
Kongenitální fibróza zevních okohybných svalů u české rodiny a její molekulárně genetická příčina
Congenital fibrosis of the extraocular muscles in a Czech family and its molecular genetic cause
Aim: Congenital fibrosis of the extraocular muscles (CFEOM) is a rare autosomal dominant disorder characterized by bilateral non-progressive ophthalmoplegia and ptosis. The aim of this study was to identify the molecular genetic cause in a four-generation family with CFEOM and to describe the clinical findings in four affected and one unaffected member.
Patients and methods: All patients underwent an eye examination. Exons 2, 8, 20 and 21 of the KIF21A gene were directly examined by Sanger sequencing in the proband. Sanger sequencing was also used to test for the presence of the detected mutation in other relatives.
Results: Clinical findings were typical in all affected individuals manifesting as ptosis and severely limited vertical and horizontal eye movements with compensatory backward tilt of the head. All patients also had decreased visual acuity attributed to amblyopia and synkinetic eye movements; synergistic convergence on attempted vertical gaze and divergence in the downgaze. A known heterozygous mutation c.2860C>T; p.(Arg954Trp) in KIF21A was identified in all available affected family members with CFEOM. This mutation was not found in a grandson of the proband who had no clinical disease symptoms.
Conclusion: CFEOM is a serious disorder leading to life-long functional and often psychological problems. The molecular genetic cause in patients of Czech origin has been discovered for the first time.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
捷克家庭眼外肌的先天性纤维化及其分子遗传原因
目的:先天性眼外肌纤维化(CFEOM)是一种罕见的常染色体显性遗传疾病,其特征是双侧非进行性眼肌麻痹和上睑下垂。这项研究的目的是确定具有CFEOM的四代家庭的分子遗传原因,并描述四名受影响和一名未受影响的成员的临床发现。
患者和方法:所有患者均接受了眼科检查。通过先证者中的Sanger测序直接检查了KIF21A基因的外显子2、8、20和21。Sanger测序也被用来检测在其他亲属中检测到的突变的存在。
结果:在所有受影响的个体中,典型的临床表现为上睑下垂,垂直和水平眼运动严重受限以及头部向后倾斜。所有患者的视力均因弱视和眼球运动同步而下降;尝试垂直注视和向下注视的协同收敛。已知的杂合突变c.2860C> T;在所有可能患有CFEOM的受影响家庭成员中都发现了KIF21A中的p。(Arg954Trp)。在没有临床疾病症状的先证者的孙子中未发现此突变。
结论:CFEOM是一种严重的疾病,导致终生的功能障碍和心理问题。首次发现捷克籍患者的分子遗传原因。
关键词:先天性眼外肌纤维化–下垂–眼肌麻痹 – KIF21A
Keywords:
ptosis – ophthalmoplegia – KIF21A – congenital fibrosis of the extraocular muscles
Authors:
Ľ. Ďuďáková 1; E. Vyhnálková 2; P. Sklenka 3; P. Kuthan 3; P. Diblík 3; P. Lišková 1,3
Authors‘ workplace:
Klinika dětského a dorostového lékařství, 1. LF UK a VFN v Praze
1; Ústav biologie a lékařské genetiky, Oddělení klinické genetiky, FN Motol, Praha
2; Oční klinika 1. LF UK a VFN v Praze
3
Published in:
Cesk Slov Neurol N 2019; 82(5): 561-566
Category:
Original Paper
doi:
https://doi.org/10.14735/amcsnn2019561
Overview
Cíl: Kongenitální fibróza zevních okohybných svalů (congenital fibrosis of the extraocular muscles; CFEOM) je vzácné autozomálně dominantní onemocnění charakterizované oboustrannou neprogresivní oftalmoplegií a ptózou. Cílem práce bylo popsat klinické projevy a určit molekulárně genetickou příčinu u čtyř postižených a jednoho nepostiženého člena čtyřgenerační rodiny s CFEOM.
Soubor a metodika: Všichni jedinci podstoupili oční vyšetření. U probandky jsme provedli pomocí Sangerova sekvenování skríning exonů 2, 8, 20 a 21 genu KIF21A. Stejnou metodou jsme testovali přítomnost zjištěné mutace u dalších příbuzných.
Výsledky: Klinické nálezy byly typické u všech postižených jedinců. Konkrétně byla přítomna ptóza a nehybnost bulbů ve vertikále i v horizontále kompenzovaná záklonem hlavy při přímém pohledu, snížení zrakové ostrosti přičítané amblyopii a paradoxní synkinézy. V genu KIF21A jsme nalezli v heterozygotním stavu již dříve popsanou mutaci c.2860C>T; p.(Arg954Trp) u všech dostupných postižených členů rodiny s CFEOM. U vnuka probandky, který byl klinicky bez příznaků onemocnění, nebyla tato mutace nalezena.
