
Mutagenní vliv vyššího věku otců u neurokardiofaciokutánního syndromu
Mutagenic effect of advanced paternal age in neurocardiofaciocutaneous syndrome
Background.
Increased frequency of chromosomal aberration in children of mothers aged 35 years and older is very well known and since 1973 it is an indication to investigate the foetal karyotype in cells obtained by invasive method (amniocentesis), because the genetic risk of severe affection is higher than the risk of necessary invasive method. Mutagenic effect of advanced paternal age is known only among geneticists (1–4). The reason is not only low absolute risk of new mutation but particularly a high number of involved genes and last not least the limited spectrum of autosomal dominant disorders without abiotrofic character. Therefore the preventive methods for elimination of this risk are very limited. Only a few of them could be recognized prenatally by noninvasive methods of prenatal diagnostics.
Methods.
Genealogical, anamnestic and clinical data of 83 patients were studied with clinical suspection on neurocardiofaciocutaneous syndrome (NCFCs) (5–7). The diagnosis has not been confirmed in 29 patients, no mutation was detected in 8 investigated genes (PTPN11, SOS1, HRAS, BRAF, RAF1, MEK1, KRAS, NRAS). In 54 patients with autosomal dominant inherited Noonan syndrome, Costello syndrome and cardiofaciocutaneous syndrome the diagnosis was confirmed on DNA level and the biological fitness was estimated for each disorder. Paternal age at conception was compared in the group of patients with familial and sporadic occurrence of Noonan and NCFC syndromes. The clinical prognosis of this disorder is represented by biological fitness of patients. Coefficient of selection is 0,6 in Noonan and LEOPARD syndromes (29 from 48). All 6 patients with Costello and cardiofaciocutaneous syndromes developed due to a new mutation.
Conclusion.
Paternal age at birth was studied in 83 children patients with autosomal dominant Neurocardiofaciocutaneous syndrome (Noonan, LEOPARD, Costello, CFC) with a high population incidence and decreased biological fitness. Due to severe congenital heart defects, failure to thrive in infancy, increased risk for malignancy and further health problems the clinical prognosis of patients in the past was not good. Therefore high mutation rate is expected until now. Identification of genes responsible for manifestation of this disorder, enables to confirm the diagnosis and to recognize inherited and de novo mutations. Genealogy and DNA analysis of PTPN11, SOS1, HRAS, BRAF, RAF1, MEK1, KRAS and NRAS were obtained in cohort of 54 patients with NCFC syndromes and their parents. There were 48 patients with Noonan and LEOPARD syndromes, in 29 cases due to mutation de novo, 19 patients inherited the mutation from one of parents. All 6 patients with Costello syndrome and CFC syndrome were affected due to new mutation. DNA analysis revealed 32 mutations in PTPN11 gene, mutation in SOS1 gene was found in 10 patients, RAF1 mutation was present in 3 patients; mutation in MEK1, KRAS and NRAS genes was present in one patient each. In Costello syndrome and CFC syndrome mutations in HRAS (4 patients) and BRAF (2 patients) genes were detected. Genealogic data allow analysing parental age in the group of patients with new mutation and inherited mutation. Paternal age at conception of patients with Noonan syndrome due to new mutation was significantly increased in comparison to the group of fathers of Noonan patients with inherited mutation – 38,4 years and 29,6 years, resp., range 28 to 55 years and 25 to 35 years, resp. Maternal age was slightly increased too, –30,9 and 27,7, resp. and range 24 to 42 years and 21 to 36 years, resp. but not significantly. The results support the mutagenic effect of paternal age, espec. autosomal dominant mutations.
Keywords:
advanced parental age – mutagenic effect – chromosomal mutations – gene mutations – manifestation of new mutations – coefficient of selection – mutation rate
:
prof. MUDr. Eva Seemanová, DrSc. 1; Martin Zenker 2
:
Oddělení klinické genetiky, Ústav biologie a lékařské genetiky 2. LF UK, Praha
1; Institut für Humangenetik der Technischen Hochschule, Magdeburg, SRN
2
:
Čas. Lék. čes. 2014; 153: 242-245
:
Original Article
Východisko.
Zvýšená pravděpodobnost vzniku chromozomálních aberací u plodů matek starších 35 let je obecně známa a již 40 let je indikací k vyšetření karyotypu plodu při invazivní metodě prenatální diagnostiky, neboť genetické riziko a jeho klinický význam je podstatně vyšší než riziko spojené s nezbytnou amniocentézou. Mutagenní vliv vyššího věku otců je znám jen genetikům (1–4). Důvodem je nepochybně řádově nižší riziko vzniku čerstvých genových mutací, velké spektrum možných mutovaných genů a jen limitovaný počet autozomálně dominantně determinovaných poruch s bezprostřední manifestací po porodu. Abiotrofický charakter některých afekcí umožňuje jen omezenou nabídku jejich prevence neinvazivními metodami prenatální diagnostiky.
Metody.
V souboru 83 pacientů s autozomálně dominantně dědičnými syndromy spektra neurokardiofaciokutánní symptomatiky (NCFCs) jsme zhodnotili genealogická data a věk rodičů, abychom ověřili podíl tohoto mutagenního faktoru na restituci populační incidence při vysokém selekčním koeficientu (0,65). Syndrom neurokardiofaciokutánního spektra (5–7) byl zvolen po identifikaci odpovědných genů, aby mohla být na molekulární úrovni ověřena diagnóza pacientů a rozlišeny formy zděděné a mutace vzniklé de novo.
Závěr.
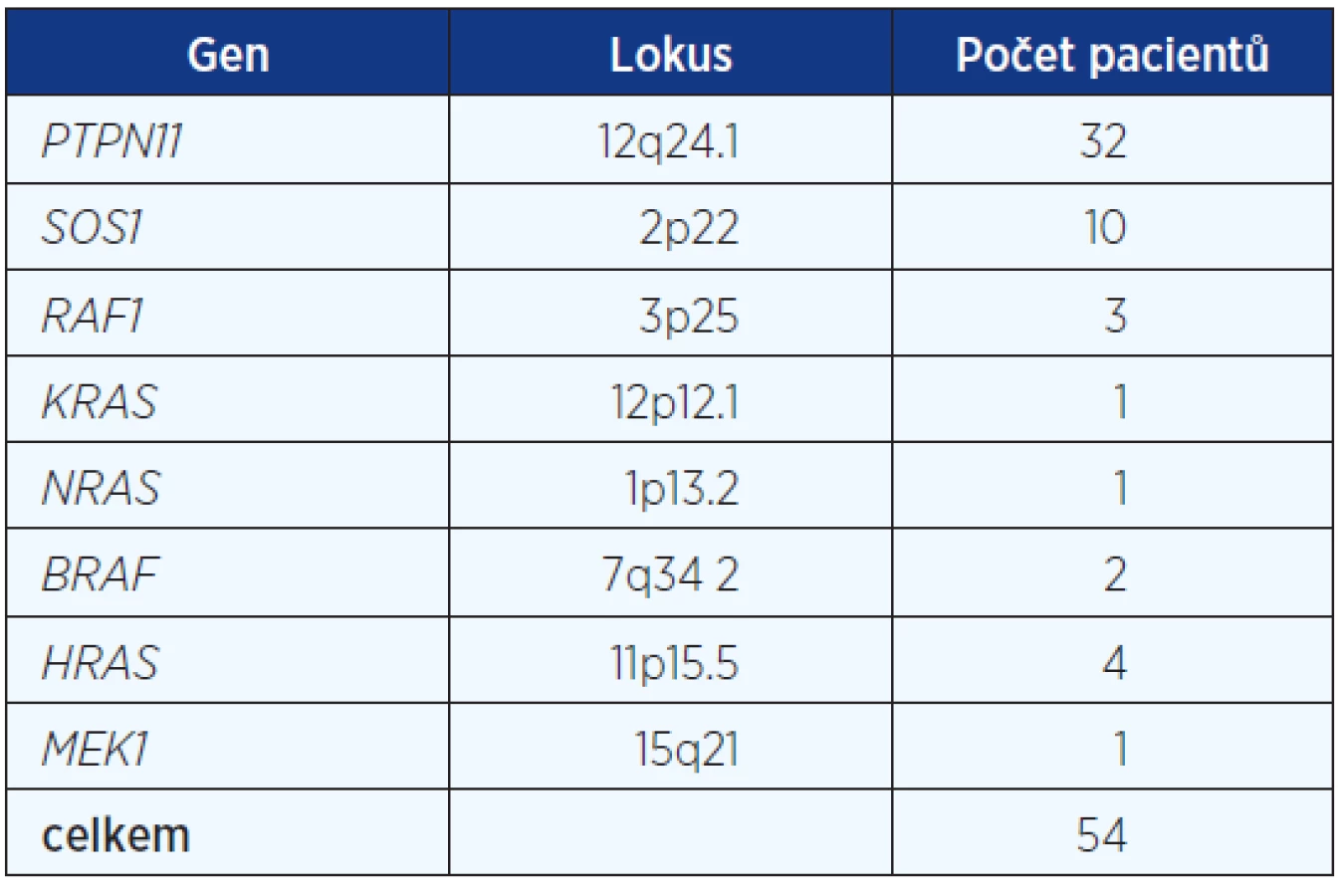
V souboru 83 pacientů s klinickou diagnózou NCFC syndromu byla u 32 detekována mutace v PTPN11 genu, u deseti mutace v SOS1 genu, u čtyř mutace v HRAS genu, u tří pacientů mutace v RAF1 genu, u dvou pacientů mutace v BRAF genu a mutace v MEK1, KRAS a NRAS genech detekována vždy u jednoho pacienta. Mutace de novo byla zjištěna u 35 pacientů a u 19 pacientů byla mutace děděna. Selekční koeficient 0,60 (29 de novo ze 48 ) byl zjištěn u pacientů s Noonanovým a LEOPARD syndromy (mutace v PTPN11, SOS1 a RAF1, KRAS a NRAS genech), syndromy Costello a kardiofaciokutánní byly ve všech šesti případech důsledkem čerstvé mutace v HRAS a BRAF genech. U 29 pacientů nemohla být diagnóza NCFC syndromu potvrzena, mutace nebyla v žádném z osmi genů detekována.
Klíčová slova:
vyšší věk rodičů při porodu dětí – mutagenní efekt, chromozomální aberace – genové mutace – manifestace čerstvých mutací – selekční koeficient – mutační poměry
Úvod
Mutace, změny genetické výbavy, mohou postihnout celou chromozomální sadu (polyploidie), jednotlivé chromozomy (numerické aberace, aneuploidie) nebo jejich části (strukturální aberace jako delece, duplikace, translokace, inverze, prstencové chromozomy) nebo jednotlivé geny (genové mutace dominantního či recesivního charakteru na autozomech či gonozomech). Mutace může postihnout pohlavní buňky (germinální mutace, předávané do dalších generací) nebo nepohlavní buňky (somatické mutace, jejichž výsledkem je mozaika, kdy organismus má nejméně dvě linie buněk různého genotypu). Časně postzygoticky vzniklé mozaiky narušují ontogenezi a projeví se malformacemi, pozdní postzygotické po ukončené ontogenezi mohou dát vznik nádoru, mohou však postihnout jen buňky pohlavních žláz – gonadální mozaiky). Mutace většinou probíhají od normální „divoké“ formy k patogenní, vzácněji obráceně (zpětné mutace jako uniparentální dizomie, izodizomie, zpětné mutace časté u syndromů chromozomální instability jako Fanconiho anémie). Mutace mohou vznikat spontánně, nebo jsou indukované vlivem mutagenů. Mutagenní efekt vyššího věku matek na vznik chromozomálních aberací jejich plodů (trizomie na 21. chromozomu – Downův syndrom, trizomie 18. chromozomu – Edwardsův syndrom, trizomie X chromozomu – superfemale syndrom, Klinefelterův syndrom) je dobře znám i laické veřejnosti, neboť je důvodem k nabídce genetické prevence starším těhotným, vyšetřením karyotypu plodu. Genetické riziko se pohybuje od 0,5 % do několika procent podle stáří ženy, a tím opravňuje nabídnout nezbytnou invazivní metodu, jejíž riziko představuje 0,3–0,5 %, k získání plodových buněk pro cytogenetické vyšetření. Normální karyotyp plodu tak dovolí vyloučit zvýšené genetické riziko numerické chromozomální čerstvě vzniklé mutace vyplývající z věku matky. Vyšší věk otců, a to také již od 35. roku výše (1–4) má mutagenní efekt především na genové mutace. Relativní riziko je vysoké, obvykle až desetinásobné (např. u achondroplazie s populační incidencí
1 : 20 000, má plod staršího otce pravděpodobnost postižení
1 : 2000, což však absolutně představuje pouze 0,5 ‰). Kromě malého absolutního rizika postižení plodu především velký počet genů, ve kterých mutace může nastat, a zatím úzký záběr molekulárně genetických metod (cílené na jednotlivé geny) nedovoluje nabídnout genetickou prevenci (vyšší riziko spojené s invazivním odběrem plodových buněk, než je riziko genetické, široké spektrum možných genů, mutacemi zasažených, fenotypická manifestace pouze autozomálně dominantních mutací non abiotrofického charakteru a v neposlednířadě i jejich lepší klinická prognóza než u chromozomálních mutací.
Studovali jsme mutagenní vliv vyššího věku otců u pacientů s čerstvě vzniklou mutací, podmiňující autozomálně dominantně dědičný syndrom Noonanové (5, 6) s vysokou populační incidencí (1 : 2000 porodů) a vysokým selekčním koeficientem 0,60. Nižší selekce je známa u LEOPARD asociace, naopak až 100 % u Costello a CFC syndromů (6, 7).
Syndrom Noonanové popsaný v roce 1963 (5) je klinicky charakterizován faciální dysmorfií (antimongoloidní oční štěrbiny, ptózou víček, epikanty, strabismus, anomálií ušních boltců), závažnou kardiální anomálií, nejčastěji progresivní valvulární pulmonální stenózou, benigní hypertrofickou kardiomyopatií, nebo defekty septa, otevřenou tepennou dučejí, anomáliemi větví a. pulmonalis). Bývá přítomen hypogenitalismus, kryptorchismus u chlapců, anomálie hrudníku (soudkovitý, pectus carinatum nebo infundibulare), neprospívání v kojeneckém věku, později růstová porucha z necitlivosti tkání k STH (somatotropní hormon). Charakteristické jsou kučeravé vlasy a snížení vlasové hranice zejména na krku, který může mít ptery-
gium nebo jen kožní řasy. Mentální vývoj je u 80 % pacientů normální, avšak ve srovnání s rodinnými předpoklady většinou horší. Řeč bývá opožděna a hlas hrubší. Pacienti mohou mít relativně větší hlavu, neurosenzorickou sluchovou poruchu, sušší kůži, drobné mamily, vysoké patro, odstálé lopatky, cubiti valgi, kyfoskoliotické změny páteře, krční přespočetná žebra, edémy na dorzech rukou a nohou z dysplazie mízních cév, atypické dermatoglyfické vzory. Klinická prognóza v minulosti nebyla příznivá, neboť jen necelá polovina pacientů zanechávala potomky (selekční koeficient 0,6). Úmrtnost na srdeční anomálie, infekce a neprospívání v kojeneckém období a později na malignity byla vysoká. I zhoršená šance nalézt partnera (neatraktivní facies, nízký vzrůst, hypogonadismus, smyslové poruchy) přispívaly ke genetické letalitě (6).
Nezanechá-li i dlouhověký jedinec potomstvo, je podíl selekce kompenzován podle Hardyho-Weinbergova zákona o rovnováze mezi mutacemi a selekcí vysokou mutační intenzitou. NCFC syndromy jsou geneticky i molekulárně heterogenní, v současnosti je známo devět genů, jejichž mutace se manifestují spektrem příznaků charakteristických pro Noonanové syndrom (6–8). Zajímal nás podíl čerstvých mutací v souboru našich pacientů a věk otce při jejich narození, resp. koncepci.
Soubor pacientů a použité metody
Soubor pacientů
V roce 1999 byl identifikován PTPN11 gen odpovědný za manifestaci fenotypu Noonanova syndromu, čímž byla otevřena cesta k potvrzení diagnózy na DNA úrovni a ověření přítomnosti mutace u rodičů pacienta pro odlišení čerstvých a děděných forem. Shromáždili jsme soubor 83 pacientů s klinickou diagnózou Noonanova, LEOPARD, Costello and CFC syndromy, u všech pacientů byl sestaven čtyřgenerační rodokmen, zaznamenána podrobná těhotenská, osobní, pracovní anamnéza a po písemném souhlasu rodin odebrán vzorek EDTA krve. Biologický materiál pacientů, jejich rodičů a příp. sourozenců jsme odeslali v rámci meziuniverzitní spolupráce na zahraniční pracoviště (v současnosti je spoluautor ředitelem IHG v Magdeburgu), neboť v té době metoda u nás nebyla dostupná. V průběhu desetileté spolupráce byly identifikovány další geny RAF1, MEK1, HRAS, NRAS, BRAF, KRAS, SOS1, RIT1 odpovědné za manifestaci fenotypu Noonanové syndromu a tyto geny s výjimkou RIT1 genu byly vyšetřeny postupně i u našich pacientů.
Z rodokmenů jsme analyzovali věky rodičů pacientů, údaje o jejich styku s mutageny v prekoncepčním období a výskyt NCFC v rodinách.
Použité metody
Genomová DNA byla extrahována z EDTA krevních vzorků. Postupně bylo provedeno v každém vzorku sekvenování osm genů (9, 10). Vyšetřeny byly geny: PTPN11 lokalizovaný na 12q34.13, SOS1 na 2p22, RAF1 na 3p25, KRAS na 12p12.1, NRAS na 1p13.2, BRAF na 7q34, HRAS na 11p15.5 a MEK1 (MAP2K1) na 15q21. Gen RIT1 byl identifikován až v roce 2013 a u našeho souboru nebyl vyšetřován.
Výsledky
Celkem jsme získali genealogická, anamnestická a klinická data a vzorky DNA od 83 pacientů s klinickou diagnózou Noonanova syndromu. Mutace v některém z odpovědných genů byla potvrzena u 54, tj. 65 %. Zastoupení jednotlivých mutovaných genů v našem souboru je uvedeno v tabulce 1.
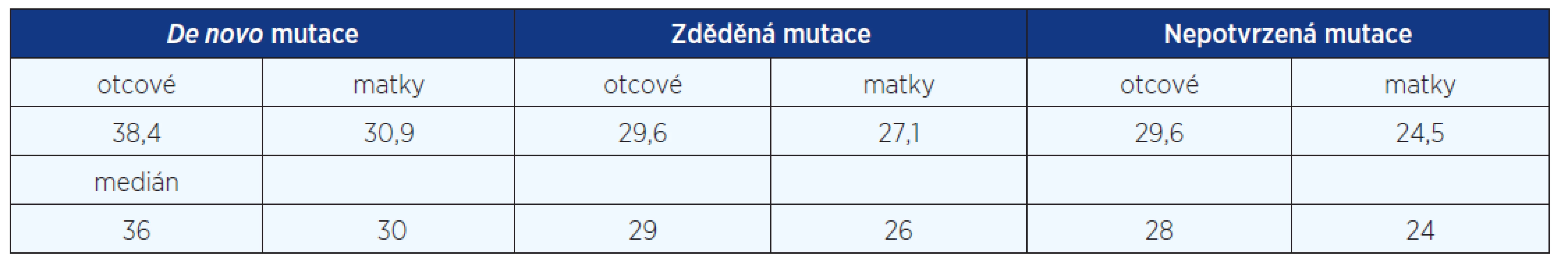
Na podkladě DNA analýzy v rodinách pacientů s potvrzenou diagnózou jsme zjistili 19 děděných mutací, tj. 35 %, 35 pacientů bylo důsledkem mutace de novo (65 %). Styk s mutageny nebyl udáván rodiči našich 57 dětských pacientů. U 26 dospělých pacientů nebylo již možné od rodičů data o styku s mutageny v prekoncepčním období získat. Analyzovali jsme věk rodičů ve skupině pacientů se zděděnou mutací a ve skupině pacientů s izolovaným, prvním výskytem afekce v rodině. Rodiče pacientů z čerstvých mutací byli starší. Rodiče pacientů s de novo mutací průměrného věku 38,4 roku (otcové) a 30,9 roku (matky), rodiče pacientů zděděných mutací 29,6 roku (otcové) a 27,1 roku (matky), rodiče pacientů se suspektní, ale nepotvrzenou diagnózou NCFC 29,6 roku (otcové) a 24,5 roku (matky). Signifikantní rozdíl byl zjištěn jen u věku otců de novo mutací oproti otcům pacientů zděděných mutací a pacientů se suspektní, ale nepotvrzenou diagnózou NCFC (P = 0,01 dle Studentova t-testu). Podíl čerstvých mutací je poměrně vysoký (fittness 0,35) a svědčí stále o závažnosti klinické prognózy pacientů se syndromem Noonanové, Costello a CFC (tab. 2).
Diskuze
Před 40 lety se naši studenti v rámci studentské vědecké činnosti zabývali vlivem věku otce při početí na vznik genových mutací a první manifestaci autozomálně dominantně dědičné neurofibromatózy von Recklinghausenovy, Marfanova syndromu, tuberózní sklerózy a gonozomálně recesivně dědičné Duchennovy muskulární dystrofie v rodině. Ověření diagnózy molekulárně genetickým vyšetřením nebylo tehdy ještě možné, a proto byly zvoleny afekce s dobře rozpoznatelným fenotypem. Z těchto studentských prací vysvítalo, že více než 50 % pacientů jsou prvními nemocnými ve své rodině, a tedy s vysokou pravděpodobností důsledkem čerstvé mutace. U autozomálně dominantně dědičných poruch byl sledován věk otce, u gonozomálně recesivních věk maternálního děda a ve všech skupinách byl zjištěn významně vyšší věk otců (resp. maternálních dědů) pacientů z čerstvé mutace oproti otcům/maternálním dědům pacientů, v jejichž rodině se afekce již dědila po generace. V posledním desetiletí je studován mutagenní vliv věku otce (1–4).
K novému zájmu o mutagenní vliv věku otců vedou změněné rodičovské zvyklosti (mnohočetná manželství s novým potomstvem) a ovšem i dostupnost exaktní diagnózy choroby molekulárně genetickým vyšetřením.
V posledním desetiletí je možné potvrdit diagnózu NCFC přímou DNA analýzou, detekcí odpovědné mutace (6–8), která tak dovolí odlišit vyšetřením rodičů i děděné mutace od čerstvě vzniklých. Vysoký podíl potvrzených mutací u pacientů s klinickou diagnózou Noonanova syndromu (65 %) svědčí o dostatečně charakteristickém fenotypu pacientů (5–7), nicméně je u nás afekce zřejmě poddiagnostikována, neboť na základě udávané populační incidence 1 : 2000 je očekáváno narození 50 pacientů ročně. V našem souboru je však v jednom roce narozeno tři až pět pacientů, tedy necelá desetina očekávaného počtu v období, kdy jsme zajišťovali molekulárně genetickou diagnostiku pacientů z celé republiky v zahraničí. Dosud vysoký podíl čerstvých mutací mezi pacienty s NCFC syndromy svědčí o trvající závažnosti klinické prognózy afekce. Ve španělské studii bylo 35 % mutací v genu PTPN11 vzniklých čerstvě (7). Geneticky letálních, selekcí vyloučených z přenosu do další generace, je v našem souboru 65 %, což lze vysvětlit zařazením pacientů s Costello a CFC syndromů, u kterých se pro závažnou klinickou prognózu (hluboká mentální deficience a vysoká dětská úmrtnost) s reprodukcí nesetkáváme.
Zvýšené genetické riziko čerstvé genové mutace v důsledku věku otců nad 35 let není dosud preventabilní medicínskými metodami. Lze mu ale snadno předcházet včasným otcovstvím. Pokud by toho osvěta docílila, kromě osobních problémů by se podařilo i zlevnit výdaje na zdravotní péči (o postižené děti i náklady na procedury prenatální diagnostiky).Poděkování patří rodinám pacientů s klinickou diagnózou Noonanova syndromu za trpělivou a vstřícnou spolupráci.
Seznam použitých zkratek
EDTA ethylen diamino-tetraacetic acid
NCFCs neurokardiofaciokutánní symptomatiky
STH somatotropní hormon
ADRESA PRO KORESPONDENCI:
prof. MUDr. Eva Seemanová, DrSc.
Oddělení klinické genetiky
Ústav biologie a lékařské genetiky 2. LF UK
V Úvalu 84, 150 06 Praha 5 – Motol
e-mail: eva.seemanova@lfmotol.cuni.cz
Sources
1. de la Rochebrochard E, Thonneau P. Paternal age and maternal age are factors for miscarriage, results of a multicentre European study. Hu Reprod 2002; 17(6): 1649–1656.
2. Wiener-Megnazi Z, Auslender R, Dirnfeld M. Advanced paternal age and reproductive outcome. Asian J Androl 2012; 14(1): 69–76.
3. Balasch J, Gratacos E. Delayed childbearing: effects on fertility and the outcome of pregnancy. Curr Opin Obstet Gynecol 2012; 24(3): 187–193.
4. Alio AP, Salihu HM, McIntosh C, August EM, Weldeselasse H, Sanchez E, Mbah AK. The effect of paternal age on fetal birth outcomes. Am J. Mens Health 2012; 6(5): 427–435.
5. Noonan JA, Elinke DA. Associated noncardiac malformations in children with congenital heart disease. J Pediatr 1963; 63 : 469–474.
6. http://OMIM org/entry
7. Ferrezo GB, Baldassarre G, Delmonaco AG, Biamano E, Bananda E, Carta C, Rossi C, Silengo MC. Clinical and molecular characterization of 40 patinets with Noonan syndrome. Eur J Med. Genet 2008; 51(6): 566–572.
8. Ezguieta B, Santorné JL, Carcavillo A, Guillen-Navarro E, Perez-Aytes A, Sanchez del Pozo J, Garcia-Minaur S, Castillo E, Alonso M, Vendrell T, Santoria A, Maroto E, Gallus L. Alterations in RAS-MAPK genes in 200 spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol 2012; 65(5): 447–455.
9. Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach G, Helter R, Horn D., Hübner CA, Korenke GC, König R, Kress W, Krüger G, Meinecke P, Mücke J, Plecko B, Rossler E, Schinzel A, Schultze A, Seemanová E, Seidel H, Spranger S, Tuysuz B, Uhring S, Wieczorek D, Kutsche K, Zenker M. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndromes. Clin Genet 2007; 73(1): 62–70.
10. Cristea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts A-E, Lepri F, Merblitz-Zahradnik T, König R, Kratz CP, Panta-leoni F, Dentici ML, Joski VA, Kucherlapati RS, Mazzanti L, Mundlos S, Patton MA, Silengo MC, Rossi C, Zampino G, Digillo C, Stuppia L, Seemanová E, Pennacchio LA, Gelb BD, Dallapiccola B, Wittinghofer A, Ahmadian MR, Tartaglia M, Zenker M. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 2010; 42(1) 27–29.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Perioperative chemotherapy in gastric cancer treatment – the surgeon’s view
- Mutagenic effect of advanced paternal age in neurocardiofaciocutaneous syndrome
- Calcium, dairy products and weight reduction
- Basic biological roles of galectins in tissue repair and tumor growth