
Rad59-Facilitated Acquisition of Y′ Elements by Short Telomeres Delays the Onset of Senescence
In humans, telomerase is expressed in the germline and stem, but is repressed in somatic cells, which limits replicative lifespan of the latter. To unleash cell proliferation, telomerase is reactivated in most human cancers, but some cancer cells employ alternative lengthening of telomeres (ALT) based on homologous recombination (HR) to escape senescence. Recombination-based telomere maintenance similar to ALT was originally discovered in budding yeast deficient in telomerase activity. Two types of telomere arrangement that depend on two genetically distinct HR pathways (RAD51- and RAD59-dependent) were identified in post-senescent survivors, but the transition to telomere maintenance by HR is poorly understood. Here, we show that one of the earliest steps of short telomere rearrangement in telomerase-negative yeast is directly related to the “short telomere rescue pathway” proposed 20 years ago by Lundblad and Blackburn, which culminates in the acquisition of subtelomeric Y′ element by shortened telomere. We found that this telomere rearrangement depends on Rad52 strand annealing activity stimulated by Rad59, thus it is distinct from Rad51-dependent Y′ amplification process observed in type I survivors. We show that continuous repair of critically short telomeres in telomerase-negative cells delays the onset of senescence and prepares the ground for telomere maintenance by HR.
Published in the journal:
Rad59-Facilitated Acquisition of Y′ Elements by Short Telomeres Delays the Onset of Senescence. PLoS Genet 10(11): e32767. doi:10.1371/journal.pgen.1004736
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004736
Summary
In humans, telomerase is expressed in the germline and stem, but is repressed in somatic cells, which limits replicative lifespan of the latter. To unleash cell proliferation, telomerase is reactivated in most human cancers, but some cancer cells employ alternative lengthening of telomeres (ALT) based on homologous recombination (HR) to escape senescence. Recombination-based telomere maintenance similar to ALT was originally discovered in budding yeast deficient in telomerase activity. Two types of telomere arrangement that depend on two genetically distinct HR pathways (RAD51- and RAD59-dependent) were identified in post-senescent survivors, but the transition to telomere maintenance by HR is poorly understood. Here, we show that one of the earliest steps of short telomere rearrangement in telomerase-negative yeast is directly related to the “short telomere rescue pathway” proposed 20 years ago by Lundblad and Blackburn, which culminates in the acquisition of subtelomeric Y′ element by shortened telomere. We found that this telomere rearrangement depends on Rad52 strand annealing activity stimulated by Rad59, thus it is distinct from Rad51-dependent Y′ amplification process observed in type I survivors. We show that continuous repair of critically short telomeres in telomerase-negative cells delays the onset of senescence and prepares the ground for telomere maintenance by HR.
Introduction
Telomeres are nucleoprotein structures found at the physical ends of chromosomes. Their terminal location defines their two main functions: protection of the chromosome ends from illegitimate repair reactions and prevention of the loss of terminal DNA due to either degradation or incomplete replication [1]. In Saccharomyces cerevisiae, the first function is accomplished primarily by Rap1, which wraps tandem telomeric DNA repeats to inhibit NHEJ [2], and recruits Rif1 and Rif2 to restrain MRX-mediated 3′ end resection [3], [4], thus limiting recruitment of HR factors and checkpoint signaling [5]. The second function is mediated by Cdc13 bound to the single-stranded G-rich 3′ overhang at the extreme terminus of a telomere. Cdc13 forms alternative complexes with either Est1 or Stn1-Ten1 to coordinate telomerase-mediated synthesis of the G-rich strand with the synthesis of the complementary strand by DNA polymerase α [6].
As in mammals, telomeres in yeast cells with reduced telomerase activity progressively shorten with each cell division until they are recognized as DNA damage and recruit Mec1 kinase that initiates irreversible G2/M arrest [7]–[9]. At the level of cell population, telomere dysfunction is manifested as crisis, when majority of the cells irreversibly arrest in G2/M [7]. Most of the cells die, but at low frequency survivors emerge, which maintain their telomeres via recombination [10], [11], implying that homologous recombination (HR) can serve as a bypass pathway to sustain viability in the absence of telomerase.
The survivors are classified in two types based on their telomere arrangement and growth characteristics [12], [13]. The type I survivors have tandem arrays of subtelomeric Y′ elements separated by short tracts of TG1–3 repeats at most chromosome ends, and also short terminal TG1–3 repeats [10]. Their growth is interrupted by frequent periods of arrest and in the competitive conditions of liquid culture they are outcompeted by the more robust type II survivors. In type II survivors, terminal TG1–3 repeats are abnormally elongated and are very heterogeneous in length. It is believed that they are established by stochastic lengthening events that likely involve rolling circle replication [14]. RAD52 is required for generation of both types of survivors. RAD51, RAD54, RAD57 are specifically required to generate type I, whereas type II survivors depend on MRX complex, RAD59 and SGS1, encoding the only RecQ helicase in yeast [13], [15], [16]. In addition, POL32 encoding a non-essential subunit of DNA polymerase δ is required for generation of both survivor types, implying involvement of the processive repair DNA synthesis in the recombination-based telomere rearrangements [17], [18]. Recently, a genome-wide screen aimed to identify telomere-length-maintenance genes that regulate telomere structure in post-senescence survivors unveil new regulators of Type I and II recombination [19]. Notably, Type I recombination was shown to depend on the helicase Pif1 and on the chromatin remodelling complex INO80.
Although genetic requirements for the formation of two types of survivors and their telomere patterns have been well characterized, much less is known about actual recombination events that lead to reorganization of the original short telomere into the patterns observed in survivors. In budding yeast, telomere shortening does not cause end-to-end chromosome fusions, as does the removal of Rap1 from telomeres [2]; instead, gene conversion increases near short telomeres indicating de-repressed recombination [20]. There is a controversy, however, whether type II recombination preferentially takes place at long or short telomeres [14], [21], [22]. Little is known about the telomere length preference of type I pathway. Early studies looking at the propagation of linear plasmids in yeast uncovered that they can recombine with the yeast chromosome ends and acquire telomere-adjacent sequences called Y′ elements [23]. Y′ elements found at many chromosome ends fall into two size classes, 6.7 (Y′-L) and 5.2 (Y′-S) kb-long, that differ by a 1.5 kb insertion/deletion [24]. Another subtelomeric sequences called X elements are present at all chromosome ends immediately proximal to either Y's or terminal TG1-3 repeats when Y′ is absent. The junction between X and Y′ elements often, but not always, contain short tracts of TG1–3 repeats [24], [25]. Importantly, only the Y′ and not X elements can be transferred on linear plasmids, and this is mediated by recombination between the terminal TG1–3 repeats added onto the plasmid ends by telomerase and the internal TG1–3 tracts present between the X and Y′ elements [23]. Pioneering work of Lundblad and Blackburn showed that est1Δ survivors arose as the result of the acquisition of Y′ elements by X-only telomeres and amplification of these elements on many chromosome ends. They proposed a model of telomere rescue via a recombination event between the terminal TG1–3 repeats of one telomere and an internal TG1–3 tract in another [10].
We have previously demonstrated using single cell analysis that Rad52-containing foci are assembled at the telomeres in a length-dependent manner in presenescent cells many generations before the onset of senescence [26]. The recruitment of recombination factors to short telomeres is in accord with increased recombinogenic activity of short telomeres observed in both yeast [20] and mammals [27]. Of note, inactivation of HR, particularly via deletion of RAD52 and RAD51 (but to a much lesser extent of RAD59), causes early decline in proliferative capacity of telomerase-negative yeast indicating that telomere maintenance most likely becomes dependent on HR soon after telomerase inactivation [28], [29]. Surprisingly, the rate of telomere shortening (population average length) is unaffected in HR-deficient yeast. All these observations raise the question of telomere recombination dynamics in presenescent cells, the mechanism of Y′ acquisition by X-only telomeres and the role of recombination proteins in maintaining telomerase-negative strains alive during presenescence. Another unresolved issue is whether a single critically short telomere is sufficient to induce cell cycle arrest. Complete loss of a single telomere causes Rad9-dependent arrest even in telomerase-proficient cells [30]. This does not seem to be the case when a very short telomere is created in telomerase negative cells [8], [28], but the fate of this abruptly shortened telomere remains obscure. In this study, we aimed to characterize the primary recombination event that takes place at short telomeres in the absence of telomerase. To this end we put together a system to simultaneously shorten modified VII-L telomere and inactivate telomerase. Bulk liquid cultures turned out to be inappropriate to address the fate of the abruptly shortened VII-L telomere, so we adapted clonal analysis. We found that the subtelomereless VII-L end acquired Y′ element in clonal populations originated from transiently arrested cells. Cloning and sequencing of the Y′ translocation junctions from multiple clones revealed that Y′ acquisition was initiated by recombination between the short terminal TG1–3 repeats at VII-L and the Rap1-bound internal TG1–3-like tracts present between X and Y′ elements on other chromosome ends. Such recombination initiates Pol32-dependent BIR, which results in non-reciprocal translocation of the entire Y′ element and terminal TG1–3 tract from the chromosome-donor onto the shortened telomere. Surprisingly, Y′ translocation events were Rad51-independent, but were instead promoted by Rad59 that stimulates Rad52 strand annealing activity. We found that the same mechanism operates at short native X-only telomeres, but it is much more efficient since translocated Y's are readily detectable in bulk liquid cultures during presenescence. In addition, sequence composition of the translocation junctions is simpler at native telomere VI-R, indicating that Y′ translocation on a native end is relatively straightforward event. We further show that RAD59 deletion compromises the efficiency of Y′ translocation on native telomere XV-L, and results in both accelerated senescence and prolonged crisis. Our results extend the model of short telomere rescue proposed by Lundblad and Blackburn more than 20 years ago [10], and they reinforce the notion that it is the overall depletion of the TG1–3 repeats on multiple chromosome ends rather than abrupt shortening of a few telomeres that defines the onset of senescence.
Results
VII-L telomere shortened beyond 60 bp becomes unstable and causes transient arrest
To address the processing of a short telomere without Y′ elements in the absence of telomerase, we employed the site-specific recombination system to induce abrupt shortening of a single telomere [31]. In this system, Cre induction causes excision of the basal portion of the telomere VII-L (TelVII-L) flanked by loxP sites (Figure 1A). In the presence of telomerase, shortened telomere is extended until its length returns to equilibrium [32]. To examine how this telomere will be processed in the absence of telomerase, we combined the abrupt shortening of TelVII-L with an inducible deletion of the plasmid-borne EST2 [33].
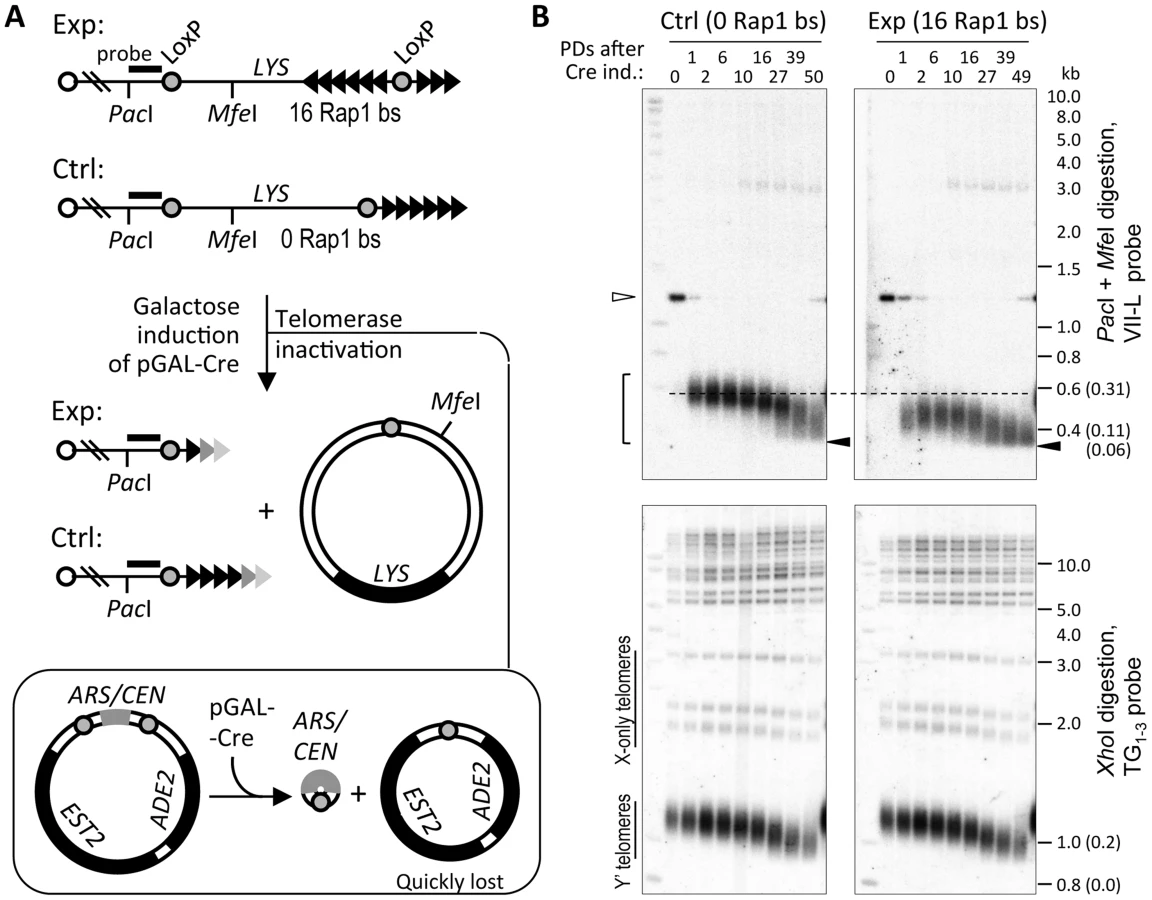
As expected, inactivation of telomerase completely abolished elongation of the TelVII-L after it was shortened via Cre-loxP recombination. Instead, its length decreased further until the bottom of the telomere length distribution reached a defined limit beyond which no shortening was observed (Figure 1B). The lower tail of the TelVII-L length distribution in the control strain also reached the same limit albeit with a delay of ∼20 population doublings (PDs) consistent with its greater initial length. We estimated that the lower limit of the TelVII-L length distribution corresponds to ∼60 bp of TG1–3 repeats (Figure S1). Since the probe anneals to the unique sequence of the terminal PacI fragment (Figure 1A), even complete loss of TG1–3 repeats should not affect hybridization signal. Thus, we reasoned that shortening of the TG1–3 tract beyond 60 bp causes the elimination of cells with critically short telomeres from the exponentially growing culture propagated via serial dilutions.
Clonal analysis of the telomerase-negative cells reveals VII-L end rearrangement consistent with Y′ element translocation
To isolate the cells undergoing cell cycle arrest due to TG1–3 tract shortening beyond the 60 bp threshold, we conducted clonal analysis of the telomerase-negative cultures at ∼15 PD after Cre induction. To this end, single cells were micromanipulated on a grid of agar, and analyzed for their ability to form microcolonies. While many cells divided regularly, at least once every 2 hours, and formed microcolonies of more than 16 cells after 8 hours on agar, a fraction of cells never divided during this time or stopped dividing at the 2- or 4-cell stage. These arrested microcolonies were marked (Figure 2A). Unexpectedly, most of the cells, which initially failed to divide, formed colonies after four days at 30°C (Figure 2A). Therefore, the majority of cells was able to overcome cell cycle arrest and resumed divisions. The fraction of arrested cells was significantly greater in the strain with shortened TelVII-L compared to the control strain (Figure 2B and Figure S2) indicating that shortening of a single telomere aggravated the effect of telomerase inactivation on cell cycle progression.
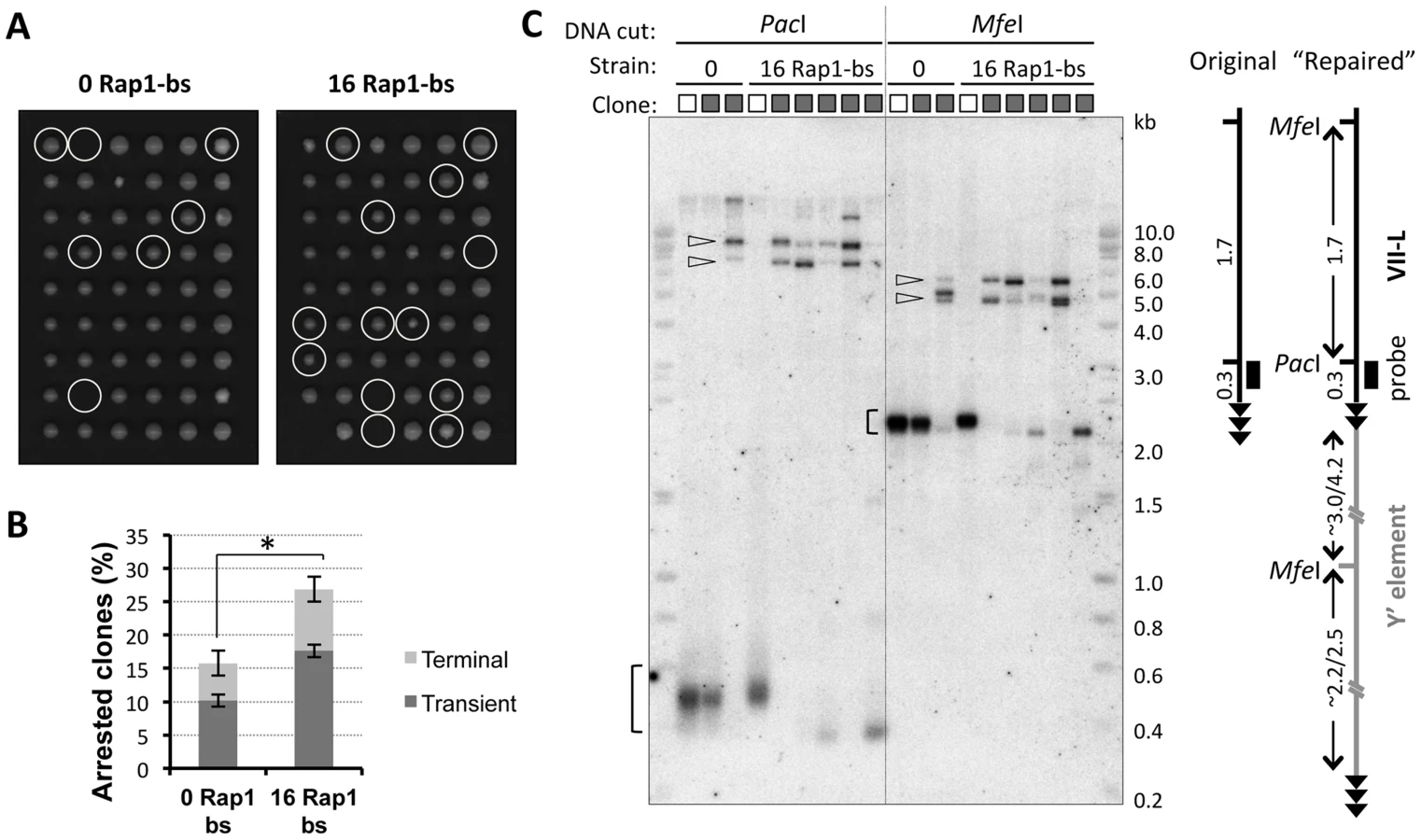
The state of the TelVII-L in clonal populations was analyzed by Southern blotting (Figure 2C). The terminal VII-L fragments with typical smeary appearance were detected in the expected size range for the control (no arrest) clones. In contrast, most of the clones that had undergone transient arrest completely lost the VII-L signal in this range. Instead, much larger fragments hybridized with VII-L-specific probe (Figure 2C), suggesting that VII-L end has been rearranged. These larger fragments grouped in two size classes after digestion with either PacI or MfeI, and were remarkably consistent with Y′ element translocation (see schematic in Figure 2C). Grouping of the fragments in two size classes could be well explained by translocation of either long (Y′-L) or short (Y′-S) version of the Y′ elements, which differ by a 1.5 kb insertion/deletion [24]. Most of the clones showed the presence of both Y′ classes. This heterogeneity is likely created due to independent acquisition of Y′ elements by TelVII-L in different cells (2- or 4-cell stage arrest), or even by two sister TelVII-L chromatids within a G2-arrested cell. Thus, we were able to isolate subclones descended from single recombination events (group A in Figure S2) by sequential micromanipulation of the cells as they came out of the arrest. Those transiently arrested clones that did not show VII-L end rearrangement (Figure 2C and Figure S2) could have either repaired it by addition of the terminal TG1–3 repeats [22] or arrested due to shortening of one of the native telomeres.
Y′ element is preferentially translocated on short telomeres
To verify the hypothesis of Y′ translocation and the kinetics of this repair process, we designed primer pairs to amplify the putative junction region between the VII-L end and the Y′ element (Figure 3A, Table S1). We analyzed the presence of the junction PCR product at 0, 10, and 50 PDs after inactivation of telomerase in the strain bearing a critically short telomere (16 Rap1-bs) and in the control strain (0 Rap1-bs) (Refer to Figure 1B). We detected the junction-specific PCR product at VII-L (Figure 1A) as early as 10 PD in liquid culture after inactivation of telomerase and telomere shortening. The intensity and heterogeneity of the junction PCR product increased dramatically by 50 PD (Figure 3B). Moreover, the junction PCR product was more intense at 10 PD in the strain with short telomere compared to that in the control strain. When the same PCR approach to detect Y′ translocation was performed on the native VI-R end, the appearance of recombined telomere followed the same kinetics in the “0” and “16 Rap1-bs” strains (see further).
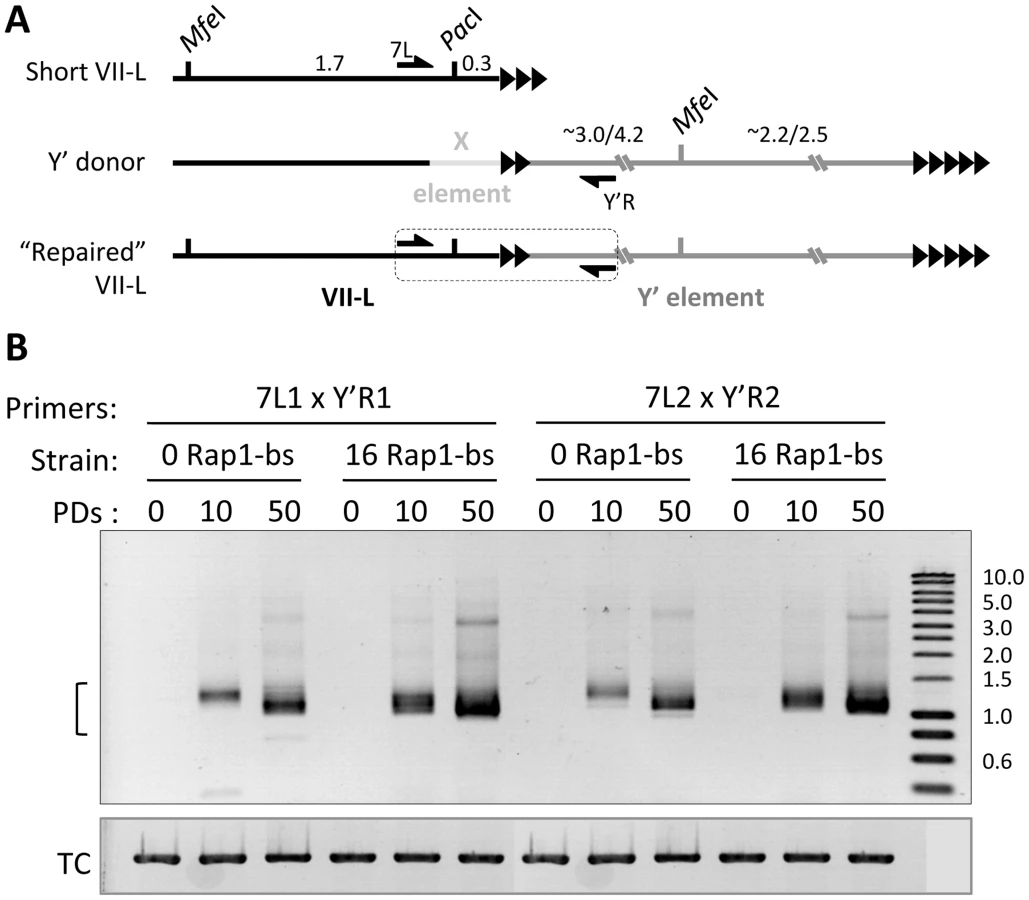
To assess the relationship between telomere length and Y′ acquisition in a more quantitative fashion, we analyzed the frequency of Y′ translocation on TelVII-L by Southern blotting in the random clones isolated from “0” and “16 Rap1-bs” strains. This analysis revealed significantly greater fraction of clones with Y′ element translocated on TelVII-L in the “16 Rap1-bs” (10/18) as compared to that in “0 Rap1-bs” (1/19) strain when the clones were isolated at 18 PD after Cre induction (Fisher's exact test P value 0.001; Figure S2). The same tendency was observed when the clones were isolated at 12 PD after Cre induction, but the frequency of Y′ translocation at this earlier time was too low for statistical evaluation (0/24 in “0 Rap1-bs” and 3/24 in “16 Rap1-bs” strain). In addition, the fraction of “16 Rap1-bs” clones with Y′ translocated on TelVII-L was significantly greater when the clones were isolated at 18 as compared to 12 PD after Cre induction (Fisher's exact test P value 0.006; Figure S2). The frequency of Y′ translocation among “0 Rap1-bs” clones was still low at these time points for statistical evaluation. We concluded that Y′ element is preferentially translocated on short telomeres; and the fraction of cells with translocated Y′ element grows with time after telomerase inactivation.
Y′ translocation is mediated by homeologous recombination between the terminal and internal TG1–3 repeats
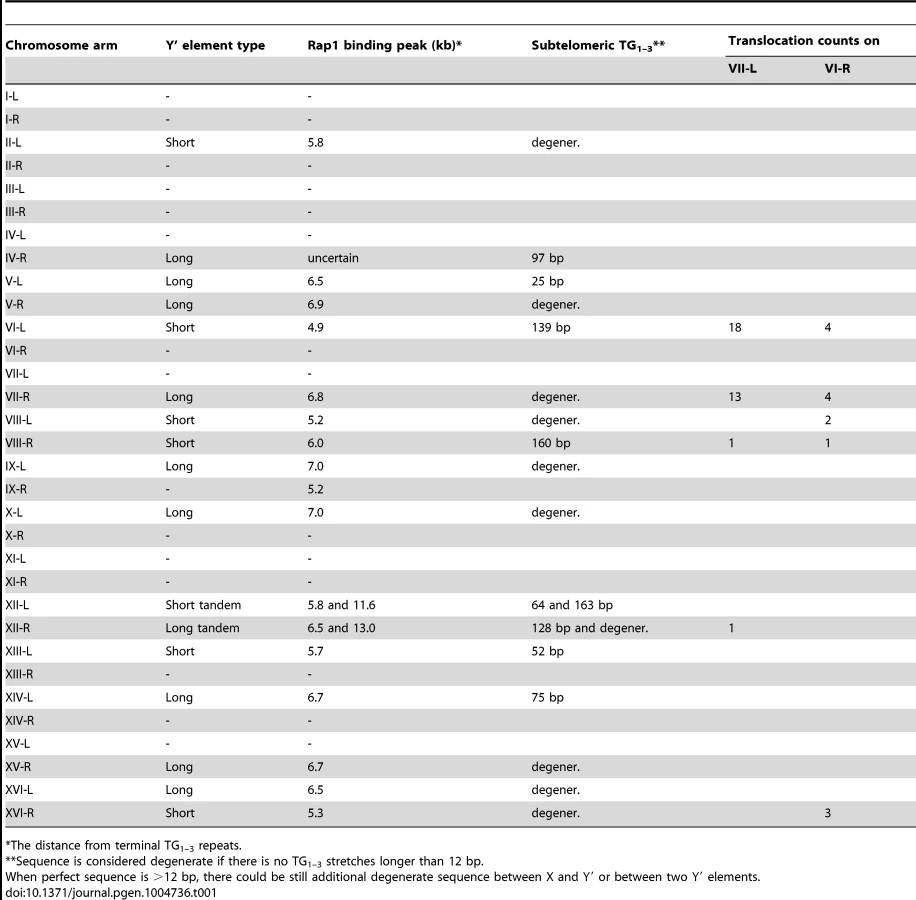
Cloning and sequencing of the junction PCR products revealed that they are in fact composed of the joint VII-L and Y′ element sequences, thereby confirming Y′ translocation on the VII-L end. Remarkably, all clones contained the TG1–3 repeats at the breakpoint between the VII-L and Y′ sequences, consistent with our assumption that Y′ translocation was initiated by VII-L terminal repeats, which recombined with internal TG1–3 tracts located between the subtelomeric X and Y′ elements of the chromosome donor. Sequence analysis of the Y′ segments of the junction clones revealed that the choice of the donor for Y′ translocation was not random: the chromosome arms VI-L and VII-R were used most frequently as the donors (Table 1).
To characterize recombination breakpoint we searched for the point of sequence divergence within TG1–3 repeats at the VII-L and Y′ junction. The exact pattern of irregular repeats newly synthesized by yeast telomerase differs between the molecules [34]. When telomere repeat sequences from clonal populations are aligned, only very distal 40–100 nt-long portions maintained by telomerase are divergent, whereas proximal regions are identical in sequence [35]. The divergent distal portion is quickly lost after telomerase inactivation. We found that TG1–3 repeats adjacent to the VII-L end shared 47±18 (SD) nt of identity before they diverged (Figures 4 and S3A), indicating that VII-L telomeres were at least that long at the time they engaged in recombination. Only in a few clones, the TG1–3 sequence past the divergence point continued without interruption by repeats found proximal to the acquired Y′ element (Figure S3B). In most of the clones, however, the repeats of the VII-L and Y′ origins were separated by divergent TG1–3 repeats (Figure 4). These intervening TG1–3 tracts varied in length and sometimes were quite long (e.g. 255 bp in the clone H10). We speculate that they might have resulted from abortive DNA repair synthesis due to rejection of homeologous heteroduplexes formed by TG1–3 repeats followed by template switch events [36]. Otherwise, the divergent regions at the junctions could be generated via recombination between two terminal TG1–3 repeat tracts, or even by residual telomerase activity during the first few PDs after Cre induction.

RAD51-independent BIR accounts for the majority of Y′ translocations on VII-L end
To get insight into the mechanism of short telomere repair, we examined its genetic requirements. To this end, we generated a set of isogenic mutants by deleting RAD52, RAD51, and RAD59 genes in a strain with the VII-L telomere modified for inducible shortening and TLC1 allele with a tetracycline-regulatable promoter (tetO2-TLC1). Upon addition of doxycycline (Dox), expression of TLC1 is tightly repressed and telomeres shorten progressively with each generation ([26] and Figure S4). We first examined the effect of gene deletions on the efficiency of Y′ translocation by semi-quantitative PCR assay. Telomerase was inactivated by addition of Dox and abrupt shortening of the VII-L was then induced following the scheme in Figure 5A. We analyzed junction PCR products in clonal populations of HR-proficient or mutant cells (Figure 5B,C and Figure S5). As expected, robust junction PCR products indicating Y′ translocation events were detected in the clonal populations of HR-proficient cells (Figure 5B). Weaker product was also detectable in the mixed population of cells grown in liquid culture (Bulk). RAD52 deletion nearly completely abolished Y′ translocation as judged by severe reduction of the amplified VII-L/Y′ junctions in either mixed or clonal populations (Figure 5B). Thus, Rad52 is essential for recombination between TG1–3 repeats that leads to Y′ translocation. Surprisingly, deletion of the RAD51 had little effect on the efficiency of Y′ translocation (Figure 5C, top panel), demonstrating that Rad51 may not be essential for recombination between TG1–3 repeats. In contrast, RAD59 deletion reduced both the amount and the heterogeneity of amplified VII-L/Y′ junctions (Figure 5C, midpanel) suggesting lower frequency of TG1–3 repeat recombination in the absence of Rad59. These results indicate that Rad51 filament assembly may not be required at the 3′ overhang of the short telomere, which pairing with an internal tract of the TG1–3 repeats could depend only on Rad52 strand annealing activity which is stimulated by Rad59.
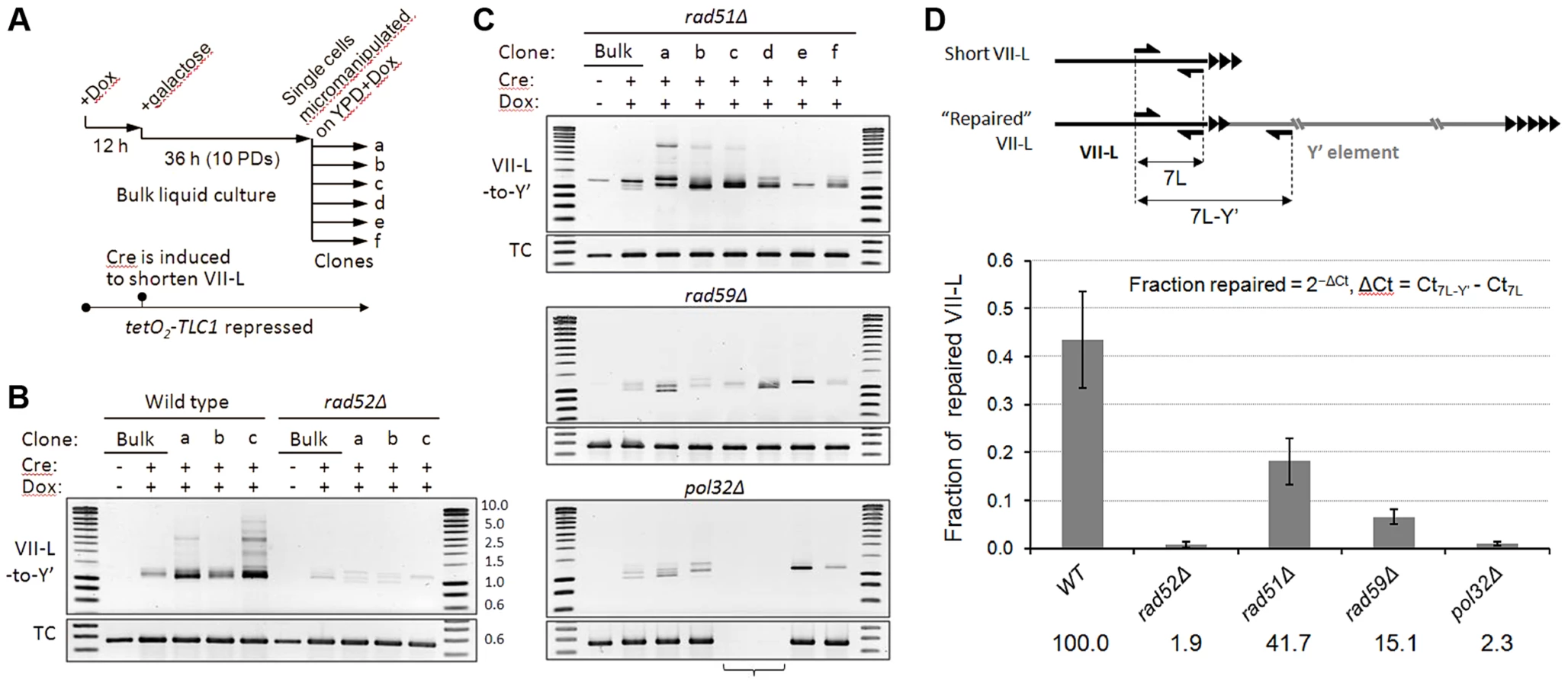
While Y′ translocation is initiated by TG1–3 repeat recombination, its completion likely depends on break induced replication (BIR), a pathway used to repair a DSB when homology is limited to its one side [37]. Deletion of POL32, encoding a nonessential subunit of Polδ required for processive DNA synthesis during BIR [38], severely reduced the efficiency of Y′ translocation as evidenced by drastically reduced junction PCR product (Figure 5C, bottom panel). Notably, the PCR at the VII-L terminus, which served as a loading control, failed for two transiently arrested pol32Δ clones (clones c and d) implying disappearance of the primer(s) site from the VII-L terminus. Therefore, we verified the integrity of the VII-L terminus in pol32Δ clones by Southern blot and found that it was indeed rearranged in both clones in a similar manner, which is different from Y′ translocation (Figure S6). We failed to detect Y′ translocation in any of the pol32Δ clones that we have analyzed by Southern blot. The junction PCR product that is reduced but still detectable in pol32Δ clones could have been generated on single strand extension products that were terminated before reaching chromosome end. Thus, we concluded that Pol32-dependent BIR is essential for the completion of Y′ translocation.
To quantify the Y′ translocation efficiencies in the aforementioned deletion mutants, we employed real-time qPCR using two pairs of primers to amplify either the total amount of VII-L or the VII-L/Y′ junctions (Figure 5D). For this analysis we used DNA extracted from random clones isolated at ∼16 PD after Cre induction which were also analyzed by Southern blotting with VII-L-specific probe (Figure S7). We performed clonal analysis because it provides an unbiased snapshot of the repair frequencies at any given time, whereas bulk liquid cultures of telomerase-negative cells are strongly affected by selection for best-growing clones. We estimated that at the time of analysis (∼40 PD after Cre) on average 43.5% of VII-L ends in wild-type clones acquired Y′ element. The fraction of “repaired” VII-L was reduced to 18.2 and 6.6% in rad51Δ and rad59Δ, respectively, whereas it was at the background level in both rad52Δ and pol32Δ clones (Figure 5D). We inferred from these results that Rad52 predominantly cooperates with Rad59 to initiate Pol32-dependent BIR which leads to Y′ translocation. Nevertheless, Rad51 also appears to contribute in this process since it reduces the efficiency of Y′ translocations by at least two fold.
Native telomeres shortened beyond critical length also acquire Y′ elements
To show that Y′ translocation is not a unique phenomenon that takes place at the modified VII-L end, but is common for eroded X-only telomeres, we used the same PCR approach to detect Y′ translocation on the native VI-R end (Figure S8A), which normally has only X element in the subtelomere region (W303 genome sequencing project, contig 00111). PCR product encompassing the junction between the VI-R end and the Y′ element was already detectable at 10 PD and increased further by 50 PD after telomerase inactivation (Figure S8B). Thus, erosion of the native telomeres in the absence of telomerase also leads to Y′ element translocations. Sequence analysis of the cloned PCR fragments confirmed that the VI-R/Y′ junction regions were predominantly amplified. However, we also identified a few clones resulted from mispriming on the X- and Y′-containing chromosomes V-R and XIII-L, which explains the background amplification before inactivation of telomerase.
As expected, all clones contained TG1–3 repeat tracts at the junction between the VI-R end and Y′ element. Thus, similar to modified VII-L, native chromosome VI-R end also acquired Y′ element as a result of recombination between the eroded terminal and internal TG1–3 repeats. The extent of TG1–3 sequence identity at the VI-R side of the junction was limited to 60±45 (mean ±SD) nt (Figure 6), the minimum length of the native telomeres at the time they recombined. The divergent repeat sequence was shorter on average (or even absent) compared to that at the junction of modified VII-L (Figure 6). Notably, among the chromosome ends that served as Y′ donors we identified a greater variety of ends (Table 1), indicating that native telomere VI-R was less selective with respect to a Y′ donor. Of note, not all junctions between the X and Y′ elements contain perfect TG1–3 repeats, but they all tend to be G-rich and are all bound by Rap1 (Table1 and Figure S9).

Rad59 promotes early acquisition of Y′ elements by native X-only telomeres
Since the native VI-R/Y′ junctions showed simpler composition indicative of possibly greater efficiency of Y′ translocation, we attempted to detect them during the senescence time course by Southern blot directly in the bulk liquid cultures. To this end, we performed standard senescence assay in liquid with est2Δ spore clones. The samples were collected for DNA extraction daily at the time of culture dilution. XhoI-digested DNA was then Southern blotted and probed for the native X-only XV-L end using subtelomeric probe. We were surprised to find that two high-molecular-weight bands characteristic of Y′L and Y'S translocations were readily detectable early on during the outgrowth of the est2Δ cultures (Figure 7A, B; top panels). The same result was obtained with the native VI-R end (not shown). As expected, the timing of the onset of Y′ translocations correlated with the initial XV-L telomere length, which differed among the spore clones. The abundance of cells with Y′ translocated on XV-L in est2Δ cultures during presenescence was consistent with the notion of greater efficiency of the Y′ element translocation on the native X-only telomere than on the modified subtelomereless VII-L end.
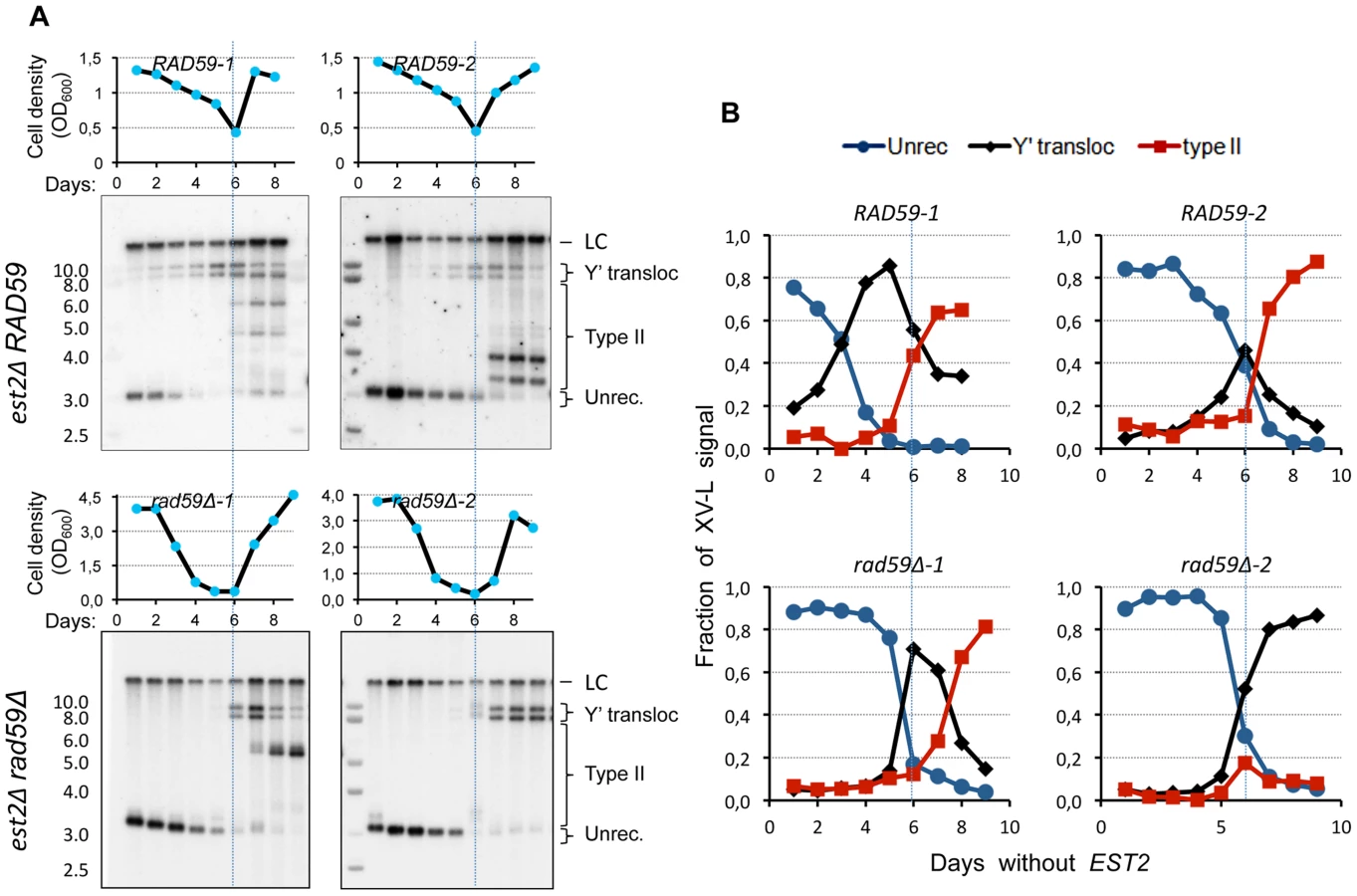
We next asked whether the RAD59 deletion, which reduced the frequency of Y′ translocation on the modified VII-L, would also compromise (or delay the onset of) Y′ translocations on the native XV-L as cells progress into senescence. Using the approach described above, we detected a substantial delay in the onset of Y′ translocations in the est2 rad59Δ relative to est2Δ clones (Figure 7). In the two representative est2Δ rad59Δ clones that are shown in Figure 7 (bottom panels), Y′ translocations were not detectable until the cells with very short XV-L telomere nearly disappeared from the population. In contrast, the cells with unrecombined short XV-L telomere and the cells which have already undergone Y′ translocation coexisted in the cultures of Rad59-proficient est2Δ clones long before they reached growth nadir. This result clearly points to the role of Rad59 in promoting Y′ translocation on the short native telomere. Note that in clone rad59Δ-1, type II pattern is seen after 8 days without telomerase. We usually observe that in liquid cultures, 75% of the clones are type I while 25% display a mixed pattern of type I and II (see Figure S11B).
Rad59 delays the onset of crisis and promotes transition to type I at X-only telomeres
The finding of short telomere repair by Y′ translocation raised a question whether this process can delay the onset of replicative senescence. Since Y′ element translocation is facilitated by Rad59 at X-only telomeres, we assayed the effect of RAD59 deletion on the onset of proliferative decline in the absence of telomerase. To this end we performed standard senescence assays using multiple est2Δ and est2Δ rad59Δ clones. Clones lacking Rad59 lost proliferative capacity slightly earlier and exhibited fivefold lower cell densities at the nadir of growth (Figure 8A). This observation indicates that Rad59-facilitated repair of critically short telomeres contributes to sustain cell proliferation particularly when a population of telomerase-negative cells approaches growth nadir. Consistent with this notion, we found by ChIP-qPCR that Rad59 associates with telomeres during presenescence. Although Rad59 association varies considerably among the telomeres, it may peak abruptly (up to 25-fold increase) at certain X-only telomeres when culture approaches growth nadir (Figure S10A). On average, Rad59 association with terminal and internal (between X and Y′ elements) TG1–3 repeats increases many fold as telomerase-negative cells progress into senescence (Figure S10B), highlighting the role of Rad59 in the conversion of X-only into Y′ telomeres.

We next reasoned that Y′ element translocation could be the initial step of type I telomere pattern formation at X-only telomeres. This premise seemingly contradicts established genetic requirements for survivor formation since type I survivors are predominantly obtained in cells lacking Rad59 [12], [13], [29]. The latter is due to the fact that Rad59 deletion greatly impedes type II. However, this does not exclude a possibility that Rad59 also facilitates transition to type I, particularly at X-only telomeres, although it is not strictly required. To reveal such a role of Rad59, we had to prevent the dominant type II pathway. This was performed by deleting SAE2 whose deletion strongly inhibits type II formation (Figure S11). We therefore compared the efficiency of type I survivor formation in est2Δ sae2Δ and est2Δ sae2Δ rad59Δ mutants (Figure 8B). We found that the triple mutant clones spent longer time in crisis suggesting reduced efficiency of type I survivor formation.
Y′ element acquisition and its amplification have distinct genetic requirements
The involvement of RAD59 in Y′ translocation raised a question whether the other genes of the type II survivor pathway may also contribute to Y′ acquisition. To this end, we compared the kinetics of Y′ acquisition by the native telomere XV-L between est2Δ and sgs1Δ est2Δ cells. We chose to delete SGS1, another gene required for type II survivors to arise [13], [15], [16], [19], because unlike the genes encoding the subunits of MRX complex it does not cause short telomeres, which would complicate comparative analysis of Y′ translocation kinetics since short telomeres acquire Y′ faster. We found that although est2Δ and sgs1Δ est2Δ clones started to senesce with XV-L telomere of comparable size, there was a substantial delay in Y′ translocation on XV-L in sgs1Δ est2Δ compared to est2Δ cells (Figure 9). Therefore, Sgs1 also appear to promote Y′ element acquisition. As expected, XV-L telomere converted to type I pattern by the time sgs1Δ est2Δ cells generated survivors. Remarkably, recombination between Y′ elements increases in sgs1Δ mutants [39], thus the inhibitory effect of SGS1 deletion on Y′ translocation further highlights the mechanistic difference between largely RAD51-independent Y′ acquisition and RAD51-dependent Y′ amplification, the two steps of type I survivor formation.

Since we found that short telomere repair is associated with transient arrest, we asked whether the checkpoint function is required for Y′ translocation. To address it, we deleted MEC1, which is required for G2/M arrest in telomerase-deficient cells [40] and also mediates type II recombination [41]. As expected, mec1Δ sml1Δ mutant clones exhibited flatter senescence profiles indicative of defective cell cycle arrest in response to telomere shortening and delayed senescence as reported previously [8], and yet Y′ translocation was not affected in any of the three clones analyzed (Figure 9). We concluded that checkpoint function is not required for Y′ acquisition by X-only telomeres. This is in contrast to deleting RAD59 and SGS1, which both substantially delay Y′ acquisition.
Discussion
Many studies from several laboratories have characterized the genetic pathways contributing to survivor formation [42]. In this study we demonstrated that a single telomere that was experimentally shortened in the absence of telomerase can acquire during presenescence Y′ element along with the terminal TG1–3 repeats from other chromosome ends which terminal repeats are still sufficiently long. Sequencing of the Y′ translocation junctions clearly evidenced that Y′ translocation was initiated by recombination between the short terminal TG1–3 repeats of the TelVII-L and the internal TG1–3 tracts located between the X and Y′ elements on certain native chromosome ends thereby confirming the model of Lundblad and Blackburn [10]. In addition, our results suggest that there is a continuous repair of the shortest telomeres that delays erosion into the subtelomere and allow the cell to escape DNA damage checkpoint activation. We showed that Y′ translocation was entirely RAD52-dependent, and was further promoted by RAD59. In contrast, RAD51 deletion had rather modest effect on the efficiency of Y′ translocation (two fold reduction), and Y′ translocations on VII-L end were detectable in rad51Δ clones. Therefore, Rad51-ssDNA filament formation and strand invasion do not seem to be obligatory for TG1–3 repeat recombination; instead, it could be accomplished by Rad52 strand annealing activity that is stimulated by Rad59. This mechanism bears strong similarities with the “short tract homology” recombination that has been described in the other context [43]. It remains possible that Rad51 affects Y′ translocations indirectly via its involvement in the recombination among the Y′ elements themselves [44]. Another question that rises with respect to Rad51-independent recombination between TG1–3 repeats is how the G-strand overhang anneals to the internal TG1–3 repeats, which are normally double-stranded. Possibly, this single-strand annealing occurs during subtelomere replication when the single strand regions are exposed [45], particularly when the replication forks are posed at the internal TG1–3 sequences [46].
In this study, we showed that such recombination events also occurred at the native telomeres VI-R and XV-L indicating that these rearrangements are physiological in nature. We found that modified subtelomereless VII-L and native X-only telomeres behave surprisingly different with respect to the efficiency of Y′ acquisition. Low efficiency of Y′ translocation on the modified VII-L end might be explained by its poor clustering with the other telomeres due to absence of subtelomeric elements. Indeed, intranuclear localization of the truncated TelVII-L is insensitive to the absence of either Sir3 or Yku [47], whereas these proteins participate in targeting native telomeres to the nuclear periphery [48], [49]. Poor clustering and failure to localize to nuclear compartments which favour recombination could be responsible for low efficiency of Y′ translocation and may explain why single experimentally shortened telomere accelerates senescence [8], [28].
Our results suggest that the Rad59-facilitated recombination between the terminal and internal TG1–3 repeats could be responsible for initial spreading of certain Y′ elements on X-only telomeres. We demonstrated that disappearance of the bands corresponding to X-only telomeres from Southern blots as telomerase-negative cultures progress into crisis is indeed a consequence of Y′ element acquisition that is promoted by Rad59. One can, therefore, envisage that the clones that arise via Y′ translocations are the precursors of type I survivors. The homogenization of the subtelomeric sequences due to preferential translocation of a certain class of Y′ elements can further promote perhaps more efficient Rad51-dependent BIR initiated within Y′ elements resulting in their amplification that is observed in type I survivors [10]. Remarkably, the strains that harbour Y′ elements at all chromosome ends due to previous history as a telomerase-defective survivor form survivors more readily when rendered telomerase-negative again [50]. This observation suggests that spreading of the Y′ elements to all ends could be one factor that limits the rate of type I survivor formation. Consistent with this proposition, we found that Rad59 accelerates type I survivor formation when type II survivor pathway is inhibited.
The initial step of Y′ acquisition involves heteroduplex formation by TG1–3 repeats that contain mismatches and is likely recognized by mismatch-repair proteins [51], [52]. It is therefore possible that the homeologous heteroduplexes formed during Rad59-dependent single-strand annealing are often rejected, and this may lead to reiterative rounds of repair synthesis which could effectively provide a way of TG1–3 tract expansion in the absence of telomerase. Indeed, we have identified a few clones with substantially long TG1–3 tracts at the breakpoint of recombination, which could have resulted from reiterative repair synthesis. We could speculate that once very long internal tracts are generated, they can be excised as circles via intramolecular recombination events (perhaps in a few steps) [14], [53]. The ultimate escape mechanism would be achieved via conversion to type II survivors, which are thought to amplify their terminal repeats via rolling circle replication. This could explain the apparent contradiction between the fact that circular DNA molecules are efficiently generated only when telomeric repeat tracts are abnormally elongated [54] and that only short telomeres engage in type II recombination [14].
Interestingly, we found that Rap1 was strongly enriched at the internal TG1–3 tracts located between the X and Y′ elements of the donor telomeres (Figure S9 and Table 1, see also [55]). Rap1 at these internal TG1–3 tracts was not relocalized upon critical shortening of telomeres [55]. It is conceivable that these internal Rap1 binding sites are also bound by the Sir proteins [56]. Notably, there is no homology between the modified TelVII-L and the Y′ donor chromosome end outside of the TG1–3 repeat tracts. Nevertheless, the apparent efficiency of Rad51-independent single strand annealing that relies exclusively on short homeologous TG1–3 repeat sequences is rather high, which might be due to spatial clustering of the telomeres in the yeast nucleus [57]. Indeed, for double-strand break repair, it has been recently shown that proximity of the donor sequence promotes homologous recombination [58]. The choice of the Y′ donor does not appear random. Whether this preference reflects the proximity between the chromosome ends in the nucleus or is influenced by other factors is currently unknown. Of note, among all chromosome ends, the VI-L end harbours one of the longest (139 bp-long) internal TG1–3 tract between the X and Y′ elements; and VII-R end is located at approximately the same distance from the centromere as the experimental VII-L end on the opposite arm of the same chromosome, which is consistent with Rabl configuration [59]. It has been also reported that Rap1 had the intrinsic ability to locally distort telomeric double-stranded DNA provoking local conformational changes characteristic of single strand [60]. Therefore, subtelomeric Rap1 binding could favour homologous recombination by creating local structures that are amenable to annealing with single-stranded overhang. In addition, Rap1 contains four putative SIMs, and thus, it may potentially recruit SUMOylated Rad52 and Rad59 to TG1–3 tracts.
Although our study focuses on short telomere repair by Y′ translocation, the BIR events leading to only terminal TG1–3 tract extension are also possible in telomerase-negative yeast. These events are readily detectable by Southern blot in survivors [14], [22] since they often result in large increase of terminal TG1–3 tract length, but this is not the case in pre-senescent cells. Direct sequencing of the terminal repeats showed that they do occasionally get extended in pre-senescent cells, although there is a controversy of whether the short or long tracts are preferentially extended [21], [22]. In any case, recombination between terminal repeats cannot indefinitely sustain proliferation of telomerase-negative cells, whereas Y′ translocation leads toward type I telomere formation, which likely requires additional changes such as chromatin structure alteration [19].
In summary, we have characterized a repair mechanism that acts upon short X-only telomeres in budding yeast lacking telomerase during presenescence. This repair mechanism does not require Rad51 and depends on annealing between short homeologous sequences which is stimulated by Rad59. Unlike a single unrepairable DSB, a single critically short telomere is not immediately lethal for a cell in the absence of telomerase as long as there is a reserve of long telomeres on other chromosome ends. Remarkably, Rad51 independence of the short telomere rescue pathway points to yet another problem caused by telomerase inactivation which resolution depends on Rad51-dependent HR, since deletion of RAD51 is known to cause early loss of viability of telomerase-negative cells.
Materials and Methods
Strains and plasmids
All yeast strains used in this study were from the W303 background (see genotypes in Table S2). Strains used to analyze telomere rearrangement in the absence of telomerase were derivatives of the YAB892 and YAB893, which have 0 and 16 Rap1-binding sites, respectively, flanked by loxP sites at the modified telomere VII-L [31]. To obtain Tet-Off TLC1 derivatives, these strains were crossed to the tetO2-TLC1 strain. The inducible EST2 deletion derivatives (double Cre-loxP) were generated by first transforming the cells with the pDS381 plasmid [33] carrying EST2 gene linked to the ADE2 marker and the loxP-flanked ARS/CEN region, and then replacing the endogenous EST2 locus with KANr cassette. All other gene disruptions were carried out by PCR-based methods resulting in the replacement of targeted loci with the TRP1 marker.
Inducible shortening of the TelVII-L and inactivation of telomerase
To induce genome-integrated GALp-CRE for simultaneous TelVII-L shortening and loss of the plasmid-borne EST2, overnight cultures growing in SC medium lacking lysine and adenine and containing 2% raffinose were diluted (1∶20) into complete YEP medium containing 2% galactose. After 24 hours of induction the cells were diluted into YPD to prevent genome damage by Cre at non-specific sites. To inhibit telomerase in the Tet-off TLC1 strains, the cultures grown in SC medium lacking lysine and containing 2% raffinose were supplemented with doxycycline (10 µg/µl), 12 hours before induction of TelVII-L shortening by switching cells to galactose for the next 24 hours as described above. The doxycycline concentration was maintained constant throughout the experiments in all media.
Microcolony assay and isolation of the clones with rearranged VII-L end
To determine the ability of individual telomerase-negative cells to form microcolonies, the double Cre-loxP cells were micromanipulated onto a grid of YPD agar at 36 h after induction of Cre with galactose in liquid culture. Alternatively, the Tet-off TLC1 derivatives were micromanipulated onto the YPD agar freshly supplemented with Dox (20 µg/µl) at 24 h after Cre induction and 48 h after repression of TLC1 with Dox. The microcolony formation was monitored by counting the number of cells in each grid position at 2, 4, and 8 h after micromanipulation. The plates were incubated for additional 4 days to allow formation of visible colonies. The colonies which exhibited growth delay at the time or soon after micromanipulation were chosen for VII-L end analysis. In addition, the colonies with deeply nibbled edges and typically smaller size were included regardless of the growth delay. The clones that exhibited robust expansion and formed large colonies with smooth edges were used as controls.
Southern blot analysis of the VII-L and bulk telomeres
To determine the state of the VII-L end before and after induction of Cre expression, genomic DNA was digested simultaneously with MfeI and PacI. The resulting fragments were separated by 0.9% agarose gel electrophoresis, transferred on Hybond N+, and hybridized with 32P-labeled 252 bp-long probe that was generated by PCR (see Table S1 for primers sequences) using YAB892 genomic DNA as a template. The rearrangement of the VII-L end in clonal populations recovered from transient arrest was analyzed similarly, except for two aliquots of DNA were digested separately with either MfeI or PacI. To determine the length of native telomeres, XhoI-digested yeast DNA was subjected to 0.8% agarose gel electrophoresis and hybridized with a 32P-labeled (TG1–3)n probe. The probes were labeled by random priming using Klenow fragment exo-, and all hybridizations were performed in Church buffer at 58°C.
PCR across the translocation junction
To amplify the regions encompassing putative Y′ translocation junctions at either modified VII-L or native VI-R ends, the chromosome end-specific primers were designed to anneal ∼500 bp away from terminal TG1–3 repeats, and the Y′ element-specific primer sites were chosen in the centromere-proximal region that is conserved among all Y′ elements (Table S1). The Y′ sequence alignments are available from Ed Louis at http://www2.le.ac.uk/colleges/medbiopsych/research/gact/resources/yeast-telomeres. Analytical PCR was performed using Phusion DNA Polymerase (Thermo Scientific) in 1xHF buffer and 1 ng/µl genomic DNA purified via phenol-chlorophorm extraction. Primers were used at 500 nM, and the annealing temperature was set at 3°C above the Tm of the least stable primer.
Cloning and sequencing of the translocation junction regions
For cloning, PCR across the junction was performed using Taq DNA polymerase to produce fragments with 3′ A overhangs, and the purified product was inserted into the pCR2.1 vector via one-step TA cloning (Invitrogen). The inserts were sequenced from both ends using M13F-20 and M13-26REV primers by Sanger method (at Beckman Coulter). As a rule, the read going through the TG-rich strand failed at the junction, so the entire sequence was assembled from both reads.
Rad59 ChIP-qPCR
Rad59 was 13xMyc epitope–tagged at the C-terminus using one-step PCR. ChIP was performed as previously described [61]. Briefly, chromatin was cross-linked with 1% formaldehyde and sonicated to an average 200- to 500-bp DNA fragment size. After clarifying centrifugation, soluble chromatin was incubated with mouse anti-Myc tag monoclonal antibodies (9E10) and immunocomplexes were bound to magnetic Dynabeads Protein G (Novex). Following successive washes in standard solutions, Rad59-Myc bound chromatin was eluted from beads and incubated at 68°C to reverse crosslinks. DNA purified from the immunoprecipitates and inputs was quantified by real-time qPCR using chromosome end-specific primers listed in Table S1. The enrichment of the telomere-specific sequences bound by Rad59 was normalized to input and an unaffected GAL2 locus.
Rap1 ChIP-on-chip
Rap1 ChIP experiments (duplicates) were performed with anti-Rap1 antibodies kindly provided by David Shore (University of Geneva). Rap1 ChIP and input DNA samples were hybridized to Nimblegen S. cerevisiae high density tiling arrays that were designed by us in collaboration with Frédéric Devaux (Ecole Normale de Paris) and Nimblegen to cover the entire genome. They contain 50 nt-long oligonucleotides separated by ∼15 nt-long gaps. The chip covers both strands of S. cerevisiae genome. Hybridizations and data analysis were performed by Nimblegen (Roche NimbleGen). Rap1 peaks were visualized with the signalMap software (Nimblegen) or with the Integrative Genomics Viewer (Broad Institute). The genome-wide Rap1 binding profiles were consistent with the recent published studies [55].
Supporting Information
Zdroje
1. PalmW, de LangeT (2008) How Shelterin Protects Mammalian Telomeres. 42: 301–334 doi:10.1146/annurev.genet.41.110306.130350
2. PardoB, MarcandS (2005) Rap1 prevents telomere fusions by nonhomologous end joining. 24: 3117–3127 doi:10.1038/sj.emboj.7600778
3. BonettiD, MartinaM, ClericiM, LucchiniG, LongheseMP (2009) Multiple Pathways Regulate 3′ Overhang Generation at S. cerevisiae Telomeres. 35: 70–81 doi:10.1016/j.molcel.2009.05.015
4. XueY, RushtonMD, MaringeleL (2011) A novel checkpoint and RPA inhibitory pathway regulated by Rif1. 7: e1002417 doi:10.1371/journal.pgen.1002417
5. Ribeyre C, Shore D (2012) Anticheckpoint pathways at telomeres in yeast: 1–8. doi: 10.1038/nsmb.2225.
6. ChurikovD, CordaY, LucianoP, GéliV (2013) Cdc13 at a crossroads of telomerase action. 3: 39 doi:10.3389/fonc.2013.00039
7. IJpmaAS, GreiderCW (2003) Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. 14: 987–1001 doi:10.1091/mbc.02-04-0057
8. AbdallahP, LucianoP, RungeKW, LisbyM, GéliV, et al. (2009) A two-step model for senescence triggered by a single critically short telomere. 11: 988–993 doi:10.1038/ncb1911
9. HectorRE, RayA, ChenB, ShtofmanR, BerknerKL, et al. (2012) Mec1p associates with functionally compromised telomeres. 121: 277–290 doi:10.1007/s00412-011-0359-0
10. LundbladV, BlackburnEH (1993) An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73: 347–60.
11. Teng S, Zakian V (1999) Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Molecular and cellular biology.
12. TengSC, ZakianVA (1999) Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. 19: 8083–8093.
13. ChenQ, IjpmaA, GreiderCW (2001) Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. 21: 1819–1827 doi:–10.1128/MCB.21.5.1819–1827.2001
14. TengSC, ChangJ, McCowanB, ZakianVA (2000) Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. 6: 947–952.
15. HuangP, PrydeFE, LesterD, MaddisonRL, BortsRH, et al. (2001) SGS1 is required for telomere elongation in the absence of telomerase. 11: 125–129.
16. JohnsonFB, MarciniakRA, McVeyM, StewartSA, HahnWC, et al. (2001) The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. 20: 905–913 doi:10.1093/emboj/20.4.905
17. McEachernMJ, HaberJE (2006) Break-induced replication and recombinational telomere elongation in yeast. 75: 111–135 doi:10.1146/annurev.biochem.74.082803.133234
18. LydeardJR, JainS, YamaguchiM, HaberJE (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. 448: 820–823 doi:10.1038/nature06047
19. HuY, TangH, LiuN, TongX, DangW, et al. (2013) Telomerase-Null Survivor Screening Identifies Novel Telomere Recombination Regulators. PLoS Genet 9: e1003208 doi:10.1371/journal.pgen.1003208
20. McEachernMJ, IyerS (2001) Short telomeres in yeast are highly recombinogenic. 7: 695–704.
21. ChangM, DittmarJC, RothsteinR (2011) Long telomeres are preferentially extended during recombination-mediated telomere maintenance. 18: 451–456 doi:10.1038/nsmb.2034
22. FuX, DuanY, LiuY, CaiC, MengF, et al. (2014) Telomere recombination preferentially occurs at short telomeres in telomerase-null type II survivors. 9: e90644 doi:10.1371/journal.pone.0090644
23. DunnB, SzauterP, PardueML, SzostakJW (1984) Transfer of yeast telomeres to linear plasmids by recombination. 39: 191–201.
24. LouisEJ, HaberJE (1992) The structure and evolution of subtelomeric Y′ repeats in Saccharomyces cerevisiae. Genetics 131: 559–74.
25. Louis E, Naumova E, Lee A, Naumov G (1994) The chromosome end in yeast: its mosaic nature and influence on recombinational dynamics. Genetics.
26. KhadarooB, TeixeiraMT, LucianoP, Eckert-BouletN, GermannSM, et al. (2009) The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. 11: 980–987 doi:10.1038/ncb1910
27. MorrishTA, GreiderCW (2009) Short telomeres initiate telomere recombination in primary and tumor cells. 5: e1000357 doi:10.1371/journal.pgen.1000357
28. Fallet E, Jolivet P, Soudet J, Lisby M, Gilson E, et al.. (2014) Length-dependent processing of telomeres in the absence of telomerase.: gkt1328. doi: 10.1093/nar/gkt1328.
29. LeS, MooreJK, HaberJE, GreiderCW (1999) RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. 152: 143–152.
30. SandellLL, ZakianVA (1993) Loss of a yeast telomere: arrest, recovery, and chromosome loss. 75: 729–739.
31. BianchiA, ShoreD (2007) Early replication of short telomeres in budding yeast. 128: 1051–1062 doi:10.1016/j.cell.2007.01.041
32. MarcandS, BrevetV, GilsonE (1999) Progressive cis-inhibition of telomerase upon telomere elongation. 18: 3509–3519 doi:10.1093/emboj/18.12.3509
33. CohenH, SinclairDA (2001) Recombination-mediated lengthening of terminal telomeric repeats requires the Sgs1 DNA helicase. 98: 3174–3179 doi:10.1073/pnas.061579598
34. ShampayJ, BlackburnEH (1988) Generation of telomere-length heterogeneity in Saccharomyces cerevisiae. 85: 534–538.
35. FörstemannK, HössM, LingnerJ (2000) Telomerase-dependent repeat divergence at the 3′ ends of yeast telomeres. 28: 2690–2694.
36. SugawaraN, IraG, HaberJE (2000) DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. 20: 5300–5309.
37. LlorenteB, SmithCE, SymingtonLS (2008) Break-induced replication: what is it and what is it for? 7: 859–864.
38. WilsonMA, KwonY, XuY, ChungW, ChiP, et al. (2013) Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. 502: 393–396 doi:10.1038/nature12585
39. WattPM, HicksonID, BortsRH, LouisEJ (1996) SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144: 935–45.
40. EnomotoS, GlowczewskiL, BermanJ (2002) MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Molecular biology of the cell 13: 2626–38 doi:10.1091/mbc.02-02-0012
41. TsaiY, TsengS, ChangS, LinC, TengS (2002) Involvement of replicative polymerases, Tel1p, Mec1p, Cdc13p, and the Ku complex in telomere-telomere recombination. Molecular and cellular biology 22: 5679–87.
42. WellingerRJ, ZakianVA (2012) Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. 191: 1073–1105 doi:10.1534/genetics.111.137851
43. IraG, HaberJE (2002) Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. 22: 6384–6392.
44. LouisEJ, HaberJE (1990) Mitotic recombination among subtelomeric Y′ repeats in Saccharomyces cerevisiae. Genetics 124: 547–59.
45. MottC, SymingtonLS (2011) RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. 10: 408–415 doi:10.1016/j.dnarep.2011.01.007
46. MakovetsS, HerskowitzI, BlackburnEH (2004) Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. 24: 4019–4031.
47. ThamWH, WyitheJS, Ko FerrignoP, SilverPA, ZakianVA (2001) Localization of yeast telomeres to the nuclear periphery is separable from transcriptional repression and telomere stability functions. 8: 189–199.
48. FerreiraHC, LukeB, SchoberH, KalckV, LingnerJ, et al. (2011) The PIAS homologue Siz2 regulates perinuclear telomere position and telomerase activity in budding yeast. 13: 867–874 doi:10.1038/ncb2263
49. NagaiS, DavoodiN, GasserSM (2011) Nuclear organization in genome stability: SUMO connections. 21: 474–485 doi:10.1038/cr.2011.31
50. LundbladV (2002) Telomere maintenance without telomerase. 21: 522–531 doi:10.1038/sj.onc.1205079
51. SpellRM, Jinks-RobertsonS (2003) Role of mismatch repair in the fidelity of RAD51- and RAD59-dependent recombination in Saccharomyces cerevisiae. 165: 1733–1744.
52. RizkiA, LundbladV (2001) Defects in mismatch repair promote telomerase-independent proliferation. 411: 713–716 doi:10.1038/35079641
53. NatarajanS, McEachernMJ (2002) Recombinational telomere elongation promoted by DNA circles. 22: 4512–4521.
54. LiB, LustigAJ (1996) A novel mechanism for telomere size control in Saccharomyces cerevisiae. 10: 1310–1326.
55. PlattJM, RyvkinP, WanatJJ, DonahueG, RickettsMD, et al. (2013) Rap1 relocalization contributes to the chromatin-mediated gene expression profile and pace of cell senescence. 27: 1406–1420 doi:10.1101/gad.218776.113
56. SmithCD, SmithDL, DeRisiJL, BlackburnEH (2003) Telomeric protein distributions and remodeling through the cell cycle in Saccharomyces cerevisiae. 14: 556–570 doi:10.1091/mbc.E02-08-0457
57. SchoberH, KalckV, Vega-PalasMA, Van HouweG, SageD, et al. (2008) Controlled exchange of chromosomal arms reveals principles driving telomere interactions in yeast. 18: 261–271 doi:10.1101/gr.6687808
58. AgmonN, LiefshitzB, ZimmerC, FabreE, KupiecM (2013) Effect of nuclear architecture on the efficiency of double-strand break repair. 15: 694–699 doi:10.1038/ncb2745
59. BystrickyK, HeunP, GehlenL, LangowskiJ, GasserSM (2004) Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. 101: 16495–16500 doi:10.1073/pnas.0402766101
60. GilsonE, MüllerT, SogoJ, LarocheT, GasserSM (1994) RAP1 stimulates single- to double-strand association of yeast telomeric DNA: implications for telomere-telomere interactions. 22: 5310–5320.
61. LucianoP, CoulonS, FaureV, CordaY, BosJ, et al. (2012) RPA facilitates telomerase activity at chromosome ends in budding and fission yeasts. The EMBO journal 31: 2034–46 doi:10.1038/emboj.2012.40
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2014 Číslo 11
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- An RNA-Seq Screen of the Antenna Identifies a Transporter Necessary for Ammonia Detection
- Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation
- Functional Diversity of Carbohydrate-Active Enzymes Enabling a Bacterium to Ferment Plant Biomass
- Regularized Machine Learning in the Genetic Prediction of Complex Traits