Závěr: CFEOM je závažné onemocnění, které vede celoživotně k funkčním a často i psychickým obtížím. Příčina onemocnění na úrovni genu byla u pacientů českého původu s touto vzácnou chorobou určena poprvé.
Klíčová slova:
kongenitální fibróza okohybných svalů – ptóza – oftalmoplegie – KIF21A
Úvod
Kongenitální fibróza zevních okohybných svalů (congenital fibrosis of the extraocular muscles; CFEOM), v české literatuře také označovaná jako vrozená stacionární zevní oftalmoplegie nebo syndrom vrozené fibrózy zevních okohybných svalů, je neprogresivní onemocnění charakterizované okohybnou poruchou projevující se ptózou, neschopností pohledu vzhůru a většinou i omezením pohledu do stran [1,2]. Tyto obtíže vedou k vynucenému držení hlavy se zvednutou bradou. Dalším charakteristickým znakem jsou anomální synkinézy, nejčastěji při pokusu o pohled vzhůru, kdy se oči stáčejí do konvergence [3]. Popsán byl i fenomén Marcuse Gunna – mandibulopalpebrální synkinéza [4,5].
Kongenitální fibróza zevních okohybných svalů není asociována s žádnými dalšími neurologickými či jinými abnormalitami [6,7]. Post mortem vyšetření [7] a MR [8,9] u jedinců s CFEOM typu 1 (CFEOM1) prokázaly hypoplazii n. oculomotorius variabilně asociovanou s absencí n. abducens u pacientů se synkinézami, nerozdělení axonů do horní větve, a tím i ztrátu odpovídajících motorických neuronů, abnormální inervaci v orbitě a atrofii horního a vnitřního přímého a zvedače horního víčka.
Prevalence onemocnění je odhadována 1 postižený na 230 tisíc obyvatel [10]. Výskyt onemocnění byl popsán celosvětově bez významnějších regionálních rozdílů [11]. Nejčastěji se CFEOM vyskytuje na podkladě mutací v genu KIF21A (OMIM *608283, kinesin family member 21A), asociovanými s typem 1 (CFEOM1; OMIM #135700) [12]. CFEOM typ 2 (CFEOM2, OMIM #602078) je způsoben mutacemi v genu PHOX2A (OMIM *602753, paired like homeobox 2A) [13] a CFEOM typ 3a (CFEOM3A; OMIM #600638) vzniká v důsledku mutací v genu TUBB3 (OMIM *602661, tubulin beta-3) [14]. Historicky byly na základě klinických nálezů rozlišovány i další typy jako CFEOM1A a CFEOM3B, které jsou však nyní zahrnuty pod CFEOM1. Typ CFEOM3C byl popsán pouze v jedné třígenerační rodině a spojován s balancovanou nebo nebalancovanou translokací chromozomů 2 a 13 [15]. V literatuře lze dále najít i zmínku o CFEOM spojené s abnormalitami mozku, která vzniká na podkladě mutací v genu TUBB2B (OMIM *612850). Toto onemocnění je ale v současné době vedeno pod samostatným názvem „komplexní kortikální dysplazie s dalšími abnormalitami mozku“ (CDCBM7, OMIM #610031). V jedné rodině byl dále popsán tzv. Tukelův syndrom, někdy nazývaný i CFEOM4 (OMIM %609428). U 6 postižených jedinců se kromě neprogresivní oftalmoplegie a ptózy vyskytovala i oligodaktylie a oligosyndaktylie. Gen proto toto onemocnění zůstává neznámý, byl mapován do oblasti dlouhého ramínka chromozomu 21 [16].
Typy CFEOM1 a CFEOM3 se dědí autozomálně dominantně, CFEOM2 je autozomálně recesivní onemocnění. Zatímco CFEOM1 a CFEOM2 je plně penetrantní choroba, u CFEOM3A byly popsány případy nositelů patogenních mutací bez manifestace známek onemocnění [11]. Klinicky jsou jednotlivé typy CFEOM od sebe velmi špatně rozlišitelné. Všechny vykazují od narození různou míru zevní oftalmoplegie, u typu 3 ale nemusí být přítomna ptóza, typ 2 a 3 může vykazovat normální primární postavení oka [11].
Soubor a metodika
Rodina s okohybnou poruchou byla odeslána na konziliární vyšetření na Ústav biologie a lékařské genetiky FN Motol, kde byla provedena detailní genealogická analýza vč. zakreslení rodokmenu. Oftalmologické vyšetření podstoupili pacienti na Oční klinice 1. LF a VFN v Praze, která je součástí Evropské referenční sítě pro vzácná onemocnění oka (ERN-EYE). Nejlepší korigovaná zraková ostrost byla stanovena na Snellenových optotypech.
