
Nové možnosti laboratórnej diagnostiky ochorení spojených s tvorbou amyloidov
New possibilities of laboratory diagnostics of diseases associated with amyloid formation
Many neurodegenerative diseases are defined by the aggregation and accumulation of the specific pathological protein in the CNS, leading to irreversible and fatal changes of the tissues. However, due to high clinical and epidemiological heterogeneity, a definitive ante-mortem diagnosis is very difficult to perform. The definitive diagnosis is confirmed by neuropathological evaluation made only at autopsy. Hope for early and accurate laboratory diagnostics of these diseases within a patient’s life represents methods based on the detection of seeding activity of pathological proteins. An example is a highly specific and ultrasensitive new method called Real-Time Quaking-Induced Conversion (RT-QuIC) assay. Originally, RT-QuIC was developed for the diagnosis of prions showing 92–97% sensitivity and 100% specificity. In our laboratory, we were able to detect prions in 39 brain samples, corresponding 24 cerebrospinal fluid samples, and in 38 skin samples of patients with Creutzfeldt-Jakob disease using RT-QuIC. Lately, the use of the RT-QuIC method for detection of pathological protein a-synuclein, which accumulates during Parkinson’s disease or dementia with Lewy bodies, and tau protein which is characteristic for Alzheimer’s disease or corticobasal degeneration, was described. This review aims to elucidate the diagnosis of neurodegenerative diseases and its recent approaches using RT-QuIC.
Keywords:
brain tract – comparative anatomy – Nervous system – corticospinal tract – optic chiasm – evolution
Authors:
S. Galušková 1; T. Moško 1; P. Dušek 2; R. Matěj 3; K. Holada 1
Authors‘ workplace:
Ústav imunologie a mikrobiologie, 1. LF UK v Praze
1; Neurologická klinika, 1. LF UK a VFN v Praze
2; Oddělení patologie a národní, referenční laboratoř TSE-CJN, Thomayerova nemocnice, Praha
3
Published in:
Cesk Slov Neurol N 2021; 84/117(4): 334-340
Category:
Review Article
doi:
https://doi.org/10.48095/cccsnn2021334
Overview
Rada neurodegeneratívnych ochorení je definovaná agregáciou a akumuláciou špecifického patologického proteínu v CNS, čo vedie k nezvratným a fatálnym zmenám tkaniva. Avšak kvôli vysokej klinickej a epidemiologickej heterogenite je definitívna ante-mortem diagnostika týchto ochorení obtiažna. Definitívne potvrdenie diagnózy poskytuje až neuropatologické vyšetrenie mozgového tkaniva. Nádej pre skorú a presnú laboratórnu diagnostiku týchto ochorení za života pacienta predstavujú metódy založené na detekcii konformáciu konvertujúcej aktivity patologických proteínov. Príkladom je nová, vysoko špecifická a senzitívna diagnostická metóda Real-Time Quaking-Induced Conversion (RT-QuIC). Pôvodne bola vyvinutá na diagnostiku priónových ochorení vykazujúc 92–97 % senzitivitu a 100 % špecificitu. V našom laboratóriu sme pomocou RT-QuIC detegovali prióny v 39 vzorkách mozgu, 24 korešpondujúcich vzorkách moku a v 38 vzorkách kože pacientov s Creutzfeldt-Jakobovou chorobou. Najnovšie bolo opísané využitie RT-QuIC metódy na detegovanie patologických proteínov a-synukleínu, ktorý sa hromadí pri Parkinsonovej chorobe alebo pri demencii s Lewyho telieskami a tau proteínu, charakteristického pre Alzheimerovu chorobu a kortikobazálnu degeneráciu. Predložený text sa zameriava na oboznámenie problematiky diagnostiky neurodegeneratívnych ochorení pomocou RT-QuIC metódy.
Klíčová slova:
mozková dráha – srovnávací anatomie – nervový systém – kortikospinální dráha – chisama opticum – evoluce
Amyloidy predstavujú agregáty patologicky zložených proteínov, pre ktoré je charakteristická morfológia fibríl so sekundárnou štruktúrou beta-skladaného listu. Tie vznikajú nesprávnym zložením natívneho proteínu, stratou fyziologickej funkcie a jeho následnou agregáciou spojenou s narušením buniek alebo orgánov. V súčasnosti je známych vyše 40 proteínov, ktoré tvoria patogénne amyloidné fibrily spôsobujúce amyloidózy [1]. Spomedzi týchto proteínov majú zvláštne postavenie prióny, pretože štúdium ich infekčných vlastností umožňuje lepšie porozumenie mechanizmov patogenézy iných neurodegeneratívnych ochorení.
Priónové ochorenia alebo prenosné spongiformné encefalopatie sú smrteľné neurodegeneratívne ochorenia, pri ktorých dochádza ku konformačnej zmene bunkového priónového proteínu na dominantnú patologickú formu, ktorá sa agreguje v nervovom tkanive. Tieto procesy vedú k spongiformným zmenám a k hromadeniu amyloidných plakov v mozgovom tkanive, ktoré je spojené s jeho nezvratným poškodením [2].
Prionózy sa v populácii vyskytujú ako sporadické, genetické alebo získané ochorenia. Najbežnejšia, Creutzfeldt-Jakobova choroba (CJD), má incidenciu 1 – 2 prípady na milión ľudí ročne. Až v 85 % prípadoch sa vyskytuje ako spontánne sporadické ochorenie (sCJD), ktoré sa môže prejaviť v rôznych formách. Fenotyp ochorenia je výrazne ovplyvnený metionín/valín polymorfizmom na kodóne 129 génu pre priónový proteín PRNP. Ďalší faktor, ovplyvňujúci výsledný fenotyp sCJD, je glykotyp patogénneho priónového proteínu (typ 1 a 2), ktorý sa hromadí v mozgu počas ochorenia. Glykotypy sa rozlišujú na základe elektroforetickej migrácie, keď po štiepení proteinázou K vznikajú rozličné fragmenty 19 kDa a 21 kDa priónového proteínu [3,4]. Kombináciou faktorov rozlišujeme 6 typov CJD, a to MM1, MM2, VV1, VV2, MV1 a MV2. Získané dáta naznačujú, že heterozygotnosť 129M/V pre gén PRNP predstavuje najodolnejšiu formu pre vznik sCJD. Okrem toho, genetickou analýzou bolo zistené, že takmer vo všetkých prípadoch s variantnou formou CJD (vCJD) bola prítomná iba homozygotná forma (129M/M) CJD. Heterozygotná forma 129M/V bola zistená u dvoch pacientov s formou vCJD [5].
Geneticky podmienené priónové ochorenia zahŕňajú familiárnu formu CJD (gCJD), familiárnu fatálnu insomniu (FFI) a Gerstmann-Sträussler-Scheinkerov (GSS) syndróm, ktoré sa vyskytujú u pacientov s priónovými ochoreniami s prevalenciou 10 – 15 %. Genetické priónové ochorenia podliehajú autozomálne dominantnej dedičnosti, spôsobené mutáciou génu pre priónový proteín PRNP, ktorý sa nachádza na chromozóme číslo 20. Táto mutácia zvyšuje pravdepodobnosť konverzie neagregovaného bunkového priónového proteínu (cellular prion protein; PrPC) na jeho patogénnu formu (scrapie prion protein; PrPSc) [6]. V súčasnosti bolo identifikovaných viac ako 60 typov mutácií, ale u niektorých sa predpokladá, že ich penetrancia a patogenita je veľmi nízka [7]. Mutácia PRNP E200K bola po prvýkrát identifikovaná u pacientov zo Slovenska (prevažne na území Oravy, Kysúc a Rožňavy), kde výskyt tejto mutácie dosahuje 69 % s 54–59 % penetranciou [8]. Jednu z najväčších skupín s mutáciou E200K predstavuje izraelská komunita Židov líbyjského pôvodu. V ČR boli doposiaľ zaznamenané mutácie gCJD E200K, D178N a R208H a mutácia P102L, ktorá je asociovaná s GSS syndrómom. Mutácia asociovaná s FFI ochorením nebola v ČR doposiaľ identifikovaná [9,10].
Získané / transmisívne prionové ochorenia sa vyskytujú u veľmi nízkeho počtu pacientov, avšak poukazujú na infekčný potenciál priónov. V roku 1950 bolo prvýkrát prenosné priónové ochorenie – kuru opísané u kmeňa Fore v Papua Novej Guinei, kde sa šírilo prostredníctvom kanibalistických rituálov [11]. Medzi získané ochorenia zaraďujeme aj vCJD, ktoré vzniká vystavením sa priónomi bovinnej spongiformnej encefalopatie (BSE) hlavne konzumáciou kontaminovaného jedla. V literatúre sú okrem toho opísané iatrogénné prenosy (iCJD) vystavením sa priónmi infikovaným materiálom [12]. Prvý takýto prípad bol reportovaný v roku 1974 po transplantácií rohovky. Odvtedy bolo zaznamenaných vyše 450 prípadov iCJD. Väčšina z nich sa týkala recipientov rastového hormónu hypofýzy a pri transplantácií štepu dura mater [13]. Na území Slovenska a ČR nebol doposiaľ zaznamenaný žiadny iatrogénny prenos priónového ochorenia. Navyše, od roku 2007 platí v oboch krajinách povinnosť testovania mozgových homogenátov darcov rohoviek na prítomnosť PrPSc [8].
Doporučený postup diagnostiky priónových ochorení podľa WHO a Centers for Diseases Control and Prevention (CDC) je založený na prítomnosti klinických príznakov, a to progresívnej demencie, myoklonu, cerebelárneho syndrómu, akinetického mutizmu a pyramidálnych (zvýšené reflexy, spasticita) a extrapyramidálnych (parkinsonizmus, dystonia) znakov. Pomocné vyšetrovacie metódy zahŕňajú EEG, MR mozgu, proteínovú analýzu cerebrospinálneho moku (cerebrospinal fluid; CSF), kde sa sleduje prítomnosť proteínu 14-3-3 a tau [14,15]. Špecificita a senzitivita týchto metód je limitovaná – hlavne v ranných štádiách ochorenia, kedy sú koncentrácie proteínov v CSF veľmi nízke. Zlatý štandard definitívnej diagnostiky priónových ochorení tvorí post-mortem detekcia prítomnosti patogénneho priónového proteínu pomocou imunohistochemických metód a western blotu, ktoré ale detegujú iba proteáza rezistentnú formu PrP [16]. Tieto metódy nedetegujú formy priónových ochorení, ktoré sú čiastočne alebo úplne citlivé voči štiepeniu proteázami, ako napr. variabilne proteáza-senzitívna prionopatia (VPSPr) [3,17].
Vzhľadom k neobvyklej stavbe priónovej infekčnej častici a k tomu, že prióny nevyvolávajú systémovú imunitnú reakciu v organizme, bola diagnostika klasickými sérologickými a molekulovými metódami nemožná. Značný pokrok bol dosiahnutý až po objavení metód replikácie priónov in vitro [18]. Nové senzitívne testy ako Protein Misfolding Cyclic Amplification (PMCA), Standard Quaking-Induced Conversion (S-QuIC) alebo Amyloid Seeding Assay (ASA) sú založené na detekcii tzv. „prión konvertujúcej aktivity“. Prión konvertujúca aktivita je definovaná ako schopnosť patologického priónového proteínu, ktorý sa nachádza v pacientovej vzorke, indukovať konverziu bunkového neagregovaného PrPC na jeho agregovanú formu. Práve prítomnosť patologického PrP predstavuje jediný známy špecifický marker priónových ochorení [19]. Avšak vyššie spomínané metódy sú limitované nízkou robustnosťou, obťažným štandardizovaním, sonikáciou, zložitým vyhodnocovaním výsledkov a dĺžkou protokolu. Navyše, produkt reakcie pri PMCA metóde je vysoko infekčný [20,21]. Nová metóda, Real-Time Quaking-Induced Conversion (RT-QuIC), obchádza viacero limitujúcich faktorov a predstavuje novú nádej v diagnostike neurodegeneratívnych ochorení ante-mortem [22].
Real-Time Quaking-Induced Conversion
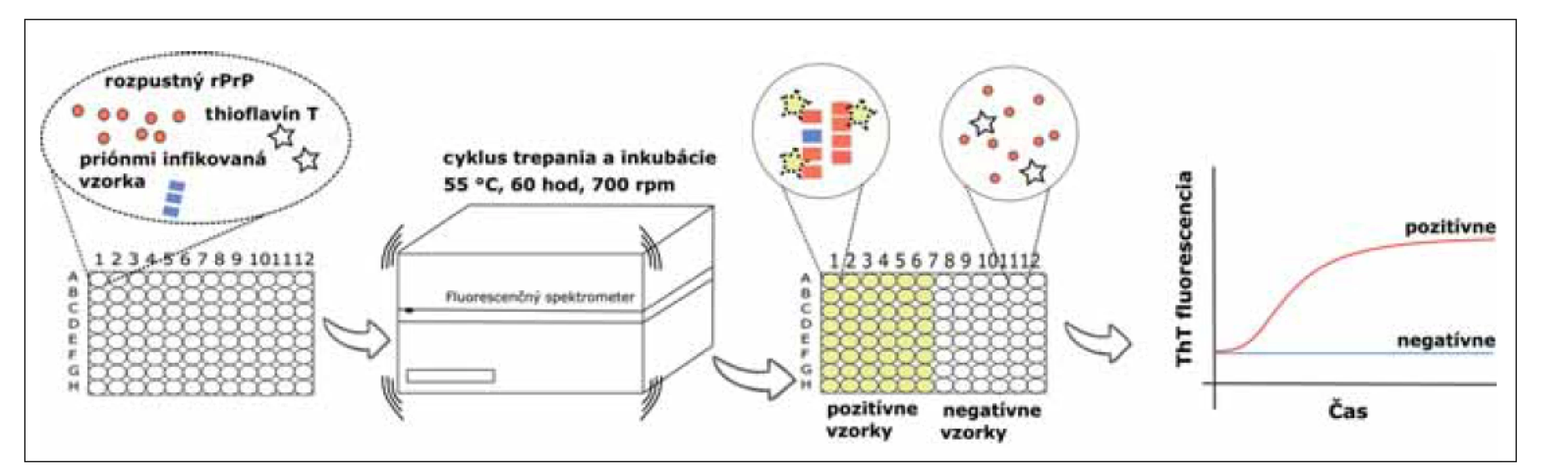
Krátko po PMCA a S-QuIC metódach bola predstavená RT-QuIC, ktorá je takisto založená na detekcii schopnosti patogénneho priónového proteínu indukovať konverziu rozpustného monomerného rekombinantného prionového proteínu (rPrP) do pozmenenej polymérnej konformácie, ale v reálnom čase. Proteáza K senzitívny rPrP, exprimovaný v bunkách Escherichia coli, sa používa ako substrát pre amplifikáciu patologickej formy PrP bohatej na štruktúry beta skladaného listu. Striedanie cyklov intenzívneho trepania a inkubácie zvyšuje efektivitu polymerizácie rPrP. Tieto novo vzniknuté konformačné štruktúry, amyloidy, majú vysokú afinitu k fluorescenčnému farbivu thioflavín T (ThT), ktorý je prítomný v reakčnej zmesi. Intenzita fluorescencie je zaznamenávaná v reálnom čase a je priamo úmerná množstvu vznikajúcich proteínových agregátov (obr. 1). Na rozdiel od PMCA metódy, produkt RT-QuIC nie je infekčný [23,24].
rPrP – rekombinantný prionovej proteín; ThT – Thioflavín T
Fig. 1. Real-Time Quaking-Induced Conversion assay. The reaction mix, which contains substrate (soluble rPrP), fluorescent dye ThT
and buffers, is loaded into a black 96-well plate. After that, an infectious sample is added into each well in quadruplicate as a source
of pathogenic prion protein. The reaction is incubated at 55 °C, for 60 hours with cycles of intensive shaking (700 rpm, 1 min) and
incubation (1 min). Aggregated rPrP has a high affinity to ThT which can be seen as the change of fluorescence intensity. This change
is observed in real time.
rPrP – recombinant prion protein; ThT – Thioflavin T
RT-QuIC predstavuje efektívnu metódu v skorej diagnostike neurodegeneratívnych priónových ochorení, vykazujúc 92 – 97 % senzitivitu a 100 % špecificitu pri testovaní vzoriek mozgovomiechového moku od žijúcich pacientov [25]. V súčasnosti prebehlo klinické testovanie, pri ktorom detegovali PrPSc vo vzorkách CSF a epitelu z čuchového bulbu (bulbus olfactorius). Testovania sa zúčastnilo šesť laboratórií, ktoré dokázali v zaslepenej štúdií detegovať priónové ochorenie s 99 % zhodou [26]. Na rozdiel od ostatných spomínaných metód, ktoré využívajú prión konvertujúcu aktivitu, RT-QuIC dokáže detegovať všetky subtypy CJD a to s vysokou citlivosťou vo veľmi nízkych koncentráciách priónov (femtogramoch). Viaceré štúdie dokazujú, že RT-QuIC je schopný detegovať priónové ochorenia ante-mortem nielen zo vzoriek mozgovomiechového moku, ale taktiež zo vzoriek biopsie kože [27] a vzoriek steru čuchovej sliznice [28]. V našom laboratóriu sme zaviedli metódu RT-QuIC po prvýkrát v rámci Českej a Slovenskej republiky. Celkovo bolo v pilotnej retrospektívnej štúdii otestovaných 39 vzoriek mozgových homogenátov a 24 korešpondujúcich vzoriek CSF [29]. V nadväzujúcej retrospektívnej štúdii bolo otestovaných 38 vzoriek CSF a korešpondujúcich vzoriek kože s definitívnou diagnózou priónových ochorení odobratých post-mortem. Ako kontrolné vzorky bolo použitých 24 vzoriek CSF a koží od pacientov s inými neurodegeneratívnymi ochoreniami a 6 vzoriek od pacientov s inými ochoreniami ako neurodegeneratívne. Prión konvertujúcu aktivitu sa nám podarilo detegovať vo všetkých post-mortem vzorkách mozgu (obr. 2), CSF a kože (obr. 3) pacientov s definitívnou diagnózou CJD.
RT-QuIC – Real-Time Quaking-Induced Conversion; sCJD – sporadická Creutzfeldt-Jakobova choroba; ThT – Thioflavín T
Fig. 2. Prion seeding activity shown in different dilutions of a brain homogenate of a patient with definite sCJD (MM1) using the
RT-QuIC assay. The assay is able to detect the presence of prion seeding activity even after a 5x10–9 dilution, which is added into the
reaction mix. The final dilution of a brain homogenate is equivalent to a 1x10–10 dilution.
RT-QuIC – Real-Time Quaking-Induced Conversion; sCJD – sporadic Creutzfeldt-Jakob disease; ThT – Thioflavin T

CSF – cerebrospinálna tekutina; RT-QuIC – Real-Time Quaking-Induced Conversion; sCJD – sporadická Creutzfeldt-Jakobova choroba; ThT –
Thioflavín T
Fig. 3. Prion seeding activity in CSF and skin samples obtained post-mortem from the same patient with sCJD (MM1) diagnosis analyzed
using RT-QuIC assay. CSF samples were analyzed undiluted and the skin samples were diluted 10x.
CSF – cerebrospinal fluid; RT-QuIC – Real-Time Quaking-Induced Conversion; sCJD – sporadic Creutzfeldt-Jakob disease; ThT – Thioflavin T

Protokol pre RT-QuIC je optimalizovaný tak, aby sa redukovala možnosť vzniku spontánnych agregátov, a tým pádom nedochádzalo ku falošne pozitívnym výsledkom. V praxi to znamená rozdiel medzi kinetikou spontánnej tvorby amyloidných fibríl a tvorby indukovanou patogénnym PrP prítomným vo vzorke. Orrú et al definovali optimálne podmienky, pri ktorých je reakcia najefektívnejšia, bez falošne pozitívnych alebo negatívnych reakcií [30]. Medzi faktory ovplyvňujúce priebeh a výsledok reakcie patrí predovšetkým teplota, intenzita trepania, prítomnosť a koncentrácia dodecylsulfátu sodného, solí a pH, pri ktorej reakcia prebieha [23].
Najdôležitejšou a najkritickejšou súčasťou RT-QuIC reakcie zostáva rozpustný rPrP. Jeho príprava a purifikácia vo forme vhodnej pre RT-QuIC test je náročná, ako časovo, tak aj metodicky. Práve kvalita substrátu je dôležitá pri minimalizovaní tvorby spontánnych agregátov počas reakcie. Rekombinantný PrP, vhodný ako substrát do reakcie, je zatiaľ komerčne nedostupný a laboratóriá sú odkázané na jeho produkciu vo vlastnej réžií.
Vplyv rPrP ako substrátu
Keď sa v roku 1997 podarilo tímu, pod vedením Wüthricha, vyprodukovať bakteriálne exprimovaný rekombinantný priónový proteín [31] v neagregovanej forme, znamenalo to veľký pokrok v štúdiu štruktúry priónového proteínu. Kvalita substrátu (rozpustného neagregovaného proteínu) zásadne ovplyvňuje špecificitu RT-QuIC reakcie. Substrát musí byť dostatočne stabilný, aby vydržal purifikáciu, skladovanie a dobu, počas ktorej prebieha RT-QuIC reakcia, avšak jeho natívna štruktúra nesmie byť priveľmi stabilná, aby došlo ku konverzii počas reakcie [32].
Pôvod primárnej sekvencie rPrP substrátu je dôležitý pri diagnostike priónových ochorení spôsobených rôznymi typmi ľudských priónov. Najbežnejšie používané substráty sú rPrP škrečka zlatého (Mesocricetus auratus) a to buď skrátený (rHaPrP 90–231) alebo celý (rHaPrP 23–231), ľudský (rHuPrP 23–231) a myší (rMoPrP 23–231) [33]. Predpokladá sa, že rHaPrP 90–231 ako substrát urýchľuje reakciu, vďaka chýbajúcemu flexibilnému N-terminálnemu koncu proteínu. Konformácia natívneho rHaPrP je rýchlejšie destabilizovaná, a tým pádom konverzia na amyloidnú štruktúru prebehne rýchlejšie [30]. Avšak pre niektoré formy ochorenia, ako je napr. GSS syndróm s mutáciou A117V, F198S a H187R génu pre priónový proteín PRNP, dlho nebol identifikovaný efektívny substrát [34]. S použitím rPrP hrdziaka lesného (Myodes glareolus) dokázali Orrú et al zvýšiť citlivosť RT-QuIC metódy voči viacerým genetickým formám CJD, vrátane GSS syndrómu [33,35].
Protokol na purifikáciu podľa autorov Orrú et al, ktorý je aplikovaný v našom laboratóriu s malými úpravami, predstavuje overený spôsob izolácie a purifikácie rPrP. Plazmidy, ktoré nesú sekvenciu pre priónový proteín sú klonované do bakteriálnych buniek druhu Escherichia coli Rosetta (DE3). Autoindukčným expresným systémom je priónový proteín exprimovaný a následne purifikovaný z inklúznych teliesok, ktoré sú rozpustené v guanidín hydrochloride (GndHCl). Izolácia proteínu prebieha pomocou afinitnej chromatografie s využitím Ni-NTA kolóny za denaturačných podmienok GndHCl [27]. Denaturovaný proteín je na kolóne spätne zložený do natívnej konformácie cez klesajúci lineárny gradient GndHCl. Na konci je rPrP vymývaný imidazolom, centrálny pík je zachytený do skúmavky, dialyzovaný a v alikvótoch zamrazený.
Využitie RT-QuIC u ostatných neurodegeneratívnych ochoreniach
Neurodegeneratívne ochorenia predstavujú v súčasnosti čoraz väčšiu záťaž v starnúcej populácii rozvinutých zemí sveta. Medzi najčastejšie sa vyskytujúce ochorenia patria Alzheimerova choroba, tauopatie frontotemporálna demencia a kortikobazálna degenerácia (corticobasal degeneration; CBD) a synukleopatie, kde patrí Parkinsonova choroba (Parkinson disease; PD), demencia s Lewyho telieskami (dementia with Lewy bodies; DLB), progresívna supranukleárna paralýza (PSP) a multisystémová atrofia (MSA). Z patologického hľadiska zdieľajú tieto ochorenia spoločný znak a to hromadenie abnormálneho proteínu v CNS vo forme amyloidu. Tieto depozity abnormálneho proteínu sú identifikované v mozgovom tkanive až post-mortem na základe molekulárnych a neuropatologických nálezov [36].
Synukleopatie predstavujú skupinu neurodegeneratívnych ochorení, pri ktorých sa v nervovom tkanive zhlukujú fibrilárne agregáty a-synukleínu. Tieto depozity sa môžu nachádzať v tele nervových buniek ako Lewyho telieska, dystrofické neurity alebo ako gliové cytoplazmatické inklúzie [37]. Tauopatie sa odlišujú tým, že počas progresie choroby dochádza k hromadeniu tau proteínu v CNS. Detekcia tau proteínu je ovplyvnená hlavne jeho diverzitou. U ľudí je rozlišovaných 6 izoforiem tau proteínu vznikajúcich alternatívnym zostrihom mRNA, ktoré sa odlišujú počtom väzobných miest. Tri izoformy tau proteínu majú tri väzobné miesta pre mikrotubuly (3R) a ďalšie tri izoformy majú až štyri miesta (4R) [38].
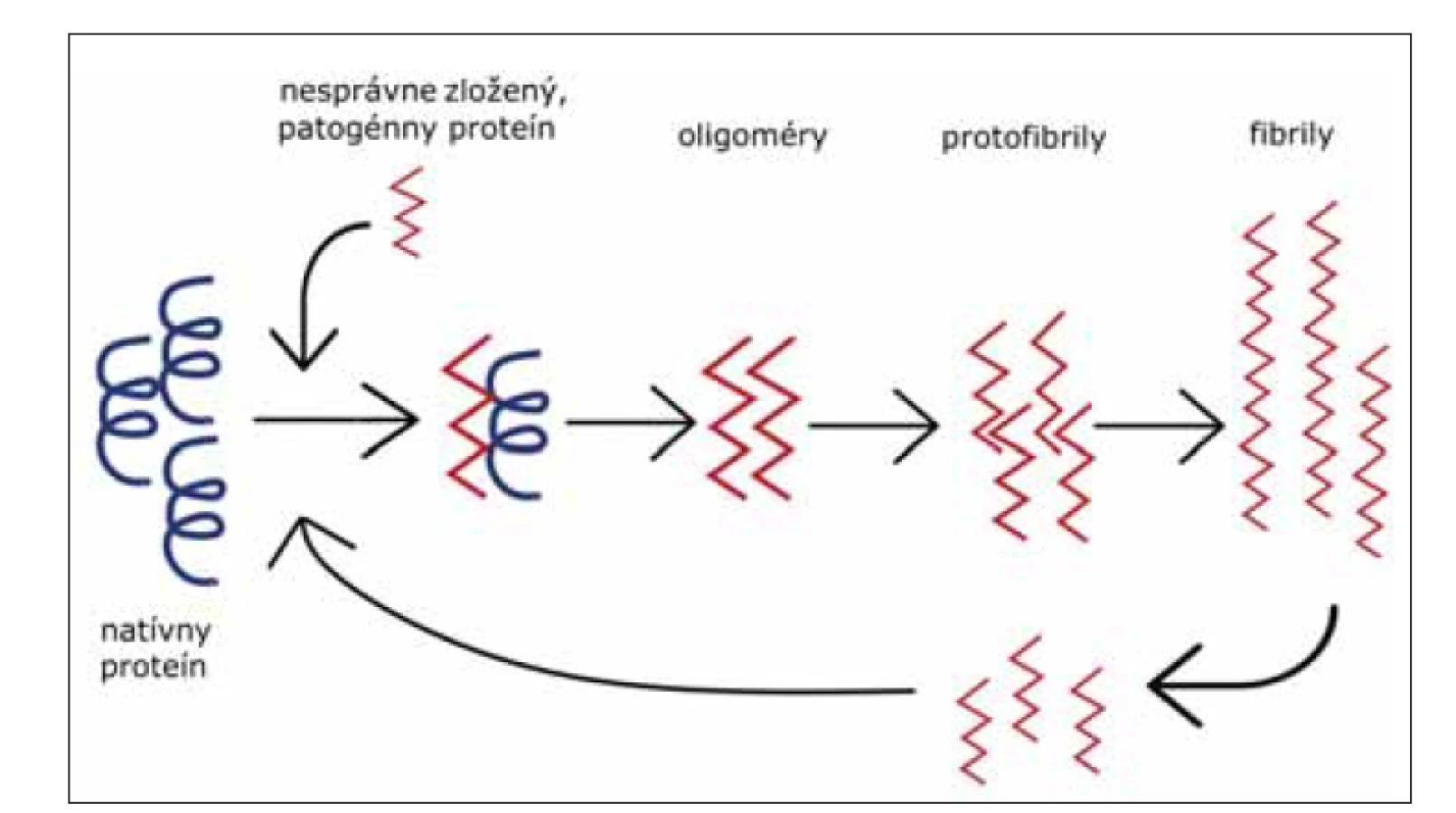
V súčasnosti, dôkazy ukazujú na fakt, že mechanizmus propagácie abnormálneho priónového proteínu v mozgu je podobný s mechanizmom propagácie abnormálnych foriem proteínov u iných neurodegeneratívnych ochorení. Tento mechanizmus pozostáva z templátom riadeného nesprávneho zloženia proteínu, oligomerizácie a akumulácie patogénneho proteínu (obr. 4) [39].
Fig. 4. Template associated conformational change of protein to its pathogenic form. Misfolded
protein (red), mostly consists of beta-sheets, binds to the native protein (black) and
induces its conversion to a pathogenic form. Foremost, small oligomers are generated,
and later protofibrils from which amyloid fibrils (aggregated forms of pathogenic protein)
originate. Breakage of fibrils into smaller particles leads to the amplification of conversion.
Táto mechanistická podobnosť s priónovými ochoreniami umožňuje využitie modifikovanej RT-QuIC metódy aj v diagnostike ostatných neurodegeneratívnych ochorení, s dôrazom na zlepšenie intra-vitam diagnostiky na začiatku klinickej fázy ochorenia [40]. Skorá diagnostika môže výrazne ovplyvniť efekt liečiv, tak isto ako kvalitu života pacienta pred nástupom klinických symptómov. Výsledky štúdií potvrdili schopnosť RT-QuIC v detekcii agregačnej aktivity a-synukleínu a tau proteínu. Fairfoul et al úspešne detegovali prítomnosť abnormálneho a-synukleínu vo vzorkách CSF s 95 % senzitivitou a 100 % špecificitou pri pacientoch s DLB a PD [41]. Garrido et al detegovali agregáciu a-synukleínu pomocou RT-QuIC metódy vo vzorkách CSF s diagnózou PD s mutáciou na géne Leucine-rich repeat kinase 2 (LRRK2) so 40 % úspešnosťou a dokonca v 20 % vzoriek bezpríznakových nosičov tejto mutácie [42]. Okrem toho, viac ako polovica vzoriek, ktorá bola odobratá klinicky diagnostikovaným pacientom s MSA (9/11) a PD (10/18), vykazovala RT-QuIC pozitívny výsledok [40]. Detekcia patogénneho tau proteínu je ovplyvnená hlavne jeho variabilnou štruktúrou a biochemickou charakteristikou v priebehu ochorenia jednotlivých tauopatií [43]. Saijo et al s využitím modifikovaného 3R tau fragmentu ako substrátu ukázali, že RT-QuIC metóda dokáže detegovať a rozlíšiť Pickovu chorobu od ostatných tauopatií na základe hromadenia prevažne 3R filamentov v nervovom tkanive. Taktiež dokázali detegovať konvertujúcu aktivitu tau proteínu pri Alzheimerovej chorobe, kedy sa v nervovom tkanive hromadí v rovnakých koncentráciách 3R a 4R tau proteín [21,44]. V najnovšej štúdií bol modifikovaný protokol s využitím 4R tau substrátu. Táto forma tau proteínu dokáže determinovať ochorenia, pri ktorých sa hromadí 4R tau proteín, konkrétne pri PSP alebo CBD [45].
Tieto výsledky dokazujú a podporujú hypotézu, že konvertujúca aktivita nielen patogénneho priónového proteínu, ale aj agregácia a-synukleínu a tau proteínu je detekovateľná vo vzorkách odobratých za života pacientov. Navyše, vysoká špecificita a senzitivita RT-QuIC metódy umožňuje diagnostiku neurodegeneratívnych ochorení v skorých štádiách choroby. Métoda je v súčastnosti v ČR dostupná iba na experimentálne využitie a dalšie štúdie sú potrebné na potvrdenie robustnosti RT-QuIC pre diagnostické účely.
Konflikt záujmov
Autori deklarujú, že v súvislosti s predmetom práce nemajú žiadny konflikt záujmov.
Grantová podpora
Podporené z grantového programu Ministerstva zdravotnictví ČR s registračným číslom AZV NV-18-04-00179 a Všeobecnej fakultnej nemocnice v Prahe s registračným číslom GIP-20-L-13-212. Všetky práva podľa predpisu na ochranu duševného vlastníctva sú vyhradené.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Mgr. Soňa Galušková
Ústav imunologie a mikrobiologie
1. LF UK
Studničkova 7
128 00 Praha
e-mail: sona.galuskova@lf1.cuni.cz
Přijato k recenzi: 4. 3. 2021
Přijato do tisku: 28. 6. 2021
Sources
1. Riek R, Eisenberg DS. The activities of amyloids from a structural perspective. Nature 2016; 539 (7628): 227–235. doi: 10.1038/nature20416.
2. Prusiner SB. The prion diseases. Brain pathology 1998; 8 (3): 499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x.
3. Ascari LM, Rocha SC, Gonçalves PB et al. Challenges and advances in antemortem diagnosis of human transmissible spongiform encephalopathies. Front Bioeng Biotechnol 2020; 8 : 1228. doi: 10.3389/fbioe.2020.585 896.
4. Monari L, Chen SG, Brown P et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A 1994; 91 (7): 2839–2842. doi: https: //doi.org/10.1073/pnas.91.7.2839.
5. Kobayashi A, Teruya K, Matsuura Y et al. The influence of PRNP polymorphisms on human prion disease susceptibility: an update. Acta Neuropathol 2015; 130 (2): 159–170. doi: 10.1007/s00401-015-1447-7.
6. Bernardi L, Bruni AC. Mutations in prion protein gene: pathogenic mechanisms in C-Terminal vs. N-Terminal domain, a review. Int J Mol Sci 2019; 20 (14): 3606. doi: 10.3390/ijms20143606.
7. Kim MO, Takada LT, Wong K et al. Genetic PrP prion diseases. Cold Spring Harb Perspect Biol 2018; 10 (5): a033134. doi: 10.1101/cshperspect.a033134.
8. Gdovinová Z. Creutzfeldt-Jakobova choroba. Cesk Slov Neurol N 2013; 76/109 (2): 138–154.
9. Mancuso M, Siciliano G, Capellari S et al. Creutzfeldt-Jakob disease with E200K PRNP mutation: a case report and revision of the literature. Neurol Sci 2009; 30 (5): 417–420. doi: 10.1007/s10072-009-0118-7.
10. Rohan Z, Parobková E, Johanidesová S et al. Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou. Cesk Slov Neurol N 2013; 76/109 (3): 300–306.
11. Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review ofthese less common human transmissible spongiform encephalopathies. J Clin Neurosci 2001; 8 (5): 387–397. doi: 10.1054/jocn.2001.0919.
12. Holada K, Simák J, Vostal JG. Transmission of BSE by blood transfusion. Lancet 2000; 356 (9243): 1772. doi: 10.1016/S0410-6736 (05) 71968-1.
13. Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull 2003; 66 (1): 255–265. doi: 10.1093/bmb/66.1.225.
14. Brown P, Brunk C, Budka H et al. WHO manual for surveillance of human transmissible spongiform encephalopathies, including variant Creutzfeldt-Jakob disease. World Health Organization 2003. [online]. Available form URL: https: //www.who.int/bloodproducts/TSE-manual2003.pdf.
15. CDC’s diagnostic criteria for Creutzfeldt-Jakob disease (CJD), 2018. [online]. Available from URL: https: //www.cdc.gov/prions/cjd/diagnostic-criteria.html.
16. Panigaj M, Glier H, Wildova M et al. Expression of prion protein in mouse erythroid progenitors and differentiating murine erythroleukemia cells. PLoS One 2011; 6 (9): e24599. doi: 10.1371/journal.pone.0024599.
17. Collins SJ, Sanchez-Juan P, Masters CL et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129 (9): 2278–2287. doi: 10.1093/brain/aw 1159.
18. Orrú CD, Caughey B. Prion seeded conversion and amplification assays. Top Curr Chem 2011; 305 : 121–133. doi: 10.1007/128_2011_184.
19. Colby DW, Zhang Q, Wang S et al. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A 2007; 104 (52): 20914–20919. doi: 10.1073/pnas.0710152105.
20. Castilla J, Saá P, Hetz C et al. In vitro generation of infectious scrapie prions. Cell 2005; 121 (2): 195–206. doi: 10.1016/j.cell.2005.02.011.
21. Kraus A, Saijo E, Metrick MA. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol 2019; 137 (4): 585–598. doi: 10.1007/s00401-018-1947-3.
22. Orrú CD, Wilham JM, Vascellari S et al. New generation QuIC assay for prion seeding activity. Prion 2012; 6 (2): 147–152. doi: 10.4161/pri.19430.
23. Atarashi R, Satoh K, Sano K et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011; 17 (2): 175. doi: 10.1038/nm.2294.
24. Saijo E, Groveman BR, Kraus A et al. Ultrasensitive RT-QuIC seed amplification assays for disease-associated tau, a-synuclein, and prion aggregates. Methods Mol Biol 2019; 1873 : 19–37. doi: 10.1007/978-1-4939-8820-4_2.
25. Candelise N, Baiardi S, Franceschini A et al. Toward an improved early diagnosis of neurodegenerative diseases: the emerging role of in vitro conversion assays for protein amyloids. Acta Neuropathol Commun 2020; 8 (1): 1–16. doi: 10.1186/s40478-020-009900-x.
26. Orrú CD, Groveman BR, Foutz A et al. Ring trial of 2nd generation RT-QuIC diagnostic tests for sporadic CJD. Ann Clin Transl Neurol 2020; 7 (11): 2262–2271. doi: 10.1002/acn3.51219.
27. Mammana A, Baiardi S, Rossi M et al. Detection of prions in skin punch biopsies of Creutzfeldt-Jakob disease patients. Ann Clin Transl Neurol 2020; 7 (4): 559–564. doi: 10.1002/acn3.51000.
28. Orrú CD, Bongianni M, Tonoli G et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med 2014; 371 (6): 519–529. doi: 10.1056/NEJMoal315200.
29. Moško T, Galušková S, Matěj R et al. Detection of prions in brain homogenates and CSF samples using a second-generation RT-QuIC assay: a useful tool for retrospective analysis of archived samples. Pathogens 2021; 10 (6): 750. doi: 10.3390/pathogens10060750.
30. Orrú C, Hughson A, Groveman B et al. Factors that improve RT-QuIC detection of prion seeding activity. Viruses 2016; 8 (5): 140. doi: 10.3390/v8050140.
31. Zahn R, von Schroetter C, Wüthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett 1997; 417 (3): 400–404. doi: 10.1016/S0014-5793 (97) 01330-6.
32. Orrù CD, Groveman BR, Hughson AG et al. RT-QuIC assays for prion disease detection and diagnostics. Methods Mol Biol 2017; 1658 : 185–203. doi: 10.1007/978-1-4939-7244-9_14.
33. Orrú CD, Groveman BR, Raymond LD et al. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog 2015; 11 (6): e1004983. doi: 10.1371/journal.ppat.1004983.
34. Nonno R, Di Bari MA, Cardone F et al. Efficient transmission and characterization of Creutzfeldt–Jakob disease strains in bank voles. PLoS Pathog 2006; 2 (2): e12. doi: 10.1371/journal.ppat.0020012.
35. Cheng K, Sloan A, Waitt B et al. Altered rPrP substrate structures and their influence on real-time quaking induced conversion reactions. Protein Expr Purif 2018; 143 : 20–27. doi: 10.1016/j.pep.2017.10.007.
36. Erkkinen MG, Kim MO, Geschwind MD. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol 2018; 10 (4): a033118. doi: 10.1101/cshperspect.a033118.
37. Adler CH, Beach TG, Hentz JG et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014; 83 (5): 406–412. doi: 10.1212/WNL.0000000000000641.
38. Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 2006; 36 (10): 652–660. doi: 10.1111/j.1445-5994.2006.011 53.x.
39. Soto C, Pritzkow S. Protein misfolding, aggregation and conformational strains in neurodegenerative diseases. Nat Neurosci 2018; 21 (10): 1332–1340. doi: 10.1038/s41593-018-0235-9.
40. De Luca CMG, Elia AE, Portaleone SM et al. Efficient RT-QuIC seeding activity for a-synuclein in olfactory mucosa samples of patients with Parkin - son’s disease and multiple system atrophy. Transl Neurodegener 2019; 8 (1): 24. doi: 10.1186/s40035-019-01 64-x.
41. Fairfoul G, McGuire LI., Pal S et al. Alpha-synuclein RT-Qu IC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 2016; 3 (10): 812–818. doi: 10.1002/acn3.338.
42. Garrido A, Fairfoul G, Tolosa ES et al. α-synuclein RT-QuIC in cerebrospinal fluid of LRRK2-linked Parkinson’s disease. Ann Clin Transl Neurol 2019; 6 (6): 1024–1032. doi: 10.1002/acn3.772.
43. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol 2013; 12 (6): 609–622. doi: 10.1016/S1474-4422 (13) 70090-5.
44. Saijo E, Ghetti B, Zanusso G et al. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol 2017; 133 (5): 751–765. doi: 10.1007/s00401-017-1692-z.
45. Saijo E, Metrick MA, Koga S et al. 4-repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol 2020; 139 (1): 63–77. doi: 10.1007/s00401-019-02080-2.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2021 Issue 4
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Poruchy čichu po COVID-19 – diagnostika, význam a léčba
- CANVAS – nově identifikovaná genetická příčina ataxie s pozdním nástupem. Popis prvních diagnostikovaných pacientů v České republice
- Proč se dráhy kříží? Základní principy uspořádání mozku obratlovců
- COVID-19 asociovaná myelitida – kazuistika vzácné komplikace závažné SARS-CoV-2 infekce