
Loss of the p53/p63 Regulated Desmosomal Protein Perp Promotes Tumorigenesis
Dysregulated cell–cell adhesion plays a critical role in epithelial cancer development. Studies of human and mouse cancers have indicated that loss of adhesion complexes known as adherens junctions contributes to tumor progression and metastasis. In contrast, little is known regarding the role of the related cell–cell adhesion junction, the desmosome, during cancer development. Studies analyzing expression of desmosome components during human cancer progression have yielded conflicting results, and therefore genetic studies using knockout mice to examine the functional consequence of desmosome inactivation for tumorigenesis are essential for elucidating the role of desmosomes in cancer development. Here, we investigate the consequences of desmosome loss for carcinogenesis by analyzing conditional knockout mice lacking Perp, a p53/p63 regulated gene that encodes an important component of desmosomes. Analysis of Perp-deficient mice in a UVB-induced squamous cell skin carcinoma model reveals that Perp ablation promotes both tumor initiation and progression. Tumor development is associated with inactivation of both of Perp's known functions, in apoptosis and cell–cell adhesion. Interestingly, Perp-deficient tumors exhibit widespread downregulation of desmosomal constituents while adherens junctions remain intact, suggesting that desmosome loss is a specific event important for tumorigenesis rather than a reflection of a general change in differentiation status. Similarly, human squamous cell carcinomas display loss of PERP expression with retention of adherens junctions components, indicating that this is a relevant stage of human cancer development. Using gene expression profiling, we show further that Perp loss induces a set of inflammation-related genes that could stimulate tumorigenesis. Together, these studies suggest that Perp-deficiency promotes cancer by enhancing cell survival, desmosome loss, and inflammation, and they highlight a fundamental role for Perp and desmosomes in tumor suppression. An understanding of the factors affecting cancer progression is important for ultimately improving the diagnosis, prognostication, and treatment of cancer.
Published in the journal:
Loss of the p53/p63 Regulated Desmosomal Protein Perp Promotes Tumorigenesis. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001168
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001168
Summary
Dysregulated cell–cell adhesion plays a critical role in epithelial cancer development. Studies of human and mouse cancers have indicated that loss of adhesion complexes known as adherens junctions contributes to tumor progression and metastasis. In contrast, little is known regarding the role of the related cell–cell adhesion junction, the desmosome, during cancer development. Studies analyzing expression of desmosome components during human cancer progression have yielded conflicting results, and therefore genetic studies using knockout mice to examine the functional consequence of desmosome inactivation for tumorigenesis are essential for elucidating the role of desmosomes in cancer development. Here, we investigate the consequences of desmosome loss for carcinogenesis by analyzing conditional knockout mice lacking Perp, a p53/p63 regulated gene that encodes an important component of desmosomes. Analysis of Perp-deficient mice in a UVB-induced squamous cell skin carcinoma model reveals that Perp ablation promotes both tumor initiation and progression. Tumor development is associated with inactivation of both of Perp's known functions, in apoptosis and cell–cell adhesion. Interestingly, Perp-deficient tumors exhibit widespread downregulation of desmosomal constituents while adherens junctions remain intact, suggesting that desmosome loss is a specific event important for tumorigenesis rather than a reflection of a general change in differentiation status. Similarly, human squamous cell carcinomas display loss of PERP expression with retention of adherens junctions components, indicating that this is a relevant stage of human cancer development. Using gene expression profiling, we show further that Perp loss induces a set of inflammation-related genes that could stimulate tumorigenesis. Together, these studies suggest that Perp-deficiency promotes cancer by enhancing cell survival, desmosome loss, and inflammation, and they highlight a fundamental role for Perp and desmosomes in tumor suppression. An understanding of the factors affecting cancer progression is important for ultimately improving the diagnosis, prognostication, and treatment of cancer.
Introduction
Carcinomas, or cancers of epithelia, comprise approximately 90% of all human cancers [1]. Cancers of the stratified epithelia, such as the skin and the tissues of the head and neck, are common and can display poor prognosis in advanced stages [2], [3]. Elaborating the normal architecture and homeostatic mechanisms of stratified epithelia, and how these are perturbed in cancer progression and metastasis, can provide significant insight into carcinoma development.
The genesis and maintenance of stratified epithelia, such as the epidermis, requires the coordinated regulation of proliferation, adhesion, migration, and differentiation [4]. Progenitor cells of the basal, inner layer of the epidermis renew the epidermis by initially proliferating, then exiting the cell cycle, detaching from the basement membrane, migrating, and differentiating to form the upper layers of the skin. The cells of stratified epithelia require a great deal of plasticity to permit division and migration of cells during the differentiation process while still providing a protective barrier to prevent dehydration and infection. This plasticity relies on the modulation of various intercellular adhesion junctions, including tight junctions, adherens junctions and desmosomes [5]. While adherens junctions generally help to maintain cell-cell adhesion in epithelial tissues, desmosomes have been found to be particularly important for endowing epithelia with the strength needed to withstand mechanical stress, via anchorage to the intermediate filament network [5], [6]. The crucial nature of desmosomes in maintaining the integrity of stratified epithelia is underscored by the observation that mutation or inactivation of several desmosomal proteins is linked to human epithelial diseases [7].
The importance of dysregulated adhesion in cancer has been best understood from analysis of adherens junctions. Adherens junctions promote intercellular adhesion through homotypic interactions between transmembrane E-cadherin molecules at the plasma membrane [8], [9] and connections to the actin cytoskeleton via alpha-catenin and beta-catenin [10], [11]. In human cancers, inactivation of E-cadherin, through mutation, promoter methylation, or transcriptional repression, is associated with epithelial to mesenchymal transition (EMT), progression from adenoma to carcinoma, and the acquisition of metastatic potential [12]–[16]. The importance of this inactivation is underscored by functional studies in mouse models showing that inhibition of E-cadherin in certain tumor-prone strains facilitates invasion and metastasis [17]. In addition, compound mutant mice lacking both E-cadherin and p53 in the mammary epithelium exhibit accelerated tumor development and increased metastasis relative to mice lacking only p53 [18]. Loss of p120-catenin, a key regulator of E-cadherin stability, also promotes neoplasia, in the salivary gland and the skin [19]–[21]. Similarly, specific deletion of Alpha-catenin in the mouse epidermis results in squamous cell carcinoma development [21], [22].
While a variety of studies have implicated inactivation of adherens junctions in tumor development and progression, the contribution of desmosome loss to carcinogenesis remains largely unexplored. Desmosome complexes form when the desmosomal cadherins, desmogleins and desmocollins, participate in heterotypic interactions that bring the plasma membranes of adjacent cells in close apposition [23]–[25]. The cytoplasmic tails of these cadherins interact with plakoglobin [26], [27] and plakophilins [28]–[30], which connect to the intermediate filament cytoskeleton via desmoplakin [31]–[33]. An impediment to studying desmosomes in a genetic cancer model has been the high frequency of embryonic or perinatal lethality observed in various knockout mice lacking desmosomal components, precluding long-term tumor studies [34]. Additionally, correlative studies examining expression patterns of desmosomal components during human cancer progression have yielded conflicting results. Several studies have suggested that downregulation of desmosome components, including desmoglein 3, desmoglein 2, plakoglobin, and desmoplakin, occurs during the progression of a variety of cancers in humans and is often correlated with and predictive of tumor metastasis [35]–[38]. In contrast, other studies have documented the overexpression of desmosome components during the progression of diverse cancers, and this pattern is associated with poor prognosis [39]–[41]. The use of tractable genetic systems is therefore critical for unraveling the contribution of desmosomes to cancer development.
The Perp tetraspan membrane protein was originally identified as a transcriptional target of the p53 tumor suppressor upregulated during apoptosis [42]. Subsequent analysis of Perp knockout mice revealed an additional function for Perp as a target of the p53-related transcription factor, p63, involved in maintaining epithelial integrity by promoting desmosomal cell-cell adhesion [43]. Perp−/ − mice display postnatal lethality accompanied by dramatic blisters throughout their stratified epithelia, including the oral mucosa and skin. Electron microscopy and biochemical analyses established that the blistering phenotype observed in the Perp-deficient epithelia is accounted for by both a reduction in desmosome number and compromised desmosome complex formation. Immunogold electron microscopy demonstrating localization of Perp to desmosomes conclusively established a clear role for Perp as a novel critical component of the desmosome in the skin and other stratified epithelia.
Here, we aim to characterize the consequence of Perp-deficiency for UVB-induced squamous cell carcinoma (SCC) development in the skin, where Perp plays a pivotal role in cell-cell adhesion and tissue homeostasis. Using conditional Perp knockout mice to selectively ablate Perp expression in stratified epithelia, we reveal an important role for Perp as a tumor suppressor in this model for human skin cancer. These results provide definitive genetic evidence that loss of a desmosome component can in fact promote tumorigenesis in vivo, thereby enhancing our understanding of the factors driving tumor progression.
Results
Loss of Perp promotes tumorigenesis
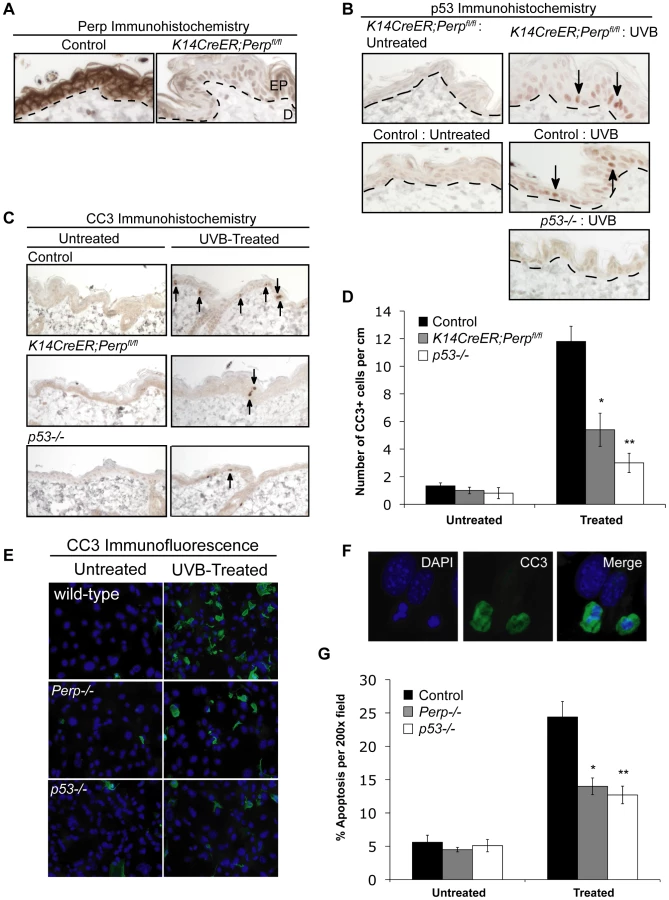
To characterize the function of Perp during tumorigenesis, we examined its role in cancer of the epidermis, where it is critical for tissue integrity and homeostasis through its role in desmosomal cell-cell adhesion [43]. We examined the tumor predisposition of Perp-deficient mice using a well-defined model for squamous cell carcinoma (SCC) development in which mice are exposed to chronic UVB irradiation [44]. This model provides an accurate mimic of human SCC development, which is similarly driven by UVB irradiation. As Perp constitutive null mice die postnatally, we utilized conditional Perp knockout mice (Perpfl/fl; fl = floxed) expressing a tamoxifen-inducible K14CreER transgene to drive tissue-specific deletion of the Perp locus in the epidermis [45], [46]. Immunofluorescence confirmed that Perp expression was successfully ablated in the epidermis of the majority of these mice 4 weeks after tamoxifen injection (Figure 1A). To induce SCC development, tamoxifen-treated 10-week old control and Perpfl/fl mice expressing a K14CreER transgene were exposed to chronic treatments (2.5 kJ/m2) of UVB irradiation three times weekly for 30 weeks (Figure 1B). Interestingly, Kaplan-Meier analysis revealed that mice lacking Perp in the epidermis developed SCCs with reduced average latency (32 wks) compared to control mice (51 wks; Figure 1C). In addition, the average number of SCCs per K14CreER;Perpfl/fl mouse was far greater than in control animals (Figure 1D). The prominent early tumor development and increased tumor number in Perp-deficient mice compared to controls suggest that Perp loss promotes tumor initiation. Histological analyses to grade the SCCs according to cellular morphology, invasiveness into the dermis, and overall architecture revealed that SCCs arising in K14CreER;Perpfl/fl mice had a greater propensity to progress to a poorly differentiated stage than tumors arising in control mice, suggesting that Perp loss may also contribute to tumor progression (Figure 1E, 1F). Despite the presence of invasive tumors, however, no metastases were apparent in the liver or lungs of mice from either cohort (data not shown). Together, these findings indicate that Perp is a key suppressor of skin carcinogenesis and provide the first in vivo demonstration that genetic loss of a desmosomal component can lead to accelerated carcinoma development, facilitating both tumor initiation and progression.

Perp is an important mediator of UVB-induced apoptosis
To understand the basis for the tumor development driven by Perp-deficiency, we sought first to determine whether Perp is an important mediator of p53-induced apoptosis in the skin in response to ultraviolet light. Perp plays a cell-type-specific role in p53-mediated apoptosis, being dispensable for apoptosis in fibroblasts but essential for apoptosis of thymocytes and embryonic neurons in response to DNA damage signals [47]. To examine Perp's role in p53-dependent apoptosis in keratinocytes, 6-week old K14CreER;Perpfl/fl and control mice were injected with tamoxifen (which ablated Perp expression in K14CreER;Perpfl/fl mice; Figure 2A), and 4 weeks later, mice were exposed to one dose of 2.5 kJ/m2 of UVB radiation. We confirmed that p53 is induced 24 hours after UVB treatment in the epidermis of both wild-type and K14CreER;Perpfl/fl mice (Figure 2B). Apoptotic indices were then determined by quantifying the number of cleaved Caspase 3-positive cells in the epidermis. Minimal apoptosis was detected in untreated skin of mice of all genotypes (Figure 2C, 2D). While robust apoptosis was observed in the epidermis of wild-type mice in response to UVB radiation, p53 null mice displayed significantly attenuated levels of apoptosis (Figure 2C, 2D). Interestingly, analysis of the epidermis of Perp-deficient mice also revealed diminished apoptosis levels, to an extent nearly equivalent to p53 loss (Figure 2C, 2D). We confirmed these findings by assaying apoptosis in differentiated keratinocytes in vitro, through analysis of both cleaved Caspase 3 positivity and classical apoptotic nuclear morphology and condensed chromatin by DAPI staining [48], [49]. We found that Perp−/− keratinocytes displayed defective apoptosis in response to UVB, similar to p53−/− keratinocytes (Figure 2E–2G). Together, these results demonstrate that Perp plays an important role in UVB-induced apoptosis in both the skin in vivo and keratinocytes in vitro. As p53 inactivation is proposed to promote UVB-induced carcinogenesis by allowing inappropriate survival of cells sustaining UVB-induced damage [50], enhanced survival of Perp-deficient cells after exposure to ultraviolet light could similarly enable tumor initiation.
Desmosome components, but not adherens junction components, are selectively downregulated during tumorigenesis
As a key desmosomal constituent in the epidermis, Perp loss could also promote cancer through effects on cell-cell adhesion. In constitutive Perp knockout mice, Perp loss does not abrogate desmosome formation, but leads to decreased numbers of desmosomes and reduced stability of desmosomal components [43]. Simple immunofluorescence analysis of desmosome proteins in Perp−/− newborn skin or K14CreER;Perpfl/fl adult mouse skin does not show perturbed membrane localization of desmosomal components ([43], Figure 1A, Figure 3A), and thus it is necessary to use a biochemical assay to show that desmosomes are functionally compromised upon Perp loss. This solubility assay is based on the fact that properly formed desmosomal complexes can only be solubilized by chaotropic agents, whereas improperly assembled desmosomal components can be solubilized by the nonionic detergent Triton X-100 [51]. We found that the desmosomal constituents Desmoglein 1/2 and Plakoglobin displayed enhanced Triton X-100-solubility in skin from K14CreER;Perpfl/fl mice compared to skin from control mice (Figure 3B), confirming that acute deletion of Perp leads to impaired desmosome function similar to that observed in constitutive Perp−/− mice [43].

To determine whether Perp ablation might facilitate tumorigenesis by promoting complete desmosome loss, tumors from Perp-deficient mice were analyzed for the expression of Desmoglein 1/3 and Plakoglobin. Both the percentage of epithelial cells expressing each marker at the plasma membrane and the intensity of staining were measured. Staining for each antigen in tumors was categorized as high (>70%), medium (30–70%), or low (<30%) level expression. Analysis of the tumors in the K14CreER;Perpfl/fl mice revealed that the majority of lesions expressed low levels of desmosome components, suggesting that desmosome dissolution had occurred (Figure 3C, 3D). Thus, complete desmosome destabilization can occur during tumorigenesis.
To assess whether this loss of desmosomal component expression reflected a general change in differentiation status of the cells during tumorigenesis, such as Epithelial to Mesenchymal Transition (EMT) [52], we stained tumors for markers of adherens junctions. Interestingly, most cells in the tumors displayed robust membrane staining for both E-cadherin and Beta-catenin, suggesting that adherens junctions remained intact (Figure 3C, 3D). The observation that adherens junctions are maintained suggests that desmosome loss does not promote tumorigenesis through a general trans-differentiation mechanism, but rather through a more specific mechanism related to changes caused by complete desmosome-deficiency. Accordingly, staining of tumors arising in the Perp-deficient mice for Smooth Muscle Actin and Keratin 8 revealed a lack of expression of these markers (data not shown). Together, these data indicate that Perp loss can facilitate desmosome downregulation and that direct loss of desmosomes contributes to tumor development, but in a manner distinct from that of adherens junction dysfunction.
We also sought to establish whether desmosome destabilization occurred during SCC development in control animals by staining tumors from these mice for desmosomal proteins. Whereas non-lesional skin in the control mice exhibited normal expression of Perp, Desmoglein 1/3, and Plakoglobin (data not shown), all of the tumors from these mice displayed low level expression of one or more desmosomal components (Figure 3E). Interestingly, in this case, Desmoglein 1/3 appeared to be lost most commonly. Since desmosomes were intact at the beginning of the study, these data further suggest that targeted downregulation of the desmosome is an active characteristic of tumor development, and that weakened desmosomal adhesion, as in the case of Perp-deficiency, facilitates this downmodulation. In addition, analysis of adherens junction components in control tumors revealed intact E-cadherin and Beta-catenin expression at the plasma membrane, similar to tumors in the K14CreER;Perpfl/fl mice (Figure 3E). Thus, downregulation of desmosomal constituents with maintenance of adherens junctions is a general feature of SCC development.
Perp expression is downregulated in human SCCs
Our findings suggest that desmosome loss, coupled with maintenance of adherens junctions, may represent an important stage of tumorigenesis. To determine the relevance of this observation to human SCC development, we stained a panel of samples reflecting different stages of human skin SCC development, ranging from actinic keratoses to moderately differentiated SCCs, for both PERP and E-cadherin. Each sample was scored based on the percentage of epithelial cells exhibiting membrane staining in addition to the intensity of the membrane staining in the epithelial portion of each tumor. Samples with greater than 10% of the epithelial cells expressing intense membrane staining were given a positive score, while those with less than 10% of the epithelial cells expressing either PERP or E-cadherin were given a negative score. We first noted a significant decline in the percentage of samples displaying Perp expression in the transition between AKs and SCCIS, suggesting that Perp expression is downregulated during tumor progression (p = 0.049, z-test). Interestingly, while a subset of all tumors displayed intact PERP and E-cadherin expression and another group showed complete lack of expression of both PERP and E-cadherin, the majority of tumors (57%) lacked PERP expression while retaining E-cadherin expression, akin to our observations in the mouse SCC model (Figure 4A, 4B). These findings suggest that PERP loss with retention of E-cadherin represents a significant phase of human tumorigenesis. Moreover, since the vast majority of all SCCs examined (93%) retain E-cadherin expression, and since E-cadherin loss is thought to be a late event in tumorigenesis, our data suggest a temporal sequence whereby PERP loss occurs before E-cadherin loss in the progression of human SCC.

Perp ablation induces an inflammatory gene signature
To understand how Perp-deficiency might cooperate with chronic UVB exposure to promote cancer, we performed microarray analyses to identify those genes whose expression is altered upon Perp loss. Cohorts of K14CreER;Perpfl/fl and control mice were generated and skin was processed for RNA analysis two weeks post tamoxifen injection, a timepoint at which we could first detect Perp protein expression loss throughout the epidermis (Figure 5A). Using Significance Analysis of Microarrays (SAM) [53], we identified a panel of 143 genes that were differentially regulated in the K14CreER;Perpfl/fl mice compared to controls (FDR = 10%; Figure 5B). These included 51 upregulated and 92 downregulated genes. We next classified the genes changing with altered Perp status based on Gene Ontology functional annotation. The three largest statistically significantly enriched groups of genes upregulated upon Perp ablation were in the categories of metabolic process, transport, and immune system process (Figure 5C). Significantly enriched classes of genes downregulated upon Perp loss included those linked to developmental processes and cell communication (Figure 5C). To identify genes whose induction might promote cancer, we examined those genes that were most highly upregulated in the absence of Perp. Interestingly, upon examination of the list of genes induced 3-fold or greater in the absence of Perp, we discovered that the most highly induced were several inflammation-related genes (Figure 5D). These included: Interleukin 1 Family member 6 (Il1f6), an inflammatory cytokine sufficient to induce an inflammatory response when expressed in keratinocytes of transgenic mice [54]; S100a9, a calcium binding protein with cytokine-like function in inflammation and cancer [55], [56]; Chitinase 3-like 1 (Chi3l1), a mammalian chitinase involved in enhancing inflammation as well as angiogenesis and extracellular matrix remodeling, thereby promoting tumorigenesis [57]; and Chemokine ligand 20 (Ccl20), an established chemoattractant for subsets of lymphocytes and dendritic cells which is also known to promote tumor growth [58]–[61]. Additionally, this list included Il22ra, a class II cytokine receptor and mediator of innate immune responses [62]. Quantitative RT-PCR analysis verified that the expression of these genes is indeed significantly induced in the skin of Perp-deficient mice (Figure 5E, data not shown).

Chronic UVB exposure combined with Perp loss leads to the recruitment of immune cells to the skin
The induced inflammatory gene signature in Perp-deficient mice could reflect gene expression changes intrinsic to keratinocytes, or, alternatively, the recruitment of inflammatory cells to the skin of Perp-deficient mice. To distinguish these possibilities, we analyzed untreated K14CreER;Perpfl/fl and control skin samples for the presence of inflammatory cells. Histological staining for T-cells, mast cells, and myeloid cells indicated no difference in immune cell numbers between the Perp-ablated samples and the controls (Figure 5F–5I, data not shown), suggesting that Perp-deficiency induces an inflammatory gene expression program directly in keratinocytes rather than causing recruitment of immune cells to the skin.
The observation that Perp loss triggers the induction of genes known to promote inflammation, combined with the fact that inflammation is causally linked to cancer development [63], provides a rationale for how Perp-deficiency could contribute to cancer. We hypothesized that persistent cytokine/chemokine signaling in K14CreER;Perpfl/fl mice, combined with chronic UVB exposure, might ultimately attract immune cells, thereby promoting tumor formation. To investigate this idea, we queried the presence of inflammatory cells in the skin from a subset of control and K14CreER;Perpfl/fl mice at an intermediate timepoint in the tumor study, after 19 weeks of chronic UVB treatment, by quantifying numbers of myeloid cells, T-cells, and mast cells (Figure 6A–6F). While we did not detect any differences in the number of myeloid cells, assessed by MPO-positivity (Figure 6A, 6B; [64]), we did observe increased numbers of T-cells present throughout the skin of K14CreER;Perpfl/fl mice compared to controls (Figure 6C, 6D). Moreover, we noted a striking increase in mast cell numbers in the skin from K14CreER;Perpfl/fl mice relative to controls (Figure 6E, 6F). As mast cells have been reported to surround tumors in a variety of cancers, including SCCs [65]–[68], and because they have been shown to play a key role in promoting tumorigenesis through the secretion of factors that remodel the tumor microenvironment and stimulate angiogenesis [68]–[71], their presence in the UVB-treated Perp-deficient mouse skin suggests an additional mechanism through which Perp loss may stimulate tumorigenesis.

Discussion
The importance of disrupted cell-cell adhesion for cancer development is underscored by the observed downregulation of adherens junction components during human cancer progression and genetic experiments demonstrating tumor prone phenotypes of mice deficient for E-cadherin, Alpha-catenin, or p120-catenin, components of the adherens junction [19]–[21]. In contrast, the data regarding desmosome protein expression during human cancer progression are conflicting [35]–[41], and the contribution of desmosome dysfunction to cancer development has not been clearly established using in vivo mouse models. Here, we show that loss of the desmosomal component Perp predisposes mice to UVB-induced SCC development by enhancing both tumor initiation and progression. The effects of Perp ablation are at multiple levels, leading both to compromised apoptosis in response to ultraviolet light and loss of desmosomal adhesion (Figure 7). The defective apoptosis could allow the inappropriate survival of damaged cells, which could help initiate tumors. The exact mechanism through which Perp promotes apoptosis remains to be elucidated, but it may relate to Perp function at the desmosome, as apoptotic defects were previously reported in cells lacking either the desmosomal component desmoglein 1 or desmoglein 2 [72], [73]. In addition, compromised UVB-induced apoptosis in the epidermis has also been observed in mice lacking the p53 target gene Noxa, a member of the Bcl-2 family [74]. Our findings indicate that Noxa is insufficient to drive apoptosis in the absence of Perp, and therefore that Perp and Noxa may collaborate to cause apoptosis. In addition, we observe perturbations in desmosomal adhesion. Interestingly, the desmosome downregulation we observe in tumors occurs without adherens junction loss or other signs of EMT, highlighting a specific role for desmosome loss in tumor development. It may be that desmosome loss occurs during early stages of tumorigenesis, facilitating early cancer progression, and that adherens junctions are lost subsequently, thereby promoting invasion and metastasis phenotypes. Our analysis of human SCC samples supports the idea that PERP-deficient, E-cadherin positive samples reflect an important stage of human skin carcinogenesis.

To understand further how Perp-deficiency might enhance tumor development, we examined gene expression profiles upon Perp loss. Interestingly, several of the genes induced upon Perp inactivation are known to be involved in promoting inflammation and tumorigenesis [54]–[62]. Inflammation is a well-established causative factor in tumorigenesis [63], as evidenced by tumor-prone mouse strains deficient for specific subsets of immune or inflammatory cells exhibiting reduced tumor burdens [66], [75]. The induction of a set of inflammation-associated genes presents a plausible explanation for how Perp-deficiency can promote cancer in cooperation with chronic UVB damage. Indeed, we found that Perp-deficiency, in conjunction with chronic UVB exposure, led to the infiltration of T-cells and mast cells. Mast cells can clearly promote tumorigenesis [69]–[71], and their accumulation in the UVB-treated, Perp-deficient epidermis provides another mechanism through which Perp loss can stimulate cancer development (Figure 7).
Some of the previous controversy about whether desmosomal components promote or inhibit cancer may relate to differences in the contribution of desmosomes in different contexts. Depending on the tissue type, the genetic lesions already accrued, and the tissue microenvironment, desmosome-deficiency may have different effects. Indeed, certain proteins, such as Tgf-ß1 and E2f1, can have either pro - or anti-tumorigenic roles depending on the exact setting [76], [77]. Consistent with this notion, skin carcinogenesis experiments in which Perp-deficient mice were treated with DMBA/TPA showed that Perp loss actually hindered the development of papillomas, suggesting that Perp enables the formation of this type of tumor [78]. Importantly, however, loss of the p53 tumor suppressor in this model also reduces papilloma formation, suggesting that this may represent an atypical route to tumorigenesis [79]. Therefore, in the study described here, we sought to analyze Perp function in an accurate model for human cancer, by treating mice with UVB, the causative factor for SCC of the skin.
Our studies also provide insight into mechanisms of p53-mediated tumor suppression in skin cancer. The relevance of p53 in skin cancer development is highlighted by the observations that p53 is mutated in at least 90% of human SCCs [80] and that p53 null mice display an enhanced predisposition to UVB-triggered skin cancer [44], [81]. While p53's ability to drive apoptosis in response to ultraviolet light has been shown to limit SCC formation [82], the molecular pathways underlying p53's tumor suppressor properties are incompletely understood. p53 is a transcriptional activator, but the genes mediating p53 tumor suppressor function have been unclear, as none of the mouse strains deficient for p53 target genes exhibits a spontaneous tumor predisposition [83]. Instead, analysis of target genes in specific contexts may reveal key functions as mediators of p53 tumor suppressor function. This notion is exemplified by studies of the apoptotic target gene Puma, which is important for p53 tumor suppression in the setting of Eμ-myc driven B-cell lymphomas [84]. Our studies have provided important novel insight into pathways of p53 tumor suppression by suggesting that Perp is a critical mediator of p53 tumor suppressor function in UVB-induced SCC development. In addition, although the role of p63 in cancer has been more controversial, Perp loss could also potentially explain how tumors might arise in the absence of p63.
Non-melanoma skin cancer is one of the most common malignancies in the US [85]. Fortunately, identifying SCC lesions before they progress into poorly differentiated tumors is aided both by facile detection and increased awareness of the consequences of chronic sun exposure. However, this is not the case for other stratified epithelia-derived cancers such as head and neck or esophageal cancers, which have poor survival rates [86]. Our findings may provide a framework to better understand how these more deadly diseases progress. Indeed, the loss of desmosomal component expression with maintenance of adherens junction expression observed in the K14CreER;Perpfl/fl mouse tumors and in human skin SCCs was recapitulated in samples derived from humans with head and neck SCCs (data not shown). The idea that desmosome loss may precede adherens junction loss could have important clinical implications. While E-cadherin status can provide a useful prognostic indicator for a variety of epithelial cancers, loss of this marker is associated with late-stage tumor progression [16], [87], [88]. Identifying markers like PERP that are altered earlier during tumorigenesis could potentially enhance diagnosis, grading, and prognostication, leading to more informed choices of therapy. The increased frequency of advanced tumors in the Perp-deficient mice relative to controls supports the idea that human tumors lacking PERP may ultimately progress more aggressively, and thus that PERP status may provide a useful diagnostic or prognostic marker. Indeed, previously published expression profiling studies suggest that PERP may be one in a group of key predictors for patient treatment response in esophageal cancers [89]. Further investigation of the potential diagnostic and prognostic value of PERP expression in human cancers represents an exciting avenue to pursue.
Materials and Methods
Ethics statement
All animal studies were approved by the Stanford University Administrative Panel on Laboratory Animal Care and were performed in strict accordance with IACUC guidelines.
Tumor study
Keratin-14CreERT2 mice were bred to Perpfl/fl conditional mice and kept on a 129/Sv; C57BL/6 mixed background [46]. At 6 weeks of age, 0.1 mg of tamoxifen (Sigma Chemical Corp., St. Louis, MO) diluted first in ethanol then in corn oil was administered to mice for 5 consecutive days via intraperitoneal injection. Four weeks post-injection, mice began chronic UVB treatments (2.5 kJ/m2, three times a week, for 30 weeks). Mice were shaved on a weekly basis and treated using Kodacel-filtered FS40 sunlamps. Mice were placed 5 in a cage and allowed to roam freely during treatment. Cages were rotated along the shelf below the light bulbs before each treatment to compensate for uneven distribution of energy along the bulbs. Mice were monitored for tumor incidence by visual inspection.
Immunofluorescence/immunohistochemistry
Tissue samples were fixed overnight in 10% formalin, processed, and embedded using standard procedures. Samples were deparaffinized, rehydrated, and unmasked using Trilogy (Cell Marque, Rocklin, CA) in a pressure cooker for 15 minutes according to the manufacturer's instructions. Samples were then rinsed in phosphate buffered saline (PBS) and blocked in PBS containing 5% normal goat serum (Sigma Chemical Corp.), 2.5% bovine serum albumin (Sigma Chemical Corp.), and 0.01% Triton X-100 (Fisher Scientific, Pittsburgh, PA). Sections were incubated in primary antibody overnight at 4°C, rinsed in PBS with 0.01% Tween-20 (Fisher Scientific), incubated with secondary antibody and 1 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Chemical Corp.) for 1 hr at 37°C, and washed in PBS. Samples were mounted with Mowiol (EMD Chemicab, Gibbstown, NJ). Fluorescence images were examined using a Leica DM6000B microscope (Leica Microsystems, Bannockburn, IL), and images were acquired using a Retiga Exi Camera (Q imaging, Surrey, British Columbia, Canada) and Image Pro 6.2 software from Media Cybernetics (Silver Spring, MD).
Keratinocyte culture and apoptosis assays
Keratinocytes were derived from P.05–P1.5 mouse skin as described [43]. Cells were grown on collagen/fibronectin-coated dishes and maintained in an undifferentiated state by growing the cells in low calcium EMEM (Lonza, Basel, Switzerland) containing 0.05 mM calcium, 8% dialyzed FCS, and antibiotics. Cells were then differentiated for 24 hrs in the same media as the undifferentiated cells, except the calcium concentration was raised to 2 mM. For immunofluorescence, keratinocytes were grown on collagen/fibronectin-coated glass coverslips. For UVB treatment, media was removed and cells were washed once in PBS, then treated with 1 kJ/m2 UVB radiation using a Kodacel filter (Eastman Kodak, Rochester, NY) to block residual UVC rays. After 48 hours, cells were fixed in cold methanol for 20 minutes at −20°C. Cells were stained with rabbit anti-cleaved Caspase 3 antibodies (Cell Signaling, Beverly, MA), followed by staining with FITC-anti-rabbit antibodies (Vector Laboratories, Burlingame, CA) and DAPI and mounting using Mowiol.
In vivo apoptosis assays
Cohorts of K14CreER;Perpfl/fl mice were generated, and at 6 weeks of age mice were injected with tamoxifen as described above. Four weeks later the dorsal skin of mice was shaved. Mice were placed underneath a Kodacel filter and allowed to roam freely in their cage during UVB treatment. Half of the dorsal skin was exposed to a one time dose of 2.5 kJ/m2 of UVB irradiation while the other half was blocked. 24 hours later the dorsal skin of the mice was collected and immunostained for cleaved Caspase 3. Apoptosis, indicated by cleaved Caspase 3-positivity, was quantified in at least 2–3 cm of skin per mouse.
Antibodies
Primary antibodies against Perp [43], Desmoglein 1 (Santa Cruz Biotechnology, Santa Cruz, CA), Plakoglobin (clone 11E4; Invitrogen), Keratin 14 (Covance, Princeton, NJ), Alpha-tubulin (Sigma Chemical Corp.), E-cadherin (Invitrogen), GAPDH (Fitzgerald Industries, Acton, MA), Plakoglobin (1408; gift of K. Green), Smooth Muscle Actin (Santa Cruz Biotechnology), Desmoglein 1/3 (clone 32-2B; gift of D. Garrod), Desmoglein 1/2 (4B2; gift of K. Green), cleaved Caspase 3 (Cell Signaling), p53 (Vector Laboratories), Beta-catenin (BD Biosciences, San Jose, CA), Desmoplakin (11-5F; gift of D. Garrod), CD3 (Dako, Denmark), and MPO (AbCam, Cambridge, MA) were used in this study. Secondary antibodies used were: FITC-anti-mouse and FITC-anti-rabbit (Vector Laboratories) and TRITC-anti-chicken, HRP-anti-mouse, and HRP-anti-rabbit (Jackson ImmunoResearch, West Grove, PA).
Solubility assays
For the solubility assay, skin samples were frozen and ground up, resuspended in a 0.1% Triton X-100-based solution, and nutated for 1 hr at 4°C [43]. The insoluble pellet was lysed in 9M urea buffer. Samples were then analyzed using conventional western blotting protocols.
Human tissue microarrays
Archival paraffin embedded tissue blocks were retrieved from the Dermatopathology Section of the Department of Pathology and Dermatology, University of California, San Francisco and from outside pathology laboratories. Tumor-bearing regions from paraffin-embedded, formalin-fixed tissue samples were identified by a dermatopathologist using routine hematoxylin and eosin stained sections. Tissue microarrays comprising 0.6 mm cores of skin actinic keratoses, skin carcinomas in situ, skin SCCs, and adjacent normal tissue samples were constructed. Sections (5 µm) were cut and placed onto Superfrost plus slides (Fisher Scientific) [90]. 23 AKs, 20 SCCIS, and 160 SCCs were analyzed.
Microarray analysis
RNA was isolated from the dorsal skin of control and K14CreER;Perpfl/fl mice two weeks post-tamoxifen injection using Trizol (Invitrogen), according to the manufacturer's protocol. RNA samples were processed at the Stanford University Pan Facility and analyzed using Affymetrix Mouse Genome 430 2.0 expression arrays. Probe-level data were processed using BRB-ArrayTools (Biometric Research Branch of the National Cancer Institute) based on RMA (Robust Multichip Average) for background adjustment, normalization and expression summarization. Class comparison analysis was conducted using SAM (Significance Analysis of Microarrays) with an FDR (False Discovery Rate) of 10% [53]. Gene function categorization based on GO (Gene Ontology) was carried out using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) classification system.
Quantitative reverse transcription PCR
Skin from control and K14CreER;Perpfl/fl mice was isolated two weeks after tamoxifen injections. RNA was isolated using Trizol (Invitrogen), and 1 µg RNA was reverse transcribed using Moloney Murine Leukemia Virus reverse transcriptase (Invitrogen) and random primers. PCR was performed in triplicate using SYBR green (SA-Biosciences, Frederick, MA) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems, Foster City, CA), and results were computed relative to a standard curve made with cDNA pooled from all samples. Values were first normalized to β-actin and then represented relative to untreated wild-type samples. Five mice per genotype were analyzed. Primers used were as follows: β-actin 5′-TCC TAG CAC CAT GAA GAT CAA GAT C-3′, Rev 5′-CTG CTT GCT GAT CCA CAT CTG-3′, S100a9 5′-GGA AGG AAG GAC ACC CTG AC-3′, Rev 5′-CCA GGT CCT CCA TGA TGT C-3′, Il1f6 5′-CTG TTC TGC ACA AAG GAT GG-3′, Rev 5′-GCT GCA GAC TCA AAT GTA GAG G-3′, Il22ra 5′-GGA CAC CCC GCT TCA CTC-3′, Rev 5′-ATT TGG CAA CTC TGG AGG AC-3′, Chi3l1 5′-AGC AGT ATT TCT CCA CCC TGA T-3′, Rev 5′-CGC TGA GCA GGA GTT TCT CT-3′, and Ccl20 5′-AAC TGG GTG AAA AGG GCT GT-3′, Rev 5′-GTC CAA TTC CAT CCC AAA AA-3′.
Zdroje
1. CooperGM
1995 Oncogenes Boston Jones and Bartlett Publishers xv, 384
2. AlamM
RatnerD
2001 Cutaneous squamous-cell carcinoma. N Engl J Med 344 975 983
3. HunterKD
ParkinsonEK
HarrisonPR
2005 Profiling early head and neck cancer. Nat Rev Cancer 5 127 135
4. FuchsE
RaghavanS
2002 Getting under the skin of epidermal morphogenesis. Nat Rev Genet 3 199 209
5. GreenKJ
GaudryCA
2000 Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 1 208 216
6. YinT
GreenKJ
2004 Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol 15 665 677
7. ChidgeyM
2002 Desmosomes and disease: an update. Histol Histopathol 17 1179 1192
8. NoseA
NagafuchiA
TakeichiM
1988 Expressed recombinant cadherins mediate cell sorting in model systems. Cell 54 993 1001
9. NagafuchiA
ShirayoshiY
OkazakiK
YasudaK
TakeichiM
1987 Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature 329 341 343
10. OzawaM
BaribaultH
KemlerR
1989 The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J 8 1711 1717
11. RimmDL
KoslovER
KebriaeiP
CianciCD
MorrowJS
1995 Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A 92 8813 8817
12. VleminckxK
VakaetLJr
MareelM
FiersW
van RoyF
1991 Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 66 107 119
13. FrixenUH
BehrensJ
SachsM
EberleG
VossB
1991 E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 113 173 185
14. BatlleE
SanchoE
FranciC
DominguezD
MonfarM
2000 The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2 84 89
15. MoodySE
PerezD
PanTC
SarkisianCJ
PortocarreroCP
2005 The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8 197 209
16. SchipperJH
FrixenUH
BehrensJ
UngerA
JahnkeK
1991 E-cadherin expression in squamous cell carcinomas of head and neck: inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res 51 6328 6337
17. PerlAK
WilgenbusP
DahlU
SembH
ChristoforiG
1998 A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392 190 193
18. DerksenPW
LiuX
SaridinF
van der GuldenH
ZevenhovenJ
2006 Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10 437 449
19. DavisMA
ReynoldsAB
2006 Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev Cell 10 21 31
20. Perez-MorenoM
SongW
PasolliHA
WilliamsSE
FuchsE
2008 Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci U S A 105 15399 15404
21. VasioukhinV
BauerC
DegensteinL
WiseB
FuchsE
2001 Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell 104 605 617
22. KobielakA
FuchsE
2006 Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A 103 2322 2327
23. ChitaevNA
TroyanovskySM
1997 Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J Cell Biol 138 193 201
24. TselepisC
ChidgeyM
NorthA
GarrodD
1998 Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci U S A 95 8064 8069
25. SyedSE
TrinnamanB
MartinS
MajorS
HutchinsonJ
2002 Molecular interactions between desmosomal cadherins. Biochem J 362 317 327
26. MathurM
GoodwinL
CowinP
1994 Interactions of the cytoplasmic domain of the desmosomal cadherin Dsg1 with plakoglobin. J Biol Chem 269 14075 14080
27. TroyanovskySM
TroyanovskyRB
EshkindLG
LeubeRE
FrankeWW
1994 Identification of amino acid sequence motifs in desmocollin, a desmosomal glycoprotein, that are required for plakoglobin binding and plaque formation. Proc Natl Acad Sci U S A 91 10790 10794
28. HatzfeldM
HaffnerC
SchulzeK
VinzensU
2000 The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol 149 209 222
29. ChenX
BonneS
HatzfeldM
van RoyF
GreenKJ
2002 Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem 277 10512 10522
30. BonneS
GilbertB
HatzfeldM
ChenX
GreenKJ
2003 Defining desmosomal plakophilin-3 interactions. J Cell Biol 161 403 416
31. KowalczykAP
HatzfeldM
BornslaegerEA
KoppDS
BorgwardtJE
1999 The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J Biol Chem 274 18145 18148
32. KowalczykAP
BornslaegerEA
BorgwardtJE
PalkaHL
DhaliwalAS
1997 The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol 139 773 784
33. BornslaegerEA
CorcoranCM
StappenbeckTS
GreenKJ
1996 Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell-cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J Cell Biol 134 985 1001
34. GreenKJ
SimpsonCL
2007 Desmosomes: new perspectives on a classic. J Invest Dermatol 127 2499 2515
35. YashiroM
NishiokaN
HirakawaK
2006 Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer 42 2397 2403
36. RoepmanP
WesselsLF
KettelarijN
KemmerenP
MilesAJ
2005 An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 37 182 186
37. PapagerakisS
ShabanaAH
PollockBH
PapagerakisP
DepondtJ
2009 Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum Pathol 40 1320 1329
38. DepondtJ
ShabanaAH
Florescu-ZorilaS
GehannoP
ForestN
1999 Down-regulation of desmosomal molecules in oral and pharyngeal squamous cell carcinomas as a marker for tumour growth and distant metastasis. Eur J Oral Sci 107 183 193
39. FurukawaC
DaigoY
IshikawaN
KatoT
ItoT
2005 Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res 65 7102 7110
40. ChenYJ
ChangJT
LeeL
WangHM
LiaoCT
2007 DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene 26 467 476
41. KurzenH
MunzingI
HartschuhW
2003 Expression of desmosomal proteins in squamous cell carcinomas of the skin. J Cutan Pathol 30 621 630
42. AttardiLD
ReczekEE
CosmasC
DemiccoEG
McCurrachME
2000 PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev 14 704 718
43. IhrieRA
MarquesMR
NguyenBT
HornerJS
PapazogluC
2005 Perp is a p63-regulated gene essential for epithelial integrity. Cell 120 843 856
44. JiangW
AnanthaswamyHN
MullerHK
KripkeML
1999 p53 protects against skin cancer induction by UV-B radiation. Oncogene 18 4247 4253
45. IndraAK
LiM
BrocardJ
WarotX
BornertJM
2000 Targeted somatic mutagenesis in mouse epidermis. Horm Res 54 296 300
46. MetzgerD
LiM
ChambonP
2005 Targeted somatic mutagenesis in the mouse epidermis. Methods Mol Biol 289 329 340
47. IhrieRA
ReczekE
HornerJS
KhachatrianL
SageJ
2003 Perp is a mediator of p53-dependent apoptosis in diverse cell types. Curr Biol 13 1985 1990
48. HakemR
HakemA
DuncanGS
HendersonJT
WooM
1998 Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94 339 352
49. JozaN
SusinSA
DaugasE
StanfordWL
ChoSK
2001 Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410 549 554
50. ZieglerA
JonasonAS
LeffellDJ
SimonJA
SharmaHW
1994 Sunburn and p53 in the onset of skin cancer. Nature 372 773 776
51. BornslaegerEA
GodselLM
CorcoranCM
ParkJK
HatzfeldM
2001 Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci 114 727 738
52. KalluriR
WeinbergRA
2009 The basics of epithelial-mesenchymal transition. J Clin Invest 119 1420 1428
53. TusherVG
TibshiraniR
ChuG
2001 Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98 5116 5121
54. BlumbergH
DinhH
TruebloodES
PretoriusJ
KuglerD
2007 Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med 204 2603 2614
55. GebhardtC
NemethJ
AngelP
HessJ
2006 S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol 72 1622 1631
56. SalamaI
MalonePS
MihaimeedF
JonesJL
2008 A review of the S100 proteins in cancer. Eur J Surg Oncol 34 357 364
57. EurichK
SegawaM
Toei-ShimizuS
MizoguchiE
2009 Potential role of chitinase 3-like-1 in inflammation-associated carcinogenic changes of epithelial cells. World J Gastroenterol 15 5249 5259
58. BeiderK
AbrahamM
BeginM
WaldH
WeissID
2009 Interaction between CXCR4 and CCL20 pathways regulates tumor growth. PLoS One 4 e5125 doi:10.1371/journal.pone.0005125
59. HasanL
MazzucchelliL
LiebiM
LisM
HungerRE
2006 Function of liver activation-regulated chemokine/CC chemokine ligand 20 is differently affected by cathepsin B and cathepsin D processing. J Immunol 176 6512 6522
60. PunjV
MattaH
SchamusS
YangT
ChangY
2009 Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: role of viral FLICE inhibitory protein K13-induced NF-kappaB activation. Blood 113 5660 5668
61. Ben-BaruchA
2006 The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev 25 357 371
62. O'SheaJJ
MurrayPJ
2008 Cytokine signaling modules in inflammatory responses. Immunity 28 477 487
63. GrivennikovSI
GretenFR
KarinM
2010 Immunity, inflammation, and cancer. Cell 140 883 899
64. PinkusGS
PinkusJL
1991 Myeloperoxidase: a specific marker for myeloid cells in paraffin sections. Mod Pathol 4 733 741
65. RibattiD
VaccaA
NicoB
CrivellatoE
RoncaliL
2001 The role of mast cells in tumour angiogenesis. Br J Haematol 115 514 521
66. CoussensLM
RaymondWW
BergersG
Laig-WebsterM
BehrendtsenO
1999 Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13 1382 1397
67. Ch'ngS
WallisRA
YuanL
DavisPF
TanST
2006 Mast cells and cutaneous malignancies. Mod Pathol 19 149 159
68. MeiningerCJ
1995 Mast cells and tumor-associated angiogenesis. Chem Immunol 62 239 257
69. MaltbyS
KhazaieK
McNagnyKM
2009 Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta 1796 19 26
70. BashkinP
RazinE
EldorA
VlodavskyI
1990 Degranulating mast cells secrete an endoglycosidase that degrades heparan sulfate in subendothelial extracellular matrix. Blood 75 2204 2212
71. VlodavskyI
EldorA
Haimovitz-FriedmanA
MatznerY
Ishai-MichaeliR
1992 Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis 12 112 127
72. NavaP
LaukoetterMG
HopkinsAM
LaurO
Gerner-SmidtK
2007 Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol Biol Cell 18 4565 4578
73. DusekRL
GetsiosS
ChenF
ParkJK
AmargoEV
2006 The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem 281 3614 3624
74. NaikE
MichalakEM
VillungerA
AdamsJM
StrasserA
2007 Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol 176 415 424
75. AndreuP
JohanssonM
AffaraNI
PucciF
TanT
2010 FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 17 121 134
76. CuiW
FowlisDJ
BrysonS
DuffieE
IrelandH
1996 TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 86 531 542
77. PierceAM
Schneider-BroussardR
Gimenez-ContiIB
RussellJL
ContiCJ
1999 E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol 19 6408 6414
78. MarquesMR
HornerJS
IhrieRA
BronsonRT
AttardiLD
2005 Mice lacking the p53/p63 target gene Perp are resistant to papilloma development. Cancer Res 65 6551 6556
79. KempCJ
DonehowerLA
BradleyA
BalmainA
1993 Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74 813 822
80. BenjaminCL
UllrichSE
KripkeML
AnanthaswamyHN
2008 p53 tumor suppressor gene: a critical molecular target for UV induction and prevention of skin cancer. Photochem Photobiol 84 55 62
81. BruinsW
ZwartE
AttardiLD
IwakumaT
HoogervorstEM
2004 Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Mol Cell Biol 24 8884 8894
82. MelnikovaVO
AnanthaswamyHN
2005 Cellular and molecular events leading to the development of skin cancer. Mutat Res 571 91 106
83. LozanoG
ZambettiGP
2005 What have animal models taught us about the p53 pathway? J Pathol 205 206 220
84. HemannMT
ZilfouJT
ZhaoZ
BurgessDJ
HannonGJ
2004 Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci U S A 101 9333 9338
85. ArmstrongBK
KrickerA
2001 The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63 8 18
86. 2009 Detailed Guide: Esophagus Cancer. American Cancer Society
87. KashiwagiS
YashiroM
TakashimaT
NomuraS
NodaS
2010 Significance of E-cadherin expression in triple-negative breast cancer. Br J Cancer 103 249 255
88. MellLK
MeyerJJ
TretiakovaM
KhramtsovA
GongC
2004 Prognostic significance of E-cadherin protein expression in pathological stage I-III endometrial cancer. Clin Can Res 10 5546 5553
89. LuthraR
WuTT
LuthraMG
IzzoJ
Lopez-AlvarezE
2006 Gene expression profiling of localized esophageal carcinomas: association with pathologic response to preoperative chemoradiation. J Clin Oncol 24 259 267
90. HarradineKA
RiddK
SaunierEF
ClermontFF
Perez-LosadaJ
2009 Elevated cutaneous Smad activation associates with enhanced skin tumor susceptibility in organ transplant recipients. Clin Cancer Res 15 5101 5107
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2010 Číslo 10
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance