
Comparative Genome Structure, Secondary Metabolite, and Effector Coding Capacity across Pathogens
The genomes of five Cochliobolus heterostrophus strains, two Cochliobolus sativus strains, three additional Cochliobolus species (Cochliobolus victoriae, Cochliobolus carbonum, Cochliobolus miyabeanus), and closely related Setosphaeria turcica were sequenced at the Joint Genome Institute (JGI). The datasets were used to identify SNPs between strains and species, unique genomic regions, core secondary metabolism genes, and small secreted protein (SSP) candidate effector encoding genes with a view towards pinpointing structural elements and gene content associated with specificity of these closely related fungi to different cereal hosts. Whole-genome alignment shows that three to five percent of each genome differs between strains of the same species, while a quarter of each genome differs between species. On average, SNP counts among field isolates of the same C. heterostrophus species are more than 25× higher than those between inbred lines and 50× lower than SNPs between Cochliobolus species. The suites of nonribosomal peptide synthetase (NRPS), polyketide synthase (PKS), and SSP–encoding genes are astoundingly diverse among species but remarkably conserved among isolates of the same species, whether inbred or field strains, except for defining examples that map to unique genomic regions. Functional analysis of several strain-unique PKSs and NRPSs reveal a strong correlation with a role in virulence.
Published in the journal:
Comparative Genome Structure, Secondary Metabolite, and Effector Coding Capacity across Pathogens. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003233
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003233
Summary
The genomes of five Cochliobolus heterostrophus strains, two Cochliobolus sativus strains, three additional Cochliobolus species (Cochliobolus victoriae, Cochliobolus carbonum, Cochliobolus miyabeanus), and closely related Setosphaeria turcica were sequenced at the Joint Genome Institute (JGI). The datasets were used to identify SNPs between strains and species, unique genomic regions, core secondary metabolism genes, and small secreted protein (SSP) candidate effector encoding genes with a view towards pinpointing structural elements and gene content associated with specificity of these closely related fungi to different cereal hosts. Whole-genome alignment shows that three to five percent of each genome differs between strains of the same species, while a quarter of each genome differs between species. On average, SNP counts among field isolates of the same C. heterostrophus species are more than 25× higher than those between inbred lines and 50× lower than SNPs between Cochliobolus species. The suites of nonribosomal peptide synthetase (NRPS), polyketide synthase (PKS), and SSP–encoding genes are astoundingly diverse among species but remarkably conserved among isolates of the same species, whether inbred or field strains, except for defining examples that map to unique genomic regions. Functional analysis of several strain-unique PKSs and NRPSs reveal a strong correlation with a role in virulence.
Introduction
The filamentous ascomycete genus Cochliobolus (anamorph Bipolaris/Curvularia) is comprised of more than forty closely related, often highly aggressive, pathogenic species with particular specificity to their host plants. All members of the genus known to cause serious crop diseases fall in a tight phylogenetic group suggesting that a progenitor within the genus gave rise, over a relatively short period of time (<20 MYA, Ohm et al., [1]) to the series of distinct species [2], each distinguished by unique pathogenic capability to individual types of cereal (Table 1). Aggressive members include the necrotrophic corn pathogens Cochliobolus heterostrophus and Cochliobolus carbonum, the oat pathogen, Cochliobolus victoriae, the rice pathogen, Cochliobolus miyabeanus, the sorghum pathogen, Bipolaris sorghicola, the sugarcane pathogen, Bipolaris sacchari, and the hemibiotrophic generalized cereal and grass pathogen, Cochliobolus sativus.

All of the known Cochliobolus pathogens are classified as necrotrophs, except for C. sativus, which, although previously classified as such, has more recently been described as a hemibiotroph [3]. Many necrotrophic Cochliobolus spp. and related taxa (e.g., Pyrenophora tritici-repentis, Stagonospora nodorum) are notorious for their ability to evolve novel, highly virulent, races producing Host Selective Toxins (HSTs) and their concomitant capacity to cause diseases on cereal crops that were bred, inadvertently, for susceptibility to the HST-producing pathogen [4], [5]. For example, in 1970, race T, a previously unseen race of C. heterostrophus (Bipolaris maydis) caused a major epidemic [Southern Corn Leaf Blight (SCLB)], destroying more than 15% of the maize crop [6]. Before 1970, C. heterostrophus was known as an endemic pathogen (race O) of minor economic importance, first described in 1925 [7]. Subsequent research over the ensuing four decades since 1970 has demonstrated that the epidemic was triggered by the unfortunate confluence of complex DNA recombination events in both the fungal pathogen and the plant host. Race T is genetically distinct from race O in that it possesses an extra 1.2 Mb of DNA [8], [9] encoding genes for biosynthesis of the polyketide secondary metabolite, T-toxin, an HST essential for high virulence [4]. These genes are missing in race O. On the plant side, the presence in Texas male sterile cytoplasm (Tcms) maize, of a hybrid mitochondrial gene called T-urf13, composed of segments of two mitochondrial and one chloroplast gene, is essential for susceptibility. Tcms corn does not need to be detasseled to prevent self-crossing because it is male sterile, a desirable trait for breeders producing hybrid seed. The resulting popularity of Tcms maize was disastrous from the perspective of pathogen attack, however, as it served as a monoculture of susceptible germplasm [10], [11].
Similarly, C. victoriae (Bipolaris victoriae), causal agent of Victoria Blight, produces the chlorinated cyclic pentapeptide HST, victorin, rendering it highly virulent to oats carrying the dominant Vb allele [12]. The fungus caused widespread destruction (20 states) in the 1940's on oat varieties containing the recently introduced Pc-2 gene for resistance to crown rust caused by Puccinia coronata [13]. Like the C. heterostrophus T-toxin/Tcms case, the monoculture of Victoria oats carrying Pc-2 was the perfect milieu for attack by C. victoriae producing victorin, which elicits Pc-2-dependent Programmed Cell Death (PCD). Recent work with Arabidopsis identified a resistance-like protein responsible for susceptibility to C. victoriae and victorin [14], [15]. This work is seminal in demonstrating fungal HSTs can target resistance proteins to promote disease.
In contrast to the dominant plant host genes required for susceptibility to C. heterostrophus and C. victoriae, susceptibility to Northern Corn Leaf Spot caused by C. carbonum (Bipolaris zeicola) is conferred by a homozygous recessive maize gene(s) [16], [17]. C. carbonum race 1 produces the cyclic-tetrapeptide HST, HC-toxin, which is specifically active against corn with the genotype hmhm, as is the fungus itself [4], [18], [19]. Hm1 and Hm2 encode carbonyl reductases which inactivate the toxin [16]; hmhm lines, cannot inactivate the toxin, and are therefore sensitive. The site of action of HC-toxin in susceptible corn is histone deacetylase; it is hypothesized that HC-toxin acts to promote infection of maize of genotype hm1hm1 by inhibiting this enzyme, resulting in accumulation of hyperacetylated core (nucleosomal) histones. This then alters expression of genes encoding regulatory proteins involved in plant defense [20], [21]. C. carbonum races 2 and 3 do not produce the toxin.
C. miyabeanus (Bipolaris oryzae) is the causal agent of Brown Spot of rice which contributed, along with a cyclone and tidal waves, to the Bengal rice famine of 1942/1943 that resulted in starvation of more than two million people [22]. To date, no HST has been associated with virulence, although C. miyabeanus culture filtrates can suppress plant phenol production [23].
C. sativus (Bipolaris sorokiniana) causes diseases of roots (Common Root Rot), leaves (Spot Blotch), and spikes (known as black point or kernel blight) of cereals (mainly barley and wheat) [24], [25], but also attacks many grasses, including switch grass (Panicum virgatum L.) [3], [26], [27] and Brachypodium distachyon (S. Zhong, unpublished). Three C. sativus pathotypes (0, 1 and 2) have been described [28], based on differential virulence patterns on three barley genotypes (ND5883, Bowman, and NDB112). Pathotype 0 isolates show low virulence on all three barley genotypes. Pathotype 1 isolates show high virulence on ND5883 but low virulence on other barley genotypes. Pathotype 2 isolates show high virulence on Bowman but low virulence on ND5883 and NDB112. Genetic analysis and molecular mapping indicates that a single locus, VHv1, controls high virulence of the pathotype 2 isolate ND90Pr on Bowman [29], [30], however, the exact nature of the gene(s) was unknown before this study (see Results). More recently, a new pathotype, highly virulent on NDB112, the most durable spot blotch resistance source in barley [31], has been found in North Dakota [32] and Canada [33].
Setosphaeria turcica (Exserohlium turcicum, Helminthosporium turcicum), a hemibiotrophic vascular leaf pathogen, is a member of the closest genus to Cochliobolus (see Figure 1 in Ohm et al., [1]), and causes Northern Leaf Blight (NLB), a major disease of maize and sorghum in the US and internationally [34]. To date, at least four races of S. turcica have been identified based on their differential virulence performance on maize carrying resistance genes known as Ht [35], [36]. In recent work, Martin et al. [34], identified new resistance genes (named St) in both maize and sorghum. The connections between the Ht and St resistance genes are unclear at this point.
Until recently, it was assumed that necrotrophs use brute force methods (e.g., arsenals of cell wall degrading enzymes, HSTs) to invade and kill host tissues and do not subtly manipulate the host during infection, as do biotrophs with their arsenal of effectors [37]. Several lines of evidence, from studies of the Dothideomycete, necrotrophic wheat pathogens, Pyrenophora tritici-repentis [38] and Stagonospora nodorum, [39] indicate that, like biotrophs, these pathogens secrete protein effectors (in this case also called HSTs) that interact with host targets in a gene-for-gene manner. Unlike biotrophs, however, interaction of the fungal effector and host protein results in susceptibility, rather than resistance. The above-mentioned research on C. victoriae, further indicates mechanistic overlap of pathogenic lifestyle and has major implications regarding the challenge plants face in defending themselves against both necrotrophs and biotrophs [37], [40]. Recent studies involving Arabidopsis have revealed that victorin-induced PCD requires a host NB-LRR-type resistance protein [15], [41]. Thus a protein with canonical resistance protein structure is required for susceptibility. These observations point toward victorin, subverting effector triggered defenses against biotrophs, such as P. coronata (see above), to promote susceptibility to a necrotroph.
Here we provide a comparative analysis of genome similarities and differences among Cochliobolus and Setosphaeria pathogens, with particular emphasis on strain and species-unique sequences, secondary metabolism genes, and genes encoding small secreted proteins. Identification of these key structural genomic and molecular differences is the first step in understanding species-specificity and how closely related necrotrophic and hemibiotrophic pathogens cause disease. As proof of concept, we offer several examples of how comparative approaches pinpoint virulence associated, species-specific regions of interest. The Joint Genome Institute (JGI) has sequenced five strains of C. heterostrophus (three race T and two race O strains) and four additional species of Cochliobolus, including C. carbonum, C. victoriae, C. miyabeanus, and C. sativus and one strain of S. turcica. Add to this that host genome sequences (corn, rice, barley and B. distachyon) for six of these pathogens are available and one has the information base for dissecting both sides of the interaction mechanism going forward.
Results
Genome statistics
Five strains of C. heterostrophus, one strain of C. sativus, and one strain each of C. victoriae, C. carbonum, C. miyabeanus, and S. turcica were sequenced by JGI (Table 2, Table 3). Two C. heterostrophus strains and one strain each of C. sativus and S. turcica were fully sequenced as described in Materials and Methods, while the remaining genomes were sequenced using Illumina and assembled de novo using Velvet, as described in Materials and Methods. The highly inbred race O lab strain C5 was used as the reference sequence for all comparisons, as it is the most complete, consisting of only 68 scaffolds.


Overall sequence assembly and annotation statistics are presented in Table 2 and Table 3. All Cochliobolus genomes are in the 32–38 Mb range with an estimated gene content of 11,700–13,200. The S. turcica genome is ∼43 Mb with ∼11,700 genes. Overall gene content and genome organization are highly similar within this group of fungi. In contrast, comparative analysis of C. heterotrophus, C. sativus and S. turcica in the context of 14 more distantly related Dothideomycetes genomes described elsewhere (Ohm et. al., [1]) revealed significant variation.
Mapping scaffolds to the genetic maps
C. heterostrophus: A genetic map with 125 RFLP markers was constructed previously [8] using C. heterostrophus race O field strain Hm540 (sequenced herein) and race T C-strain B30.A3.R.45 (same backcross series as strains C5 and C4, sequenced herein [42], [43]) as parents. RFLP sequences were used to refine the C. heterostrophus race O strain C5 physical assembly and link it to the genetic RFLP map (Figure 1, Text S1). The interconnected genetic and physical maps allowed comparisons of physical and genetic distance, which was found to be ∼13 kb/cM on average (Figure S1). Correlations were also made between previously estimated chromosome sizes based on CHEF gel analysis [9], and physical size based on sequence assemblies (Tables S1, S2).

Based on RFLP map data, parental field isolate Hm540 was reported to lack the dispensable chromosome present in the other parental strain (B30.A3.R.45) used to build the map [8]. Comparisons of all five sequenced C. heterostrophus strains, using the Mauve alignment tool [44], supported this observation and revealed that all sequenced strains carried the previously recognized ‘B’ or dispensable chromosome (which corresponds to strain C5 scaffold 16) (Figure S2). Race O chromosomes 6 and 12 are of interest because counterparts are reciprocally translocated in race T and the high-virulence conferring Tox1 locus, encoding genes for biosynthesis of T-toxin, maps genetically to both breakpoints [9]. Comparison of the race O and race T assemblies provided some clues as to the physical locations of these breakpoints, but the exact positions remain elusive, due to structural complexity associated with these regions [45]. Additional details regarding linkage of the C. heterostrophus physical assembly to the genetic map are available in Text S1.
C. sativus: Before mapping sequenced scaffolds to the previously constructed C. sativus genetic map [30] 121 polymorphic simple sequence repeat (SSR) markers were identified in the assembly sequences of the ND90Pr and ND93-1 parents, as described in Materials and Methods. Then, sequences of the SSRs and other markers were used to assign sequenced scaffolds to the updated map. Thirty of these linkage groups contained SSR markers and were found to be associated with 16 scaffolds, summing to 29.32 Mb. Seven linkage groups were unassigned (Figure S3). The two AFLP marker sequences (E-AG/M-CG-121 and E-AG/M-CA-207), cosegregating with the VHv1-associated high virulence of C. sativus pathotype 2 on cultivar Bowman [30], were used as blast queries against the ND90Pr genome assembly. E-AG/M-CG-121 mapped to scaffold 5, while E-AG/M-CA-207 mapped to scaffold 40 (Figure S3). Details of the construction, linking, and analysis of the C. sativus map are available in Text S1.
Single nucleotide polymorphisms (SNPs) between the reference C. heterostrophus C5 strain and other strains
SNPs in C. heterostrophus strains
Genome assemblies were aligned, pairwise, to the C. heterostrophus race O C5 reference using MUMmer [46] to analyze SNP frequencies between strains and species and to infer whole-genome similarity. C. heterostrophus race T strain C4, which belongs to the same inbred laboratory strain series as the C5 reference [42] contained 1,584 SNPs (Table 4, Table S3). In contrast, the three C. heterostrophus field strains, race O strain Hm540, race T strains Hm338, and PR1x412 had 50,864, 30,624, and 33,552 SNPs, respectively, (∼20–30× more than strain C4) (Table 4, Table S3). More SNPs were identified for Hm540, the only race O field isolate, than for the race T field strains Hm338 and PR1x412, despite the reference C5 strain being race O. This supports previous RFLP data which indicated that race O field strain Hm540 was more diverse than any field isolate examined [8] and the hypothesis that race T arose once and more recently than race O [9], [47], [48].

For all C. heterostrophus strains, 30–31.5 Mb (95–97%) of the assembled basepairs (bp) could be aligned, although only 40–80% of the scaffolds could be aligned. Thus, 3–5% of each genome could not be aligned and corresponding sequences were located on small, difficult to assemble contigs.
SNPs in C. sativus strains
For the two C. sativus genomes, ND90Pr and ND93-1, 86.6% of the higher quality ND90Pr genome could be aligned to 96.6% of the ND93-1 genome, yielding 60,448 SNPs (Table 5). The relative similarity between these two strains is comparable to that seen between C. heterostrophus strains.

SNPs in Cochliobolus species
C. victoriae, C. carbonum, C. miyabeanus, and C. sativus species had 2,083,899, 2,059,993, 2,110,786, and 1,981,616 SNPs, respectively, when aligned to C. heterostrophus reference strain C5, ∼54× fold more than comparisons within species (Table 4, Table S3). In each case, 24–25 Mb (75%) of assembled bps could be aligned to the reference, indicating that, in addition to the much higher quantity of SNPs than between C. heterostrophus strains, a full quarter of each genome was not present in C. heterostrophus. Although C. sativus has a wider host range than any of the other species, there is no compelling evidence that it is less related to C. heterostrophus than the other species. All species are estimated to have arisen relatively recently (<20MYA ago, see Figure 1 in Ohm et al., [1]). Analysis of the mating type (MAT) regions (Text S1, Figure S4, Table S4) yielded similar patterns, except that within the C. heterostrophus species, the most similar MAT flanking regions were those of the same mating type, regardless of whether the strain was race O or race T, or an inbred or field strain. C. carbonum and C. victoriae are capable of crossing with each other [48] and thus would be predicted to be closely related and to have fewer SNPs between them than between either of them and other species. Indeed, when C. victoriae was aligned to C. carbonum, 292,216 SNPs were identified, roughly 10-fold fewer than when Cochliobolus species are compared to the C. heterostrophus reference, yet 10-fold more than are present when C. heterostrophus field strains are compared to the C. heterostrophus C5 reference strain (Table 6, Table S3). Thus, the SNP data are in line with the notion that C. victoriae and C. carbonum are closely related and that C. victoriae may have arisen from a C. carbonum isolate [49]. For comparison, aligning C. miyabeanus or C. sativus to the C. carbonum genome yields 2,078,277 and 2,022,068 SNPs, respectively (Table 6, Table S3), in the range seen when comparisons are made with C. miyabeanus, C. carbonum, C. victoriae, or C. sativus to C. heterostrophus C5 (Table 4, Table S3).

SNP calls generated in all genome comparisons were inflated for A to G, T to C, C to T, and G to A transitions, the most common type of mutation. In all comparisons made, including C5 to C4, these transitions were 5–10 times more abundant than other changes (Table S3).
SNPs between C. heterostrophus and S. turcica
We could align only 16.12% (6.9 Mb) of the S. turcica genome to the C. heterostrophus reference genome, as indicated in Table 4. Full details are available in Table S3.
Summary
Inbred C. heterostrophus C4 strain has far fewer SNPs (1,584), than C. heterostrophus field strains, when compared to the C. heterostrophus C5 reference. The numbers of SNPs in the C. heterostrophus field strains are comparable to each other, with Hm540, a race O isolate, containing the most (50,864) when compared to race O C. heterostrophus C5. The largest numbers of SNPs are between C. heterostrophus C5 and the additional Cochliobolus species (1.9–2.1 million) and these are ∼54 fold higher than within species SNPs (Table 4, Table S3). The C. sativus strains are as similar to each other as different C. heterostrophus strains are to one another, while C. carbonum and C. victoriae demonstrate a level of relatedness in between that seen among isolates within a species and across species.
Identification of strain - and species-specific genomic regions
To begin to identify regions in the C. heterostrophus C5 assembly not represented in other strains, we first mapped gaps in the reference assembly (thick vertical black bars, Figure 2). A single gap was present in the assembly of 9 of 16 chromosomes and we speculate that these gaps correspond to centromeric regions. We then mapped sequence reads of all Cochliobolus genomes in this study to the C. heterostrophus C5 reference, identifying regions in the C5 reference that were not present in the query genome (Table S5). The sets of C5 genomic regions that were absent in a given query were combined to determine genomic regions unique and/or conserved at different taxonomic levels.

Individual reference-unique region counts were recorded for each of the C. heterostrophus strains. There were 609 areas of the C5 assembly unique (C4 reads did not map there) to the C5 genome when C4 reads were mapped to it. For C. heterostrophus strains Hm540, PR1x412, and Hm338, there were 3,279, 4,383 and 1,480 such regions, respectively. When only regions greater than 5,000 bp were considered, there were 19 when C4 was used as the query, and 33, 75, and 30, when PR1x412, Hm540, and Hm338 were used as the queries, respectively. Many of the gaps associated with Hm540 mapped to reference C5 scaffold 16 corresponding to the dispensable B chromosome, which is absent in Hm540 (Figure S2).
The C5 reference-unique regions were then combined and filtered to identify conserved genomic regions at the strain or species level, where regions were unique in one type of comparison, but not in others (Table S5). We designated “inbred C strain”-specific regions as gaps found when all field strains were aligned to C5 but not when C4 was aligned to C5, race O specific regions as gaps found when all race T strains were aligned to C5 but not when race O strain Hm540 was aligned to race O strain C5, and C. heterostrophus-specific regions as gaps found when all Cochliobolus species were aligned to C5, but not when any C. heterostrophus strain was aligned.
A total of 28,556 bp is missing from all C. heterostrophus field strains, yet present in the C4 assembly (Table S5). This ∼30 kb of DNA represents sequence uniquely conserved in the inbred C strains. There are at least six fungal-specific Zn2Cys6 transcription factors (ID# 1019013, 1020538, 1021058, 1021066, 1100349, 1100899) present in this C strain unique cache that may signify “early action” strain diversification. Zn2Cys6 transcription factors were among the most abundant predicted domains in the C. heterostrophus gene catalogue [1].
There were almost no race O specific regions identified (sequence found only in C5 and Hm540); only 11 regions, summing to 4,309 bp, were identified (Table S5). Our working hypothesis is that the essential difference between race O and race T is the 1.2 Mb of Tox1 race T DNA (not in race O C5 and therefore not able to be aligned). Both race O only regions (scaffold 12, 732532–734880, and scaffold 19, 441888–443521) contain a single protein each (ID# 59063, 34937) with no conserved domains or predicted function.
Most significantly, at the species level, a total of 11.76 Mb DNA was missing from all non-C. heterostrophus Cochliobolus genomes analyzed, yet found in all field strains of C. heterostrophus. Only 1.6 Mb of this was found in pieces larger than 5 kb. Most of the sequence that separates C. heterostrophus from other species, therefore, is not the result of large wholesale insertions or deletions of DNA, but from a more piecewise gain and loss.
Summary
By mapping query reads to the reference C. heterostrophus C5 assembly, we generated relative coverage maps for each strain and species. Intersecting these coverage maps as present or absent in different genome sets gives us insight into short-term strain evolution. 28,556 bp of sequence was found in both of the C. heterostrophus C strains, but none of the C. heterostrophus field strains, while only 4,309 bp was uniquely conserved in race O strains and 11.7 Mb was uniquely conserved in C. heterostrophus. This sequence is what makes Cochliobolus heterostrophus unique from other Cochliobolus species.
Secondary metabolism: Nonribosomal peptide synthetases
Nonribosomal peptide synthetases (NRPSs), found in fungi and bacteria, are multimodular megasynthases that catalyze biosynthesis of small bioactive peptides (NRPs), including virulence determinants, such as HSTs, via a thiotemplate mechanism independent of ribosomes [50], [51], [52], [53], [54]. NRPSs synthesize peptides using sets of core domains, known as modules, which consists of three domains: 1) an adenylation (AMP) domain which recognizes and activates the substrate via adenylation with ATP, 2) a thiolation (T) or peptidyl carrier protein (PCP) domain which binds the activated substrate to a 4′ - phosphopantetheine (PP) cofactor via a thioester bond and transfers the substrate to 3) a condensation (C) domain which catalyzes peptide bond formation between adjacent substrates on the megasynthase complex. NRPSs can be mono-, bi-, or multi-modular and core domains in any particular multimodular enzyme may be most closely related to one another or to a domain from a different NRPS.
The suites of NRPS encoding genes (NPS) in the C. heterostrophus C4 and C5 genomes were identified and annotated previously [55], [56], [57]. To address degree of conservation and evolutionary relationships of NRPS proteins in our subject species in order to make inferences about function, we used the fungal AMP-binding (AMP) domain Hidden Markov Model (HMM) developed by Bushley and Turgeon [57] to identify individual AMP domains in the additional strains of C. heterostrophus and other Cochliobolus species, plus S. turcica. Phylogenetic trees were built to develop a comparative NRPS AMP domain inventory and included the known C. heterostrophus C5 AMP domains as a reference (Table S6).
Conservation of known C. heterostrophus NRPSs
First, using the highly curated set of C. heterostrophus NRPSs (Table 7, left column), we determined if all 28 AMP domains of all 14 C. heterostrophus C5 reference NRPS proteins were present in each genome analyzed. For the C. heterostrophus genomes, all C5 reference AMP domains and thus all complete NRPS proteins were present (Table 7, Figure 3, for full phylogenetic trees see Figure S5A, S5B, and for master inventories, see Table S6), with the single exception of bimodular NPS5, which was absent from race T strain PR1x412. Thus, there is almost complete conservation of NRPSs at the species level.

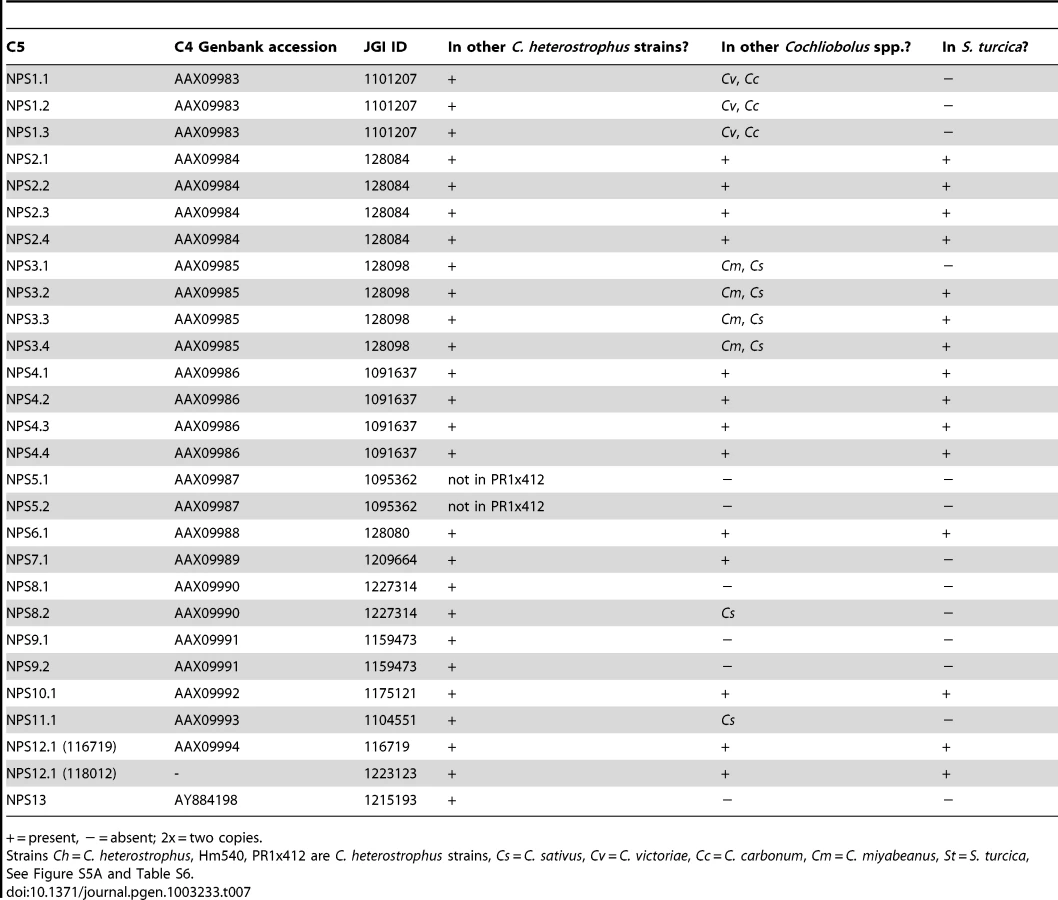
At the genus level, seven of the 14 NRPS proteins (and thus genes) in C. heterostrophus reference strain C5 were conserved across all species (Table 7, Figure 3, for full phylogenetic trees see Figure S5A, S5B). When conservation of NRPSs was considered across all Cochliobolus spp. and the related maize pathogen S. turcica, six of the 14 NRPS proteins in C. heterostrophus reference strain C5 were completely conserved across all species (NPS2, 4, 6, 19, and 12, and 12-like, Table 7). The phylogenetic profile (Figure 4) of the highly conserved NPS2 protein is an example of complete conservation. Note that all four NPS2 AMP domains are found in all species. NPS2 biosynthesizes the hexa-peptide siderophore, ferricrocin, and an evolutionary mechanism by which a four AMP domain NRPS can generate a six component metabolite has been proposed by us [58], [59]. In cross genome evaluation of conservation of NRPSs (see Table S18 in [1]), this was one of only two NRPSs conserved across 18 Dothideomycete genomes.

