
CANVAS – nově identifikovaná genetická příčina ataxie s pozdním nástupem. Popis prvních diagnostikovaných pacientů v České republice
CANVAS – a newly identified genetic cause of late-onset ataxia. Description of the first cases in the Czech Republic
Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) is an autosomal recessive neurodegenerative disease from hereditary ataxias with a late onset that affects the cerebellum, sensory nerves, and the vestibular system. Its genetic cause was discovered in 2019 and since 2020, diagnostic molecular genetic testing has also been available. According to the existing literature, it seems that CANVAS is a major cause of late onset hereditary ataxia. Out of the first nine examined patients with the suspected disease, we confirmed four cases of CANVAS; three of the patients are described in detail in this manuscript.
Keywords:
Cerebellar ataxia – neuropathy – vestibulopathy – hereditary ataxias – CANVAS
Authors:
M. Danková 1; Z. Mušová 2; J. Jeřábek 1; J. Paulasová; Schwabová 1,3; A. Zumrová 3; E. Vyhnálková 2; S. Skalská 4; R. Mazanec 1; M. Vyhnálek 1
Authors‘ workplace:
Centrum hereditárních ataxií, Neurologická, klinika 2. LF UK a FN Motol, Praha
1; Centrum hereditárních ataxií, Ústav, biologie a lékařské genetiky, 2. LF UK a FN Motol, Praha
2; Centrum hereditárních ataxií, Klinika, dětské neurologie 2. LF UK, a FN Motol, Praha
3; Neurologická klinika 1. LF UK a VFN, v Praze, Praha
4
Published in:
Cesk Slov Neurol N 2021; 84/117(4): 397-402
Category:
From Clinical Praxis
doi:
https://doi.org/10.48095/cccsnn2021397
Overview
Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) je autozomálně recesivní neurodegenerativní onemocnění z okruhu hereditárních ataxií s pozdním začátkem postihující mozeček, senzitivní nervy a vestibulární systém. Jeho genetická příčina byla odhalena v roce 2019 a od roku 2020 je dostupné také diagnostické molekulárně genetické vyšetření. Dle dosavadní literatury se ukazuje, že CANVAS je nejčastější příčinou hereditární ataxie s pozdním nástupem. Ve skupině prvních devíti vyšetřených pacientů s podezřením na toto onemocnění jsme zachytili čtyři případy CANVAS, tři pacienty podrobně popisujeme v této práci.
Klíčová slova:
cerebelární ataxie – neuropatie – vestibulopatie – hereditární ataxie – CANVAS
Úvod
Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) je autozomálně recesivní neurodegenerativní onemocnění z okruhu hereditárních ataxií s pozdním začátkem postihující mozeček, senzitivní nervy a vestibulární systém [1].
I když se CANVAS zdá být významnou příčinou ataxie s pozdním nástupem, byla jeho genetická podstata odhalena až v roce 2019.
Molekulárně genetickým podkladem je expanze repetitivní sekvence AAGGG na obou alelách v intronu 2 genu RFC1 (replication factor complex subunit 1) na chromozomu 4p [2]. Zajímavostí je, že se u normálních i patologických alel objevují i polymorfní repetice (tj. variace). Heterozygotní přenašeči jsou zdrávi. Při autozomálně recesivní dědičnosti je související reprodukční riziko pro pár přenašečů 25 % pro rozvoj onemocnění u každého dítěte bez rozdílu pohlaví.
Zpočátku bylo onemocnění popsáno jako klinický syndrom charakterizovaný postupně progredujícím mozečkovým syndromem, senzitivní axonální neuropatií a bilaterální vestibulopatií [3–7]. V návazné práci se ukázalo, že CANVAS je v rámci dědičných ataxií onemocněním častým, dle některých autorů může být příčinou až u 22 % případů cerebelárních ataxií s pozdním začátkem (late-onset) vedených jako sporadické [2], s odhadovanou frekvencí heterozygotního nosičství jedné expandované alely AAGGG v evropské populaci u 0,7 % zdravých kontrol [2]. Odhadovaná prevalence onemocnění je 1 na 20 000 [8]. Pro srovnání odhadovaná frekvence FXTAS (syndrom tremoru/ataxie asociovaného s fragilním X chromozomem) je v mužské populaci 1 na 4 900, nicméně dominujícím klinickým příznakem tohoto onemocnění je třes a jen část pacientů má přítomnou ataxii [9]. V roce 2020 byl publikován mezinárodní soubor, v rámci kterého bylo cíleně geneticky vyšetřeno 363 pacientů a u 105 z nich byl CANVAS potvrzen [8].
Kromě výše zmíněné trias je zajímavá přítomnost chronického kašle, který byl zjištěn u více než třetiny pacientů s CANVAS a předchází ostatní příznaky až o několik desítek let. Předpokládá se, že podkladem je postižení neuronálních okruhů ovládajících kašlací reflex [2].
V molekulárně genetické laboratoři Ústavu biologie a lékařské genetiky FN Motol a v Centru hereditárních ataxií FN Motol, které je součástí Evropské referenční sítě pro vzácná a neurologická onemocnění (ERN-RND), jsme v červnu 2020 v rámci pilotní studie vyšetřili přítomnost expanze (AAGGG) n v genu RFC1 u prvních devíti českých pacientů a potvrdili mutaci u čtyř z nich.
V této práci podrobně prezentujeme klinické projevy u tří z potvrzených případů CANVAS a princip molekulárně genetické diagnostiky tohoto onemocnění. Znalost klinického průběhu může mít podstatný význam pro běžnou klinickou praxi neurologa vzhledem k předpokládanému vysokému výskytu tohoto syndromu mezi pacienty s pozdním nástupem ataxie.
Soubor
Ke genetickému vyšetření bylo z Centra pro hereditární ataxie FN Motol indikováno celkem sedm pacientů s klinickým podezřením na CANVAS. Dva z nich (dále pacientka 1 a pacient 2) splňovali kompletní kritéria CANVAS (senzorická neuropatie, mozečkový syndrom a vestibulární areflexie) a u obou se onemocnění prokázalo, další dva pacienti vykazovali vestibulární hyporeflexii/areflexii v kombinaci s ataxií a tři pacienti měli izolovanou vestibulární areflexii nejasné etiologie. Všichni byli vyšetřeni klinicky, absolvovali vestibulární vyšetření, vyšetření EMG a zobrazovací vyšetření mozku.
Třetí potvrzená pacientka (dále pacientka 3) byla odeslána na oddělení klinické genetiky FN Motol z neuromuskulární ambulance Neurologické kliniky VFN k dalšímu vyšetření pro senzitivní neuropatii nejasné etiologie a vzhledem ke klinicky dominující cerebelární ataxii byla neurogenetikem indikována k vyšetření CANVAS. Čtvrtý potvrzený pacient byl k vyšetření indikován genetiky z jiného pracoviště na základě cíleného podezření na CANVAS, podrobná klinická data nemáme v tuto chvíli k dispozici.
Molekulárně genetické vyšetření
Molekulárně genetické stanovení diagnózy CANVAS u pacienta umožňuje nález bialelické expanze (AAGGG) n v genu RFC1 pomocí repeat primed polymerase chain reaction (RP-PCR). Tato amplifikační reakce využívá trojici primerů: první primer se váže do různých míst repetice AAGGG a na svém konci nese úsek nekomplementární k lidské genomové DNA, kterým naváže druhý primer. Fluorescenčně značený třetí primer se váže vně repetice. Po hybridizaci prvního primeru do repetice AAGGG probíhá amplifikace pomocí druhého a třetího primeru ve specifických podmínkách [2]. RP-PCR vede ke vzniku různě dlouhých fragmentů, které se liší o délku jedné AAGGG repetice (5 nukleotidů), přičemž preferenčně se amplifikují kratší fragmenty. Reakce je vyhodnocena kapilární elektroforézou s fluorescenční detekcí fragmentů. Expanze vytváří charakteristický profil amplikonů, jejich množství s narůstající délkou postupně klesá. Metoda neurčí délku expanze, jen odhalí její přítomnost. Délku normálních alel přesně určí standardní PCR s použitím dvou primerů (jeden je fluorescenčně značený) umístěných vně repetice. Detekce produktů a přesné určení jejich délky probíhá v kapilární elektroforéze. U jedinců s bialelickou expanzí (AAGGG) n nejsou normální alely přítomny. Diagnostiku může komplikovat polymorfismus v délce normálních alel a dále existence různých nukleotidů uvnitř repetice jak u normálních, tak u patologických alel – u normálních alel: nejčastěji (AAAAG) 11, dále (AAAAG) 12–200 a (AAAGG) 40–100; u patologických expanzí: nejčastěji (AAGGG) n, vzácně (ACAGG) n či (AAAGG) 10–25 (AAGGG) exp (AAAGG) 4–6 [10]. Proto je důležité expanzi (AAGGG) n zjištěnou u pacienta ověřit u jeho relevantních příbuzných (rodiče, potomstvo).
Výsledky
Z devíti dosud molekulárně geneticky vyšetřených pacientů jsme potvrdili bialelickou expanzi repetitivní sekvence v genu RFC1 u čtyř z nich. Jednalo se o dva pacienty s kompletně vyjádřeným syndromem CANVAS (pacientka 1 a pacient 2) a dále pacientku s dominující ataktickou neuropatií (pacientka 3), u čtvrtého potvrzeného pacienta nejsou bližší klinická data k dispozici. Jde o první případy geneticky verifikovaného onemocnění CANVAS v ČR.