Po podepsání informovaného souhlasu byl z leukocytů venózní krve izolován genetický materiál (DNA) kitem AutoGen Flex STAR (AutoGen, Holliston, MA, USA). Vzhledem k tomu, že klinický nález odpovídal onemocnění CFEOM, které je v literatuře v naprosté většině případů spojováno s mutacemi pouze ve 4 exonech (2, 8, 20 a 21) genu KIF21A [17,18], provedli jsme Sangerovo sekvenování polymerázové řetězové reakce produktů těchto exonů vč. přilehlých intronových oblastí. Amplifikace proběhla pomocí dříve publikovaných primerů [19] a vlastní sekvenace pak na kapilárním sekvenátoru ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Jako referenční sekvenci jsme použili NM_001173464.1.
Patogenita zjištěných sekvenčních variant byla ověřována na základě studia literatury, pomocí různých predikčních softwarových nástrojů a vyhodnocením frekvencí nalezených změn v populační databázi gnomAD [20], která soustřeďuje exomová data od více než 120 tisíc jedinců. Frekvence mutací specifické pro českou populaci byla získána z Národního centra lékařské genomiky [21].
Výsledky
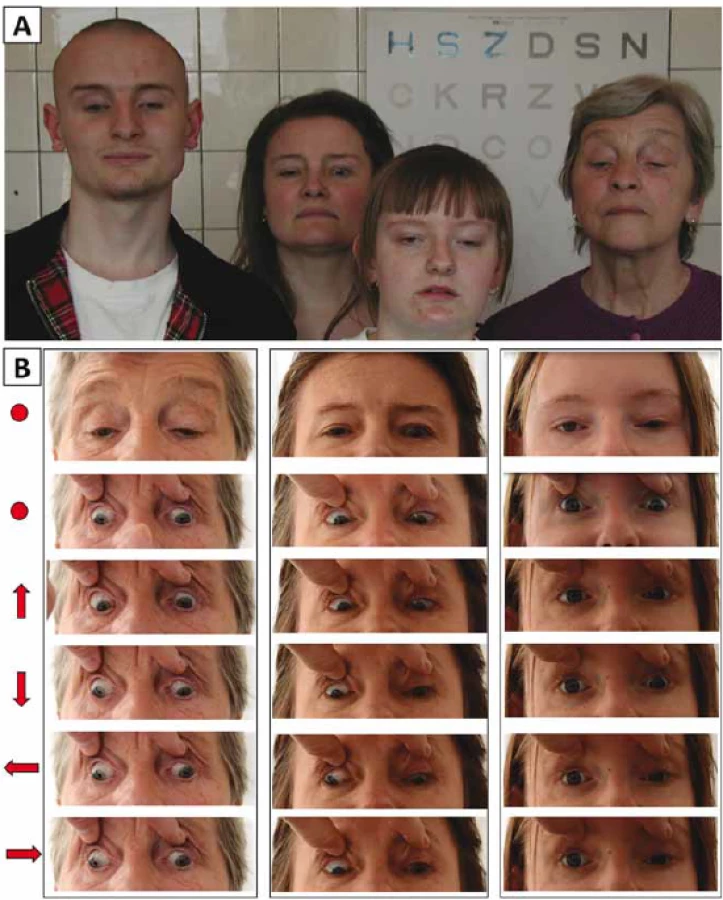
Na první pohled nás zaujalo u všech čtyř rodinných příslušníků vynucené držení hlavy se zvednutou bradou (tzv. kompenzační postavení se záklonem) (obr. 1A).
Fig. 1. Documentation of eye movement abnormality in 3 members of one family with
congenital fi brosis of the extraocular muscles. (A) Four examined family members looking
straight ahead with a marked compensatory backward tilting of the head (B) Different
gaze positions are indicated by arrows; primary gaze position (with and without
passive lid elevation) is indicated with a dot.
Až na ženu II:3 prodělali všichni vyšetřovaní v dětství operaci ke korekci postavení bulbů (tab. 1). Anamnesticky bylo u muže IV:1 již při narození pravé oko stočeno nazálně do té míry, že nebyla vidět rohovka. I přes chirurgické zákroky byl u všech postižených jedinců vizus snížen na podkladě amblyopie vzniklé v souvislosti s abnormálním postavením bulbů. U ženy IV:2 navíc došlo ke snížení zrakové ostrosti pravděpodobně i v souvislosti s oboustrannou těžkou myopií a astigmatizmem, brýlová korekce na jejím pravém oku činila –9,5 D = –1,0 cyl ax 41° a na levém oku –11,5 D = –1,25 cyl ax 102°. U ostatních tří příslušníků nebyly refrakční vady závažné (tab. 1), nicméně je třeba dodat, že přesnější změření pomocí autorefraktometru bylo nespolehlivé pro nemožnost pohledu vpřed.
Tři postižení jedinci měli oboustrannou ptózu víček, z toho dva asymetrickou. U probandky (III:3; obr. 2A) byla ptóza přítomna pouze na oku pravém, vlevo byla naopak zjištěna retrakce víčka. V souladu s diagnózou CFEOM bylo u všech vyšetřených jedinců pozorováno omezení pohybu očí ve vertikále i horizontále. Dále byla přítomna paradoxní synkinéza obou, event. pouze slabšího oka projevující se jako konvergence při pokusu o pohled vzhůru a divergencí při pohledu dolů s výjimkou muže IV:1 (obr. 1B, tab. 1). Je třeba ale zdůraznit, že tyto nálezy jsou ovlivněny i opakovanými operacemi ke korekci šilhání.
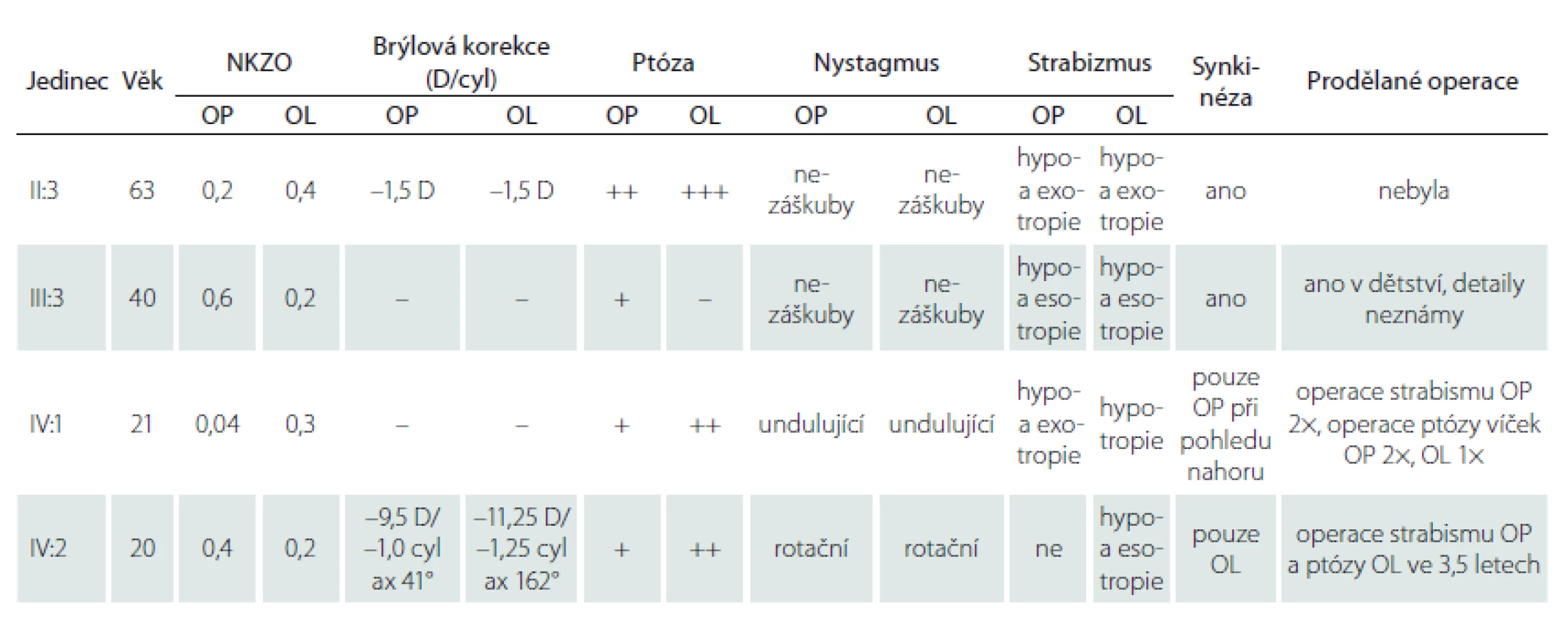
ax – axis; cyl – cylindr; D – dioptrie; NKZO – nejlepší korigovaná zraková ostrost; OL – oko levé; OP – oko pravé
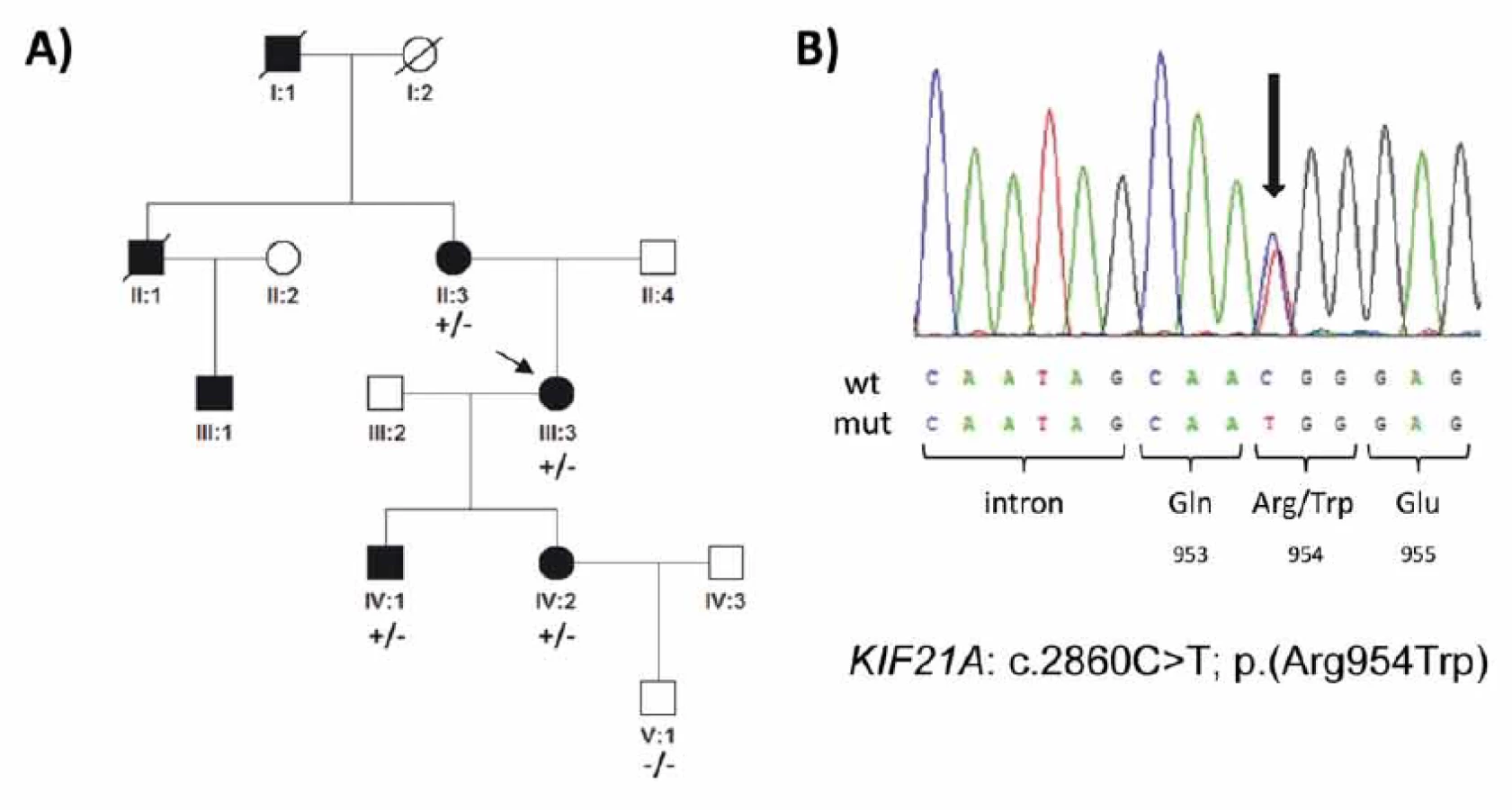
Fig. 2. Genetic fi ndings in a family with congenital fibrosis of the extraocular muscles. (A) Pedigree of the family and segregation of the
heterozygous mutation c.2860C>T. The proband is indicated by an arrow. (B) Sequence chromatogram; the mutation (arrow) causes
an arginine to tryptophan amino acid substitution at codon 954. Aff ected females are represented by black circles and males by black
squares; mutation status in tested subjects is shown +/− for those who are heterozygous for the identifi ed mutation and −/− for those
who do not carry the pathogenic variant.
Základní klinické oční nálezy všech čtyř vyšetřených členů rodiny jsou shrnuty v tab. 1, rodokmen ukazuje obr. 2A.
U tří členů rodiny (II:3, IV:1, IV:2; obr. 2A) nebylo anamnesticky přítomno žádné další závažné onemocnění. 41letá probandka (III:3; obr. 2A) měla vrozenou srdeční vadu – bikuspidální aortální chlopeň s rozvojem významné aortální stenózy a dilatací ascendentní aorty. Ve věku 38 let u ní byla pro symptomatologii anginy pectoris III. stupně (podle Kanadské kardiologické společnosti) nutná náhrada aortální chlopně mechanickou protézou a ascendentní aorty cévní protézou. U dcery (IV:2; obr. 2A) byl kardiologický nález v normě, vč. echokardiografie, žena trpěla ale lehkou smíšenou nedoslýchavostí vlevo. Matka (II:3; obr. 2A) a syn probandky (IV:1; obr. 2A) kardiologem vyšetřeni nebyli, nicméně ve věku 63 a 21 let nevykazovali žádné známky závažnější poruchy, která by byla velmi pravděpodobně i s ohledem na rodinnou anamnézu zachycena. U ženy IV:2, naposledy vyšetřené ve věku 21 let, byla dále zjištěna insuficience kardie, mírná neocerebelární symptomatika bilaterálně a hraniční hyperreflexivita dolních končetin. Všechny tyto nálezy ale byly vyhodnoceny jako náhodná koincidence s diagnózou CFEOM, neboť v literatuře nejsou u pacientů s tímto onemocněním popisovány.
