na toto téma je recentní celonárodní německá studie zkoumající efekt tacrolimu a IVIg u RE [79]. Bohužel ani v této multicentrické studii nedostačovaly počty pacientů k zhodnocení relativního přínosu těchto jednotlivých léčebných strategií. Zdá se, že imunologická léčba může zpomalit progresivní nárůst invalidity; její efekt na refrakterní epileptické záchvaty se však nejeví jako dostatečný. Nejúčinnější léčbou RE proto zůstává funkční hemisférektomie. Imunoterapie však může být velmi přínosná u ojedinělých případů RE u dospělých [81].
Febrile Infection‑ Related Epilepsy Syndrome
Autoimunitní mechanizmy byly zvažovány též u onemocnění sdružených pod souhrnným názvem Febrile Infection ‑ Related Epilepsy Syndrome (FIRES). Jedná se naštěstí o velmi vzácné onemocnění dětského věku, charakterizované refrakterními epileptickými záchvaty (často v podobě SE) rozvinutými v návaznosti na předchozí nespecifické febrilní onemocnění. Pokud se podaří úspěšně zvládnout akutní fázi onemocnění, přetrvávají u pacientů závažné neurologické následky a zpravidla též refrakterní epilepsie. V největší sérii 12 pacientů se nepodařilo identifikovat přítomnost žádné z dosud známých protilátek [82]. MR mozku může v časné fázi prokázat zvýšený signál mediotemporálně na T2-v.o. a FLAIR (nelze vyloučit, že v důsledku samotného SE). Efekt imunomodulace u FIRES je minimální. U téměř poloviny pacientů byl zaznamenán efekt ketogenní diety na frekvenci záchvatů [75]. Etiopatogenetické nejasnosti panují též u syndromu NORSE (New‑Onset Refractory SE).
Autoimunitně podmíněné extrapyramidové syndromy – encephalitis lethargica
Mezi AIE podle nových informací patří též encephalitis lethargica, popsaná v roce 1916 Constantinem von Economo (v Čechách podrobně studovaná prof. Ladislavem Syllabou a prof. Kamilem Hennerem). Dale ve své práci z roku 2004 prokázal, že jde o postreptokokovou AIE s protilátkami proti dosud neidentifikovaným cílům v oblasti bazálních ganglií [83]. Mezi další autoimunitně podmíněné choroby s postižením bazálních ganglií lze řadit dnes vzácnou Sydenhamovu choreu, PANDAS (Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal infection) a nově definovaný syndrom akutních extrapyramidových a psychiatrických příznaků spojených s protilátkami proti dopaminovým D2 receptorům [84]. Stručná charakteristika těchto syndromů je uvedena v tab. 1.
Závěr
AIE představují velmi pestrou a intenzivně se rozvíjející oblast neurologie. Znalost jednotlivých syndromů z této skupiny umožňuje identifikovat nejen pacienty, u nichž může správná léčba přispět k úplné úzdravě, ale i nemocné, u kterých neurologické příznaky vedou k včasnému odhalení okultního tumoru. Doufáme, že tato minimonografie vzbudí u kliniků zájem o nové poznatky na tomto poli a poskytne informace potřebné k diagnostice a léčbě AIE dle současných mezinárodních standardů.