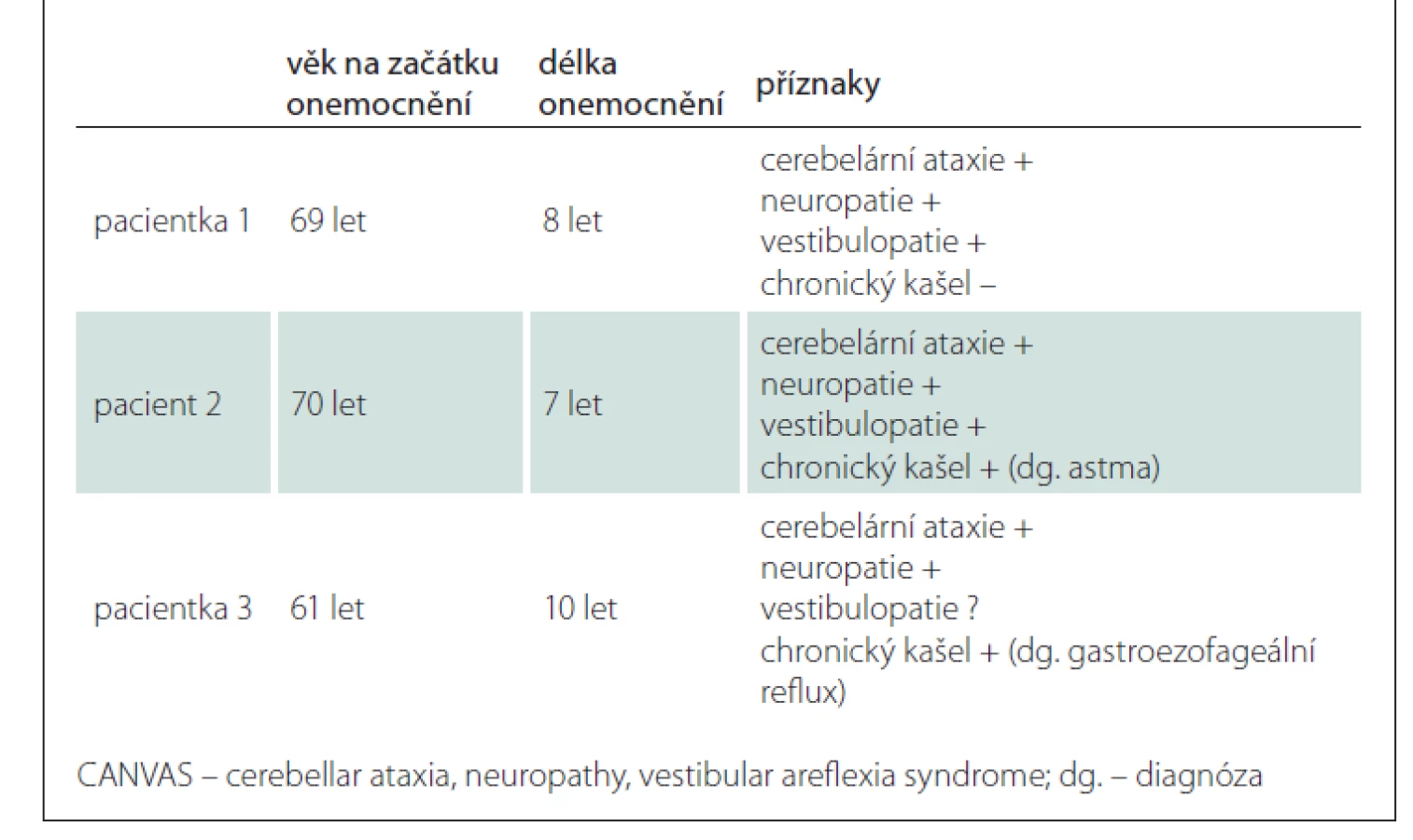
Pacientka 1
Pacientka (77 let) byla sledována pro chronickou antrální gastritidu, eufunkční strumu, v neurologické péči dispenzarizovaná pro migrénu a periodické pohyby ve spánku. Chronický kašel negovala, onkologicky se neléčila. Rodinná anamnéza stran neurologických onemocnění byla negativní (otec zemřel na komplikace spojené s karcinomem tlustého střeva v 63 letech, matka v 80 letech „na stáří“, potíže s hybností či rovnováhou neměli, sourozence pacientka nemá, jeden syn ročník 1975 je zdráv).
Podle ambulantního neurologa byla prvním příznakem porucha rovnováhy trvající od roku 2013 (69 let), již od věku 66 let však byly přítomny parestézie dolních končetin (DK). Hlavní stížností pacientky byla zhoršená stabilita chůze, zejména ve tmě. Do roku 2018 zvládala chůzi bez opory, nyní využívá při chůzi trekingové hole nebo lehkou oporu druhé osoby. Při chůzi či jiném pohybu udávala oscilopsie, rozmazání obrazu. Pacientku dále obtěžovaly parestézie, pocit chladu DK a „těžkých“ nohou, tyto obtíže byly zmírněny po nasazení pregabalinu.