V genu KIF21A byla v exonu 21 zjištěna u probandky (III:3; obr. 2A) v heterozygotním stavu mutace c.2860C>T; p.(Arg954Trp) (obr. 2B). Tato mutace není přítomna v databázi v gnomAD s daty od více než 120 000 zdravých jedinců a v minulosti byla popsána jako nejčastější příčina CFEOM [13]. Stejnou mutaci jsme našli dále u postiženého syna (IV:1), dcery (VI:2) a u matky probandky (II:3), mutace nebyla nalezena u nepostiženého vnuka probandky (V:1; obr. 2A). Další rodinní příslušníci neprojevili o genetické testování zájem.
Diskuze
V rámci této práce jsme u pacientů českého původu s onemocněním CFEOM poprvé provedli molekulárně genetické testování a detekci kauzální mutace. Stejně jako v námi studované rodině, zvláště pokud se CFEOM vyskytuje u většího počtu rodinných příslušníků, bývá klinický obraz poruchy natolik charakteristický, že stanovení diagnózy by nemělo představovat větší obtíže. Detekcí kauzální mutace byla diagnóza definitivně potvrzena.
U všech vyšetřených dominovala na první pohled zřejmá ptóza a kompenzační postavení hlavy. Onemocnění se v rodině přenášelo zcela jednoznačně autozomálně dominantně. Nález byl natolik charakteristický, že jsme v zájmu pacientů neprovedli ani EMG ani test pasivní dukce, při kterém by došlo k traumatickému postižení spojivky. Všichni pacienti vykazovali snížení zrakové ostrosti, které bylo přičítáno amblyopii. Zajímavý byl nález těžké myopie a astigmatizmu u 20leté ženy. Závažnější refrakční vady nebývají s tímto onemocněním běžně spojovány [1]. U jedné ženy se vyskytovala i řada dalších systémových nálezů vč. závažné srdeční vady. V literatuře lze najít zmínku o více než 25 rodinách CFEOM1 na podkladě stejné mutace, jako byla pozorována v námi studované rodině. Souvislost s kardiologickým nálezem nebyla u členů s tímto onemocněním popsána [12,22,23]. Z tohoto důvodu se domníváme, že u probandky se jedná spíše o náhodný nález než o variabilně asociovaný znak CFEOM1.
Přestože je klinický obraz onemocnění velmi typický, vzhledem k tomu, že se jedná o vzácné onemocnění, může činit diferenciální diagnostika v běžné praxi obtíže. Uvažovat lze o jiných onemocnění manifestujících se jako chronická progresivní zevní oftalmoplegie, buď na podkladě mitochondriálního onemocnění nebo v rámci autozomálně dominantní okulofaryngeální dystrofie. Podobný klinický obraz může vzniknout i u endokrinní orbitopatie nebo u myastenie gravis. Hlavním diferenciálně diagnostickým znakem CFEOM je kongenitální manifestace, která se u ostatních klinických jednotek nevyskytuje, a přestože některé znaky nemusí být vyjádřeny plně, podstatné je, že se po celý život nemění. Ptóza víček a porucha hybnosti, hlavně vertikální, je patrná ihned po narození, detailnější posouzení hybnosti, přítomnost synkinéz a kompenzační držení hlavy lze posoudit až po ukončení senzomotorického vývoje, tj. od 6. měsíce věku [5]. Napomoci může i rodinná anamnéza, pokud lze v rodokmenu vystopovat přenos z otce na syna, je mitochondriální etiologie vyloučena.
Mutace c.2860C>T v heterozygotním stavu byla popsána i jako de novo vzniklá [24]. V tomto případě je pak pacient v rodině jediný postižený, bez pozitivní rodinné anamnézy pro danou chorobu. Mutace se však na další generace přenáší s 50% pravděpodobností.
V české literatuře jsme našli popis klinických nálezů onemocnění CFEOM u 9 pacientů z 3 rodin [5]. Vzhledem k anamnestickým údajům nejstarší členky námi vyšetřené rodiny je pravděpodobné, že ona a její otec byly do této kohorty před několika desítkami let také zařazeni.
Kongenitální fibróza zevních okohybných svalů se řadí do skupiny vrozených onemocnění s primární poruchou inervace okohybných svalů. Již od 19. století se vedly spory o to, zda u těchto poruch je primární příčina neurogenní nebo myogenní. Neurogenní etiologii podporovaly různé synkinézy, pro myogenní svědčily histologicky prokázané fibrózy některých svalů [25,26]. Dnes je jasné, že primární je porucha inervace některých svalů. Soudí se, že u CFEOM dochází k abnormalitám růstu axonů a jejich správného směřování do cílového svalu.
Protein KIF21A je během vývoje i po jeho dokončení exprimován v řadě tkání, nejvíce pak v nervovém systému [27]. Mutace c.2860C>T v exonu 21 genu KIF21A, kterou jsme prokázali v heterozygotním stavu, byla opakovaně u pacientů s CFEOM1 z různých populací popsána, odhaduje se, že jejími nositeli je až 70 % pacientů s tímto onemocněním [17,18]. Tato patogenní varianta, stejně jako další popsané, narušuje tvorbu homodimérů a heterodimerů s proteinem KIF21B a tím autoinhibiční mechanizmus mezi motorickou doménou a regulační oblastí KIF21A, což následně vede k její hyperaktivaci [28].
Selektivita postižení u CFEOM1 nebyla dosud uspokojivě vysvětlena. Soudí se, že u všech typů CFEOM je postižen proces modulace buněčné signalizace, dynamiky cytoskeletu. Příčina selektivní vulnerability pouze některých nervů se vysvětluje rozdíly v neuronálních populacích [29].
Léčba CFEOM je chirurgická, nicméně vzhledem k těžkým vazivovým změnám okohybných svalů a jejich anomální inervaci nebývá příliš úspěšná. Rodiče postižených dětí i pacienti samotní si především přejí zmírnění vynuceného držení hlavy pro ptózu a hypotropii očí. Pacienti z námi vyšetřené rodiny také prodělali v minulosti několik zákroků zaměřených na korekci ptózy a hypotropie bez výraznějšího efektu, jak bylo zjištěno při našem vyšetření a také dle subjektivního hodnocení pacientů. Výsledný stav operovaných příslušníků se příliš nelišil od matky probandky, která žádné operace neprodělala. Vedle těžkých vazivových změn okohybných svalů bývá úskalím výkonu riziko expoziční keratitidy či vředu z lagoftalmu při chybějícím Bellově fenoménu. Vzhledem k variabilitě postižení jednotlivých svalů vyžaduje indikace a výběr vhodného chirurgického výkonu vysloveně individuální přístup [5,30]. Pacient by měl být odeslán ke zkušenému oftalmologovi, který posoudí možnosti jak konzervativní (předpis brýlové korekce), tak i chirurgické léčby (operace ptózy víček, úprava strabizmu).
K pacientům s CFEOM diagnózou je tedy třeba přistupovat individuálně, ať již z hlediska možné chirurgické léčby, tak i v rámci genetického poradenství. Protože onemocnění působí často celoživotní funkční, psychické a společenského potíže, lze v případě stanovení molekulárně genetické příčiny zvážit i preimplantační nebo prenatální diagnostiku. V literatuře byla již preimplantační diagnostika z této indikace dokumentována [31,32].
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Petra Lišková, MD., Ph.D.
Klinika dětského a dorostového lékařství
a Oční klinika 1. LF UK a VFN v Praze
Ke Karlovu 2
128 00 Praha
e-mail: petra.liskova@lf1.cuni.cz
Přijato k recenzi: 24. 4. 2019
Přijato do tisku: 13. 8. 2019
Tato práce vznikla za podpory projektů Univerzity Karlovy UNCE 204064, PROGRES Q26 a SVV 260367/ 2017.
Sources
1. Sener EC, Lee BA, Turgut B et al. A clinically variant fibrosis syndrome in a Turkish family maps to the CFEOM1 locus on chromosome 12. Arch Ophthalmol 2000; 118(8): 1090–1097. doi: 10.1001/ archopht.118.8.1090.
2. Engle EC, Kunkel LM, Specht LA et al. Mapping a gene for congenital fibrosis of the extraocular muscles to the centromeric region of chromosome 12. Nat Genet 1994; 7(1): 69–73. doi: 10.1038/ ng0594-69.
3. Lim KH, Engle EC, Demer JL. Abnormalities of the oculomotor nerve in congenital fibrosis of the extraocular muscles and congenital oculomotor palsy. Invest Ophthalmol Vis Sci 2007; 48(4): 1601–1606. doi: 10.1167/ iovs.06-0691.
4. Yamada K, Hunter DG, Andrews C et al. A novel KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn jaw-winking phenomenon. Arch Ophthalmol 2005; 123(9): 1254–1259. doi: 10.1001/ archopht.123.9.1254.
5. Otradovec J. Klinická neurooftalmologie. 1. vyd. Praha: Grada Publishing 2003.
6. Heidary G, Engle EC, Hunter DG. Congenital fibrosis of the extraocular muscles. Semin Ophthalmol 2008; 23(1): 3–8. doi: 10.1080/ 08820530701745181.
7. Engle EC, Goumnerov BC, McKeown CA et al. Oculomotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Ann Neurol 1997; 41(3): 314–325. doi: 10.1002/ ana.410410306.
8. Miao W, Man F, Wu S et al. Brain abnormalities in congenital fibrosis of the extraocular muscles type 1: a multimodal MRI imaging study. PLoS One 2015; 10(7): e0133473. doi: 10.1371/ journal.pone.0133473.
9. Kim JH, Hwang JM. Hypoplastic oculomotor nerve and absent abducens nerve in congenital fibrosis syndrome and synergistic divergence with magnetic resonance imaging. Ophthalmol 2005; 112(4): 728–732. doi: 10.1016/ j.ophtha.2004.12.006.
10. Reck AC, Manners R, Hatchwell E. Phenotypic heterogeneity may occur in congenital fibrosis of the extraocular muscles. Br J Ophthalmol 1998; 82(6): 676–679. doi: 10.1136/ bjo.82.6.676.
11. Whitman M, Hunter DG, Engle EC. Congenital fibrosis of the extraocular muscles. In: Adam MP, Ardinger HH, Pagon RA et al (eds.). GeneReviews. Seattle: University of Washington 1993.
12. Yamada K, Andrews C, Chan WM et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet 2003; 35 : 318–321.
13. Nakano M, Yamada K, Fain J et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet 2001; 29(3): 315–320. doi: 10.1038/ ng744.
14. Tischfield MA, Baris HN, Wu C et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010; 140(1): 74–87. doi: 10.1016/ j.cell.2009.12.011
15. Aubourg P, Krahn M, Bernard R et al. Assignment of a new congenital fibrosis of extraocular muscles type 3 (CFEOM3) locus, FEOM4, based on a balanced translocation t(2;13) (q37.3;q12.11) and identification of candidate genes. J Med Genet 2005; 42(3): 253–259. doi: 10.1136/ jmg.2004.021899.
16. Tukel T, Uzumcu A, Gezer A et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. J Med Genet 2005; 42(5): 408–415. doi: 10.1136/ jmg.2004.026138.
17. Khan AO, Shinwari J, Omar A et al. Lack of KIF21A mutations in congenital fibrosis of the extraocular muscles type I patients from consanguineous Saudi Arabian families. Mol Vis 2011; 17 : 218–224.
18. Chan WM, Andrews C, Dragan L et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet 2007; 8 : 26. doi: 10.1186/ 1471-2156-8-26.
19. Ali Z, Xing C, Anwar D et al. A novel de novo KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Mobius syndrome. Mol Vis 2014; 20 : 368–375.
20. gnomAD browser. [online]. Available from URL: https:/ / gnomad.broadinstitute.org/ .
21. Národní centrum lékařské genomiky. [online]. Dostupné z URL: https:/ / ncmg.cz/ .
22. Rudolph G, Nentwich M, Hellebrand H et al. KIF21A variant R954W in familial or sporadic cases of CFEOM1. Eur J Ophthalmol 2009; 19(4): 667–674.
23. Li ND, Zhao J, Wang LM et al. R954 mutations in KIF21A gene in Chinese patients with congenital fibrosis of extraocular muscles. Zhonghua Yan Ke Za Zhi 2012; 48(12): 1077–1082.
24. Yang X, Yamada K, Katz B et al. KIF21A mutations in two Chinese families with congenital fibrosis of the extraocular muscles (CFEOM). Mol Vis 2010; 16 : 2062–2070.
25. Harley RD, Rodrigues MM, Crawford JS. Congenital fibrosis of the extraocular muscles. Trans Am Ophthalmol Soc 1978; 76 : 197–226.
26. Sudiwala S, Knox SM. The emerging role of cranial nerves in shaping craniofacial development. Genesis 2019; 57(1): e23282. doi: 10.1002/ dvg.23282.
27. Desai J, Velo MP, Yamada K et al. Spatiotemporal expression pattern of KIF21A during normal embryonic development and in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Gene Expr Patterns 2012; 12(5–6): 180–188. doi: 10.1016/ j.gep.2012.03.003.
28. Bianchi S, van Riel WE, Kraatz SH et al. Structural basis for misregulation of kinesin KIF21A autoinhibition by CFEOM1 disease mutations. Sci Rep 2016; 6 : 30668. doi: 10.1038/ srep30668.
29. Whitman MC, Engle EC. Ocular congenital cranial dysinnervation disorders (CCDDs): insights into axon growth and guidance. Hum Mol Genet 2017; 26(R1): R37–R44. doi: 10.1093/ hmg/ ddx168.
30. Magli A, de Berardinis T, D’Esposito F et al. Clinical and surgical data of affected members of a classic CFEOM I family. BMC Ophthalmol 2003; 3 : 6.
31. Hlavata L, Dudakova L, Trkova M et al. [Preimplantation genetic diagnosis and monogenic inherited eye diseases]. Cesk Slov Oftalmol 2016; 72(5): 167–171.
32. De Rycke M, Belva F, Goossens V et al. ESHRE PGD Consortium data collection XIII: cycles from January to December 2010 with pregnancy follow-up to October 2011. Hum Reprod 2015; 30(8): 1763–1789. doi: 10.1093/ humrep/ dev122.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 5
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Léčba nespavosti při neuropatické bolesti
- Kompresivní neuropatie jako nemoc z povolání
- Změna struktury paraspinálních svalů u pacientů s chronickými nespecifickými bolestmi dolní části zad
- Endoskopické operace výhřezu bederních meziobratlových plotének – první zkušenosti