Použité zkratky
AIE autoimunitní encefalitidy
AMPA α ‑ amino ‑ 3 - hydroxy ‑ -5 - metyl ‑ 4 - isoxazol propionát
anti‑AMPA protilátky proti glutamátovým AMPA receptorům
anti‑amphi-physin protilátky proti amphiphysinu
anti‑CASPR2 protilátky proti Contactin Associated Proteinu 2
anti-CRMP5/ CV2 protilátky proti Collapsin Response Mediator Proteinu 5
anti‑GABABR protilátky proti GABAB receptorům
anti‑GABAAR protilátky proti GABAA receptorům
anti‑GAD protilátky proti dekarboxyláze kyseliny γ ‑ aminomáselné
anti‑Hu protilátky anti‑Hu (ANNA1)
anti‑LGI1 protilátky proti Leucine ‑ rich Glioma Inactivated proteinu 1
anti‑Ma2 protilátky anti‑Ma2
anti‑NMDAR protilátky proti NMDA glutamátovým receptorům
anti‑Ri protilátky anti‑Ri
anti‑TPO protilátky proti tyroidální peroxidáze (antimikrozomální protilátky)
anti‑Yo protilátky anti‑Yo
anti‑VGKC protilátky proti komplexu proteinů sdružených s napěťově řízenými draslíkovými kanály
β ‑ hCG lidský choriový gonadotropin
CASPR2 Contactin Associated Protein 2
CFM cyklofosfamid
CPS parciální komplexní epileptické záchvaty
CPSE komplexní parciální status epilepticus
CRMP5/ CV2 Collapsin Response Mediator Protein 5
CT počítačová tomografie
DPPX/ DPP6 Dipeptidylpetidase‑like Protein 6
DM diabetes mellitus
EDB Extreme Delta Brush vzorec v EEG
EEG elektroencefalografie
ELISA Enzyme ‑ Linked Immunosorbent Assay
EPC Epilepsia Partialis Continua
FBDS faciobrachiální dystonické záchvaty
18FDG ‑ PET pozitronová emisní tomografie s využitím 18fluoro‑deoxyglukózy
FIRES Fever ‑ Induced Refractory Epilepsy Syndrome
FLAIR Fluid ‑ Attenuated Inversion Recovery
GABA γ ‑ aminomáselná kyselina
GAD glutamát dekarboxyláza
GS zvýšená senzitivita vůči glutenu
HLA Human Leukocyte Antigen
HSE herpetická encefalitida
HSV Herpes Simplex Virus
IVIg intravenózní imunoglobuliny
KS kortikosteroidy
LE limbická encefalitida
LGI1 Leucine ‑ rich Glioma Inactivated protein 1
MR magnetická rezonance
MoCA Montreal Cognitive Assessment
MMSE Mini Mental Status Examination
NLE neparaneoplastická limbická encefalitida
NMDA N ‑ metyl ‑ D ‑ aspartát
NMO Neuromyelitis Optica (m. Devic)
OCB oligoklonální IgG pásy
PCR Polymerase Chain Reaction
PERM progresivní encefalomyelitida s rigiditou a myoklonem
PLE paraneoplastická limbická encefalitida
PLEX plazmaferéza
PS klasický paraneoplastický syndrom
PNP ‑ Ab dobře charakterizované onkoneurální protilátky
RE Rasmussenova encefalitida
RIA radioimunoesej
RTX rituximab
SCLC malobuněčný karcinom plic
SE Status Epilepticus
SIADH syndrom inadekvátní sekrece ADH
sGTCS sekundárně generalizované tonicko‑klonické záchvaty
SOX ‑ 1 Sex determining region Y ‑ box 1
SPS Stiff ‑ Person Syndrom
T2 - v.o. T2 vážené sekvence magnetické rezonance
TLE epilepsie temporálního laloku
TPO tyroidální peroxidáza
UPV umělá plicní ventilace
VGKC napěťově řízené draslíkové kanály
Poděkování
Velký dík za pomoc při přípravě článku patří doc. MU Dr. Petru Marusičovi, Ph.D. Za odbornou spolupráci v oblasti laboratorní diagnostiky autoimunitních encefalitid autoři děkují RNDr. Jitce Hanzalové.
MUDr. David Krýsl, Ph.D.
Klinisk Neurofysiologi
Sahlgrenska Universitetssjukhuset
Blå Stråket 5
413 45 Göteborg
Sweden
e-mail: dkrysl@gmail.com
Přijato k recenzi: 14. 8. 2014
Přijato do tisku: 18. 11. 2014
Recenzenti
MUDr. Zuzana Libá, Ph.D.
doc. MUDr. Petr Marusič, Ph.D.
doc. MUDr. Pavel Štourač, Ph.D.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. David Krýsl, Ph.D.

Po ukončení studia na 3. lékařské fakultě Univerzity Karlovy v Praze (2003) nastoupil David Krýsl na Neurologickou kliniku 2. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice v Motole, Praha. Na tomto pracovišti následně strávil 10 let, nejprve jako sekundární lékař a od roku 2010 jako vedoucí lékař. Během působení ve FN v Motole jej zaujala především epileptologie, současně však i problematika roztroušené sklerózy a autoimunitních onemocnění CNS. V roce 2011 inicioval ve spolupráci s RNDr. Jitkou Hanzalovou v Likvorové laboratoři Ústavu imunologie a Neurologické kliniky 2. LF UK a FN v Motole vyšetřování protilátek proti membránovým a synaptickým antigenům metodikou nepřímé imunofl uorescence. Díky vstřícnosti a podpoře obou pracovišť bylo postupně možné nabídnout vyšetřování těchto protilátek i kolegům z dalších nemocnic v České republice i v zahraničí (Slovenská republika). Koncepce vyšetřování protilátek ve FN v Motole od počátku zahrnuje též konzultační činnost, které se MUDr. Krýsl věnoval. Souběžně pokračoval v postgraduálním studiu v oboru fyziologie a patofyziologie člověka, které v roce 2013 ukončil obhajobou dizertační práce na téma „Funkční důsledky epileptických záchvatů a hypoxicko-ischemického poškození CNS“. Paralelní zájem o základní výzkum se odráží i v publikovaných článcích, jejichž je MUDr. Krýsl autorem či spoluautorem. Publikace se zabývají mj. změnami propustnosti hematoencefalické bariéry v důsledku jednotlivých záchvatů a fokální ischemie, změnami excitability mozku v důsledku ischemie anebo důsledky jednotlivých epileptických záchvatů na prostorové učení. MUDr. Krýsl je také spoluautorem několika přehledových článků a kapitol v odborné literatuře zabývajících se akutními symptomatickými epileptickými záchvaty, diagnostikou a léčbou status epilepticus a využitím EEG v intenzivní péči. Od roku 2014 pracuje David Krýsl jako epileptolog na Oddělení klinické neurofyziologie Univerzitní nemocnice Sahlgrenska v Göteborgu, Švédsko. Nadále úzce spolupracuje se svou mateřskou klinikou, zejm. na projektech týkajících se
Sources
Literatura doporučená ke studiu je označena hvězdičkou
1.* Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F et al. Immunopathology of autoantibody‑associated encephalitides: clues for pathogenesis. Brain 2012; 135(5): 1622 – 1638. doi: 10.1093/ brain/ aws082.
2. Albert ML, Austin LM, Darnell RB. Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol 2000; 47(1): 9 – 17.
3.* Darnell RB. The importance of defining the paraneoplastic neurologic disorders. N Engl J Med 1999; 340(23): 1831 – 1833.
4. Kishitani T, Matsunaga A, Yoneda M. The biomarker and treatment in Hashimoto‘s encephalopahty. Nihon Rinsho 2013; 71(5): 893 – 897.
5.* Saiz A, Blanco Y, Sabater L, Gonzalez F, Bataller L, Casamitjana R et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008; 131(10): 2553 – 2563. doi: 10.1093/ brain/ awn183.
6. Kohler W, Ehrlich S, Dohmen C, Haubitz M, Hoffmann F, Schmidt S et al. Tryptophan immunoadsorption for the treatment of autoimmune encephalitis. Eur J Neurol 2014. doi: 10.1111/ ene.12389.
7. Antozzi C, Granata T, Aurisano N, Zardini G, Confalonieri P, Airaghi G et al. Long‑term selective IgG immuno ‑ adsorption improves Rasmussen‘s encephalitis. Neurology 1998; 51(1): 302 – 305.
8. Hayashi A, Nakamagoe K, Ohkoshi N, Hoshino S, Shoji S. Double filtration plasma exchange and immunoadsorption therapy in a case of stiff ‑ man syndrome with negative anti‑GAD antibody. J Med 1999; 30(5 – 6): 321 – 327.
9. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long‑term outcome in patients with anti‑NMDA receptor encephalitis: an observational cohort study. The Lancet Neurol 2013; 12(2): 157 – 165. doi: 10.1016/ S1474 ‑ 4422(12)70310 ‑ 1.
10.* Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004; 75(8): 1135 – 1140.
11.* Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008; 7(4): 327 – 340. doi: 10.1016/ S1474 ‑ 4422(08)70060 ‑ 7.
12.* Šťourač P, Ambler Z. Paraneoplastické neurologické syndromy – základní charakteristika, klasifikace, etiopatogeneze a diagnostika. Neurol Prax 2013; 14(1): 9 – 12.
13.* Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol 2011; 18(1): 19. doi: 10.1111/ j.1468 ‑ 1331.2010.03220.x.
14. Mathew RM, Vandenberghe R, Garcia ‑ Merino A, Yamamoto T, Landolfi JC, Rosenfeld MR et al. Orchiectomy for suspected microscopic tumor in patients with anti‑Ma2‑associated encephalitis. Neurology 2007; 68(12): 900 – 905.
15. Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50(1): 73 – 78.
16.* Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice ‑ Gordon R et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010; 9(8): 776 – 785. doi: 10.1016/ S1474 ‑ 4422(10)70137 ‑ X.
17. Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B et al. Clinical analysis of anti‑Ma2‑associated encephalitis. Brain 2004; 127(8): 1831 – 1844.
18. Overeem S, Dalmau J, Bataller L, Nishino S, Mignot E, Verschuuren J et al. Hypocretin‑1 CSF levels in anti‑Ma2 associated encephalitis. Neurology 2004; 62(1): 138 – 140.
19.* Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav 2007; 10(4): 529 – 538.
20. Haberlandt E, Bast T, Ebner A, Holthausen H, Kluger G, Kravljanac R et al. Limbic encephalitis in children and adolescents. Arch Dis Child 2011; 96(2): 186 – 191. doi: 10.1136/ adc.2010.183897.
21. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9(1): 67 – 76. doi: 10.1016/ S1474 ‑ 4422(09)70324 ‑ 2.
22. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti‑GAD‑associated neurologic disorders. Neurology 2011; 76(9): 795 – 800. doi: 10.1212/ WNL.0b013e31820e7b8d.
23. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65(4): 424 – 434. doi: 10.1002/ ana.21589.
24. Najjar S, Pearlman D, Najjar A, Ghiasian V, Zagzag D, Devinsky O. Extralimbic autoimmune encephalitis associated with glutamic acid decarboxylase antibodies: an underdiagnosed entity? Epilepsy Behav 2011; 21(3): 306 – 313. doi: 10.1016/ j.yebeh.2011.03.038.
25. Lopez ‑ Sublet M, Bihan H, Reach G, Dupont S, Didelot A, Mourad JJ et al. Limbic encephalitis and type 1 diabetes with glutamic acid decarboxylase 65 (GAD65) autoimmunity: improvement with high‑dose intravenous immunoglobulin therapy. Diabetes Metab 2012; 38(3): 273 – 275. doi: 10.1016/ j.diabet.2012.02.005.
26. Dalakas MC. The role of IVIg in the treatment of patients with stiff person syndrome and other neurological diseases associated with anti‑GAD antibodies. J Neurol 2005; 252 (Suppl 1): I19 – I25.
27. Thompson PD. The stiff ‑ man syndrome and related disorders. Parkinsonism Relat Disord 2001; 8(2): 147 – 153.
28. Josephs KA, Silber MH, Fealey RD, Nippoldt TB, Auger RG, Vernino S. Neurophysiologic studies in Morvan syndrome. J Clin Neurophysiol 2004; 21(6): 440 – 445.
29.* Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012; 72(2): 241 – 255. doi: 10.1002/ ana.23577.
30. Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al. Paraneoplastic anti‑N ‑ methyl ‑ D ‑ aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61(1): 25 – 36.
31.* Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population‑based prospective study. Lancet Infect Dis 2010; 10(12): 835 – 844. doi: 10.1016/ S1473 ‑ 3099(10)70222 ‑ X.
32.* Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M et al. Anti‑NMDA ‑ receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008; 7(12): 1091 – 1098. doi: 10.1016/ S1474 ‑ 4422(08)70224 ‑ 2.
33. Ito Y, Abe T, Tomioka R, Komori T, Araki N. Anti‑NMDA receptor encephalitis during pregnancy. Rinsho Shinkeigaku 2010; 50(2): 103 – 107.
34. Hacohen Y, Wright S, Waters P, Agrawal S, Carr L, Cross H et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry 2013; 84(7): 748 – 755. doi: 10.1136/ jnnp ‑ 2012 ‑ 303807.
35. Irani SR, Vincent A. NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep 2011; 11(3): 298 – 304. doi: 10.1007/ s11910 ‑ 011 ‑ 0186 ‑ y.
36. Agrawal S, Vincent A, Jacobson L, Milford D, Gupta R, Wassmer E. Successful treatment of antiN ‑ methyl ‑ d ‑ aspartate receptor limbic encephalitis in a 22 – monthold child with plasmapheresis and pharmacological immunomodulation. Arch Dis Child 2010; 95(4): 312. doi: 10.1136/ adc.2009.164889.
37. Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S et al. Anti‑N ‑ methyl ‑ D ‑ aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009; 66(1): 11 – 18. doi: 10.1002/ ana.21756.
38. Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS et al. N ‑ methyl ‑ D ‑ aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‑paraneoplastic disorder of both sexes. Brain 2010; 133(6): 1655 – 1667. doi: 10.1093/ brain/ awq113.
39. Le Foll J, Pelletier A. Psychiatric symptoms of a paraneoplastic anti‑N ‑ methyl ‑ D ‑ aspartate receptor encephalitis: a case report. Encephale 2010; 36(2): 166 – 171. doi: 10.1016/ j.encep.2009.06.008.
40. Consoli A, Raffin M, Laurent C, Bodeau N, Campion D, Amoura Z et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case ‑ control study. Schizophr Res 2012; 137(1 – 3): 151 – 158. doi: 10.1016/ j.schres.2012.02.012.
41. Casanova ‑ Gracia N, Banzo ‑ Arguis C, Sanz ‑ Asin P, Zapata ‑ Usabel M, Jordana ‑ Vilanova N, Cortina ‑ Lacambra MT. Encephalitis associated to anti‑NMDA receptor antibodies: a description of two cases in the child/ youth population. Rev Neurol 2012; 54(8): 475 – 478.
42. Borlot F, Santos ML, Bandeira M, Liberalesso PB, Kok F, Lohr A jr et al. Anti‑N ‑ methyl D ‑ aspartate receptor encephalitis in childhood. J Pediatr (Rio J) 2012; 88(3): 275 – 278. doi:10.2223/ JPED.2172.
43. Uchino A, Iizuka T, Urano Y, Arai M, Hara A, Hamada J et al. Pseudo ‑ piano playing motions and nocturnal hypoventilation in anti‑NMDA receptor encephalitis: response to prompt tumor removal and immunotherapy. Intern Med 2011; 50(6): 627 – 630.
44. Chia PL, Tan K, Foo D. Profound Sinus Node Dysfunction in anti‑N ‑ methyl ‑ D ‑ aspartate receptor limbic encephalitis. Pacing Clin Electrophysiol 2011; 36(3): e90 – e92. doi: 10.1111/ j.1540 ‑ 8159.2011.03154.x.
45. Nazif TM, Vazquez J, Honig LS, Dizon JM. Anti‑N ‑ methyl ‑ D ‑ aspartate receptor encephalitis: an emerging cause of centrally mediated sinus node dysfunction. Europace 2012; 14(8): 1188 – 1194. doi: 10.1093/ europace/ eus014.
46. Liba Z, Sebronova V, Komarek V, Sediva A, Sedlacek P. Prevalence and treatment of anti‑NMDA receptor encephalitis. Lancet Neurol 2013; 12(5): 424 – 425. doi: 10.1016/ S1474 ‑ 4422(13)70070 ‑ X.
47. Sadalage G, Karim A, Jacob S. Autoimmune encephalitis screen – a review of rapid diagnostic screening in 600 patients over 5 years. J Neurol Neurosurg Psychiatry 2013; 84(11): e2.
48. Fujita K, Yuasa T, Takahashi Y, Tanaka K, Sako W, Koizumi H et al. Antibodies to N ‑ methyl ‑ D ‑ aspartate glutamate receptors in Creutzfeldt ‑ Jakob disease patients. J Neuroimmunol 2012; 251(1 – 2): 90 – 93. doi: 10.1016/ j.jneuroim.2012.06.010.
49. Prüss H, Finke C, Holtje M, Hofmann J, Klingbeil C, Probst C et al. N ‑ methyl ‑ D ‑ aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol 2012; 72(6): 902 – 911. doi: 10.1002/ ana.23689.
50. Leypoldt F, Titulaer MJ, Aguilar E, Walther J, Bonstrup M, Havemeister S et al. Herpes simplex virus ‑ 1 encephalitis can trigger anti‑NMDA receptor encephalitis: case report. Neurology 2013; 81(18): 1637 – 1639. doi: 10.1212/ WNL.0b013e3182a9f531.
51. Kirkpatrick MP, Clarke CD, Sonmezturk HH, Abou ‑ Khalil B. Rhythmic delta activity represents a form of nonconvulsive status epilepticus in anti‑NMDA receptor antibody encephalitis. Epilepsy Behav 2011; 20(2): 392 – 394. doi: 10.1016/ j.yebeh.2010.11.020.
52. Johnson N, Henry C, Fessler AJ, Dalmau J. Anti‑NMDA receptor encephalitis causing prolonged nonconvulsive status epilepticus. Neurology 2010; 75(16): 1480 – 1482. doi: 10.1212/ WNL.0b013e3181f8831a.
53. Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti‑NMDA receptor encephalitis. Neurology 2012; 79 (11): 1094 – 1100. doi: 10.1212/ WNL.0b013e3182698cd8.
54. Chanson JB, Diaconu M, Honnorat J, Martin T, De Seze J, Namer IJ et al. PET follow‑up in a case of anti‑NMDAR encephalitis: arguments for cingulate limbic encephalitis. Epileptic Disord 2012; 14 (1): 90 – 93. doi: 10.1684/ epd.2012.0486.
55.* Dalmau J, Lancaster E, Martinez ‑ Hernandez E, Rosenfeld MR, Balice ‑ Gordon R. Clinical experience and laboratory investigations in patients with anti‑NMDAR encephalitis. Lancet Neurol 2011; 10(1): 63 – 74. doi: 10.1016/ S1474 ‑ 4422(10)70253 ‑ 2.
56. Eker A, Saka E, Dalmau J, Kurne A, Bilen C, Ozen H et al. Testicular teratoma and anti‑N ‑ methyl ‑ D ‑ aspartate receptor‑associated encephalitis. J Neurol Neurosurg Psychiatry 2008; 79(9): 1082 – 1083. doi: 10.1136/ jnnp.2008.147611.
57. Ishiura H, Matsuda S, Higashihara M, Hasegawa M, Hida A, Hanajima R et al. Response of anti‑NMDA receptor encephalitis without tumor to immunotherapy including rituximab. Neurology 2008; 71(23): 1921 – 1923. doi: 10.1212/ 01.wnl.0000336648.43562.59.
58. Tumbi A, Gilani A, Scarff JR, Kaur G, Lippmann S. Anti‑N ‑ methyl ‑ D Encephalitis. Innov Clin Neurosci 2011; 8(9): 24 – 25.
59. Finke C, Kopp UA, Pruss H, Dalmau J, Wandinger KP, Ploner CJ. Cognitive deficits following anti‑NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry 2012; 83(2): 195 – 198.
60. Dalmau J. Limbic encephalitis and variants related to neuronal cell membrane autoantigens. Rinsho Shinkeigaku 2008; 48(11): 871 – 874.
61. Cantiniaux S, Azulay JP, Boucraut J, Pouget J, Attarian S. Stiff man syndrome: clinical forms, treatment and clinical course. Rev Neurol (Paris) 2006; 162(8 – 9): 832 – 839.
62. Barker RA, Revesz T, Thom M, Marsden CD, Brown P. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry 1998; 65(5): 633 – 640.
63. Espay AJ, Chen R. Rigidity and spasms from autoimmune encephalomyelopathies: stiff ‑ person syndrome. Muscle Nerve 2006; 34(6): 677 – 690.
64. Spitz M, Ferraz HB, Barsottini OG, Gabbai AA. Progressive encephalomyelitis with rigidity: a paraneoplastic presentation of oat cell carcinoma of the lung. Case report. Arq Neuropsiquiatr 2004; 62(2B): 547 – 549.
65. Gouider ‑ Khouja N, Mekaouar A, Larnaout A, Miladi N, Ben Khelifa F, Hentati F. Progressive encephalomyelitis with rigidity presenting as a stiff ‑ person syndrome. Parkinsonism Relat Disord 2002; 8(4): 285 – 258.
66. Gazulla Abio J, Benavente Aguilar I, Capablo Liesa JL. Progressive encephalomyelitis with rigidity. Clinical and electrophysiological aspects. Neurologia 2001; 16(2): 85 – 88.
67. Cooke WT and Smith WT. Neurological disorders associated with adult coeliac disease. Brain 1966; 89(4): 683 – 722.
68. Hadjivassiliou M, Davies ‑ Jones GA, Sanders DS, Grunewald RA. Dietary treatment of gluten ataxia. J Neurol Neurosurg Psychiatry 2003; 74(9): 1221 – 1224.
69. Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology 1958; 8(6): 435 – 445.
70. Rasmussen T. Further observations on the syndrome of chronic encephalitis and epilepsy. Appl Neurophysiol 1978; 41(1 – 4): 1 – 12.
71. Dulac O. Rasmussen‘s syndrome. Curr Opin Neurol 1996; 9(2): 75 – 77.
72. Bauer J, Elger CE, Hans VH, Schramm J, Urbach H, Lassmann H et al. Astrocytes are a specific immunological target in Rasmussen‘s encephalitis. Ann Neurol 2007; 62(1): 67 – 80.
73.* Bien CG and Elger CE. Epilepsia partialis continua: semiology and differential diagnoses. Epileptic Disord 2008; 10(1): 3 – 7. doi: 10.1684/ epd.2008.0161.
74. Terra ‑ Bustamante VC, Machado HR, dos Santos Oliveira R, Serafini LN, Souza ‑ Oliveira C, Escorsi ‑ Rosset Set al. Rasmussen encephalitis: long‑term outcome after surgery. Childs Nerv Syst 2009; 25(5): 583 – 589. doi: 10.1007/ s00381 ‑ 008 ‑ 0795 ‑ 1.
75. Nabbout R. Autoimmune and inflammatory epilepsies. Epilepsia 2012; 53 (Suppl 4): 58 – 62. doi: 10.1111/ j.1528 ‑ 1167.2012.03614.x.
76. Armangue T, Petit ‑ Pedrol M, Dalmau J. Autoimmune Encephalitis in Children. J Child Neurol 2012; 27(11): 1460 – 1469. doi: 10.1177/ 0883073812448838.
77. Caraballo RH, Fortini S, Cersosimo R, Monges S, Pasteris MC, Gomez M et al. Rasmussen syndrome: an argentinean experience in 32 patients. Seizure 2013; 22(5): 360 – 367. doi: 10.1016/ j.seizure.2013.02.003.
78. Feasby T, Banwell B, Benstead T, Bril V, Brouwers M, Freedman M et al. Guidelines on the use of intravenous immune globulin for neurologic conditions. Transfus Med Rev 2007; 21 (Suppl 1): S57 – S107.
79. Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia 2013; 54(3): 543 – 550. doi: 10.1111/ epi.12042.
80. Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO. Plasmapheresis in Rasmussen‘s encephalitis. Neurology 1996; 46(1): 242 – 246.
81. Leach JP, Chadwick DW, Miles JB, Hart IK. Improvement in adult ‑ onset Rasmussen‘s encephalitis with long‑term immunomodulatory therapy. Neurology 1999; 52(4): 738 – 742.
82.* van Baalen A, Hausler M, Plecko‑Startinig B, Strautmanis J, Vlaho S, Gebhardt B et al. Febrile infection‑related epilepsy syndrome without detectable autoantibodies and response to immunotherapy: a case series and discussion of epileptogenesis in FIRES. Neuropediatrics 2012; 43(4): 209 – 216. doi: 10.1055/ s ‑ 0032 ‑ 1323848.
83.* Dale RC, Church AJ, Surtees RA, Lees AJ, Adcock JE, Harding B et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain 2004; 127(1): 21 – 33.
84. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TKet al. Antibodies to surface dopamine ‑ 2 receptor in autoimmune movement and psychiatric disorders. Brain 2012; 135(11): 3453 – 3468. doi: 10.1093/ brain/ aws256.
85. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al. Paraneoplastic anti‑N ‑ methyl ‑ D ‑ aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61(1): 25 – 36.
86. Petit ‑ Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen and analysis of the effects of antibodies. Lancet Neurol 2014; 13(3): 276 – 286.
87. Corsellis JA, Goldberg GJ,Norton AR. „Limbic encephalitis“ and its association with carcinoma. Brain 1968; 91(3): 481 – 496.
88. Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50(1): 73 – 78.
89. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L et al. Antibodies to Kv1 potassium channel ‑ complex proteins leucine ‑ rich, glioma inactivated 1 protein and contactin‑associated protein‑2 in limbic encephalitis, Morvan‘s syndrome and acquired neuromyotonia. Brain 2010; 133(9): 2734 – 2748. doi: 10.1093/ brain/ awq213.
90. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9(1): 67 – 76. doi: 10.1016/ S1474 ‑ 4422(09)70324 ‑ 2.
91. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu Het al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65(4): 424 – 434. doi: 10.1002/ ana.21589.
92. Giometto B, Nicolao P, Macucci M, Tavolato B, Foxon R,Bottazzo GF. Temporal ‑ lobe epilepsy associated with glutamic ‑ acid ‑ decarboxylase autoantibodies. Lancet 1998; 352(9126): 457.
93. Turner MR, Irani SR, Leite MI, Nithi K, Vincent A, Ansorge O. Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology 2011; 77(5): 439 – 443. doi: 10.1212/ WNL.0b013e318227b176.
94. Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm („stiff ‑ man“ syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin 1956; 31(15): 421 – 427.
95. Boronat A, Gelfand JM, Gresa ‑ Arribas N, Jeong HY, Walsh M, Roberts K et al. Encephalitis and antibodies to dipeptidyl ‑ peptidase‑like protein‑6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013; 73(1): 120 – 128. doi: 10.1002/ ana.23756.
96. Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology 1958; 8(6): 435 – 445.
97. Aron AM, Freeman JM, Carter S. The natural history of Sydenham‘s chorea. Review of the literature and long‑term evaluation with emphasis on cardiac sequelae. Am J Med 1965; 38 : 83 – 95.
98. Swedo SE, Leonard HL, Garvey M, Mittleman B, Allen AJ, Perlmutter S et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry 1998; 155(2): 264 – 271.
99. Dale RC, Brilot F. Autoimmune basal ganglia disorders. J Child Neurol 2012; 27(11): 1470 – 1481. doi: 10.1177/ 0883073812451327.
100. Dale RC, Church AJ, Surtees RA, Lees AJ, Adcock JE, Harding B et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain 2004; 127(1): 21 – 33.
101. Cooke WT, Smith WT. Neurological disorders associated with adult coeliac disease. Brain 1966; 89(4): 683 – 722.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 1
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Protokol diagnostiky a léčby hyponatremie a hypernatremie v neurointenzivní péči
- Mini‑Mental State Examination – česká normativní studie
- Autoimunitní encefalitidy
- Asymptomatická spondylogenní komprese krční míchy