Při prvním vyšetření v Centru hereditárních ataxií v roce 2015 (ve věku 71 let) byly patrné velmi lehký neocerebelární syndrom (lehká dystaxie a jemný intenční tremor horních končetin [HK]), lehká posturální instabilita (neschopnost stoje na jedné noze, obtíže s tandemovou chůzí, bez pádů), areflexie L2/S2 oboustranně, známky oboustranné vestibulopatie (pozitivní Head Impulse Test). Neurologický nález za posledních 5 let lehce progredoval – objevila se mírná dysartrie, zhoršila se stabilita stoje a chůze (stoj spojný s titubacemi, v Rombergově zkoušce tendence k pádu, chůze o širší bázi, otočky rozfázované), ataxie hodnocená škálou SARA (Scale for the Assessment and Rating of Ataxia) [11] byla 8,5/40 (mírná ataxie).
Vzhledem k anamnéze bylo pomýšleno na pomalu progredující neurodegenerativní onemocnění. Na MR mozku byly patrny pouze ojedinělé gliové změny frontálně bilaterálně, bez patrné atrofie mozečku. Elektromyografie (EMG) prokázala těžkou axonální senzitivní neuronopatii, bez postižení motorických vláken. Vyšetření elektronystagmografické odhalilo vestibulo-cerebelární postižení (dysrytmii optokinetického nystagmu, poruchu vizuo-vestibulární interakce) a těžkou periferní vestibulární kalorickou hyporeflexii potvrzenou video Head Impulse Testem a testem dynamické zrakové ostrosti. Posturograficky jsme verifikovali 3 Hz tremor, typický pro postižení mozečku a jeho drah. Laboratorní výsledky vč. skríningu zahrnujícího hormony štítné žlázy, vitamín B12, kyselinu listovou, vitamín E a paraneoplastické protilátky byly negativní.
Molekulárně-genetické vyšetření spinocerebelárních ataxií (SCA) typu 1, 2, 3, 6, 7, 8, 12, 17, dentato-rubro-palidoluysiánská atrofie (DRPLA), SCA28 (gen AFG3L2) a Friedreichova ataxie (FRDA; gen FXN) bylo s negativním výsledkem. Vzhledem k naplněným klinickým kritériím pro CANVAS (kombinace senzitivní neuropatie, mozečkové ataxie a vestibulární areflexie) jsme doplnili vyšetření genu RFC1, které prokázalo přítomnost expanze repetitivní sekvence (AAGGG) n bez přítomnosti normální alely.
Pacient 2
Pacient narozen v roce 1941, léčený pro arteriální hypertenzi s aterosklerózou (vč. karotid) a astma bronchiale s kašlem. Bratr pacienta trpěl obdobnou pomalu progredující poruchou rovnováhy od 65 let a zemřel ve věku 80 let. Pacient udával, že určité poruchy rovnováhy měla ve stáří i jeho matka, která zemřela také v 80 letech. Rodiče matky a ostatní sourozenci pacienta byli zdrávi. Má dva syny a dceru, kteří jsou zdrávi.
Poruchu rovnováhy pacient vnímá od 70 let. Od začátku onemocnění popisuje chůzi charakteru opilosti zhoršující se ve tmě, při pohybu udává mírné oscilopsie, nyní po 8 letech trvání obtíží však chodí bez opory. Obtíže charakteru zhoršení citlivosti končetin či parestezií neguje.
V neurologickém nálezu v roce 2016 byly patrné lehká dysartrie, oboustranný pohledový nystagmus, oboustranně pozitivní Head Impulse Test, dystaxie s převahou na DK, areflexie L2/S2 bilaterálně, porucha vibračního čití na DK, lehká posturální instabilita s titubacemi ve stoji spojném a v Rombergově zkoušce. Během tří let se zvýraznily pohledový nystagmus a posturální instabilita, stoj a chůze byly s rozšířenou bází, bez opory, v Rombergově zkoušce výrazná instabilita, při posledním klinickém vyšetření v 2019 byla hodnota škály SARA 11/40.
Vzhledem k průběhu onemocnění a rodinné anamnéze bylo pomýšleno na dědičné pomalu progredující neurodegenerativní onemocnění. Na MR mozku byly patrné nespecifické gliové změny supratentoriálně v bílé hmotě, lehká difuzní atrofie mozku, bez výraznější akcentace v oblasti zadní jámy. EMG vyšetření prokázalo chybění senzitivních akčních potenciálů při zachovaných sumačních svalových akčních potenciálech na HK i DK. Nález byl hodnocen jako senzitivní ganglionopatie. Elektronystagmografie odhalila narušení sledovacích očních pohybů a optokinetického nystagmu, dále pak těžkou oboustrannou vestibulární kalorickou hyporeflexii až areflexii potvrzenou video Head Impulse Testem a testem dynamické zrakové ostrosti. Posturograficky nález odpovídal poruše vestibulocerebelární, bez přítomnosti 3 Hz tremoru. Vyšetření hormonů štítné žlázy, vitamínu B12, kyseliny listové, vitamínu E byla negativní, vyšetření mozkomíšního moku bylo s normálním nálezem.
Molekulárně-genetické vyšetření SCA typu 1, 2, 3, 6, 7, 8, 12, 17, DRPLA, SCA28 a FRDA (gen FXN) bylo s negativním výsledkem. Pacient klinicky splňoval všechna kritéria pro syndrom CANVAS, proto jsme doplnili vyšetření genu RFC1, které prokázalo přítomnost expanze repetitivní sekvence (AAGGG) n na obou alelách genu.
Pacientka 3
Narozena v roce 1946, chronicky sledována pro arteriální hypertenzi a dyslipidemii, chronickou thyroiditidu, depresivní syndrom, horní dyspeptický syndrom s refluxem a kašlem. Pacientka byla po excizi bazaliomu. Matka pacientky zemřela v 76 letech v důsledku gangrény, otec měl poruchu rovnováhy a zemřel v 82 letech na cévní mozkovou příhodu. Sestra pacientky trpěla Ménièrovou chorobou, zemřela v 86 letech, bratr pacientky narozen v 1939 je zdráv. Pacientka má dvě zdravé dcery ve věku 47 a 50 let.
Pacientka byla vyšetřena v neuromuskulární ambulanci Neurologické kliniky 1. LF VFN na doporučení ambulantního neurologa pro postupně vzniklou a pomalu progredující poruchu rovnováhy od 61 let. Rovnováha se výrazně horší ve tmě a večer, kdy udává až pocit opilosti, přes den jsou obtíže mírnější. Oscilopsie neguje. Až do 73 let chodila bez opory, od 75 let chodila venku s hůlkou, po bytě sice nadále bez opory, ale již opakovaně upadla. Parestézie, bolesti DK negovala, od 72 let ji obtěžoval pocit „studených“ nohou.
V 71 letech v neurologickém nálezu nebyla pozorována dysartrie ani nystagmus a dominovala senzitivní propriocepční ataxie s těžkou poruchou vibračního čití s maximem akrálně na DK, reflexy C5/8 i L2/S2 byly výbavné oboustranně, byl pozitivní Rombergův příznak, chůze s lehce rozšířenou bází. Během 4 let se zhoršily pallhypestezie – nově přítomny i na HK akrálně, přidaly se lehká dysartrie a jemný pohledový horizontální nystagmus.
Elektromyografické vyšetření v roce 2018 prokázalo nevýbavnost senzitivních neurogramů, motorické neurogramy HK i DK byly v normě. Metabolický skríning vč. hladiny vitaminu B12, kyseliny listové byl v normě, vyloučena byla také neurosyfilitida. MR mozku v roce 2020 odhalila leukoaraiózu (která se také mohla podílet na poruše chůze), středně těžkou mozkovou atrofii a mírnou atrofii vermis mozečku. MR hrudní a lumbosakrální páteře byla bez známek útlaku nervových struktur. Vestibulární vyšetření nebylo provedeno.
V 71 letech věku proběhlo cílené molekulárně-genetické vyšetření Friedreichovy ataxie (gen FXN). Vzhledem k výrazné progresi poruchy rovnováhy bylo v 72 letech věku doplněno i vyšetření tzv. SCA panelu (SCA typu 1, 2, 3, 6, 7, 8, 12, 17, DRPLA a SCA28) s negativním výsledkem. Vzhledem k průběhu onemocnění a EMG nálezu bylo zvažováno onemocnění z okruhu hereditárních neuropatií. Pacientka byla doporučena k dalšímu vyšetření na naše pracoviště, kde na základě kombinace progredující poruchy rovnováhy a senzitivní neuropatie bylo cíleně pomýšleno na CANVAS. Cílené vyšetření genu RFC1 následně prokázalo přítomnost expanze repetitivní sekvence (AAGGG) n na obou alelách genu.
V tab. 1 uvádíme základní údaje a přehled příznaků výše popsaných pacientů.
Diskuze
Představili jsme první tři případy geneticky ověřeného onemocnění CANVAS, které klinicky asociuje cerebelární ataxii, neuropatii a bilaterální vestibulopatii. Jedná se o ataxii/neuropatii s pozdním nástupem a pomalým průběhem.
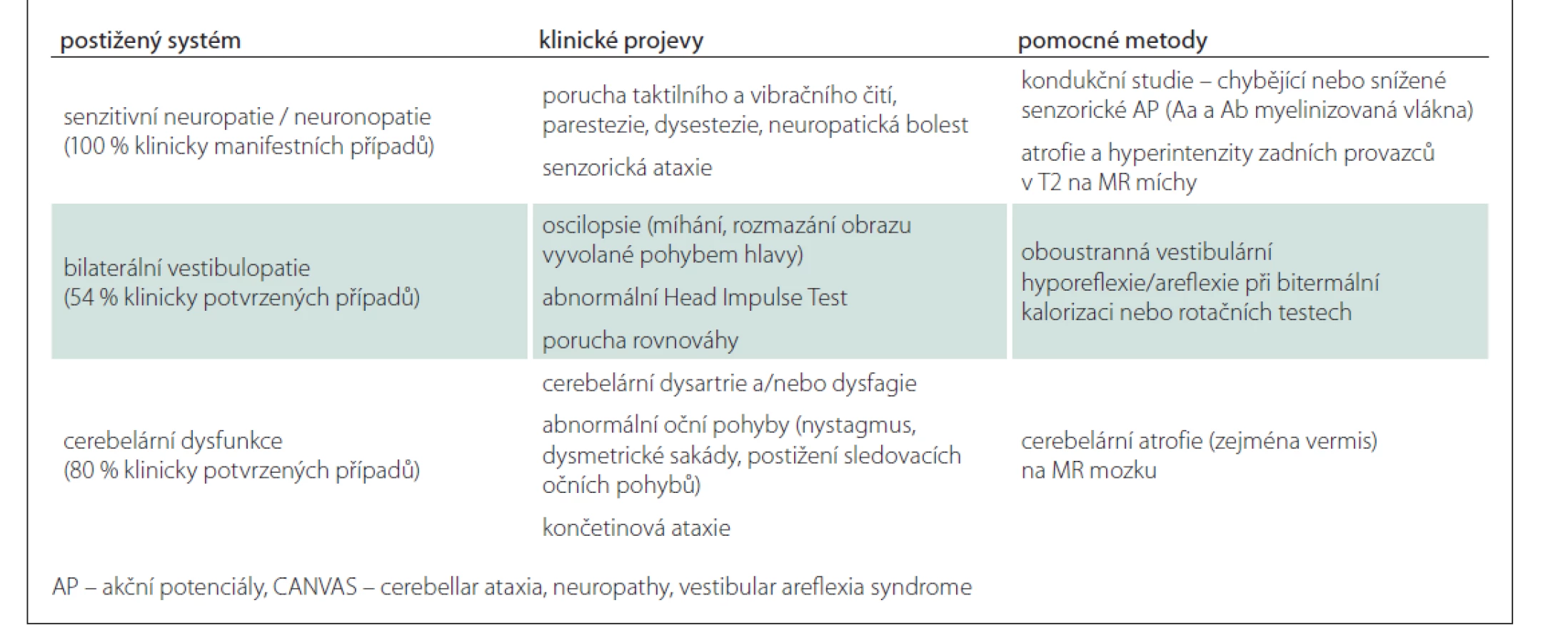
Průměrný věk nástupu onemocnění je 52 let (v rozpětí 19–76 let) – nejdříve se objevují distálně senzorické obtíže a ataxie chůze, až později se mohou přidat oscilopsie, případně dysartrie, dysfagie. Senzorická neuropatie byla popsána u všech pacientů, známky vestibulární dysfunkce u více než 80 % z nich (asi třetina však bývá asymptomatická), 60 % pacientů má i mozečkový syndrom. Izolované mozečkové nebo vestibulární varianty nebyly zatím pozorovány. Na rozdíl od ostatních ataxií není u CANVAS přítomen parkinsonský či pyramidový syndrom ani kognitivní deficit [8].
Z anamnézy našich pacientů i literatury vyplývá, že mozečkový syndrom je často předcházen příznaky senzorické neuropatie, u části pacientů je pak neuropatie hlavním a dominujícím příznakem a ataxie nemusí být přítomna vůbec. Možné jsou i čistě neuropatické formy. Jak je zmíněno výše, výskyt izolovaného vestibulárního či mozečkového syndromu bez neuropatie nebyl dosud popsán [8].
Naši pacienti měli začátek příznaků ve věku 65–70 let, v souladu s publikovanými údaji předcházely ataxii příznaky senzitivní neuropatie (onemocnění postihuje nejdříve senzorické neurony, až později vestibulární systém a mozeček). V souladu s literaturou měli naši pacienti relativně pozvolný průběh, i po 8–14 letech od rozvoje prvních příznaků byli všichni schopni chůze bez opory či s malou oporou. Onemocnění je autozomálně recesivně dědičné, v našem souboru byla u pacienta číslo 2 suspektní pozitivní rodinná anamnéza u bratra, nicméně pacient udával výskyt poruch rovnováhy ve stáří i u jeho matky. Vzhledem ke způsobu náboru pacientů z našeho pracoviště měli všichni naši pacienti potvrzenou bilaterální vestibulopatii. Pacient referovaný z neuromuskulární ambulance nebyl vestibulárně vyšetřen.
U obou pacientů, kteří splňovali kompletní klinická kritéria CANVAS, byla diagnóza CANVAS na úrovni DNA analýzy potvrzena. Naopak z kohorty pacientů s vestibulární areflexií či s ataxií s pozdním nástupem bez přítomnosti neuropatie jsme nepotvrdili žádného pacienta s tímto onemocněním. To je ve shodě s literaturou udávající 100% prevalenci senzitivní neuropatie u CANVAS.
Elektromyografie prokázala u pacientů s CANVAS charakteristickou poruchu pouze senzorických akčních potenciálů, která je přítomna u 100 % případů (chybění u 57 % pacientů na HK a 83 % na DK, u zbylých 43 a 17 % pak snížení jejich amplitudy). Naopak motorické akční potenciály jsou výbavné u téměř 90 % pacientů [8]. Tento nález byl potvrzen u všech našich pacientů s CANVAS. Diferenciálně diagnosticky je z hlediska EMG nutné odlišit obdobný nález u pacientů s Friedreichovou ataxií.
Zajímavým publikovaným příznakem CANVAS je přítomnost dráždivého kašle, který je přítomen u 60 % pacientů, objevuje se často již v časném stadiu či představuje prodromální příznaky přítomné již několik desítek let před vznikem prvních neurologických obtíží [2]. Jako příčina se zvažuje neurodegenerativní postižení neuronálního okruhu kašlacího reflexu [8]. U našich pacientů se kašel vyskytoval u případu 2, nicméně nebyl důvod zpochybňovat diagnózu astmatu.
Vzhledem k tomu, že gen RFC1 kóduje subjednotku replikačního faktoru C, který se účastní procesu replikace a oprav DNA, byl u pacientů s CANVAS také zkoumán výskyt onkologických onemocnění, ten se však nelišil od ostatních pacientů s ataxií [8].
V tab. 2 uvádíme klinická kritéria syndromu CANVAS asociovaného s repeatovou expanzí RFC1 a jejich prevalenci [2].
Jak již bylo zmíněno, na molekulární úrovni je onemocnění podmíněno bialelickou expanzí AAGGG repetitivní sekvence v intronu genu RFC1. Neobvyklý typ repetitivní sekvence a navíc její umístění v intronu, tj. nekódující části genu, byly příčinou takto pozdního odhalení standardně používanými molekulárně genetickými metodami (pokusy zachytit variantu metodou celoexomového sekvenování nebyly úspěšné).
Zajímavé je, že na rozdíl od klasických onemocnění s expanzí repetitivní sekvence (např. Huntingtonova choroba nebo SCA2 s opakováním CAG repeatu v kódující oblasti kauzálních genů), nebyla u CANVAS nalezena klinická souvislost mezi délkou expanze a věkem nástupu/tíží onemocnění. Rovněž není zatím známo, zda je mutace při přenosu do další generace stabilní. Z klinického hlediska je důležité připomenout, že vzhledem k malému počtu dětí v českých rodinách jsou onemocnění s autozomálně recesivní dědičností často považována za nehereditární, tedy sporadická. Ze známých dědičných ataxií se nabízí určitá paralela mezi CANVAS a nejčastější autozomálně recesivně dědičnou Friedreichovou ataxií (FRDA) [12]. Obě jsou způsobené expanzí repetice v intronu, která leží v Alu elementu, avšak u CANVAS dochází kromě změny délky repetice i ke změně sekvence a též patogenní mechanizmus obou chorob je rozdílný. U FRDA dochází ke snížení genové exprese a ztrátě funkce proteinu frataxinu [12].
Gen RFC1 kóduje velkou podjednotku replikačního faktoru C, který je nutný pro správnou funkci DNA polymerázy při replikaci a opravách DNA. Avšak přesný mechanismus, kterým autozomálně recesivní expanze repetitivní sekvence v tomto genu vede ke ztrátě Purkyňových buněk, vestibulárních nervů či neuronů ganglií dorzálních kořenů, dosud není jasný – nebylo totiž potvrzeno, že by docházelo k redukci exprese genu na úrovni mRNA či proteinu, nebo ke ztrátě genové funkce (tzv. loss of function) [2]. Patologická expanze v genu RFC1 by však mohla narušit sekvenci, a tím i funkci elementu AluSx3, kterého je součástí. Elementy Alu hrají rozhodující roli při tvorbě nervových sítí a epigenetické regulaci biochemických procesů v celém CNS. Dysregulace elementů Alu může narušit mitochondriální funkce, což má za následek zhoršení dostupnosti energie, buněčný stres a smrt neuronu [13]. Někteří autoři zvažují i možný dopad patologické expanze v genu RFC1 na strukturu chromatinu a související tkáňově specifické změny [2].
Závěr
Představili jsme první tři molekulárně geneticky potvrzené případy CANVAS v ČR zachycené pomocí nově zavedené molekulárně genetické metodiky na našem pracovišti.
Na základě literatury i naší pilotní studie je možné, že se jedná o poměrně časté onemocnění a dokonce o nejčastější dědičnou příčinu poruchy rovnováhy ve stáří. Předpokládáme, že vzhledem k pozdnímu nástupu, benignímu průběhu a multifaktoriální etiologii poruch rovnováhy (neuropatie, mozečkový syndrom a vestibulopatie), může být velká část pacientů vedena pod diagnózou neuropatie či nespecifické závratě a poruchy rovnováhy způsobené multisenzorickým deficitem ve stáří. Obdobný klinický a EMG nález může být i u prodromální fáze Friedreichovy ataxie, pro kterou je ale typický začátek ve 2. dekádě a nástup po 50. roce věku je vzácný.
Vzhledem k autozomálně recesivní dědičnosti a k malému množství sourozenců v českých rodinách předpokládáme, že bude převažovat sporadický výskyt onemocnění u jednotlivců, což může mylně vést k podezření na nehereditární etiologii obtíží. Genetické vyšetření CANVAS je nyní výzkumně dostupné na Ústavu biologie a lékařské genetiky FN Motol. Molekulárně genetické vyšetření doporučujeme indikovat v případě chronicko-progresivní senzitivní neuropatie nejasné etiologie, zejména dominují-li poruchy rovnováhy, či pokud pacienti uvádějí rozmazané vidění při pohybu (oscilopsie). Podezření posiluje výskyt více případů mezi sourozenci. V případě klinického podezření na CANVAS doporučujeme se cíleně ptát na chronický kašel, který je v rámci neurologických diagnóz pro CANVAS patognomonický.
Grantová podpora
Práce byla podpořena Grantovou agenturou UK – projekt číslo 608218 a projekty MZ ČR – RVO, FN v Motole 00064203, IPE 2. LF UK No. 699012. Centrum hereditárních ataxií je součástí evropské sítě specializovaných pracovišť pro vzácná onemocnění ERN-RND European Reference Network for Rare Neurological Diseases – Projekt číslo 739510.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem práce nemají žádný konflikt zájmů.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bio medicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for bio medical papers.
MUDr. Michaela Danková
Centrum hereditárních ataxií
Neurologická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha
e-mail: dankova6@gmail.com
Přijato k recenzi: 24. 3. 2021
Přijato do tisku: 8. 7. 2021
Sources
1. Szmulewicz DJ, Waterston JA, Macdougall HG et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 2011; 1233 (1): 139–147. doi: 10.1111/j.1749-6632.2011.06 158.x.
2. Cortese A, Simone R, Sullivan R et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019; 51 (4): 649–658. doi: 10.1038/s41588-019-0372-4.
3. Migliaccio AA, Halmagyi GM, McGarvie LA et al. Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 2004; 127 (2): 280–293. doi: 10.1093/brain/awh030.
4. Szmulewicz DJ, Roberts L, McLean CA et al. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract 2016; 6 (1): 61–68. doi: 10.1212/CPJ.000 0000000000215.
5. Wu TY, Taylor JM, Kilfoyle DH et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia „CANVAS“ syndrome. Brain 2014; 137 (10): 2649–2656. doi: 10.1093/brain/awu 196.
6. Cazzato D, Bella ED, Dacci P et al. Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome: a slowly progressive disorder with stereotypical presentation. J Neurol 2016; 263 (2): 245–249. doi: 10.1007/s00415-015-7951-9.
7. Infante J, García A, Serrano-Cárdenas KM et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) with chronic cough and preserved muscle stretch reflexes: evidence for selective sparing of afferent Ia fibres. J Neurol 2018; 265 (6): 1454–1462. doi: 10.1007/s00415-018-88 72-1.
8. Cortese A, Tozza S, Yau WY et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 2020; 143 (2): 489–490. doi: 10.1093/brain/awz418.
9. Hantash FM, Goos DM, Crossley B et al. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genet Med 2011; 13 (1): 39–45. doi: 10.1097/GIM.0b013e3181fa9fad.
10. Cortese A, Reilly MM, Houlden H. RFC1 CANVAS/Spectrum Disorder. [online]. GeneReviews 2020 Nov 25. Available from: https: //www.ncbi.nlm.nih.gov/books/NBK564656/.
11. Subramony SH. SARA – a new clinical scale for the assessment and rating of ataxia. Nat Clin Pract Neurol 2007; 3 (3): 136–137. doi: 10.1038/ncpneuro0426.
12. Campuzano V, Montermini L, Moltò MD et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996; 271 (5254): 1423–1427. doi: 10.1126/science.271.5254.1423.
13. Larsen PA, Hunnicutt KE, Larsen RJ et al. Warning SINEs: alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosom Res 2018; 26 (1–2): 93–111. doi: 10.1007/s10577-018-9573-4.
14. Agrawal Y, Van De Berg R, Wuyts F et al. Presbyvestibulopathy: diagnostic criteria Consensus document of the classification committee of the Bárány Society. J Vestib Res 2019; 29 (4): 161–170. doi: 10.3233/VES-190672.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2021 Issue 4
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Metamizole vs. Tramadol in Postoperative Analgesia
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
Most read in this issue
- Poruchy čichu po COVID-19 – diagnostika, význam a léčba
- CANVAS – nově identifikovaná genetická příčina ataxie s pozdním nástupem. Popis prvních diagnostikovaných pacientů v České republice
- Proč se dráhy kříží? Základní principy uspořádání mozku obratlovců
- COVID-19 asociovaná myelitida – kazuistika vzácné komplikace závažné SARS-CoV-2 infekce