Discontinuously distributed and expanded NRPSs
Of those NRPS proteins for which all C. heterostrophus AMP domains are not conserved across all species (Table 7), three, NPS1, NPS3, and NPS13 are of particular note as they are expanded discontinuously, with multiple homologs for some, but not all AMP domains of these proteins in different species (Figure 5, Figure 6). We hypothesize that this group of NRPSs is a spawning ground for AMP domain diversity. On the whole protein level, the complete C. heterostrophus NPS1 (trimodular) and NPS3 (tetramodular) domain sets are either present or absent in other species. NPS1 is intact in C. victoriae and C. carbonum, while NPS3 is intact in C. miyabeanus and C. sativus, but absent from the other genomes (Figure 5, Table 7). Monomodular C. heterostrophus NPS13 is found only in C. heterostrophus (Figure 5, Table 7). NPS1, NPS3 and NPS13 protein AMP domains, as noted above, are expanded discontinuously resulting in a suite of novel proteins we call ‘NPS1/NPS3/NPS13 expanded’ (Figure 5). These new proteins may be mono - or multi-modular. C. victoriae has three such proteins, C. carbonum and C. miyabeanus have one each, and C. sativus has seven (Table 8, Figure 5). A C. heterostrophus NPS13-related module is present in tetramodular form in C. carbonum, C. victoriae, and C. miyabeanus, and in trimodular form in C. sativus. Both the monomodular NPS13, and tetramodular NPS13 related protein, are found in the C. heterostrophus Hm540 strain.



As shown in Figure 6, C. heterostrophus NPS1 AMP2 groups with C. heterostrophus NPS3 AMP2 and AMP4 orthologs, with 99% bootstrap support. The NPS1 and NPS3 AMP2 and AMP4 expanded group (New_1, 2, 3, Figure 3) is most closely related to NPS3 AMP2 (100% bootstrap support). C. heterostrophus NPS1 AMP1 and AMP3 group with C. heterostrophus NPS3 AMP 1 and AMP3 orthologs (NPS1 AMP1, NPS3 AMP 1 and AMP3 group together with 93% bootstrap support). The NPS1 and NPS3 AMP1 and AMP3 expanded group (New_7, Figure 3) is nested within, but without bootstrap support. S. turcica has seven total NPS1/3/13 associated AMP domains. Four cluster with NPS3 AMP3, two with NPS3 AMP2, and one with NPS3 AMP4 (Figure 6, Table S6). These expanded S. turcica domains belong to four different NRPSs (ID# 51661, 36641, 65284, 48467), none of which corresponds to Cochliobolus NRPSs.
Monomodular NPS13, as noted, is found only in C. heterostrophus, but there is an NPS13 expanded group of AMP domains, sister to (95% bootstrap support) C. heterostrophus NPS13. C. sativus possesses additional NPS13 expanded domains, always co-occurring with NPS1 and NPS3 expanded domains (Figure 5, Figure 6).
Species-unique NRPSs
In initiating our survey, we hypothesized that this category of NRPS would be most likely to include candidates associated with virulence functions, given the unique or spotty distribution signatures of HSTs. We define an NRPS as unique when no other Cochliobolus species has all of the orthologous AMP domains in identical whole-protein organization. The reference, C. heterostrophus, has 14 NRPSs, three of which (NPS5, NPS8 and NPS9) are unique to this species but found in both race O and T (Table 7, Table 8, Table S6). None of these three has an obvious role in virulence [55]. Of the other Cochliobolus species, C. miyabeanus has the fewest total (11) NRPSs and no unique ones, while C. sativus has the most (25), 14 of which are unique (Table 8) and include seven belonging to the NPS1/NPS3/NPS13 expanded group (and an eighth unique NRPS, ID# 358216) (Table 8, Figure 5). When the AMP domains from the 25 NRPSs identified in C. sativus isolate ND90Pr (pathotype 2) were used as blast queries to identify orthologs in C. sativus isolate ND93-1 (pathotype 0), five (ID# 130053, 140513, 104448, 115356, and 350779) were not present in the latter and thus are unique to ND90Pr.
C. carbonum and C. victoriae have 20 and 18 total NRPSs, respectively. Six are unique to C. carbonum (Table 8). None of these is in the NPS1/NPS3/NPS13 expanded group. One of the C. carbonum unique NRPSs is HTS1, responsible for HC-toxin biosynthesis. It has long been recognized that HTS1 is only found in race 1 of this species and not in any other Cochliobolus species [60] and our genome survey confirms this. Our NRPS survey, however, identified an ortholog in S. turcica (see section below). Four other novel C. carbonum NRPS AMP domains group in the 11 AMP domain Tolypocladium inflatum SimA clade for biosynthesis of cyclosporin, suggesting C. carbonum as a possible source of a cyclosporin-type molecule (Table 8, Figure S5). C. victoriae has five unique NRPSs, including two from the NPS1/NPS3/NPS13 expanded group (Figure 5, Table 8). One of the C. victoriae unique NRPSs is on n1179 (g7087) and is an ortholog of the gene for gliotoxin. Another C. victoriae unique AMP domain (on node 572, Figure S5A) is incomplete but has a match to a bimodular S. turcica NRPS (ID #97841). The fifth unique AMP, on node 3108 (Figure S5A), groups with the NRPS for aminoadipate reductase (lysine biosynthesis), but without bootstrap support.
For S. turcica, eight (ID# 36641, 48467, 51661, 65284, 99043, 155102, and 54477, 97841, 99181 Table S6) NRPSs were unique, not occurring in any Cochliobolus genome. One of these, 99181, clusters with varying support with PesA, a Metarhizium anisopliae NRPS producing an unknown product. Four of S. turcica's unique NRPSs (ID# 36641, 48467, 51661, 65284) contained NPS1/NPS3/NPS13 expansion AMP domains as noted above (Table S6).
NRPSs showcasing the value of the phylogenomic approach in pinpointing candidate virulence determinants
Case Study 1: NRPSs unique to C. sativus pathotype 2, isolate ND90Pr determine virulence on barley cultivar Bowman. Previous genetic studies have indicated that a single locus (VHv1) in C. sativus pathotype 2 isolate ND90Pr controls high virulence on barley cv. Bowman [30]. Two AFLP markers E-AG/M-CG-121 and E-AG/M-CA-207 that co-segregate with the VHv1 locus in ND90Pr [30] mapped to scaffold 5 (Figure 7A) and 40 (Figure S3), respectively, in the genome assembly. The VHv1 region (distal end of scaffold 5) carrying the E-AG/M-CG-121 marker includes 43 predicted genes (Table S7), plus many repetitive elements (Figure 7A, Table S8). None of the 43 genes was found in the C. sativus pathotype 0 isolate ND93-1 genome. Two of the genes in the region encode NRPS (ID# 115356, 140513), mentioned above as unique to isolate ND09Pr. 115356 is on a branch by itself in the NPS1/NPS3/NPS13 expansion group on the NRPS AMP tree (Figure 6, Figure 5, double asterisks, Table 8) and is not found in any of the other Cochliobolus species, in S. turcica, or in Genbank. NRPS ID# 140513, also unique to the VHv1 region (Figure 7), maps to a C. sativus-specific clade we call ‘New_8’, consisting of seven AMP domains (Figure 3, asterisk) corresponding to three additional C. sativus- unique NRPSs. One of these is ID# 130053 with three AMPs on scaffold 25. Based on proximity in the genome assembly, we believe a second unique AMP, in protein ID# 49884, also on scaffold 25, is actually a fourth AMP domain of 130053. The remaining NRPS grouping in the New_8 clade is ID# 104448.

Differences in NPS gene content and pathogenicity phenotype of closely related C. sativus strains allowed us to identify candidates for functional analyses. We hypothesized that, since the VHv1 region is unique to isolate ND90Pr and contains two NRPSs (ID#140513, 115356) unique to C. sativus, one or both of these might be responsible for high virulence on barley. We conducted real time PCR (Table S9) on infected barley leaves and demonstrated that expression levels of the genes corresponding to 140513 and 115356 in the VHv1 region, and also of the genes corresponding to unique proteins 130053 and 49884 described above were up-regulated 12 hours post inoculation (Figure 7B), while the gene corresponding to protein ID# 350779, which maps in the Gliotoxin clade (Figure S5A, Table S9), was not. Deletion of the gene corresponding to protein 115356 indicates that, indeed, it is involved in the high virulence of ND90Pr on cv. Bowman, as the mutant is significantly reduced in virulence to the host, compared to the wild-type strain (Figure 7C, Figure S7). Thus, our comparative approach to analyzing secondary metabolite core proteins (NRPSs) led to the identification of a unique genomic region in C. sativus pathotype 2 isolate ND90Pr associated with high virulence on barley cv. Bowman that carries NRPSs which, when functionally manipulated, impacted virulence.
Case Study 2: Among Cochliobolus species, the NRPS HTS1, which biosynthesizes the tetrapeptide HST HC-toxin, is unique to C. carbonum race 1 and has been demonstrated previously to be required for pathogenicity to hmhm maize [4], [18], [19]. HTS1, however, is present in S. turcica and other fungi. Given the thorough documentation of HTS1 as a pathogenicity determinant, functional analyses were not necessary to cement the connection between its unique signature within the genus and its role as a virulence determinant. With the wider genomic resources reported here, we found orthologs of all four HTS1 AMP domains in S. turcica (ID# 29755) (Figure 8A). Manning et al., [61] also report orthologs in P. tritici repentis (ID# 12015) and Wight and Walton have found an ortholog in Alternaria jesenkae and demonstrated, furthermore, that the isolate makes HC-toxin [62]. In addition, there are HTS1 orthologs (APS1, Acc#: ACZ66258) in the Sordariomycete, Fusarium incarnatum/semitectum, that biosynthesize a different metabolite, apicidin [63].

In C. carbonum race 1 strain, SB111, the original strain in which the HTS1 locus was described, the structural organization of the cluster of genes encoding enzymes for HC-toxin production is complex and includes two copies of most genes, in two clusters residing in an ∼600 kb region. The organizations of the S. turcica and P. tritici-repentis clusters are similar to each other, but different from that described for C. carbonum (Figure 8). Firstly, there is no evidence that the S. turcica and P. tritici-repentis genes are duplicated. Secondly, only orthologs of C. carbonum HTS1, ToxA, ToxE, and ToxF (C. carbonum HTS1 cluster nomenclature) proteins are clustered in S. turcica and P. tritici-repentis; orthologs of C. carbonum ToxC, ToxD, and ToxG proteins are found in both genomes, but on separate scaffolds in each genome (Figure 8). HTS1, ToxA, ToxE, and ToxF orthologs are present in the F. semitectum APS1 cluster, however there is no ToxC, ToxD, and ToxG in the sequenced cluster and, as genome sequence is not available for this species, we were unable to search for these genes.
Thus the phylogenetic approach, when conducted with the suite of Cochliobolus genomes, pinpointed the NRPS, HTS1, as unique to C. carbonum race 1 and functional analyses done previously, prove that the metabolite is an HST required for pathogenicity. The inclusion of a genome from a genus sister to Cochliobolus, however, identified an ortholog in S. turcica. This, combined with reports of HTS1 orthologs in other phylogenetically scattered groups, indicates a complex genetic history.
Case Study 3: C. victoriae has a NRPS that groups with high support with the A. fumigatus NRPS for Gliotoxin biosynthesis. In addition to the NRPSs extracted from our sequenced genomes, our phylogenetic trees included NRPSs producing known products, such as the two AMP domain A. fumigatus NRPS, GliP, for gliotoxin biosynthesis and the Leptosphaeria maculans NRPS, SirP, for sirodesmin production, both epipolythiodioxopiperazine (ETP) toxins. Two C. victoriae AMP domains on node 1179 clustered with 99–100% bootstrap support with A. fumigatus GliP AMP1 and AMP2 (Figure 9, Table 8). Both were evolutionarily closer than the corresponding SirP AMP domains. Thus, our objective to determine if any of the unknown NRPSs grouped with NRPSs with characterized products yielded a C. victoriae candidate for production of gliotoxin or a related metabolite. Furthermore, examination of the neighborhood surrounding the C. victoriae bimodular NRPS, indicates that all of the genes in the A. fumigatus gene cluster [64] are present in a cluster in C. victoriae (Figure 9A).

Summary
To thoroughly understand the evolutionary history of multimodular NRPSs, AMP domains were analyzed as individuals using a combination of amino acid alignments and phylogenetic tree building. Results show that, within a species, NRPSs are highly conserved, but conservation dissipates as comparisons are made across the genus. Thus, diversity of these genes, their encoded proteins and corresponding metabolite potential, is truly enormous. Strain-unique NRPSs are primary suspects for producing small molecules conferring high virulence or host specificity. A robust example of this are the species - and strain-unique C. sativus ND90Pr NRPS proteins 115356 and 140153 which map to the unique VHv1 high virulence conferring region, for which gene deletion confirms a role in cultivar specific virulence. The phylogenetic structure of NPS1 and NPS3 enzymes suggest that corresponding genes undergo rapid duplication and expansion and could act as a cauldron for the formation of new NPS genes.
Secondary metabolism: Polyketide synthases
Polyketide synthases (PKSs), like NRPSs, are large multidomain enzymes that produce small molecules (polyketides) with functions that include HSTs. The suites of PKS encoding genes (PKS) in the C. heterostrophus C4 and C5 genomes were identified and annotated previously [65]. To address degree of conservation and evolutionary relationships of PKSs in our subject species in order to make inferences about function, we used the PFAM ketosynthase domain (KS) HMM as a query to search for orthologs in the additional strains of C. heterostrophus and other species, and the related maize pathogen, S. turcica.
Conservation of known reference strain C. heterostrophus C5 polyketide synthases
Most C. heterostrophus PKSs are conserved across all C. heterostrophus strains, although PKS16 is absent from the genome of strain Hm338 and PKS25 is absent from strain Hm540 (Table 9, Figure 10, for the full phylogenetic trees see Figure S6, and Table S10 for master inventories). PKS13 is a pseudogene found only in strain C5 (and C4). As with the NRPSs, conservation of PKSs across the Cochliobolus genus is not as high as within C. heterostrophus species and is even less when the related genus, S. turcica is considered. Seven out of the 23 PKSs in reference strain C5 are conserved in all Cochliobolus species and S. turcica (Table 9, Figure 10); the only known product of these is melanin (produced by C. heterostrophus PKS18). Otherwise, the products of conserved PKSs are unknown. Three PKSs are present in all Cochliobolus genomes, but not S. turcica (Table 9). Two PKSs are unique to C. heterostrophus, while nine are present discontinuously throughout the species examined. Blast searches using the predicted protein sequences of C. sativus isolate ND90Pr PKSs as queries against the genome sequences of the isolate ND93-1 (a pathotype 0 isolate) indicated that all PKSs predicted in ND90Pr were found in ND93-1, except one (ID# 184740).


Species-unique PKSs
As for the NRPSs, we anticipated that species-unique PKSs (PKSs possessing KS domains lacking orthologous KS domains with bootstrap support in other species) would be the likeliest candidates for virulence functions, given the well-documented roles of HSTs in virulence and their corresponding unique or spotty distribution patterns in related strains. C. heterostrophus race O has 23 PKSs and no unique ones, while C. heterostrophus race T has 25 PKSs, two of which are unique. C. victoriae, C. miyabeanus, and C. sativus have 21, 21, and 18 PKSs, respectively, and no unique ones; C. carbonum has 27 PKSs including two unique ones, and S. turcica has 27 PKSs, including 13 unique ones (Table S10).
Discontinuously distributed and expanded PKSs
There were ten clusters (designated ‘New’ 1–10) of PKS genes that did not have an ortholog in C. heterostrophus (Figure 10, Table S10). Some of these clusters, such as ‘New_2 or New_5’, had representatives of only a few species. Others such as ‘New_3 or New_6’ contained representatives of all species except C. heterostrophus. Two clusters (New_1 and New_8) were not sister to a group with a C. heterostrophus reference PKS, but contained an ortholog from a C. heterostrophus field strain (Figure 10).
The only expanded group of PKSs from the C. heterostrophus set was PKS14, which had two orthologs in C. miyabeanus (Table 9, Figure 10). Otherwise, C. heterostrophus PKSs were either conserved as single copies, discontinuously present in single copy, or C. heterostrophus unique. Expansion did not seem to occur centered around a certain set of ‘birthing reservoir’ genes, as for NPS1 and NPS3 AMP domains (Figure 6).
PKSs showcasing the value of the phylogenomic approach in pinpointing candidate virulence determinants
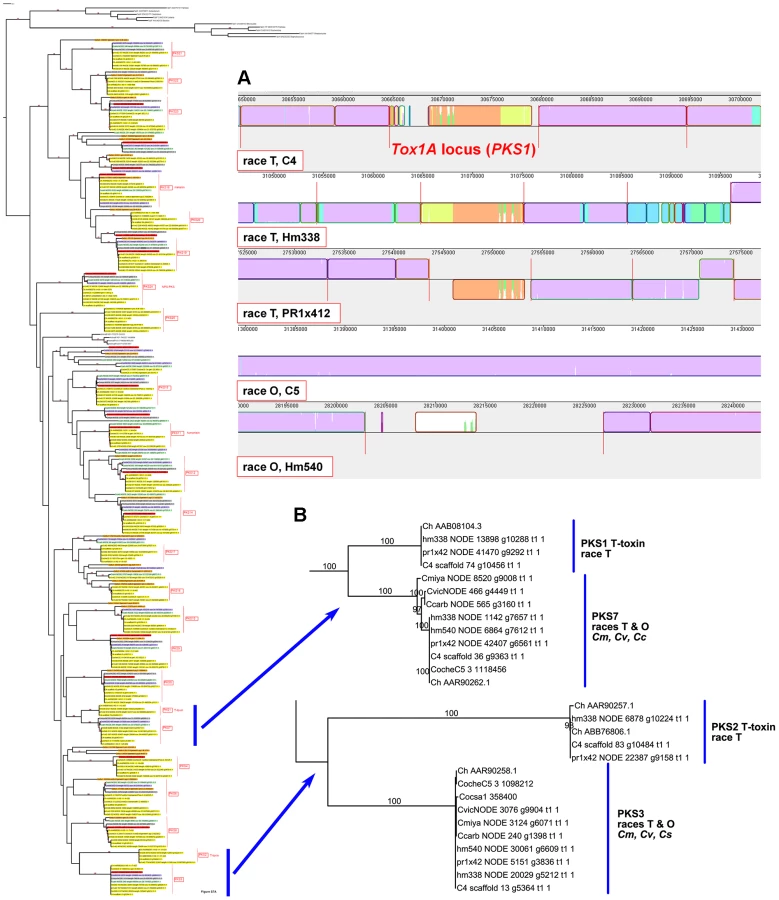
Case Study 1: All race T strains of C. heterostrophus have two PKSs not found in race O strains or any other known species. PKS1 and PKS2, genes required for biosynthesis of T-toxin, are present in all C. heterostrophus race T strains but absent from all C. heterostrophus race O strains and all Cochliobolus species examined to date (Figure 10, Figure 11A, Table 9). In previous work, we demonstrated that strains deleted for either of these two PKS genes fail to make T-toxin and are much reduced in virulence on T cytoplasm corn [45], [48], [66], [67]. This race-specific PKS example mirrors the aforementioned NRPS examples, i.e., the C. sativus ND90Pr region carrying the NRPSs 115356 and 140513 (Figure 7) and the C. carbonum region encoding HTS1 (Figure 8).
Case Study 2: A S. turcica-specific PKSs is up-regulated in planta. Three S. turcica PKSs grouped together in a S. turcica - specific clade, labeled ‘New_10’ (Figure 10, Table S10). Using real time PCR, we examined in planta expression of one of the S. turcica unique genes (ID 161586) and found that expression was increased 560-fold at three days post inoculation (Figure S8), then dipped and rose again to the same level at seven days post inoculation. Although we haven't yet deleted this gene, based on other test cases, it is tempting to couple the in planta expression pattern with a possible role in virulence.
Summary
Like NRPS proteins, the PKSs examined were either highly conserved, partially conserved, or strain unique. Some orthologs had duplicated members for some species, but this expansion did not orbit a particular set of genes such as NPS1 and NPS3. PKSs identified as strain or species-unique include characterized, as well as unknown, candidate virulence factors. The race T unique C. heterostrophus PKS genes PKS1 and PKS2 are examples of characterized unique, highly specific virulence factors. Further characterization of strain-unique PKSs, such as S. turcica ID 161586, which is highly expressed in planta (Figure S8) could reveal novel virulence factors.
Location of C. heterostrophus NPS and PKS genes
Several publications demonstrate that species/strain unique sequences tend to reside in variable regions of the genome such as in subtelomeric locations [68], [69] and dispensable chromosomes [70]. All C. heterostrophus reference strain C5 NPS and PKS genes were mapped to the assembled linkage groups (Figure 2). For NPSs, 13 of the 14 total could be mapped to one of the 16 linkage groups/chromosomes, and 6 of the 13 were <200 kb from the end of the linkage group. Two (NPS 5, 9) of the six are unique to C. heterostrophus and two (NPS1, 11) have limited distribution in Cochliobolus spp.. For PKSs, 22 of the 25 total could be mapped to one of the 16 linkage groups, 9 of the 22 were <200 kb from the end of the corresponding linkage group and one (PKS25) mapped to the B chromosome. Five (PKS 13, 16, 17, 20, 25) of the ten are unique to C. heterostrophus and one more (PKS11) has limited distribution in Cochliobolus spp.. In sum, approximately half of the NPSs and PKSs map to scaffold ends, in some cases with mapped telomeres (Figure 1). As chromosome ends are notoriously variable, this placement could indicate a mechanism for patchy phylogenetic distribution of these genes.
Note that the two PKSs involved in T-toxin production by race T are absent in race O strain C5, but map (genetically) to the breakpoints of race T chromosomes 12;6 and 6;12 which are reciprocally translocated with respect to chromosomes 6 and 12 in race O C5 [9]. Note also that PKS3, which has a phylogenetic relationship, but without bootstrap support, to PKS2 (Figure 10, Figure 11B), maps internally to race O chromosome 6 (Figure 2), and that PKS7, which is the closest (with bootstrap support) C. heterostrophus PKS to PKS1 (Figure 11B), maps to the end of unplaced scaffold 20 (Figure 2).
Small secreted proteins (SSPs)
To identify candidate effector proteins, we searched the gene catalog of each species for proteins that were cysteine rich (≥2% cysteine), small (<200 amino acids), predicted to be secreted (using Phobius [71]), and without transmembrane domains. Between 141 and 289 SSPs per genome (Table 10) were identified with C. sativus ND90Pr containing the most and C. heterostrophus Hm338 the fewest. We next conducted an all versus all blast analysis to determine if SSPs were strain or species-unique, using an 80% blast cutoff. Very few C. heterostrophus SSPs were unique to any particular strain as most could be found in at least one other C. heterostrophus field or lab strain. Using this approach, we identified between one and 21 unique SSPs (Table 10, master inventory Table S11). We found more strain-unique SSPs in the other Cochliobolus genomes, as our analysis included five C. heterostrophus strains. S. turcica and C. sativus had the most isolate-unique SSPs, containing 191 and 167 candidates, respectively. As these are the two strains thought to act as hemibiotrophs, it is interesting that they contain more SSPs, and more unique SSPs, than the necrotrophic isolates, although this is only a correlation at this point.

The C. heterostrophus C5 assembly has 180 predicted secreted proteins matching the criteria. We examined each of these in the JGI browser with respect to EST support, SNPs, and predicted functional domains. Seventy-two of these calls had absolutely no EST support, while 24 calls had incomplete EST support (ESTs matching some portion, but not all, of the gene call), leaving 84 with complete (spanning the entire gene model) EST support (Table 11). Genes in the no-EST support category are of special interest, as they may be specifically expressed in planta and thus not expressed under conditions used for preparing our EST libraries (fungus grown in vitro on a variety of complete (CM) and minimal (MM) media, and mixed into CM or MM pools). Lack of strong EST support may also suggest an erroneous gene call.
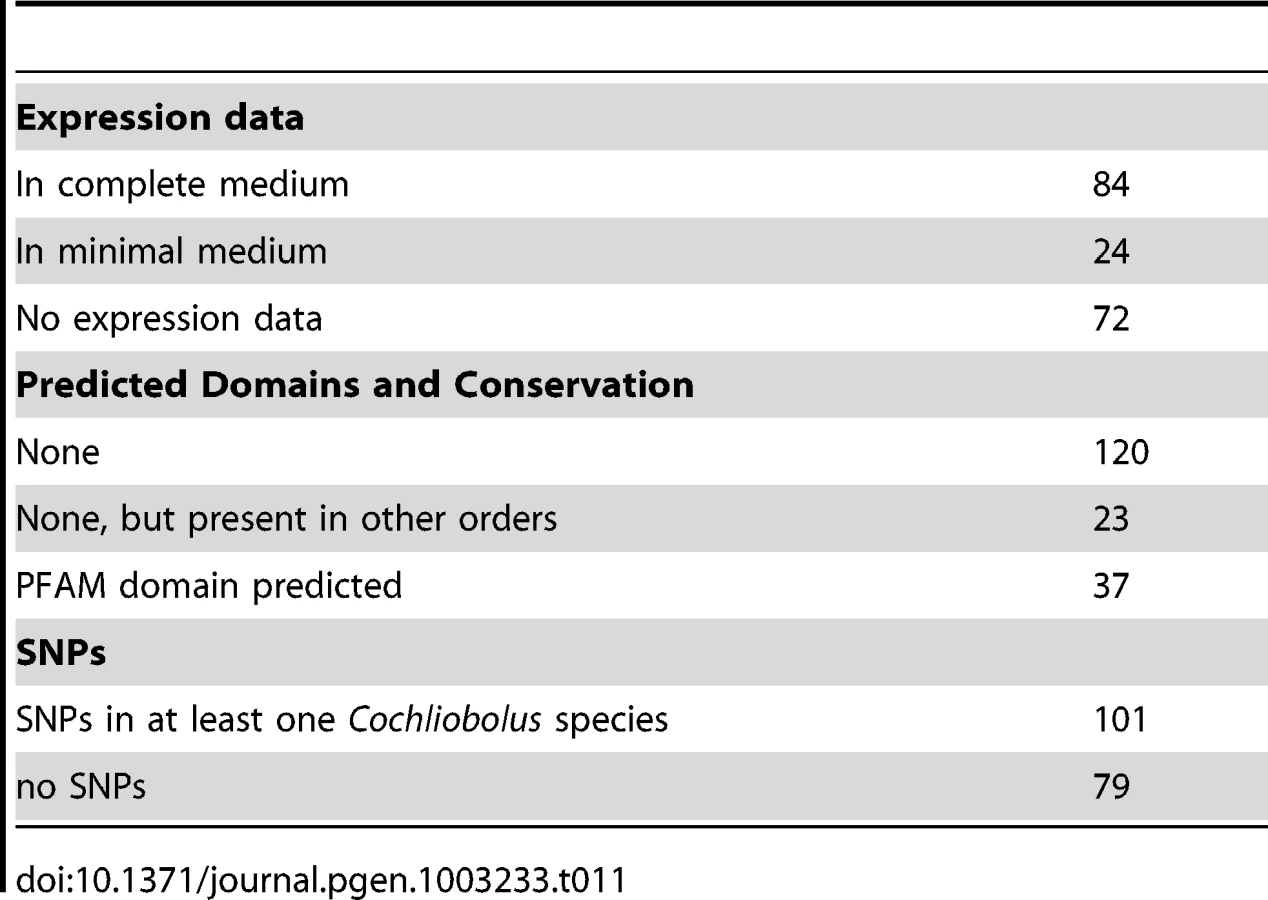
As is typical with candidate effectors, functional domain predictions were lacking, with only 37 candidates having some predicted function, generally involved in cell wall or extracellular matrix function (Table 11, Table S11). An additional 23 candidates were conserved in other fungi outside of the Dothideomycetes. The remaining 120 calls were featureless and seemingly unique to the Dothideomycetes. C. heterostrophus strain C5 SSP calls were rich in SNP calls to other Cochliobolus genomes: 101 candidate SSPs had SNPs with at least one other Cochliobolus genome.
In our all versus all blast analysis, only 6 of the 180 C. heterostrophus C5 SSPs were found in all 10 strains examined and 14 were unique to strain C5 (Table 11). The presence or absence of most SSPs did not fall into easily categorized bins such as C. heterostrophus-specific, or maize-pathogens only. Instead, SSPs were present and absent in no particular pattern across the genomes. 115 SSPs were present in at least one other species (C. victoriae, C. miyabeanus, C. carbonum, or S. turcica), with seven found in all species, and 27 in all Cochliobolus species. SSPs mapped to all scaffolds larger than S26 (Figure 2).
Unlike those in some phytopathogens, such as Leptosphaeria maculans [72], SSP encoding genes did not occur in clusters; candidates seldom were located within 10 kb of each other (Figure 2). These genes were, however, often located in or near regions we identified as C. heterostrophus species unique (Figure 2).
Discussion
The genomes of five C. heterostrophus strains, two C. sativus strains, three additional Cochliobolus species (C. victoriae, C. carbonum, C. miyabeanus) and S. turcica, a member of a close sister genus, were sequenced and compared, to identify unique genomic regions and to inventory secondary metabolism and SSP encoding genes. This dataset is distinctive in that it allows us to contrast genomes that are very closely related, yet differ in several key ways. First, our dataset includes highly related pathogens of several different host plants (corn, wheat, rice, barley, Brachypodium). Second, this set includes pathogens that are highly virulent on specific cultivars of a particular host (i.e., C. heterostrophus race T on Tcms maize, C. victoriae on Vb oats, C. carbonum on hmhm maize), as well as more generalist pathogens such as C. sativus that can cause disease on multiple hosts (barley, wheat, Brachypodium). Third, the group includes two hemibiotrophs, S. turcica and C. sativus, while the rest are necrotrophs. Fourth, we can make graduated comparisons at different levels of parental and phylogenetic relatedness as we progress from genomes of the same inbred line, species, genus, and family. We have used this final point as a probe to attempt to understand the significant genomic differences between strains that shape host choice, specificity, and lifestyle.
Structural differences across strains and species
Our whole-genome alignment data support graduated degrees of similarity at the highly inbred strain, field strain, species and genus levels. C. heterostrophus strains C4 and C5, offspring of successive backcrosses [42], were highly similar to one another, with 20 fold fewer SNPs than pairwise comparisons of reference strain C5 to C. heterostrophus field strains. This remarkably low number of SNPs highlights the power of selective inbreeding in establishing uniformity across the genome. The two C. sativus field strains, when aligned to each other, had a comparable number of SNPs to those of C. heterostrophus field strains aligned to reference C. heterostrophus strain C5. Other Cochliobolus genomes had roughly 50 fold more SNPs than C. heterostrophus field strains when aligned to the C. heterostrophus C5 reference. This level of similarity was seen when comparing any two Cochliobolus species to one another, with the exception of comparing C. victoriae to C. carbonum. These two species are capable of successfully mating, although progeny of crosses are unable to cross to each other or to their parents (Turgeon lab, unpublished). We have hypothesized that C. victoriae may have evolved from a C. carbonum strain [49]. This similarity is seen at the whole genome level, as C. victoriae and C. carbonum share an intermediary number of SNPs compared to C. heterostrophus inter - and intra-species comparisons.
Our SNP data show that approximately one quarter of the genome differs between Cochliobolus species and that only about one tenth of this is found in segments larger than 5 kb. We and others [72], [73], [74] have recently introduced the term mesosyteny [1] to describe organizational conservation between species. Genetic content is conserved across chromosomes, but not co-linearly. It seems possible that our findings here, showing that many small, scattered differences summing to significant quantitative differences (i.e., 25% dissimilar), could be the product of the same mechanisms that result in mesosyntenic patterns. Pathogens of the same host (e.g., C. carbonum and C. heterostrophus on maize) or lifestyle were not more similar to each other than those of different hosts; instead overarching genetic patterns followed phylogenetic lines. As it is estimated that the Pleosporaceae arose as a group less than 20 MYA (see Figure 1 in Ohm et al., [1]) and the genus Cochliobolus is young in the group, genome comparisons provide us with an overall picture of a timeline of how genome diversity varies with speciation.
Our intra-species SNP tallies are comparable to SNP tallies found when strains of other species are examined. For example, there were 10,495 SNPs called between two Fusarium graminearum strains [75], and a range of 13,274–188,346 SNPs called for 18 Neurospora crassa classical genetic mutants [76]. Both dataset tallies are in the same range as our C. heterostrophus field strain comparisons. With respect to SNPs in different species of the same genus, it is unusual that we were able to perform a whole-genome SNP analysis at all, without limiting our scope to coding sequence. We owe this to the very close phylogenetic relationship of these species.
Strain-, species-, and genus-specific genes for secondary metabolism
Although individual functional domains of NPS/PKS proteins can be identified bioinformatically, attempting to predict their corresponding metabolite product is challenging. The genes encoding these proteins evolve rapidly and through complex mechanisms [57], [65] and whole gene alignment methods provide misleading or unclear results when determining presence or absence of a particular NPS or PKS gene. Here, we extracted individual conserved signature catalytic domains, i.e., the AMP-binding domains from mono - or multi-modular NRPSs or the ketosynthase (KS) domain from multidomain PKSs using customized HMM models, then built alignments and phylogenetic trees with these individual units to determine the presence or absence of whole or partial NRPS and PKS proteins, and their evolutionary relationships. In our opinion this is a necessary first step towards understanding evolutionary history of the corresponding genes and the possible small molecules produced by these highly diverse proteins.
We found that within a Dothideomycete genus, in this case Cochliobolus, approximately half of the NPS and a third of the PKS genes are well conserved (present in all strains). When related S. turcica was considered these numbers dropped to a third and a fifth, respectively. The rest were found to be poorly conserved or species-unique when the highly curated C. heterostrophus gene sets were used as reference. The small molecules produced by the corresponding non-conserved proteins are largely uncharacterized, but the differences between strains and species imply that the potential for production of biochemically unique molecules is large and considerably beyond that expected for closely related strains (housekeeping genes share ∼95% identity). These findings refine our understanding of NPS and PKS genes, as very few are conserved. For example, only two NPS genes and one PKS gene were found when 18 Dothideomycete genomes were analyzed [1]. Broadly conserved secondary metabolism genes, where they have been characterized, produce small molecules that serve basic cellular functions (ferricrocin, melanin [58], [77], [78], [79]). Poorly conserved NPS and PKS genes, while still largely uncharacterized, can include those involved in host-specific high virulence.
NPS1, NPS3, and NPS13 embody the intriguing genetic origins of NRPS proteins
NPS1 and NPS3 AMP domains are discontinuously distributed and expanded across the Cochliobolus and Setosphaeria isolates sequenced and are sources of much of the NRPS diversity (Figure 5, Figure 6 and [57]). The individual AMP modules do not cluster by protein, but instead, NPS1, NPS3 and NPS13 AMP domains occur in two distinct, and mixed, groups (Figure 6). Strikingly, each Cochliobolus species possesses either a complete C. heterostrophus NPS1 or NPS3 ortholog, but never both. Furthermore, all species have one or more additional NRPS proteins consisting of NPS1/NPS3 related domains, and a NPS13 related domain that is absent from all C. heterostrophus genomes except Hm540 (Figure 5). The C. heterostrophus Hm540 genome includes all four corresponding genes: NPS1, NPS3, NPS13, and the additional NPS1/NPS3/NPS13 gene (Figure 5). The pattern of duplication and loss appears to have been very rapid to account for this distribution, and is further complicated by the presence of additional bi-, mono-, tri-, and tetra-modular proteins, particularly in C. sativus, whose AMP domains group with NPS1/NPS3/NPS13 proteins, (Figure 5, Figure 7, Figure S5).
Evolutionary origin of NPS and PKS genes
The origin of strain or species unique secondary metabolism genes is of great interest and horizontal gene transfer is a common, but not the only, explanation for their appearance [65], [80], [81], [82]. The volatility of the NPS1, NPS3 and NPS13 family raises the possibility that genes we presume are horizontally transmitted could have vertical histories obfuscated by species and strain sequence depth. We speculate that partial or whole genes encoding individual domains or whole proteins recombine and expand quickly, and, when they confer high virulence, as in the case of HSTs, can spread rapidly throughout a population. When the susceptible host allele is not present in the population, the gene is lost or not conserved in the majority, but not the entirety, of the population, as is the case for C. heterostrophus race T and genes for T-toxin production; race T is difficult to find in the field currently [43]. As we sequence more and more isolates, we might find that the T-toxin genes are present in strains of many more Dothideomycetes than we originally expected. This is certainly the case with the HC-toxin genes which is not so surprising, given that the Hm alleles are present in most plants.
Pinpointing virulence-associated secondary metabolite genes
Identifying secondary metabolites that function as virulence factors (such as HSTs) is a primary goal when studying a pathogen's genome. The impact of HSTs was realized early on because they render the producing fungi pathogenic or highly virulent to principal crops. Thus, most were characterized physiologically and genetically decades ago [4], [18], [19], [40], [83], [84], [85]. The pivotal point of our comparative analyses is the strikingly obvious observation that secondary metabolite genes, when unique to a species or strain, are likely to encode a virulence determinant. We provide several examples.
The first example is the C. heterostrophus PKS1 and PKS2 genes involved in production of the HST T-toxin. These genes reside in 1.2 Mb of DNA, not found in race O and located at the breakpoints of two race T chromosomes (12;6, 6;12), reciprocally translocated with respect to race O counterparts (chromosomes 6, 12). The T-toxin genes are not in race O or any other Cochliobolus species. Deletion of either PKS eliminates T-toxin production and drastically reduces virulence of the fungus on Tcms maize, as reported earlier [45], [67]. Known Tox1 genes, such as PKS1 are on very small scaffolds (∼25 kb) in race T strains C4, Hm338, and PR1x412 (Figure 11), which cannot be further assembled due to the repetitive and AT-rich nature of the locus. Thus the physical structure of the Tox1 locus remains elusive, but its association with a unique genomic region, however complex, is clear-cut. Although T-toxin is unique to C. heterostrophus, a closely related fungus, Didymella zeae maydis (formerly, Phyllosticta maydis, Mycosphaerella zeae maydis), produces a polyketide HST, PM toxin, with the same biological specificity as T-toxin. The central PKS for PM-toxin is the closest PKS to C. heterostrophus PKS1, but still only ∼60% identical at the amino acid level and organization of the cluster of genes required for toxin production differs; in D. zeae-maydis, the genes are present in a single tight cluster [86], [87].
The second example, examined here, is the NRPS, HTS1, for HC-toxin production. The genes for HC-toxin produced by C. carbonum, were identified two decades ago in a tour de force molecular manipulation exercise [88], [89]. A strong genomic signature attends these genes as they reside in an ∼600 kb region not found in other races of C. carbonum, or in any of the additional Cochliobolus genomes examined then or here. Two copies of a cluster of HC-toxin genes are located in this region and both copies of the core NRPS, HTS1, had to be deleted to demonstrate elimination of toxin production and reduction of virulence [88], [89]. In current investigations, we, Manning et al., [61] and Wight and Walton [62] have discovered that HC-toxin like genes are present in S. turcica (Figure 8), P. tritici-repentis and A. jesenskae, respectively. These genes are also apparent orthologs of the genes for apicidin (APS1) production by some Fusarium species [90]. Thus, genes for HC-toxin or HC-toxin-like metabolites are more broadly distributed than previously thought. In terms of amino acid identity, the S. turcica and P. tritici-repentis NRPS HTS1 proteins are 79% identical at the amino acid level, but identity drops to 39–43% when these are compared to the C. carbonum HTS1 or APS1 proteins. The S. turcica and P. tritici-repentis HTS1 orthologs lack the C-terminal condensation domain found in C. carbonum HTS1 and F. semitectum APS1, suggesting S. turcica and P. tritici-repentis make a different product. Whether or not S. turcica and P. tritici-repentis are capable of producing HC-toxin is unknown, however, it has been reported [62] that A. jesenskae does. That HC-toxin producing capability might be found in pathogens other than C. carbonum is not unreasonable, considering the maize defense gene Hm1, necessary to detoxify the toxin, is found in all grasses [91].
The third example concerns C. sativus. Two of the NPS genes unique to C. sativus ND90Pr (IDs 115356 and 140513, Figure 7) are present at the VHv1 locus associated with high virulence on cultivar Bowman. The entire VHv1 locus is absent in the low virulence isolate, ND93-1, and the two NPSs are not found in any other genomes examined here, or in Genbank. Our data show that these genes are up-regulated 12 hrs after inoculation, and that deletion of one of them, (115356), significantly reduces virulence on barley cultivar Bowman (Figure 7). Recent work on deletion of the gene encoding 4′-phosphopantetheinyl transferase provided indirect evidence that a secondary metabolite is involved in the biosynthesis of the virulence factor in ND90Pr [92]; our current work directly confirms this.
The phylogenetic location of these VHv1 NPS genes is revealing in that they are either in branches with no close sister members (ID# 140513, Figure S5A) or in the NPS1/NPS3/NPS13 expansion clade (ID# 115356, Figure 5, Figure 6). In addition to these two genes, we have evidence based on real time expression data on RNA from inoculated barley, that the C. sativus genes corresponding to protein ID#s 49884 and 130053 are also up-regulated at 12 hrs post inoculation. These genes are found in the same C. sativus specific clade (New_8, Figure 3, Figure S5A) as protein ID# 140513, and although we have not yet made mutants, our prediction is that these will also contribute to virulence.
Our analyses of the hemibiotroph, S. turcica, is in its infancy, however, as noted in the Results we have identified 13 unique PKSs, three of which (ID# 161586, 30113, 34554) grouped together in a S. turcica - specific clade, called ‘New_10’ (Figure 10, Figure S8, Table S10). As preliminary support for the importance of unique PKSs, we used real time PCR, to examine in planta expression of one of these S. turcica unique genes (161586) and found that expression was indeed increased (>500 fold) by three days post inoculation. Although we haven't yet deleted this gene, it is tempting to predict that the in planta expression pattern is indicative of a role in virulence.
One of the species-unique NRPSs in C. victoriae (on node 1179, gene #7087) is a NRPSs with two AMP domains clustering with 99–100% bootstrap support to AMP domains from the bimodular A. fumigatus NRPS, GliP, which produces the ETP toxin, Gliotoxin. Related to these NRPSs is the L. maculans NRPS, SirP, which produces sirodesmin. Candidate orthologs of these NRPSs have been reported in Chaetomium globosum, Magnaporthe oryzae, and Fusarium graminearum [93]. Gliotoxin is associated with virulence of A. fumigatus to immune-compromised patients [94]. Functional characterization of the newly discovered C. victoriae counterpart is necessary to determine the type of ETP produced and whether or not it might play a role in virulence, as Gliotoxin does. Note the entire Gliotoxin gene cluster [64] is present in C. victoriae (Figure 9). Gene knockout and screening for alteration in virulence to oats, due to victorin production, indicates no change from that of wild type (Wu, Turgeon, unpublished). This C. victoriae NRPS is not found in other Cochliobolus genomes, yet it clusters with A. fumigatus GliP, exemplifying the patchy distribution signature of most members of the NPS family of genes.
Effectors and lifestyle
Effectors are pathogen produced small secreted proteins (SSPs)/small molecules that interact with the host plant to promote disease. Effectors historically were called avirulence proteins (as their discovery hinged on association with a corresponding plant resistance gene), but we now recognize that effectors are virulence factors that aid the pathogen by specifically targeting aspects of host cell defense and recognition. Evading detection is a necessary strategy for (hemi)biotrophs, where triggering the host hypersensitive response curtails disease. Necrotrophs, on the other hand, benefit from the death of host cells, and have evolved molecules such as HSTs, like victorin which subverts function of an R gene (Lov1/Pc-2) to trigger susceptibility and plant cell death intentionally [14], [15], [95]. Protein HSTs such as P. tritici-repentis and Stagonospora nodorum ToxA are clear examples of secreted, necrotrophic, proteinaceous, host-selective virulence factors acting to effect virulence in host cells, like any other effector, but which, in the presence of a R-protein look-alike, is necessary for susceptibility [96]. The lingering question is whether or not necrotrophs utilize SSP effectors in the traditional (and difficult to identify) sense of micromanipulation of the host environment, or, instead, use effectors to trigger host cell death through abuse of (hemi)biotrophic defenses. In this regard, given our clear discovery that at least one NRPS metabolite (ID# 115356), when deleted has a much reduced phenotype reminiscent of a necrotrophic HST phenotype, we question whether C. sativus should truly be considered a hemibiotroph or a necrotroph.
On the other hand, our SSP analysis shows that C. sativus and S. turcica have an expanded SSP repertoire compared to the other species examined, which is consistent with a hemibiotroph strategy, i.e., arsenals of effectors are used to evade host detection. The repertoire of candidate effectors in necrotrophs, nevertheless, is quite large. If only a small subset of these is involved in virulence, it would mean that Cochliobolus, and perhaps other necrotrophs, use effectors more expansively than is recognized. This is a difficult question to address, and our in silico analysis requires experimental confirmation of in planta expression and secretion before we can be sure Cochliobolus species utilize protein effectors. Perhaps a strategy prioritizing species or strain unique regions would aid characterization attempts. Bearing on this point, the SSP catalogue differed markedly from secondary metabolites in their conservation across, and within, species. Only six of the 180 (3%) C. heterostrophus C5 SSPs were identified in all genomes examined (including S. turcica), unlike the 7/25 (28%) PKSs and 6/14 (43%) NRPSs. Considering C. heterostrophus genomes only, 27 of these 180 SSPs were present in each (15%), again, far fewer than the 21/25 (84%) PKS and 13/14 (93%) NRPS C. heterostrophus C5 genes conserved throughout all C. heterostrophus genomes. This indicates that, more so than secondary metabolite genes, SSP encoding genes are extraordinarily volatile in the evolutionary history of the genus.
Final thoughts
The stories of the SCLB and Victoria blight epidemics are dramatic examples of interactions between crops, whose ‘evolution’ is driven by human intervention (breeders) and their pathogens, which evolve naturally to exploit new genetic susceptibilities. Both the Tcms and Pc-2 genes were introduced into maize and oats, respectively, by breeders fewer than 30 years before the epidemic outbreaks. Specifically, Tcms was discovered in the 1940's, incorporated into elite corn inbred lines increasingly throughout the 1960's, and was present in almost all of the hybrid corn in the US by 1970. The vast monoculture of Tcms maize was the perfect host for the previously unknown race. Species of Cochliobolus spp. clearly have proven their ability to cause extraordinary crop losses. As we begin to understand the intimidating capacity for diverse production and evolution of new HSTs, we must also look for ways to apply this knowledge to our disease response strategies.
Materials and Methods
Strains
C. heterostrophus strains sequenced by JGI included inbred strains C5 (ATCC 48332, race O, MAT1-1, Tox1−) and C4 (ATCC 48331, race T, MAT1-2, Tox1+) and field strains Hm540 (geographical origin North Carolina, race O, MAT1-1, Tox1−), Hm338 (New York, race T, MAT1-2, Tox1+, ATCC 48317), and PR1x412, (a progeny of a cross between PR1C from Poza Rica, Mexico and strain 412, unknown geographical origin, race T, MAT1-1, Tox1+). In addition, the genomes of C. victoriae strain FI3 (unknown geographical origin, MAT1-2, victorin+), C. carbonum strain 26-R-13 [MAT1-1, HC-toxin+, a progeny of a cross between C. carbonum strains 2-R-6 (alb2; MAT1-1) and Five Points (unknown geographical origin, MAT1-2) performed by Dr. Steve Briggs]. C. miyabeanus strain WK1C (Wuankuei, Yulan county China, MAT1-2), C. sativus isolate ND90Pr (North Dakota, ATCC 201652, MAT1-2, pathotype 2 on barley cv. Bowman) and S. turcica strain St28A (New York, race 2,3,N, MAT1-1) were sequenced by JGI. C. sativus isolate ND93-1 (North Dakota, ATCC 201653, MAT1-1, pathotype 0 on barley cv. Bowman) was sequenced at the University of Hawaii.
Genomic resources
The highly inbred C. heterostrophus reference race O lab strain C5 was sequenced using the Sanger whole genome shotgun approach, with paired end reads and improved by manual finishing and fosmid clone sequencing (http://www.jgi.doe.gov/sequencing/protocols/prots_production.html). Four different sized libraries were sequenced: 3.1 kb, 6.8 kb, and two fosmid libraries (32.3 kb and 35.3 kb), to a total coverage of 9.95×. ESTs were generated by growing strains in complete and minimal medium under many conditions [on complete and minimal medium, on sexual reproduction plates, stress medium (-N, -Fe, etc)] and pooled as complete or minimal samples for sequencing and support of gene annotation. The genome of isogenic C. heterostrophus race T strain C4 was sequenced using Illumina technology (300 bp insert size, 2×76 bp reads to a nominal depth of 200×), assembled using Velvet [97] and AllPathsLG [98] and annotated using ESTs from C. heterostrophus strain C5. The genome of C. sativus pathotype 2 isolate ND90Pr was sequenced using a hybrid approach, which included 40 kb fosmid Sanger reads, shredded consensus from Velvet assembled Illumina data (300 bp insert size, 2×76 bp reads), Roche (454) standard and Roche (454) 4 kb insert paired ends, all assembled using Newbler [99] and annotated using C. sativus ND90Pr ESTs as described below. The genome of the second C. sativus isolate, ND93-1, was sequenced at the University of Hawaii by paired end 454 runs and assembled using Newbler. S. turcica strain 28A was sequenced using Roche (454), Sanger fosmids, and shredded consensus from Velvet assembled Illumina data; EST libraries were prepared from S. turcica strains using conditions described above for C. heterostrophus.
The JGI annotation pipeline was used to annotate C. heterostrophus strains C5 and C4, C. sativus ND90Pr, and S. turcica. For this, the assembled genomic scaffolds were masked using RepeatMasker [100]with the RepBase fungal library of 234 fungal repeats [101] and genome-specific libraries derived using [102]. Multiple sets of gene models were predicted for each assembly, and automated filtering based on homology and EST support was applied to produce a final non-redundant GeneCatalog representing the best gene model found at each genomic locus. The gene-prediction methods were: EST-based predictions with EST map (http://softberry.com) using raw ESTs and assembled EST contigs for each genome; homology-based predictions with Fgenesh+ [103] and Genewise [104], with homology seeded by BLASTx alignments of the GenBank non-redundant sequence database (NR: http://www.ncbi.nlm.nih.gov/BLAST/) to the genomic scaffolds; and ab initio predictions using Fgenesh [103]) and GeneMark [105]. Genewise models were extended to include 5′ start and/or 3′ stop codons when possible. Additional EST-extended sets were generated using BLAT-aligned [106] EST data to add 5′ UTRs, 3′UTRs, and CDS regions that were supported by ESTs but had been omitted by the initial prediction methods.
All genome annotations can be interactively accessed through MycoCosm [107], http://jgi.doe.gov/fungi.
Resequencing additional C. heterostrophus race T and race O strains and other Cochliobolus spp
Because the subject genomes are all closely related to the fully sequenced reference C. heterostrophus strain C5, additional C. heterostrophus field strains Hm540, Hm338 and PR1x412, C. victoriae, C. carbonum, and C. miyabeanus, were (re)-sequenced using using Illumina technology. DNA was randomly sheared into ∼200 bp fragments using Covaris E210 according to the manufacturer's recommendation and the resulting fragments were used to create an Illumina library. 2×76 bp reads were assembled using Velvet (version 1.1.04), [97]. There are no ESTs available for these organisms. Assembled contigs were mapped to the reference C. heterostrophus C5 for analysis of genome variation and rearrangements. Assembled reads are called ‘nodes’ (scaffolds). Overall sequence assembly and annotation statistics are presented in Table 3.
The genomes without JGI annotation pipeline gene predictions (ChHm540, ChHm338, ChPR1x412, C. carbonum, C. miyabeanus, C. victoriae) were annotated using Augustus [108].
Mapping genomes to the C. heterostrophus strain C5 assembly
Assembled genomes were mapped individually to the C5 reference strain using the nucmer program of MUMmer v 3.22 [46], a program that finds unique, exact matches to build whole genome alignments. SNPs were called and analyzed using the dnadiff wrapper on the filtered MUMmer delta files. Unassembled reads were also aligned to the C. heterostrophus C5 reference genome for low coverage analysis using maq-0.7.1. Regions of the reference genome under a depth of three aligned reads were considered “low coverage” for our analyses. Adjacent low coverage regions were merged if they were separated by less than 100 bp in order to minimize noise from mis-mapping of occasional low quality reads. After low coverage regions were identified for pairwise comparisons to C5, regions were identified that were shared as low coverage in multiple genomes: i.e., C. carbonum, C. victoriae, C. miyabeanus, and C. sativus for C. heterostrophus specific regions; C. heterostrophus Hm540, Hm338, and PR1x412 for C strain specific regions; and C4, Hm338, and PR1x412 for race O specific regions.
