
Komplexná mutačná analýza génu PAH u slovenských pacientov postihnutých fenylketonúriou
Complex Mutation Analysis of the PAH Gene in Slovak Patients Affected by Phenylketonuria
This work continues previous studies to detect molecular basis of phenylketonuria in Slovak patients. DHPLC screening for all 13 exons of the PAH gene was introduced and optimized. DNA samples from 48 subjects with biochemically proven phenylketonuria were analyzed in whom direct DNA diagnostics (detection of 11 most frequent mutations) allowed to identify mutations on only one or none PKU chromosome. In this study, 16 new mutations were detected, (Q20X, F39L, R68S, F233I, R176X, P281L, R270I, T278N, R243X, A300S, I306V, W326X, L348V, S349P, E390G, Y387X), which were not known in this population as yet. Individual mutations were allocated to the appropriate phenotypic categories of phenylketonuria.
The present results will be applied for routine diagnostic practice of PKU in this country. In addition to causative mutations, several genetic polymorphisms were detected in introns, but also in the exons of the PAH gene. Up to date, 84,3% of Slovak PKU alleles are already identified, detection of the remaining 15,7 % needs additional studies and investigational approaches.
Key words:
phenylketonuria, mutations, mutational analysis, DNA diagnostics
Authors:
E. Polák 1; A. Ficek 1; M. Baldovič 1,2; E. Feráková 1; A. Šoltýsová 1; J. Strnová 3; O. Ürge 3; L. Kovács 4; Ľ. Kádaši 1,2
Authors‘ workplace:
Katedra molekulárnej biológie PrírF UK, Bratislava
vedúci doc. RNDr. Ľ. Kádasi, DrSc.
1; Ústav molekulárnej fyziológie a genetiky SAV, Bratislava
vedúci doc. RNDr. Ľ. Kádasi, DrSc.
2; Klinika deti a dorastu Andreja Getlíka. SZU, Bratislava
prednostka doc. MUDr. K. Furková, CSc., mim. prof.
3; 2. detská klinika LFUK a DFNsP, Bratislava
prednosta prof. MUDr. L. Kovács, DrSc, MPH
4
Published in:
Čes-slov Pediat 2008; 63 (10): 528-534.
Category:
Original Papers
Overview
V predloženej práci autori nadviazali na problematiku zisťovania molekulárnej podstaty fenylketonúrie na Slovensku. Zaviedli a optimalizovali metódu DHPLC pre skríning všetkých 13 exónov PAH génu. Analyzovali DNA 48 jedincov postihnutých fenylketonúriou, u ktorých bola po priamej DNA diagnostike (zisťovanie 11 najčastejších typov mutácii v kaukazoidnej populácii) identifikovaná mutácia len na jednom PKU chromozóme alebo nebola identifikovaná žiadna mutácia. Identifikovali 16 nových mutácií (Q20X, F39L, R68S, F233I, R176X, P281L, R270I, T278N, R243X, A300S, I306V, W326X, L348V, S349P, E390G, Y387X), ktorých výskyt nebol zatiaľ v slovenskej populácii popísaný. Jednotlivé mutácie priradili k fenotypovým kategóriám fenylketonúrie a stanovili ich frekvencie.
Rutinnú diagnostiku uvedených mutácií bude možné zaviesť do DNA diagnostickej praxe. Popri kauzatívnych mutáciách autori v intrónoch, ale aj v exónoch detegovali množstvo polymorfizmov. Uvedeným postupom sa im v slovenskej populácii podarilo identifikovať 84,3 % PKU alel, identifikovanie postihnutia zvyšných 15,7 % alel si vyžaduje ďalšie práce.
Kľúčové slova:
fenylketonúria, mutácie, mutačná analýza, DNA diagnostika
Úvod
Fenylketonúria (PKU) ako aj iné formy hyperfenylalaninémie (HPA) sú primárne spôsobené deficitom dôležitého článku v metabolickej dráhe aminokyseliny fenylalanínu (Phe), hepatického enzýmu fenylalanín hydroxylázy (PAH) [1]. PAH katalyzuje v normálnej ľudskej pečeni premenu L-fenylalanínu na L-tyrozín za prítomnosti kyslíka a svojho kofaktora tetrahydrobiopterínu (BH4). Sekundárnou príčinou vzniku hyperfenylalaninémie môže byť defekt v tvorbe alebo recyklácii tohto kofaktora [2, 3, 4].
Frekvencia výskytu PKU sa mení v závislosti od jednotlivých populácií. Najvyššia incidencia ochorenia bola zaznamenaná u waleských Rómov (1 : 40) najnižšia u aškenázskych Židov (1 : 180 000). V kaukazoidnej populácii je priemerná frekvencia približne 1 : 10 000, v Českej republike je to 1 : 9000 a na Slovensku 1 : 10 000 [5, 6, 7, 8, 35].
Klinické príznaky fenylketonúrie sa začnú prejavovať od 2. mesiaca života. Patologické koncentrácie fenylalanínu a jeho metabolitov zapríčiňujú ťažkú poruchu mozgovej činnosti (oligofrénia). Trvalej mentálnej retardácii postihnutého dieťaťa sa dá predísť len včasnou dietetickou liečbou už od 1. mesiaca života. Medzi najčastejšie symptómy ochorenia patrí aj kožný ekzém, myší zápach potu a moču, nápadne svetlé vlasy, prípadne ich svetlejší odtieň v porovnaní s rodičmi alebo nepostihnutými súrodencami [3].
Podľa klinického priebehu, koncentrácie Phe v sére, tolerancie k dietetickému Phe a reziduálnej aktivity PAH sa v súčasnosti rozlišujú 4 kategórie závažnosti ochorenia [9, 10, 11, 17]:
- Klasická fenylketonúria (PKU) s najťažším priebehom sa charakterizuje úplnou absenciou aktivity alebo minimálnou aktivitou (<3 %) PAH enzýmu. Hladina Phe v sére je približne 1200 μmol/l a pacienti tolerujú menej ako 20 mg/kg Phe telesnej hmotnosti na deň.
- Mierne závažná fenylketonúria má z klinického hľadiska miernejší priebeh. Reziduálna aktivita PAH je 1–5 % a vedie k určitej tolerancii na Phe. Jeho hladina v sére sa pohybuje v rozmedzí 600–1200 μmol/l a denná tolerancia je 20–25 mg/kg na deň.
- Ľahká fenylketonúria sa charakterizuje koncentráciou Phe v rozmedzí 600–1200 μmol/l, denná tolerancia je 25–50 mg/kg na deň.
- Benígna hyperfenylalaninémia je klinicky asymptomatická, nevyžaduje eliminačnú diétu a pacienti majú len zvýšenú hladinu Phe v krvi (menej ako 600 μmol/l).
Hlavným determinantom vysvetľujúcim takúto širokú variabilitu fenotypových prejavov a klinického priebehu ochorenia je množstvo rôznych mutácií v PAH géne, a teda existencia intralokusovej alelovej heterogenity [22, 23]. Situáciu komplikuje skutočnosť, že pri PKU je veľmi častým javom výskyt zložených heterozygotov, ktorí majú dve rozličné mutantné alely v identických lokusoch obidvoch homologických chromozómov. Vzájomná korelácia týchto mutácií sa potom odzrkadlí vo fenotype. Výsledky štúdií jednoznačne poukazujú na to, že na determináciu výsledného fenotypu má väčší vplyv tá z mutácií, ktorá spôsobuje miernejšiu formu PKU [11].
Na Slovensku je zavedená priama DNA diagnostika 11 najčastejších z doteraz známych 532 PAH mutácií, pričom u slovenských PKU pacientov sa doteraz detegovalo len 7 z nich (R408W, IVS12nt1, R261Q, R158Q, R252W, IVS10nt546, Y414C). V súbore 98 slovenských PKU pacientov bola úspešnosť pri identifikácii jednotlivých mutácií génu PAH nasledovná: obidve prítomné PAH mutácie boli identifikované u 51 % pacientov, jedna PAH mutácia u 38,8 % pacientov a u 10,2 % pacientov nebola identifikovaná ani jedna mutácia v géne PAH. Celkové množstvo detegovaných PKU mutácií v slovenskej populácii predstavovalo 70,4 % [8, 28, 29, 30]. Vzhľadom na tieto skutočnosti sa predpokladal výskyt ďalších mutácií v našej populácii, avšak doterajšie metódy neboli dostačujúce na ich odhalenie. Preto sme sa rozhodli zaviesť a štandardizovať metódu DHPLC pre mutačný skríning génu PAH a pomocou nej detegovať a charakterizovať zatiaľ neidentifikované mutácie v PAH géne v súbore slovenských PKU pacientov.
Pacienti a metódy
Analýzy sa uskutočnili na súbore 48 nepríbuzných pacientov s PKU s jednou stanovenou mutáciou alebo neidentifikovanou mutáciou na oboch PKU chromozómoch. Údaje o diagnóze a klinickom priebehu pacientov boli poskytnuté z oddelení klinickej genetiky na Slovensku. Vzorky DNA sa izolovali z periférnej krvi fenol-chloroformovou extrakciou podľa štandardných protokolov a komerčným kitom Puregene (Gentra) počas obdobia od roku 1990 až po rok 2004 a boli skladované v genómovej banke DNA združeného pracoviska Katedry molekulárnej biológie Prírodovedeckej fakulty Univerzity Komenského v Bratislave a Laboratória genetiky Ústavu molekulárnej fyziológie a genetiky SAV.
Oligonukleotidové primery
Na amplifikáciu exónov 11 a 12 génu PAH sme použili primery uvedené v literatúre podľa Jeruzelska a kol. [26]. Na amplifikáciu exónov 1 až 10 a exónu 13 sme použili nami navrhnuté oligonukleotidové primery pomocou internetového softvéru Primer3 [27]. Ako referenčnú sekvenciu sme použili sekvenciu z internetovej databázy [18]. Primery sa navrhli tak, aby mali približne rovnaké Tm teploty a najmenšiu vzájomnú a vlastnú komplementaritu. Sekvencie zahrňovali intronické 5’ a 3’ oblasti génu PAH, lokalizované v oblasti približne 30–50 bp od okrajov exónu.
PCR amplifikácia
Pomocou PCR sa na automatických cykléroch amplifikovalo 13 exónov génu PAH. Objem reakčnej zmesi pre každú vzorku bol 30 µl a obsahoval: ultračistú vodu, 1-krát koncentrovaný tlmivý roztok s 1,5 mmol.l-¹ MgCl2, 0,17 mmol.l-¹ dNTP, 0,2 μmol.l-¹ každého z primerov, približne 50 ng DNA a 0,5 U Taq DNA polymerázy (Invitrogen). Pre amplifikáciu sme zvolili nasledovné podmienky: 5 minút úvodná denaturácia pri 94 ºC a vlastná amplifikácia pozostávala z 33 cyklov denaturácie pri 94 ºC po dobu 30 sekúnd, anelácie primerov v teplotnom rozmedzí od 52 do 60 ºC (v závislosti od jednotlivých primerov) 30 sekúnd a polymerizácie pri 72 ºC, 30 sekúnd. Nasledovalo 10 minút dosyntetizovanie nekompletných PCR produktov a zastavenie reakcie schladením na 4 ºC.
Detekcia heteroduplexov metódou DHPLC
Pred analýzou DHPLC (Denaturating High Performance Liquid Chromatography) sa denaturáciou a pomalou renaturáciou PCR produktov vytvorili heteroduplexy. Počiatočná denaturácia bola 5 minút pri 94 ºC a nasledovalo 80 cyklov 30-sekundového ochladzovania. Každý cyklus mal o pol stupňa menšiu teplotu. Analýza DHPLC prebehla na systéme Transgenomic WAVE 3500. Optimálne podmienky pre mutačnú analýzu každého exónu sa stanovili na základe simulovanej parciálnej denaturácie referenčnej sekvencie príslušného amplikónu pomocou softvéru Navigator® software. Pre detekciu mutácií bola každá teplotná doména PCR fragmentu analyzovaná pri špecifickej teplote, ktorá zodpovedala 80–90 % helikálnej frakcie. Amplikóny exónov 3, 5 a 7 obsahovali dve teplotné domény a preto sme ich analyzovali pri dvoch rôznych teplotách. Gradient elučného činidla bol automaticky vypočítaný softvérom pre každý amplikón a pre každú teplotu parciálnej denaturácie. Výsledné chromatogramy sa analyzovali programom Navigator® software (Transgenomic) a exportovali do formátu PDF.
Sekvenovanie
Na prečistenie PCR produktov pred aj po sekvenačnej reakcii sa použila precipitácia etanolom a octanom sodným. Pri sekvenovaní sa použila Sangerova metóda sekvenovania na automatickom sekvenátore ABI 3100 Genetic Analyser (Applied Biosystems, USA). Sekvenačná reakcia prebehla v komerčnom kite, ktorý obsahoval 12,5-krát koncentrovaný BD pufor, a 1 pmol.l-¹ BigDye. Do každej reakcie sme pridali 2 pmol.l-¹ primeru a približne 20 ng DNA.
Analýza sekvenčných dát
Pre spracovanie sekvenčných dát sa použil softvér Sequencing analysis 5.2 (Applied Biosystems). Pre identifikáciu variant v DNA sa použil softvér ChromasPro (Technelysium) a s použitím ORF finder (NCBI) sa stanovili dôsledky zmien DNA na úrovni polypeptidového reťazca proteínu.
Výsledky
Metódou DHPLC sme analyzovali všetkých 13 exónov PAH génu u všetkých 48 pacientov. Ako pozitívna kontrola pri skríningu jednotlivých exónov slúžili amplikóny so známou mutáciou, pokiaľ sa v takom exóne vyskytovala. Na základe teplotných DHPLC profilov sme identifikovali 148 rôznych heterozygotov. Sekvenčnou analýzou sme u 24 pacientov identifikovali nasledovných 16 mutácií: Q20X, F39L, R68S, F233I, R176X, P281L, R270I, T278N, R243X, A300S, I306V, W326X, L348V, S349P, E390G, Y387X. Taktiež sme identifikovali 2 polymorfizmy v exónoch (C1155G, A696G) ako aj niekoľko polymorfizmov v intrónoch génu PAH.
Polymorfizmus A696G sme detegovali u 21 pacientov v heterozygotnom stave a polymorfizmus G1155C u 11 pacientov v heterozygotnom a u 7 v homozygotnom stave.
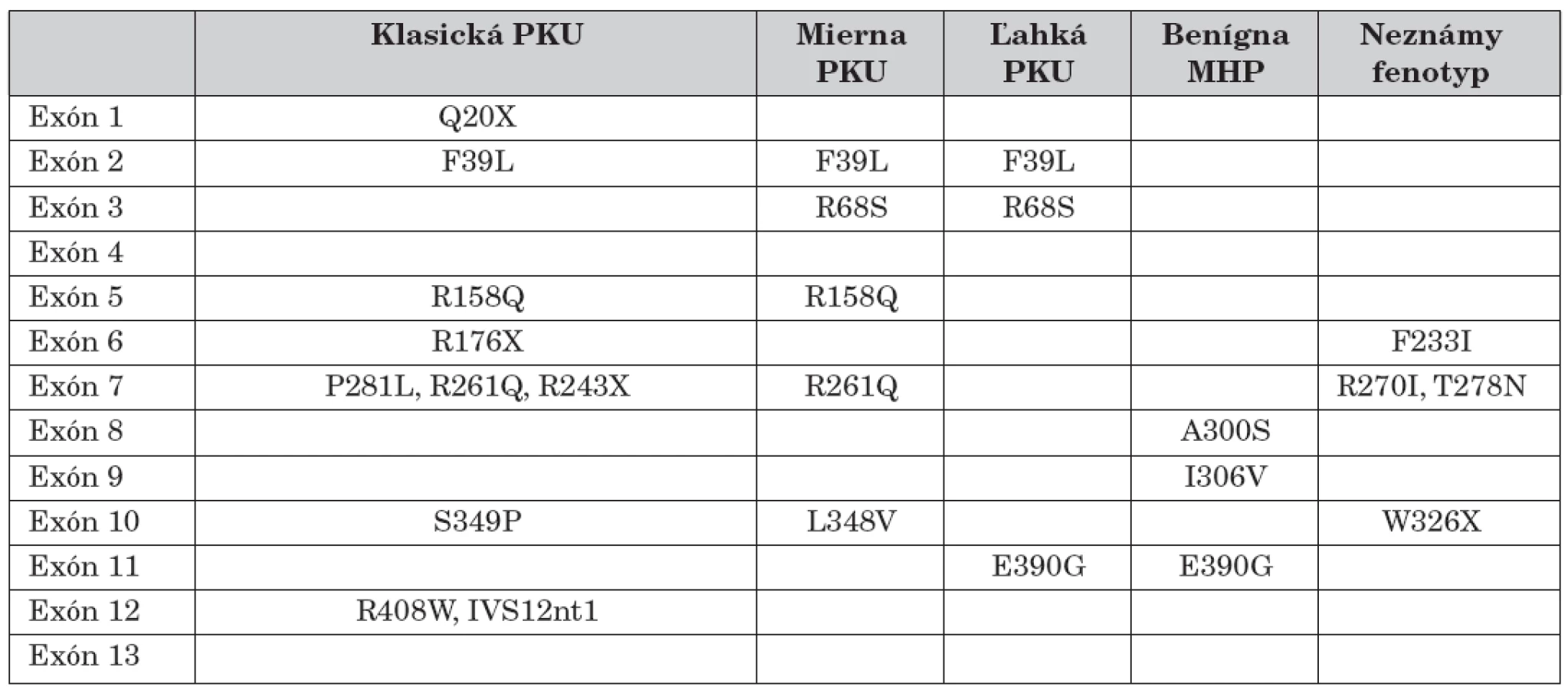
Jednotlivé identifikované mutácie, exón v ktorom sme ich identifikovali a ich asociácia s určitou fenotypovou kategóriou PKU sú znázornené v tabuľke 1.
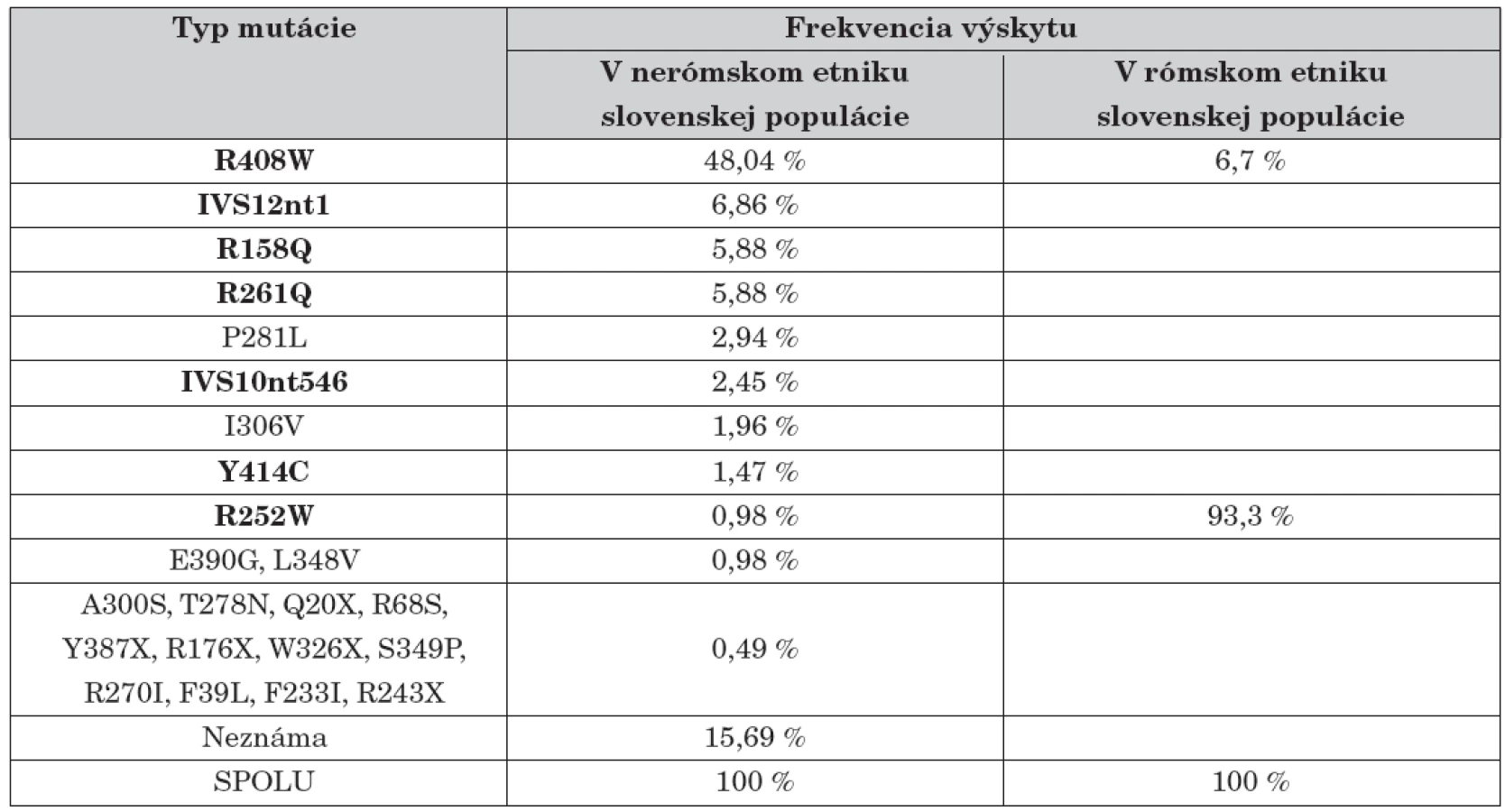
Táto práca spolu s predchádzajúcimi štúdiami dovolila stanoviť frekvenciu výskytu všetkých najčastejších identifikovaných mutácií na Slovensku, v týchto štúdiách bolo spolu analyzovaných 98 pacientov, čo znamená 196 alel (tab. 2).
Diskusia
Gén pre ľudskú fenylalanín hydroxylázu (PAH; E.C.1.14.16.1) je dlhý približne 90 kb a obsahuje 13 exónov. Bol klonovaný a lokalizovaný na distálnej časti dlhého ramienka chromozómu 12 [12, 13]. Vďaka intenzívnemu výskumu PKU vznikla v roku 1990 internetová databáza s cieľom sumarizovať informácie o PAH géne a tá sa v roku 1996 zmenila na informačnú databázu (PAH Gene Mutation Analysis Consortium Database). Dnes disponuje množstvom užitočných informácií a poskytuje veľmi komplexný a prehľadný obraz o popísaných mutáciách, haplotypoch a polymorfizmoch tohto lokusu. K poslednej aktualizácii (apríl 2008) bolo v tomto géne popísaných 532 mutácií, ich prevažná väčšina sa nachádza v exóne 7, ale ich vysoká distribúcia sa dá nájsť taktiež v exónoch 5–12 [1, 18].
Na lokuse PAH sa vyskytujú takmer všetky typy mutácií. Väčšina z nich vedie k zníženiu aktivity alebo stability enzýmu [19]. Prevládajú jednobázové substitúcie (missense resp. nonsense). Mutácie meniace zmysel (missense) sú najčastejšie zastúpené s početnosťou až 62 %. Zámena kodónu za stop kodón je výsledkom nezmyselných mutácií (nonsense). Tieto mutácie vedú k predčasnému ukončeniu translácie a tvoria približne 5 %. Jednobázové delécie, delécie kodónu alebo veľkých exónových oblastí tvoria približne 13 %. Podiel mutácií, ktoré postihujú zostrihové miesta, je okolo 12 %. Mutácie vedúce k posunu čítacieho rámca (frameshift) sú dôsledkom zostrihových mutácií alebo delécií časti génu, prípadne veľmi vzácne sa vyskytujúcich inzercií [18]. Taktiež boli identifikované mutácie, ktoré ovplyvňujú stabilitu a spracovanie mRNA [20] a veľké delécie exónov v dôsledku tzv. “preskočenia exónu” („exon-skipping effect“) [21].
V ostatných rokoch sa na Slovensku rutinne uskutočňuje určovanie jedenástich z doteraz známych 532 mutácií génu PAH. U slovenských pacientov sa doteraz identifikovalo 7 mutácií génu PAH (R408W, IVS12nt1, R261Q, R158Q, R252W, IVS10nt546, Y414C), ktoré spolu predstavujú 70,4 % všetkých mutácií u slovenských nerómskych PKU pacientov. V tejto populácii jednoznačne prevláda mutácia R408W asociovaná s haplotypom 2 [8]. Jej podiel dosahuje 48,04 %, čo je však druhý najnižší výskyt zo štátov strednej a východnej Európy. V rómskej časti slovenskej populácie sa prakticky vyskytuje len jedna mutácia R252W. Je asociovaná s haplotypom 69.3, kým v iných európskych populáciách sa stretáva iba v asociácií s iným haplotypom [25]. Tieto pozorovania jednoznačne potvrdzujú, že mutácia R252W u slovenských Rómov má odlišný pôvod ako v iných populáciách a jej vznik v tejto populácii možno vysvetliť efektom zakladateľa. Tento fenomén spolu s vysokým stupňom inbrídingu je zodpovedný aj za vysokú frekvenciu výskytu ochorenia (1 : 1000) u slovenských Rómov [25, 16] (tab. 1).
Zvýšenie účinnosti mutačnej analýzy a identifikácia doteraz neznámych mutácií si vyžadovali nový metodický prístup. Najefektívnejším spôsobom detekcie neznámych mutácií je v súčasnosti analýza heteroduplexov. DHPLC a HRM (High Resolution Melting) sú metódy využívajúce tento prístup a už boli s vysokou účinnosťou aplikované aj na skríning mutácií v géne PAH [31, 32]. V predloženej práci sme preto zaviedli metódu DHPLC pre skríning všetkých 13 exónov PAH génu u 48 slovenských PKU pacientov, u ktorých bola na lokuse PAH doteraz identifikovaná iba jedna mutácia, alebo nebola identifikovaná žiadna mutácia. Táto metóda nám pomohla vyselektovať 148 z celkového počtu 624 analyzovaných amplikónov pre následné sekvenovanie, čím sa pri detekcii mutácií značne ušetril čas aj finančné prostriedky. Skutočnosť, že boli potvrdené všetky známe mutácie (pozitívne kontroly) v jednotlivých amplikónoch, potvrdzuje citlivosť tejto metódy.
Identifikovali sme 16 nových mutácií a tým sa celkové spektrum odhalených mutácií v slovenskej populácii rozšírilo z pôvodných 70,4 % na 84,3 %. Mutácie P281L a I306V sme identifikovali s pomerne vysokou frekvenciou výskytu. Je zaujímavé, že tieto mutácie sa nevyskytujú v Českej republike [33] a pravdepodobne sú spôsobené rozdielnymi ťahmi osídľovania týchto území. Pôvod mutácie P281L je podľa Barić a kol. (2004) v juhovýchodnej Európe, čoho dôkazom je aj vysoký výskyt v krajinách stredozemia [34].
Taktiež sme dokázali mutáciu F233I, ktorá ešte nebola nájdená ani v okolitých populáciách ani v žiadnej inej populácií a taktiež nebola zatiaľ popísaná v žiadnej databáze ani publikácii. V blízkej budúcnosti by bolo vhodné uskutočniť funkčnú a štruktúrnu štúdiu PAH proteínu s touto mutáciou a registrovať ju v internetovej PAH databáze.
Na základe našej práce možno konštatovať že u 74,5 % slovenských PKU pacientov sú známe obe kauzatívne mutácie, u 19,6 % pacientov je ešte potrebné identifikovať druhú kauzatívnu mutáciu a u 5,9 % pacientov stále nie je známa ani jedna mutácia génu PAH zapríčiňujúca PKU. Spolu je v populácii slovenských PKU pacientov ešte potrebné identifikovať 15,7 % mutácií. Z tohto podielu by mali podľa našich predpokladov určitú časť tvoriť delécie [1, 18]. Keďže u 6 % analyzovaných pacientov sme nezachytili mutáciu ani na jednom PKU chromozóme, domnievame sa, že môže ísť o mutáciu v homozygotnom stave. Ostatné neidentifikované mutácie sú pravdepodobne lokalizované v intrónových oblastiach a postihujú promótor, regulačné oblasti alebo figurujú pri zostrihu mRNA PAH génu [32]. Je treba poznamenať, že kauzatívna príčina PKU môže byť aj v tvorbe alebo recyklácii PAH kofaktora tetrahydrobiopterínu. Tento prípad sa však vzťahuje len na cca 1 % príčin vzniku PKU [2].
Predošlé štúdie génu PAH naznačovali, že variabilné metabolické fenotypy PKU priamo korelujú s genotypom. V roku 1998 sa uskutočnila veľká európska multicentrická štúdia s cieľom definitívne potvrdiť tieto domnienky. Na tejto štúdii participovalo 7 európskych centier a bolo vyšetrených 686 pacientov s fenylketonúriou. U každého z nich boli identifikované obe kauzatívne mutácie. Na základe fenotypovej charakteristiky bolo 297 funkčne hemizygotných pacientov rozdelených do štyroch kategórií. Bolo identifikovaných 105 kauzatívnych mutácií priradených k jednotlivým fenotypom v príslušnej kategórii. Vychádzajúc z tohto rozdelenia, bol vypracovaný a testovaný jednoduchý model korelácie medzi genotypom a fenotypom. Očakávaný fenotyp zodpovedal skutočnému fenotypu v 79 % prípadov. Výsledok štúdie podporil existenciu silnej korelácie medzi PAH genotypom a fenotypom a potvrdil, že genotyp je hlavným určujúcim faktorom metabolického fenotypu u väčšiny pacientov s fenylketonúriou. Klasifikácia najčastejších mutácií umožnila predikciu biochemického fenotypu a tým značne uľahčila možnosť stanoviť mieru klinického poškodenia dieťaťa [11].
Na základe týchto výsledkov Guldberg a kol. (1998) navrhli systém klasifikácie PKU. Ku každej zo štyroch kategórii boli pridelené príslušné mutácie zodpovedné za tento typ ochorenia. Klasická forma PKU vzniká najčastejšie v dôsledku mutácií: R408W, IVS12nt1, IVS10nt546, G272X, R158Q, R243X. Menej vážna forma PKU bola asociovaná s mutáciami: R261Q, L348V, I65T, R261P. Mierna PKU bola charakterizovaná mutáciami: Y414C, A104D, G46S, E390G, R408Q, L48S, kým mutácie: A403V, A300S, V245A boli typické pre hyperfenylalaninémie. Fenotypové zatriedenie bolo však niekedy nepresné, pričom niektoré mutácie sa uvádzali vo viacerých fenotypových kategóriách ochorenia. Tieto nedostatky do istej miery korigovala neskoršia štúdia, ktorá sa uskutočnila na obdobnom súbore pacientov. Empirické porovnanie klinických nálezov u pacientov s touto predikciou bolo správne v jednotlivých kategóriách v rozmedzí 60 až 100 % [24] (tab. 2).
Projekt bol finančne podporovaný grantom MZ SR č. 2005/4-DFNsPBA-02 a grantom MŠ SR č. 4/2029/08.
Mgr. Emil Polák
Katedra molekulárnej biológie
Prírodovedecká fakulta UK
Mlynská dolina B2-210
842 15 Bratislava
Slovenská republika
e-mail: polakemo@gmail.com
Sources
1. Eisensmith RC, Woo SLC. Molecular basis of phenylketonuria and related hyperphenylalaninemias: Mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum. Mutat. 1992;1 : 13–23.
2. Scriver C, et al. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995 : 1015–1075.
3. Sršeň Š, Sršňová K. Základy klinickej genetiky a jej molekulárna podstata. Martin: Osveta, 2005 : 445. ISBN 80-8063-185-9.
4. Kaufman S. Phenylketonuria: biochemical mechanisms. Adv. Neurochem. 1976;2 : 1–132.
5. Tyfield LA, Meredith AL, Osborn MJ, Harper PS. Identification of the haplotype patern associated with the mutant PKU allele in the Gypsy population in Wales. J. Med. Genet. 1989;26 : 450–499.
6. Bickel H, Bachmann C, Beckers R. Neonatal mass screening for metabolic disorders. Eur. J. Pediatr. 1981;137 : 133–139.
7. Kozák L, Kuhrová V, Blažková M, Fajkusová L, Dvořáková D, Pijáčková A. Pokroky v diagnostice fenylketonurie zavedením přímé detekce mutací v PAH genu. Čas. Lék. čes. 1995;134 : 385–387.
8. Kádasi Ľ, Poláková H, Feráková E, Krivušová T, Hudecová S, Szomolayová I, Strnová J, Hruškovič I, Ferák V. PKU in Slovakia: mutation screening and haplotype analysis. Hum. Genet. 1995;95 : 112–114.
9. Gűttler, F. Hyperphenylalaninemia: Diagnosis and classification of the various types of phenylalanine hydroxylase deficiency. Acta Paediat. 1980;280 : 1–80.
10. Gűttler F, Guldberg P. The influence of mutations on enzyme activity and phenylalanine tolerance in phenylalanine hydroxylase deficiency. Eur. J. Pediatr. 1996;155: S6–S10.
11. Guldberg P, Rey F, Zschocke J, Romano V, Baudouin F, et al. A European Multicenter Study of Phenylalanine Hydroxylase Deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 1998;63 : 71–79.
12. Woo SLC, Lidsky AS, Güttler F, Chandra T, Robson K. Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. Nature 1983;306 : 151–155.
13. Lidsky AS, Law ML, Morse HG, Kao FT, Rabin M, et al. Regional mapping of the phenylalanine hydroxylase gene and the phenylketonuria locus in the human genome. Proc. Natl. Acad. Sci. USA 1985;82 : 6221–6225.
14. DiLella AG, Marvit J, Lidsky AS, Güttler F, Woo SLC. Tight linkage between a splicing mutation and a specific DNA haplotype in phenylketonuria. Nature 1986;332 : 799–803.
15. Daiger SP, Reed L, Huang SS, Zeng YT, Wang T, et al. Polymorphic DNA haplotypes at the phenylalanine hydroxylase (PAH) locus in Asian families with phenylketonuria (PKU). Am. J. Hum. Genet. 1989;45 : 319–324.
16. Kádasi Ľ. Molekulárna genetika vybraných monogénne dedičných ochorení. Bratislava: Veda, 2005 : 224. ISBN 80-224-0869-7.
17. Kayaalp E, Treacy E, Waters JP, Byck S, Nowacki P, Scriver CR. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: A metanalysis of genotype-phenotype correlations. Am. J. Hum. Genet. 1997;61 : 1309–1317.
18. http://www.pahdb.mcgill.ca
19. Erlandsen H, Patch MG, Gamez A, Straub M, Stevens RC. Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria. Pediat. 2003;112 : 1557–1565.
20. Richard I, Beckmann JS. How neutral are synonymous codon mutations? Nat. Genet. 1995;35 : 301–304.
21. Chao HK, Hsiao KJ, Su TS. A silent mutation induces exon skipping in the phenylalanine hydroxylase gene in phenylketonuria. Hum. Genet. 2001;108 : 14–19.
22. Okano Y, Eisensmith RC, Güttler F, Lichter Konecki U, et al. Molecular basis of phenotypic heterogenity in phenylketonuria. N. Engl. J. Med. 1991;324 : 1232–1238.
23. Desviat LR, Perez B, Ugarte M. Molecular basis of non-PKU hyperphenylalaninaemia in Spain: prevalence of A403V, a mutation with high residual activity. J. Inherit. Metab. Dis. 1996;19 : 227–230.
24. Gűttler F, Guldberg P. Mutation analysis anticipates dietery requirements in phenylketonuria. Eur. J. Pediatr. 2000;159 : 150–153.
25. Feráková E, Ferák V, Kadáši Ľ, Poláková H, Hejcmanová L, Pijačková A. A unique RFLP haplotype at the phenylalanine hydroxylase locus in Czechoslovak Gypsies with phenylketonuria. Funct. Develop. Morph. 1992;2 : 139–140.
26. Jeruzelska J, Matuszak R, Lyonnet S, Rey F, Rey J, et al. Genetic background of clinical homogenity of phenylketonuria in Poland. J. Med. Genet. 1993;30 : 232–234.
27. http://frodo.wi.mit.edu/
28. Búcová, D. Polymorfizmus typu STR v géne pre fenylalanín hydroxylázu a jeho využitie pri diagnostike fenylketonúrie v slovenskej populácii. Diplomová práca. Bratislava: PrírF UK, 1996.
29. Feráková E, Kádaši Ľ. Multiple origin for PKU in Slovakia. Symposium “The Origin of the Hungarian Populations and Mitochondrial Diseases”. Budapest, 1996.
30. Kojšová S. Detekcia mutácií L48S, I65T, R243X a Y414C v géne pre fenylalanín hydroxylázu v súbore slovenských pacientov s fenylketonúriou. Rigorózna práca. Bratislava: PrírF UK, 2004.
31. Bräutigam S, Kujat A, Kirst A, Seidel J, Lüleyap ÜH, Froster UG. DHPLC mutation analysis of phenylketonuria. Mol. Gen. Metab. 2003;78 : 205–210.
32. Dobrowolski SF, Ellingson C, Coyne T, Grey J, Martin R, et al. Mutations in the phenylalanine hydroxylase gene identified in 95 patients with phenylketonuria using novel systems of mutation scanning and specific genotyping based upon thermal melt profiles. Mol. Genet. Metab. 2007;10 : 12–22.
33. Kozák L, Blažková M, Kuhrová V, Pijáčková A, Ružičková Š, Šťastná S. Mutation and haplotype analysis of the phenylalanine hydroxylase alleles in classical PKU patients from Czech Republic: identification of four novel mutations. J. Med. Genet. 1997;34 : 893–898.
34. Barić I, Mardešić D, Sarnavska V, Lichter-Konecki U, Konecki DS, Trefz FK. Geographical distribution of the P281L mutation at the phenylalanine hydroxylase locus: possible origin in southeastern Europe. J. Inherit. Metab. Dis. 1994;17 : 376–377.
35. Ürge O, Strnová J, Mosendzová B. Fenylketonúria u adolescentov a dospelých na Slovensku. Čes.-slov. Pediat. 2003;58 : 423–425.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2008 Issue 10
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Henochova-Schönleinova purpura z pohľadu preventívneho podávania kortikoidov
- Hodnotenie klinických príznakov intrakraniálnej hypertenzie vo vzťahu k indikácii drenážneho výkonu u novorodencov a dojčiat s hydrocefalom
- Heterotopia žalúdočnej sliznice – literárny prehľad a naše skúsenosti
- Prader-Williho syndróm u novorodenca – dve kazuistiky