Cross genome comparisons were visualized using Mauve [44], a multiple genome alignment tool that visualizes localized collinear blocks (LCB) between genomes.
Mapping reference genome C. heterostrophus strain C5 sequenced scaffolds onto the genetic map
Cloned RFLP markers [8] were sequenced and sequences used in blast queries against the C. heterostrophus C5 assembly. Top hits were filtered using Bioperl and manually confirmed to span the entire RFLP with very high stringency to rule out markers that might exist as more than one copy. Physical and genetic distances of adjacent RFLPs mapping to the same scaffold were plotted and used to calculate an average ratio of physical to genetic distance (Figure S1, Table S2). Relative RFLP location was used to orient scaffolds along the linkage group when possible.
Mapping of sequenced scaffolds to the C. sativus genetic map
A genetic linkage map was generated previously using the mapping population derived from a cross between parental isolates ND93-1 and ND90Pr [30] and amplified fragment length polymorphism (AFLP) and RFLP markers. To add simple sequence repeat (SSR) markers to the map, the draft sequence assembly of the C. sativus isolate ND93-1 was screened for SSR loci with di - and tri-nucleotide units tandemly repeated six or more times using a Perl script (provided by Zheng Jin Tu at the University of Minnesota, St. Paul). The SSR-containing sequences from ND93-1 were aligned to the draft genome sequence of isolate (ND90Pr) of C. sativus. Only those sequences that were polymorphic between the two C. sativus parents (ND93-1 and ND90Pr) were used for primer design and tested for segregation in the mapping population used previously [30]. PCR conditions and detection of SSR markers were as previously described [109]. Map construction was performed by using MAPMAKER version 2.0 [110]. A minimum LOD value of 4.0 and a maximum theta of 0.3 were used to group all SSR markers with previously mapped AFLP, RFLP and PCR markers [30]. The Kosambi mapping function was used to calculate the map distance.
To associate linkage groups to the sequenced scaffolds of C. sativus, the sequences of mapped SSR markers were used as queries to blast against the draft genome assembly of C. sativus isolate ND90Pr and the coordinates of each SSR marker were recorded for the associated scaffold.
Identification and phylogenomic characterization of nonribosomal peptide synthetases and polyketide synthases
NRPS and PKS proteins were identified using our custom fungal AMP domain model [57] for the former and an HMM model build from C. heterostrophus KS domains plus sequences from the C-terminal and N-terminal ketosynthase (KS) Pfam domains for the latter (PF00109 and PF02801). Proteins were identified in two ways. In the first case, genome nucleotide sequences were searched using Genewise [111] and sequences extracted and concatenated by a Perl script utilizing Bioperl's searchIO system [112]. In the second case, Augustus protein models (see above) and JGI protein models (C. heterostrophus, C. sativus, S. turcica) were searched with the PKS KS and NRPS AMP HMM using HMMER 3.0 [113], and the sequences extracted and concatenated using a Perl script with Bioperl's searchIO.
AMP and KS domains were aligned, separately, with MAFFT (http://mafft.cbrc.jp/alignment/software/) and manually inspected to remove columns of poor alignment. ProtTest [114] was run on both alignments and identified the RTREVF model as the best fit for the AMP domains and the WAGF model as the best fit for the KS domain alignments, respectively. RAxML [115] using the RTREVF and WAGF models with a gamma distribution was used to infer maximum likelihood trees and bootstrap support was determined using the fast-bootstrap method with 1000 bootstrap replicates [116]. The CIPRES portal (http://www.phylo.org/sub_sections/portal/) was used for inference of phylogenetic trees.
C. sativus ND90Pr NRPS AMP domains were used as blast queries to identify AMP domain orthologs in ND93-1 with the methods above. An ND93-1 AMP was considered orthologous if it was at least 95% identical to the ND90Pr query.
Small secreted protein identification
Candidate small secreted proteins (SSP) were identified by screening the gene catalogue of each genome. Proteins smaller than 200 amino acids and containing more than 2% cysteines were searched for transmembrane domains and secretion tags using Phobius [71]. Those without transmembrane domains were retained. EST support and domain prediction for C. heterostrophus C5 SSPs was performed using the JGI portal. Cross-genome comparisons were made based on all vs. all reciprocal best hit analysis with an 80% similarity cutoff.
C. sativus EST library construction
Three EST libraries were constructed. A mycelia-only library was constructed by harvesting mycelia grown on different media including Potato Dextrose Agar (PDA), minimal medium (MM) [117], V8PDA (150 ml V8 juice, 850 ml H2O, 10 g PDA, 10 g Agar and 3 g CaCO3), and water agar (15 g agar, 1000 ml water). Mycelia were harvested after 12, 24, 48, 76 and 96 hours of growth, and time points from different media were mixed together for RNA extraction. Equal amounts of extracted RNA from each of the 5 time points was bulked to construct the mycelia only library To construct the in planta cDNA libraries, two week old barley cv. Bowman and 4 week old Brachypodium distachyon line Bd21were inoculated with conidia of ND90Pr at a concentration of 5×103/ml [118]. Inoculated plants were incubated in a humid chamber for 24 hours and moved to the greenhouse. Leaves were harvested at 6, 12, 24, 48, 72, and 96 hours after inoculation and total RNA extracted from each sample. The final in planta cDNA libraries were constructed by mixing the equal amounts of total RNA from different time points. Total RNA was isolated from all samples using the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA) and purified by treatment with DNase I (Invitrogen, Carlsbad, CA). These three libraries were sequenced by JGI.
Quantitative real-time PCR
For C. sativus, total RNA extracted as described above at six time points (6, 12, 24, 48, 72, and 96 hours) after inoculation was used for RT-PCR. The reverse transcription reaction was performed on 2 µg of total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). cDNA was diluted 20 times and used as the template for quantitative RT-PCR, which was performed with the AB7500 real time PCR system (Applied Biosystems, Foster, CA) (Table S9). For each cDNA sample, three replications were performed. Each reaction mixture (20 µl) contained 5 µl of the cDNA template, 10 µl of SYBR Green PCR Master Mix (Applied Biosystems, Foster, CA) and 0.3 µl of each primer (10 mM). All samples were normalized using RT-Actin-F and RT-Actin-R primers as a control, and values were expressed as the change in the increase/decrease of the relative levels of the control sample (M96, which is the mixture of mycelia harvested from different media including PDA, MM, V8PDA, and water agar).
For S. turcica, leaf samples with lesions were collected at five time points (3, 5, 6, 7 and 8 days) after inoculation with 9×104 spores per plant (three weeks old) and total RNA was extracted and qPCR done as described [119]. The actin gene was used as internal control using ATC1 primers [119]. The S. turcica gene primers corresponding to protein ID 161586 are listed in Table S9. Expression level was expressed as fold change versus mycelial samples harvested on Lactose Casein Agar (LCA) plates.
Transformation and gene deletion
Fungal transformation and molecular characterization of gene knockout mutants were conducted according to the methods of [120]. The split marker system [121] was used for gene deletion. The 5′ and 3′ flanking sequences of the NPS gene encoding protein ID 115356 were amplified from ND90Pr DNA using primer pairs GTCGACTGCCATCTGGAAAC/CACTGGCCGTCGTTTTACAACGTCCACTCGACAGGTCCGTAGGT and TCATGGTCATAGCTGTTTCCTGTGGTATCCACAAAGCCACAGCA/GACGAACCAGA GATGCATGA) respectively.
To verify deletion of the gene corresponding to protein ID 115356, primers CAN1-F3: AGTTGTTGGGGAGTTGTTGG and CAN1-F4: TGAGCCGTTGTCATGTATCG matching the deleted portion of the gene were used. The expected PCR product was obtained from WT DNA, but not when DNA of the deletion mutant was used as template. To further confirm that the hygromycin resistance gene replaced the target gene at the native locus, PCR was conducted using a primer located outside the 3′ flank used for gene deletion (CsNPS1-F0: GTCCTACGGCAATTGTGGAC) and a second primer (HY: GGATGCCTCCGCTCGAAGTA) located in the hygromycin resistance gene. No amplification occurred when WT DNA was used, while the expected amplicon was observed when DNA of the mutants was used as template.
Plant inoculation
For C. sativus, virulence of the mutant (ID# 115356, Figure 7C) and wild type strains was tested on barley cv. Bowman by spray inoculation as described in Fetch and Steffenson (1999), except 2×103 conidia/ml were used. Inoculated plants were incubated in a humid chamber for 18–24 hours, and then transferred to a greenhouse room (20+/−2°C).
For S. turcica, three week old W64A maize plants were sprayed with 9×104 spores per plant, and plants grown under conditions described previously for C. heterostrophus [119].
Mating type locus comparisons
MAT1-1 and MAT1-2 mating type regions were identified by blasting the corresponding known C. heterostrophus MAT sequences (MAT1-1: accession CAA48465, MAT1-2: accession CAA48464) against each genome. Regions immediately 10 kb upstream and downstream were aligned pairwise to C. heterostrophus C5 (MAT1-1) or C4 (MAT1-2) MAT regions using ProgressiveMauve [44] to generate SNP data, and as a group (with and without S. turcica for MAT1-1) for visualizing the alignment.
Data access
Genome assemblies and annotations are available via JGI Genome Portal MycoCosm (http://jgi.doe.gov/fungi, [107] and DDBJ/EMBL/GenBank under the following accessions Cochliobolus heterostrophus ATCC 48331 (race T, strain C4): AIHU00000000, Cochliobolus heterostrophus ATCC 48332 (race O, strain C5): AIDY00000000, Cochliobolus sativus ND90Pr: AEIN00000000, Setosphaeria turcica Et28A: AIHT00000000, C. sativus ND93-1:PRJNA87041, Cochliobolus carbonum 26-R-13: AMCN00000000, Cochliobolus miyabeanus ATCC 44560: AMCO00000000, Cochliobolus victoriae FI3: AMCY00000000.
Supporting Information
Zdroje
1. OhmR, FeauN, HenrissatB, SchochCL, HorwitzBA, et al. (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLoS Pathog 8: e1003037 doi:10.1371/journal.ppat.1003037.
2. BerbeeML, PirseyediM, HubbardS (1999) Cochliobolus phylogenetics and the origin of known, highly virulent pathogens, inferred from ITS and glyceraldehyde-3-phosphate dehydrogenase gene sequences. Mycologia 91 : 964–977.
3. KumarJ, SchaferP, HuckelhovenR, LangenG, BaltruschatH, et al. (2002) Bipolaris sorokiniana, a cereal pathogen of global concern: cytological and molecular approaches towards better control. Mol Plant Path 3 : 185–195.
4. YoderOC (1980) Toxins in pathogenesis. Ann Rev Phytopathol 18 : 103–129.
5. TurgeonBG, BakerSE (2007) Genetic and genomic dissection of the Cochliobolus heterostrophus Tox1 locus controlling biosynthesis of the polyketide virulence factor T-toxin. Adv Genet 57 : 219–261.
6. UllstrupAJ (1970) History of Southern Corn Leaf Blight. Plant Dis Reptr 54 : 1100–1102.
7. DrechslerC (1925) Leafspot of maize caused by Ophiobolus heterostrophus n. sp., the ascigerous stage of a Helminthosporium exhibiting bipolar germination. J Agr Res 31 : 701–726.
8. TzengTH, LyngholmLK, FordCF, BronsonCR (1992) A restriction fragment length polymorphism map and electrophoretic karyotype of the fungal maize pathogen Cochliobolus heterostrophus. Genetics 130 : 81–96.
9. KodamaM, RoseMS, YangG, YunSH, YoderOC, et al. (1999) The translocation-associated Tox1 locus of Cochliobolus heterostrophus is two genetic elements on two different chromosomes. Genetics 151 : 585–596.
10. HookerA, SmithD, LimS, BeckettJ (1970) Reaction of corn seedlings with male-sterile cytoplasm to Helminthosporium maydis. Plant Dis Reptr 54 : 708–712.
11. SmithD, HookerA, LimS (1970) Physiologic races of Helminthosporium maydis. Plant Dis Reptr 54 : 819–822.
12. LitzenbergerSC (1949) Nature of susceptibility to Helminthosporium victoriae and resistance to Puccinia coronata in Victoria oats. Phytopathology 39 : 300–318.
13. MeehanF, MurphyHC (1947) Differential phytotoxicity of metabolic by-products of Helminthosporium victoriae. Science 106 : 270–271.
14. LorangJM, Carkaci-SalliN, WolpertTJ (2004) Identification and characterization of victorin sensitivity in Arabidopsis thaliana. Mol Plant Microbe Interact 17 : 577–582.
15. LorangJM, SweatTA, WolpertTJ (2007) Plant disease susceptibility conferred by a “resistance” gene. Proc Natl Acad Sci U S A 104 : 14861–14866.
16. JohalGS, BriggsSP (1992) Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258 : 985–987.
17. MultaniDS, MeeleyRB, PatersonAH, GrayJ, BriggsSP, et al. (1998) Plant-pathogen microevolution: Molecular basis for the origin of a fungal disease in maize. Proc Natl Acad Sci USA 95 : 1686–1691.
18. WaltonJD (1987) Two enzymes involved in biosynthesis of the host-selective phytotoxin HC-toxin. Proc Natl Acad Sci 84 : 8444–8447.
19. WaltonJD (1996) Host-selective toxins: agents of compatibility. Plant Cell 8 : 1723–1733.
20. RansomRF, WaltonJD (1997) Histone hyperacetylation in maize in response to treatment with HC-toxin or infection by the filamentous fungus Cochliobolus carbonum. Plant Physiol 115 : 1021–1027.
21. WaltonJD (2006) HC-toxin. Phytochemistry 67 : 1406–1413.
22. DasguptaMK (1984) The Bengal famine, 1943 and the brown spot of rice–an inquiry into their relations. Hist Agric 2 : 1–18.
23. VidhyasekaranP, BorromeoES, MewTW (1992) Helminthosporium oryzae toxin suppresses phenol metabolism in rice plants and aids pathogen colonization. Physiological and Mol Plant Path 41 : 307–315.
24. Mathre DE (1997) Compendium of Barley Diseases. 2nd Edition APS Press, St Paul.
25. Weise MV (1987) Compendium of wheat diseases. 2nd Edition APS Press, St Paul.
26. RoaneC (2004) Graminicolous fungi of Virginia: Fungi in collections 1995–2003. Virginia J Sci 55 : 139–157.
27. GravertCE, GPM (2002) Fungi and diseases associated with cultivated switchgrass in Iowa. J Iowa Acad Sci 109 : 30–34.
28. Valjavec-GratianM, SteffensonB (1997) Pathotypes of Cochliobolus sativus on barley. Plant Dis 81 : 1275–1278.
29. Valjavec GratianM, SteffensonBJ (1997) Genetics of virulence in Cochliobolus sativus and resistance in barley. Phytopathology 87 : 1140–1143.
30. ZhongS, SteffensonBJ, MartinezJP, CiuffettiLM (2002) A molecular genetic map and electrophoretic karyotype of the plant pathogenic fungus Cochliobolus sativus. MPMI 15 : 481–492.
31. SteffensonBJ, HayesPM, KleinhofsA (1996) Genetics of seedling and adult plant resistance to net blotch (Pyrenophora teres f teres) and spot blotch (Cochliobolus sativus) in barley. Theor Appl Genet 92 : 552–558.
32. Gyawali S (2010) Association mapping of resistance to common root rot and spot blotch in barley, and population genetics of Cochliobolus sativus. Thesis NDSU, Fargo, ND.
33. GhazviniH, TekauzA (2007) Virulence diversity in the population of Bipolaris sorokiniana. Plant Dis 91 : 814–821.
34. MartinT, BirumaM, FridborgI, OkoriP, DixeliusC (2011) A highly conserved NB-LRR encoding gene cluster effective against Setosphaeria turcica in sorghum. BMC Plant Biol 11 : 151.
35. ChungCL, LongfellowJM, WalshEK, KerdiehZ, Van EsbroeckG, et al. (2010) Resistance loci affecting distinct stages of fungal pathogenesis: use of introgression lines for QTL mapping and characterization in the maize–Setosphaeria turcica pathosystem. BMC Plant Biol 10 : 103.
36. LeonardKJ, LevyY, SmithDR (1989) Proposed nomenclature for pathogenic races of Exserohilum turcicum on corn. Plant Dis 73 : 776–777.
37. SchneiderDJ, CollmerA (2010) Studying plant-pathogen interactions in the genomics era: beyond molecular Koch's postulates to systems biology. Annu Rev Phytopathol 48 : 457–479.
38. CiuffettiLM, ManningVA, PandelovaI, BettsMF, MartinezJP (2010) Host-selective toxins, Ptr ToxA and Ptr ToxB, as necrotrophic effectors in the Pyrenophora tritici-repentis-wheat interaction. New Phytol 187 : 911–919.
39. FriesenTL, FarisJD, SolomonPS, OliverRP (2008) Host-specific toxins: effectors of necrotrophic pathogenicity. Cell Microbiol 10 : 1421–1428.
40. WolpertTJ, DunkleLD, CiuffettiLM (2002) Host-selective toxins and avirulence determinants: what's in a name? Annu Rev Phytopathol 40 : 251–285.
41. SweatTA, WolpertTJ (2007) Thioredoxin h5 is required for victorin sensitivity mediated by a CC-NBS-LRR gene in Arabidopsis. Plant Cell 19 : 673–687.
42. LeachJ, LangBR, YoderOC (1982) Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. J Gen Microbiol 128 : 1719–1729.
43. Klittich C. (1985) Differences in fitness of strains of Cochliobolus heterostrophus near-isogenic for toxin production. PhD. Thesis, Iowa State Univ.
44. DarlingAE, MauB, PernaNT (2010) ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5: e11147 doi:10.1371/journal.pone.0011147.
45. InderbitzinP, AsvarakT, TurgeonBG (2010) Six new genes required for production of T-toxin, a polyketide determinant of high virulence of Cochliobolus heterostrophus to maize. Mol Plant Microbe Interact 23 : 458–472.
46. KurtzS, PhillippyA, DelcherAL, SmootM, ShumwayM, et al. (2004) Versatile and open software for comparing large genomes. Genome Biology 5: R12.
47. Yang G (1995) The molecular genetics of T-toxin biosynthesis by Cochliobolus heterostrophus. PhD Thesis, Cornell Univ.
48. YangG, RoseMS, TurgeonBG, YoderOC (1996) A polyketide synthase is required for fungal virulence and production of the polyketide T-toxin. Plant Cell 11 : 2139–2150.
49. ChristiansenSK, WirselS, YoderOC, TurgeonBG (1998) The two Cochliobolus mating type genes are conserved among species but one of them is missing in C. victoriae. Mycol Res 102 : 919–929.
50. FinkingR, MarahielMA (2004) Biosynthesis of nonribosomal peptides. Annu Rev Microbiol 58 : 453–488.
51. SieberSA, MarahielMA (2003) Learning from nature's drug factories: Nonribosomal synthesis of macrocyclic peptides. J Bacteriol 185 : 7036–7043.
52. GrunewaldJ, MarahielMA (2006) Chemoenzymatic and template-directed synthesis of bioactive macrocyclic peptides. Microbiol Mol Biol Rev 70 : 121–126.
53. SteinT, VaterJ, KruftV, OttoA, WittmannLieboldB, et al. (1996) The multiple carrier model of nonribosomal peptide biosynthesis at modular multienzymatic templates. J Biol Chem 271 : 15428–15435.
54. MootzHD, SchwarzerD, MarahielMA (2002) Ways of assembling complex natural products on modular nonribosomal peptide synthetases. ChemBioChem 3 : 490–504.
55. LeeBN, KrokenS, ChouDYT, RobbertseB, YoderOC, et al. (2005) Functional analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot Cell 4 : 545–555.
56. OideS, MoederW, KrasnoffS, GibsonD, HaasH, et al. (2006) NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell 18 : 2836–2853.
57. BushleyKE, TurgeonBG (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10 : 26.
58. OideS, KrasnoffSB, GibsonDM, TurgeonBG (2007) Intracellular siderophores are essential for ascomycete sexual development in heterothallic Cochliobolus heterostrophus and homothallic Gibberella zeae. Eukaryot Cell 6 : 1337–1353.
59. BushleyKE, RipollDR, TurgeonBG (2008) Module evolution and substrate specificity of fungal nonribosomal peptide synthetases involved in siderophore biosynthesis. BMC Evol Biol 8 : 328.
60. AhnJH, WaltonJD (1996) Chromosomal organization of TOX2, a complex locus controlling host-selective toxin biosynthesis in Cochliobolus carbonum. Plant Cell 8 : 887–897.
61. ManningV, PandelovaI, DhillonB, WilhelmL, GoodwinS, et al. (2012) Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3 In press.
62. Wight WD, Walton JD (2011) Histone deacetylase inhibitor HC-toxin from Alternaria jesenskae. Asilomar, CA. pp. abstract#385, p209.
63. JinJM, LeeS, LeeJ, BaekSR, KimJC, et al. (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol 76 : 456–466.
64. ForsethRR, FoxEM, ChungD, HowlettBJ, KellerNP, et al. (2012) Identification of cryptic products of the gliotoxin gene cluster using NMR-based comparative metabolomics and a model for gliotoxin biosynthesis. J Am Chem Soc 133 : 9678–9681.
65. KrokenS, GlassNL, TaylorJW, YoderOC, TurgeonBG (2003) Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A 100 : 15670–15675.
66. Rose MS, Yoder OC, Turgeon BG. (1996) A decarboxylase required for polyketide toxin production and high virulence by Cochliobolus heterostrophus. 8th Int. Symp. Molec. Plant-Microbe Interact. Knoxville. pp. J-49.
67. BakerSE, KrokenS, InderbitzinP, AsvarakT, LiBY, et al. (2006) Two polyketide synthase-encoding genes are required for biosynthesis of the polyketide virulence factor, T-toxin, by Cochliobolus heterostrophus. Mol Plant Microbe Interact 19 : 139–149.
68. DiohW, TharreauD, NotteghemJL, OrbachM, LebrunMH (2000) Mapping of avirulence genes in the rice blast fungus, Magnaporthe grisea, with RFLP and RAPD markers. Mol Plant Microbe Interact 13 : 217–227.
69. FarmanML (2007) Telomeres in the rice blast fungus Magnaporthe oryzae: the world of the end as we know it. FEMS Microbiol Lett 273 : 125–132.
70. AkagiY, AkamatsuH, OtaniH, KodamaM (2009) Horizontal chromosome transfer, a mechanism for the evolution and differentiation of a plant-pathogenic fungus. Eukaryot Cell 8 : 1732–1738.
71. KallL, KroghA, SonnhammerEL (2007) Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res 35: W429–432.
72. RouxelT, GrandaubertJ, HaneJK, HoedeC, van de WouwAP, et al. (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun 2 : 202.
73. HaneJK, RouxelT, HowlettBJ, KemaGH, GoodwinSB, et al. (2011) A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol 12: R45.
74. GoodwinSB, Ben M'BarekS, DhillonB, WittenbergAHJ, CraneCF, et al. (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet 7: e1002070 doi:10.1371/journal.pgen.1002070.
75. CuomoCA, GuldenerU, XuJR, TrailF, TurgeonBG, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317 : 1400–1402.
76. McCluskeyK, WiestAE, GrigorievIV, LipzenA, MartinJ, et al. (2011) Rediscovery by whole genome sequencing: classical mutations and genome polymorphisms in Neurospora crassa. G3 (Bethesda) 1 : 303–316.
77. LangfelderK, StreibelM, JahnB, HaaseG, BrakhageAA (2003) Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet Biol 38 : 143–158.
78. TsaiHF, WheelerMH, ChangYC, Kwon-ChungKJ (1999) A developmentally regulated gene cluster involved in conidial pigment biosynthesis in Aspergillus fumigatus. J Bacteriol 181 : 6469–6477.
79. PihetM, VandeputteP, TronchinG, RenierG, SaulnierP, et al. (2009) Melanin is an essential component for the integrity of the cell wall of Aspergillus fumigatus conidia. BMC Microbiol 9 : 177.
80. OliverRP, SolomonPS (2010) New developments in pathogenicity and virulence of necrotrophs. Curr Opin Plant Biol 13 : 415–9.
81. MehrabiR, BahkaliAH, Abd-ElsalamKA, MoslemM, Ben M'barekS, et al. (2011) Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev 35 : 542–554.
82. KhaldiN, CollemareJ, LebrunMH, WolfeKH (2008) Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 9: R18.
83. Turgeon BG, Lu S-W (2000) Evolution of host specific virulence in Cochliobolus heterostrophus. In Fungal Pathology, Ed Kronstad, JW; Kluwer, Dordrecht, The Netherlands: 93–126.
84. Wolpert TJ, Navarre DA, Lorang JM (1998) Victorin-induced oat cell death. In: Kohmoto K, Yoder OC, editors. Molecular Genetics of Host-Specific Toxins in Plant Disease. Dordrecht: Kluwer. pp. 105–114.
85. CiuffettiLM, TuoriRP, GaventaJM (1997) A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell 9 : 135–144.
86. YunSH, TurgeonBG, YoderOC (1998) REMI-induced mutants of Mycosphaerella zeae-maydis lacking the polyketide PM-toxin are deficient in pathogenesis to corn. Physiol Mol Plant Pathol 52 : 53–66.
87. Yun SH (1998) Molecular genetics and manipulation of pathogenicity and mating determinants in Cochliobolus heterostrophus and Mycosphaerella zeae-maydis. PhD Thesis, Cornell University.
88. PanaccioneDG, Scott-CraigJS, PocardJA, WaltonJD (1992) A cyclic peptide synthetase gene required for pathogenicity of the fungus Cochliobolus carbonum on maize. Proc Natl Acad Sci USA 89 : 6590–6594.
89. AhnJH, ChengYQ, WaltonJD (2002) An extended physical map of the TOX2 locus of Cochliobolus carbonum required for biosynthesis of HC-toxin. Fungal Genet Biol 35 : 31–38.
90. JinJM, LeeS, LeeJ, BaekSR, KimJC, et al. (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol 76 : 456–466.
91. SindhuA, ChintamananiS, BrandtAS, ZanisM, ScofieldSR, et al. (2008) A guardian of grasses: specific origin and conservation of a unique disease-resistance gene in the grass lineage. Proc Natl Acad Sci U S A 105 : 1762–1767.
92. LengYQ, ZhongSB (2012) Sfp-type 4 ′-phosphopantetheinyl transferase is required for lysine synthesis, tolerance to oxidative stress and virulence in the plant pathogenic fungus Cochliobolus sativus. Mol Plant Path 13 : 375–387.
93. GardinerDM, CozijnsenAJ, WilsonLM, PedrasMS, HowlettBJ (2004) The sirodesmin biosynthetic gene cluster of the plant pathogenic fungus Leptosphaeria maculans. Mol Microbiol 53 : 1307–1318.
94. ScharfDH, HeinekampT, RemmeN, HortschanskyP, BrakhageAA, et al. (2011) Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl Microbiol Biotechnol 93 : 467–472.
95. SweatTA, LorangJM, BakkerEG, WolpertTJ (2008) Characterization of natural and induced variation in the LOV1 gene, a CC-NB-LRR gene conferring victorin sensitivity and disease susceptibility in Arabidopsis. Mol Plant Microbe Interact 21 : 7–19.
96. FarisJD, ZhangZ, LuH, LuS, ReddyL, et al. (2010) A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci U S A 107 : 13544–13549.
97. ZerbinoDR, BirneyE (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 : 821–829.
98. GnerreS, MaccallumI, PrzybylskiD, RibeiroFJ, BurtonJN, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A 108 : 1513–1518.
99. MarguliesM, EgholmM, AltmanWE, AttiyaS, BaderJS, et al. (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437 : 376–380.
100. Smit A, Hubley R, Green PRO-, (1996–2010) RepeatMasker Open-3.0.
101. JurkaJ, KapitonovVV, PavlicekA, KlonowskiP, KohanyO, et al. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogen and Genome Res 110 : 462–467.
102. PriceAL, JonesNC, PevznerPA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21: I351–I358.
103. SalamovAA, SolovyevVV (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res 10 : 516–522.
104. BirneyE, DurbinR (2000) Using GeneWise in the Drosophila annotation experiment. Genome Res 10 : 547–548.
105. IsonoK, McIninchJD, BorodovskyM (1994) Characteristic features of the nucleotide sequences of yeast mitochondrial ribosomal protein genes as analyzed by computer program GeneMark. DNA Res 1 : 263–269.
106. KentWJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12 : 656–664.
107. GrigorievIV, NordbergH, ShabalovI, AertsA, CantorM, et al. (2011) The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res 40: D26–32.
108. StankeM, SchoffmannO, MorgensternB, WaackS (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7 : 62.
109. ZhongSB, YangBJ, AlfenasAC (2008) Development of microsatellite markers for the guava rust fungus, Puccinia psidii. Molec Ecol Res 8 : 348–350.
110. LanderES, GreenP, AbrahamsonJ, BarlowA, DalyMJ, et al. (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1 : 174–181.
111. BirneyE, ClampM, DurbinR (2004) GeneWise and genomewise. Genome Res 14 : 988–995.
112. StajichJE, BlockD, BoulezK, BrennerSE, ChervitzSA, et al. (2002) The bioperl toolkit: Perl modules for the life sciences. Genome Res 12 : 1611–1618.
113. FinnRD, ClementsJ, EddySR (2011) HMMER web server: interactive sequence similarity searching. Nucl Acids Res 39: W29–W37.
114. AbascalF, ZardoyaR, PosadaD (2005) ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 21 : 2104–2105.
115. StamatakisA (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 : 2688–2690.
116. StamatakisA, HooverP, RougemontJ (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57 : 758–771.
117. TinlineR, StaufferJ, DicksonJ (1960) Cochliobolus sativus III Effect of ultraviolet radiation. Can J Bot 38 : 275–282.
118. FetchTG, SteffensonBJ (1999) Rating scales for assessing infection responses of barley infected with Cochliobolus sativus. Plant Disease 83 : 213–217.
119. WuDL, OideS, ZhangN, ChoiMY, TurgeonBG (2012) ChLae1 and ChVel1 regulate T-toxin production, virulence, oxidative stress response, and development of the maize pathogen Cochliobolus heterostrophus. PLoS Pathog 8: e1002542 doi:10.1371/journal.ppat.1002542.
120. LengY, WuC, LiuZ, FriesenTL, RasmussenJ, ZhongS (2011) Development of transformation and RNA-mediated gene silencing systems for functional genomics of Cochliobolus sativus. Mol Plant Pathol 12 : 289–298.
121. CatlettN, LeeB-N, YoderO, TurgeonB (2003) Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newsl 50 : 9–11.
122. AhnJH, ChengYQ, WaltonJD (2002) An extended physical map of the TOX2 locus of Cochliobolus carbonum required for biosynthesis of HC-toxin. Fungal Genet Biol 35 : 31–38.
123. WirselS, HorwitzB, YamaguchiK, YoderOC, TurgeonBG (1998) Single mating type-specific genes and their 3′ UTRs control mating and fertility in Cochliobolus heterostrophus. Mol Gen Genet 259 : 272–281.
124. Turgeon B, Debuchy R, editors (2007) Cochliobolus and Podospora: Mechanisms of sex determination and the evolution of reproductive lifestyle. Washington, DC: ASM. p93–121 p.
125. Debuchy R, Turgeon BG (2006) Mating-type structure, evolution and function in Euascomycetes. In: Kües U, Fischer R, editors. The Mycota. Berlin, Heidelberg: Springer-Verlag. pp. 293–324.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2013 Číslo 1
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies