
The Streamlined Genome of spp. Relative to Human Pathogenic Kinetoplastids Reveals a Parasite Tailored for Plants
Members of the family Trypanosomatidae infect many organisms, including animals, plants and humans. Plant-infecting trypanosomes are grouped under the single genus Phytomonas, failing to reflect the wide biological and pathological diversity of these protists. While some Phytomonas spp. multiply in the latex of plants, or in fruit or seeds without apparent pathogenicity, others colonize the phloem sap and afflict plants of substantial economic value, including the coffee tree, coconut and oil palms. Plant trypanosomes have not been studied extensively at the genome level, a major gap in understanding and controlling pathogenesis. We describe the genome sequences of two plant trypanosomatids, one pathogenic isolate from a Guianan coconut and one non-symptomatic isolate from Euphorbia collected in France. Although these parasites have extremely distinct pathogenic impacts, very few genes are unique to either, with the vast majority of genes shared by both isolates. Significantly, both Phytomonas spp. genomes consist essentially of single copy genes for the bulk of their metabolic enzymes, whereas other trypanosomatids e.g. Leishmania and Trypanosoma possess multiple paralogous genes or families. Indeed, comparison with other trypanosomatid genomes revealed a highly streamlined genome, encoding for a minimized metabolic system while conserving the major pathways, and with retention of a full complement of endomembrane organelles, but with no evidence for functional complexity. Identification of the metabolic genes of Phytomonas provides opportunities for establishing in vitro culturing of these fastidious parasites and new tools for the control of agricultural plant disease.
Published in the journal:
The Streamlined Genome of spp. Relative to Human Pathogenic Kinetoplastids Reveals a Parasite Tailored for Plants. PLoS Genet 10(2): e32767. doi:10.1371/journal.pgen.1004007
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004007
Summary
Members of the family Trypanosomatidae infect many organisms, including animals, plants and humans. Plant-infecting trypanosomes are grouped under the single genus Phytomonas, failing to reflect the wide biological and pathological diversity of these protists. While some Phytomonas spp. multiply in the latex of plants, or in fruit or seeds without apparent pathogenicity, others colonize the phloem sap and afflict plants of substantial economic value, including the coffee tree, coconut and oil palms. Plant trypanosomes have not been studied extensively at the genome level, a major gap in understanding and controlling pathogenesis. We describe the genome sequences of two plant trypanosomatids, one pathogenic isolate from a Guianan coconut and one non-symptomatic isolate from Euphorbia collected in France. Although these parasites have extremely distinct pathogenic impacts, very few genes are unique to either, with the vast majority of genes shared by both isolates. Significantly, both Phytomonas spp. genomes consist essentially of single copy genes for the bulk of their metabolic enzymes, whereas other trypanosomatids e.g. Leishmania and Trypanosoma possess multiple paralogous genes or families. Indeed, comparison with other trypanosomatid genomes revealed a highly streamlined genome, encoding for a minimized metabolic system while conserving the major pathways, and with retention of a full complement of endomembrane organelles, but with no evidence for functional complexity. Identification of the metabolic genes of Phytomonas provides opportunities for establishing in vitro culturing of these fastidious parasites and new tools for the control of agricultural plant disease.
Introduction
Flagellated protists of the family Trypanosomatidae, class Kinetoplastea, infect a large variety of organisms including animals, plants and humans [1]. While African and South-American trypanosomes are responsible for sleeping sickness [2] and Chagas' disease [3], respectively, different Leishmania spp. cause visceral, cutaneous and mucocutaneous manifestations of leishmaniasis in many tropical and subtropical regions [4].
Various eukaryotes, particularly filamentous microorganisms like oomycetes and fungi have acquired the capacity to infect and grow inside the plant tissues. While some of these organisms could influence plant growth positively, in most cases they can cause major diseases in plants of economic importance [5]. The genomes of numerous of these filamentous plant pathogens have already been sequenced, unveiling an amazing variety of genome sizes and organization [6]. Certainly, a great number of these plant pathogens were molded into larger genomes by repeat-driven expansions, with the genes coding for proteins involved in host interactions located within repeat-rich regions [6]. In contrast, some filamentous plant pathogens have fairly small genomes, as a consequence of intron or gene loss, like U. maydis;(21 Mb) [7] and Albugo laibachii; (37 Mb) [8], or abridged transposon content as in Sclerotinia sclerotiorum; (38 Mb) [9].
Like fungi and oomycetes, trypanosomatids also infect plants, but using a radically different strategy to colonize and propagate inside the host [10]–[12]. Multiple insect species of the order Heteroptera act as the natural vectors of plant trypanosomatids, both in the transmission to lactiferous hosts [10], [13], and for infection by intraphloemic plant trypanosomes [14]–[16]. Phytomonas is the arbitrary genus name proposed for all trypanosomatids specific to plants [17]; however this rather restricted taxonomic description fails to fully capture the wide diversity of trypanosomatids encountered in plants, both with respect to their biological properties and their impact on the host [18]–[22]. Indeed, Phytomonas spp. infect more than 100 plant species, distributed primarily in tropical and subtropical zones, by multiplying in latex tubes, fruits and seeds or colonizing the phloem sap inside the sieve tubes. Phytomonas infection can occur without apparent pathogenicity, but conversely it can cause lethal disease in plants of substantial economic value, including the coffee tree, coconut and oil palms [10], [23]. This results in important economic losses in Latin America and the Caribbean [24]–[27]. Ten distinct subgroups of plant trypanosomatids have been defined using the internal transcribed spacer region of the ribosomal RNA locus [22]. Only Group H, encompassing the Latin American intraphloemic trypanosomatids responsible for severe wilts, can be distinguished both by rRNA markers as well as biological and serological properties [19]. A full definition of the diversity of trypanosomatids within the overarching Phytomonas genus is still outstanding.
The whole genome sequences of Trypanosoma cruzi, Trypanosoma brucei and Leishmania major were released in 2005 [28]–[31]. Since then, the genomes of several additional trypanosomatids, including several pathogens of mammals, have been completed and described [32]–[35]. These databases have provided an essential platform for investigations of basic biology and mechanisms of pathogenesis and facilitated the exploration of novel therapies.
However, to date genome level analysis of Phytomonas spp. is limited. The biology of these parasites is reasonably well described [11], [36], but little information exists on their effective control by chemicals or, most critically, on their specific adaptations to the plant host and the mechanisms underpinning pathogenesis. Moreover, few genes are available in sequence databases, and little is known about genome size, chromosomal organization and ploidy [37], [38].
We describe here the genome sequences of two plant trypanosomatids, one phloem-restricted pathogenic isolate from a diseased coconut from Guiana (HART1 from Group H) and the other a non-symptomatic latex isolate from Euphorbia (EM1 from Group D) [22]. The comparison of these two plant parasite genomes with each other and with those of other trypanosomes reveals a common simplified genome organization for the plant trypanosomes. Identification of the genes involved in Phytomonas metabolism is an important step for improving in vitro culture protocols and for development of new and better tools for the control and diagnosis of Phytomonas-mediated diseases.
Results and Discussion
General features of Phytomonas EM1 and HART1 genomes
Recently, the molecular karyotype of several different latex plant symbiont-like (i.e. not associated with apparent pathology in the host) Phytomonas isolates were analyzed by pulsed-field gel electrophoresis (PFGE), showing 21 chromosomal bands for EM1 (group D) [37]. Similar analysis performed on phloem-restricted trypanosomatids allowed the identification of 7 chromosomal bands for the Hartrot wilt pathogen isolate (HART1, Group H) [38]. A systematic genome sequencing project of these two Phytomonas isolates was initiated, as they represent two distinct phenotypes in terms of impact on the host. Both EM1 and HART1 genomes were assembled using 10× 454-technology and 0.1× Sanger reads, together with deep coverage Illumina sequencing reads for correction of sequencing errors [39] (European Nucleotide Archive accession numbers CAVQ010000001-CAVQ010001400 for EM1 and CAVR010000001-CAVR010002560 for HART1; details in Text S1). Ninety percent of the EM1 genome assembly was placed in 45 scaffolds longer than 100 kb, with one third in the size range of the Phytomonas EM1 chromosomes previously observed by PFGE [37]. In the case of EM1, the scaffold N50 (the scaffold size above which 50% of the total length of the sequence assembly can be found) was 429 kb (Figure 1A). Meanwhile, the scaffold N50 for HART1 isolate was 1.2 Mb, with 90% of the genome located in 15 of the scaffolds, again in the size range previously estimated for the HART1 chromosomes [38] (Figure 1A). These assembly statistics indicate that majority coverage of both EM1 and HART1 genomes was achieved. A striking feature of these two plant parasite genomes is their small size (18.1 Mb for HART1; 17.8 Mb for EM1), when compared to that of the human pathogenic trypanosomatids (26.3 Mb for T. brucei; 32.5 Mb for T. cruzi and 32.9 Mb for L. major) [31].

Phytomonas EM1 and HART1 are likely fundamentally diploid with some supernumerary chromosomes, with an unknown level of polymorphism between the two haplotypes [37], [38], a feature they have in common with other trypanosomatids. The massively parallel sequencing strategy provided important read depth coverage across both EM1 and HART1 assemblies (Table S1), which was used to establish ploidy for both Phytomonas genomes (details in Text S1). Median read depth analysis revealed an even depth across both Phytomonas assemblies (Figure S1), pointing towards an underlying euploidy of diploid for both Phytomonas isolates. The use of allele frequency for heterozygous single nucleotide polymorphisms (SNPs) across the scaffolds revealed a consistent diploid distribution of frequencies (Figure S2; method as described by [40]). Clusters of duplicated genes were found to be biased towards disomic scaffolds using a Monte Carlo simulation (p = 7×10−4 hypergeometric distribution). These results were similar to the distribution of multicopy genes observed in Leishmania spp. chromosomes [40], suggessting the existence of separate mechanisms for gene duplication and chromosome (scaffold) duplication in Phytomonas spp.
Nonetheless, read depth reached values greater than twofold in some cases (scaffolds 24 and 25 in Figure S1 A; scaffolds 13 and 22 in Figure S1 B), probably indicating, as for other parasite genomes, aneusomy of certain chromosomal regions (Figure S3 and S4) [40], [41]. Possible aneusomy was already envisaged for the Phytomonas HART1 isolate after study of its molecular karyotype [38]. This increase in read depth is not likely due to the amplification of specific regions of the scaffolds, since read depth was constant along the whole of both the disomic and tetrasomic regions (Figure S5).
Both assemblies were annotated using a combination of evidence (Table S2; for details, see Text S1), with the major features of the genome annotation presented in Figure 1 A. The reference annotation of the Phytomonas EM1 and HART1 genomes (European Nucleotide Archive Accession HF955061–HF955198 for EM1 and HF955199–HF955282 for HART1) harbor 6,381 and 6,451 putative protein-coding genes, covering 57.9 and 53.7% of the genome respectively (Figure 1 A). The total number of predicted genes in both Phytomonas isolates is lower than in other sequenced trypanosomatids (EuPathDB-TriTryp 4.2: T. cruzi, CL Brener Esmeraldo-like 10,342; T. cruzi CL Brener Non-Esmeraldo-like 10,834; T. brucei TREU927, 10,533; Figure 1 A), but slightly closer to the Leishmania spp. (EuPathDB-TriTryp 4.2: L. braziliensis, 8,357; L. infantum, 8,241; L. major, 8,412, Figure 1 A), as expected by the close phylogenetic relationship of Phytomonas with Leishmania [42]. Such a decrease in predicted gene numbers is the consequence of an almost complete absence of tandemly-linked duplicated genes in both Phytomonas genomes as observed when compared to other sequenced trypanosomes [43], [44]. Indeed, the genomes of T. brucei, T.cruzi and L. major contain a high percentage of repetitive genes (Figure 1A; 27% for T. cruzi, 9.6% for T. brucei and 6.7% for L. major), whereas both Phytomonas isolates only possess a very low percentage of such genes (Figure 1 A; EM1 and HART1). This is the case for the NADH-dependent fumarate reductase, arranged in several copies in the T. brucei (6 copies), T. cruzi (7 copies) and L. major genomes (4 copies) but only detected as a single-copy gene in both Phytomonas isolates (Table S3). The uniform read depth coverage observed all along the Phytomonas EM1 and HART1 scaffolds overrules a collapse of multiple tandem repeats into fewer copies during assembly as an explanation for the Phytomonas gene copy number observed (Figure S5). A small fraction of EM1 genes were observed in multiple copies on the genome: only 99 clusters of paralogous protein-coding genes (corresponding to 171 genes; for details see Methods) were identified, constituting 2.6% of the Phytomonas EM1 putative genes. Typical cases are those of the chaperonin HSP60 (32 copies (on average) in the T. cruzi CL Brener genome) and the thioredoxin peroxidase, both identified in three copies in the EM1 assembly. Excluding a multigene family (six genes) with a histone-fold domain, most of the “duplicated” genes were present in only two copies. A similar situation in which the genome was almost exclusively comprised of single-copy genes was observed in HART1, with the exception of a gene family homologous to a major surface metallopeptidase of Leishmania promastigotes [45]. The metalloprotease gp63/leishmanolysin (EC 3.4.24.36) was originally described as the most abundant surface protein of Leishmania spp, but has been subsequently demonstrated to be pan-eukaryotic. A massive expansion in the gp63 family is evident in HART1 with over 20 members, while EM1 has only two. Both expansions are lineage-specific. GP-63 has been implicated in interactions with both vertebrate and insect hosts of Leishmania, and there is preliminary evidence for it playing a role in insect interactions in P. serpens and other lower trypanosomatids [46], [47]. In P. serpens gp63 is present in many endomembrane compartments; significantly expression levels can be reduced by exposure to fetal calf serum, suggesting an ability to respond to alterations in the environment, and/or potential for degradation of specific proteins or peptides [48].
Unlike the majority of eukaryotes, mRNA transcription in trypanosomatids is polycistronic. These genomes are organized into large polycistronic transcription units (PTUs), with tens –to -hundreds of protein-coding genes arranged head-to-tail on the same DNA strand and apparently transcribed from a single upstream RNA pol II entry site, or promoter [28]–[30], [49]. This unusual gene organization was observed in both Phytomonas isolates as well, where genes are organized into 298 (EM1) and 334 (HART1) putative PTUs with an average of 21 (EM1) and 19 (HART1) genes per cistron (Figure S6).
Protein-coding genes in Phytomonas appear to lack conventional introns, similar to the structure of genes in other trypanosomatids [1], [50]. Classical cis-splicing introns are documented only in the poly(A) polymerase and an ATP-dependent DEAD/H RNA helicase genes from T. brucei, T. cruzi [51], and Leishmania spp. This striking feature is not conserved in the Phytomonas EM1 and HART1 isolates.
Contraction in both plant parasite genomes is also reflected by the short length of the intergenic regions (on average 1,140 bp for EM1; 1,280 for HART1) and a relatively low frequency of repeated sequences (0.9% and 1.2% for EM1 and HART1, respectively) (Figure 1A). No significant difference in overall gene sizes was observed between these isolates (1,614 bp and 1,507 bp on average for EM1 and HART1, respectively). These data suggest that the EM1 and HART1 genomes are compact and might lack many of the expansions of both coding and non-coding sequences that have been described for other trypanosomes [30], [43].
Members of the order Kinetoplastida display an impressive number of structural and biochemical peculiarities. The acquisition of foreign genes through lateral gene transfer is a possible explanation of the trypanosome-specific evolution of novel processes and organization [52]. A systematical search for candidate bacterial horizontal gene transfer (HGT) events (Material and Methods) allowed us to identify 87 HGT candidates in these Phytomonas isolates, all shared between the two isolates, with eight of them specific to Phytomonas (i.e. absent from Leishmania and Trypanosoma) (Table S4). Several genes of bacterial HGT origin already identified in Leishmania were also found in Phytomonas, specifically sugar kinases and other genes involved in carbohydrate metabolism, which probably reflects their life cycle in plants and phytophagous insects [52], [53]. All HGT events were common to EM1 and HART1, but a metallocarboxypeptidase of potential bacterial origin was found in only one copy in EM1 and 11 copies in HART1.
In other trypanosomatids, the tRNA genes tend to occur in clusters with a synteny often conserved among different genera (Figure S7; details in Text S1). Most of the tRNA genes predicted for EM1 and HART1 corresponded to those identified previously in T. brucei, L. major and T. cruzi (Table S5). Interestingly, Phytomonas isolates possess two tRNAs not found among the animal pathogens, and present in the plant trypanosome branch: they are Asn (ATT)-tRNA (in HART1) and Ser (GGA)-tRNA (in EM1) (Table S5, highlighted in green).
Kinetoplast DNA genome and transcriptome in Phytomonas EM1 and HART1
In all Trypanosomatidae the mitochondrial genome consists of a single network of kinetoplast (k) DNA, one of the most complex organellar genomes known. It is composed of dozens of maxicircles that carry protein-coding and mitoribosomal genes, and thousands of minicircles that encode guide (g) RNAs. The EM1 maxicircle could not be assembled, but a single maxicircle contig of 12,099 bp was recovered for HART1. A homologous 10,478-bp region was sequenced previously for Phytomonas serpens [54], and the identity over the matching region of 9,816 bp between the two Phytomonas isolates is 76.8%.
Similar to the P. serpens maxicircle, the maxicircle of HART1 is characterized by a complete absence of cytochrome c oxidase subunits I–III (COI, COII, COIII), and cytochrome b (Cyb) of the bc1 complex. Other maxicircle-encoded genes typical for trypanosomatids, 12S and 9S rRNAs, ND1 to ND5, ND7 to ND9, subunit 6 of ATP synthase (A6), ribosomal protein subunit 12 (RPS12), maxicircle unknown reading frames (MURF) 2 and 5, and unidentified cryptogenes G3 and G4, are present (Figure S8). Since PCR and limited sequencing data indicated that the same deletions are present in EM1 and in three P. serpens strains [54], these deletions likely became established at the base of the Phytomonas clade.
Some maxicircle-encoded transcripts are known to undergo extensive RNA editing via the insertion and/or deletion of four to hundreds of uridylate residues [55]. Information for the editing process is provided by hundreds of heterogeneous minicircle-encoded gRNAs. The extent of editing is reflected by the sequence identities of individual maxicircle-encoded genes. Although we lack RNA sequence data for HART1, DNA sequence alignments with other kinetoplastids allow determination of the extent of editing for a given gene (Table S6). Genes that are pan-edited in almost all trypanosomatids studied [56] (ND3, ND8, ND9, RPS12, G3, and G4) show no reduction of the edited region in HART1 as compared to P. serpens (Figure S8).
When all maxicircle-encoded genes are considered, HART1 and P. serpens are more divergent from each other than L. donovani is from. L. tarentolae, but less so than T. cruzi is from T. brucei. Furthermore, the HART1 maxicircle genes have slightly lower identity to L. tarentolae, T. brucei and T. cruzi genes, than the genes from these species have among themselves (Table S6). These facts reflect the relatively long branch of the Phytomonas clade observed in the SSU rRNA- and glycosomal GAPDH-based phylogenies and deep separation between individual branches of this clade [57], [58].
Recovered full-length kDNA minicircles differ between both Phytomonas EM1 and HART1. In HART1 the minicircles range in length from 1,626 to 1,652 bp and contain one conserved region, as does P. serpens [59]. The EM1 minicircles are longer (2,791 to 2,819 bp), and carry two conserved sequences opposite each other. These variations are not unprecedented, as the size of minicircles as well as the number of conserved regions are typically uniform within a species, but variable among species [60], [61].
Transposable elements in the Phytomonas EM1 and HART1 genomes
Extensive bioinformatics analyses have been performed for all known transposable elements (TEs) present in the trypanosomatid genomes. While both LTR-retrotransposons (also called retrotransposons) and non-LTR retrotransposons (also called retroposons) were described in the genome of T. brucei, T. congolense, T. vivax, T. cruzi, and Leishmania spp. (∼3% of nuclear genome), no transposons have been identified to date [31], [32], [62]–[67].
Significantly, there is evidence for involvement of non-autonomous TEs in the regulation of gene expression [65]. Leishmania spp. (∼2,000 copies per haploid genome), but not trypanosomes, have domesticated and expanded these small TEs, named SIDER (Short Interspersed DEgenerated Retroposon) and co-opted them as part of the gene expression machinery. All trypanosome species analysed so far contain at least one putative functional TE family of the ingi clade (Tbingi, Tvingi, Tcoingi, L1Tco, L1Tc) that may have the capability to be mobilized, but all members of the ingi clade are degenerate and non-functional in the Leishmania species sequenced to date. Two questions were considered important to address in the analysis of TEs in these Phytomonas isolates due to their relatively close phylogenetic position to Leishmania spp.: when, in the course of trypanosomatid evolution, did domestication and expansion of SIDER occur? and when was the loss of TE functionality from the ingi clade?
As observed for Leishmania spp., both Phytomonas genomes are missing potentially active ingi-like TEs, but contain a few non-functional TEs of the retroposon ingi clade. Two types of TEs belonging to the retroposon ingi clade (PhDIRE, for Phytomonas Degenerated Ingi-Related Element, and PhSIDER, Table S7) were identified, with no evidence of functional elements, since all are likely to be inactivated by the accumulation of deletions, point mutations and/or frame shifts. PhDIRE belongs to the ingi1 subclade, considered as an early diverging ingi subfamily also present in Leishmania spp., T. cruzi and T. congolense [66], as shown by phylogenetic reconstruction (Figure 2) and analysis of the conserved motif upstream of the retroposons. PhSIDERs are short elements that were probably derived from PhDIRE by deletion, as previously proposed for other potentially active ingi-like TEs [62], [65], [66], [68] (see Text S1 for details). No sequences related to other trypanosomatid TEs were detected in the Phytomonas genomes (details in Text S1).

The EM1 genome was found to contain 41 DIREs, similar to all other trypanosomes and Leishmania spp. (L. major: 52 and L. braziliensis: 65) (Table S7), however the seven SIDER copies was low in comparison to Leishmania spp. that carry around 2000 copies. Thus, the enormous expansion and domestication of SIDER in Leishmania spp. [65] is not observed in these Phytomonas isolates, and exaptation of SIDER was likely a Leishmania-specific event in the trypanosomatid lineage.
The HART1 genome is depleted of TEs. Forty-eight retroposons were identified in the EM1 genome, while two PhDIREs were found in the HART1 genome, a 24-fold difference (Table S7). Indeed, both the un-annotated contigs and the non-assembled reads showed very low coverage of PhDIRE/PhSIDER in HART1, confirming the low number of retroposons in this Phytomonas isolate.
High gene content and synteny conservation between EM1 and HART1
The majority of Phytomonas genes are shared between both isolates, as shown by independent approaches used for ortholog detection (see Materials and Methods). The combination of both Best Reciprocal Hits (BRH) and orthoMCL strategies identified 5,210 (82%) genes from EM1 with orthologs in HART1, and 5,108 (79%) genes from HART1 with counterparts in EM1, similar in gene size (Figure S9 A). The Phytomonas EM1 and HART1 orthologs were more closely related to each other than to their trypanosome orthologs with an average percentage of identity of 70.5% (Figure 3). The small nucleolar RNA (snoRNA) repertoires of HART1 and EM1 also showed higher similarity to each other than to T. brucei or L. major (Table S8).

The genes for which no orthologs could be detected by this preliminary approach are excellent candidates for understanding Phytomonas spp. behaviors. After eliminating genes for which orthologs were not detected because of annotation or assembly issues, as well as suspected annotation artifacts, 13 genes remained in EM1 and 4 in HART1 that could be confidently considered as lacking an ortholog in the other isolate (Figure S10 and Table S9, see Materials and Methods for details). The vast majority of Phytomonas genes are shared between both isolates, highlighting the high level of conservation of the gene repertoire between these two trypanosomatids.
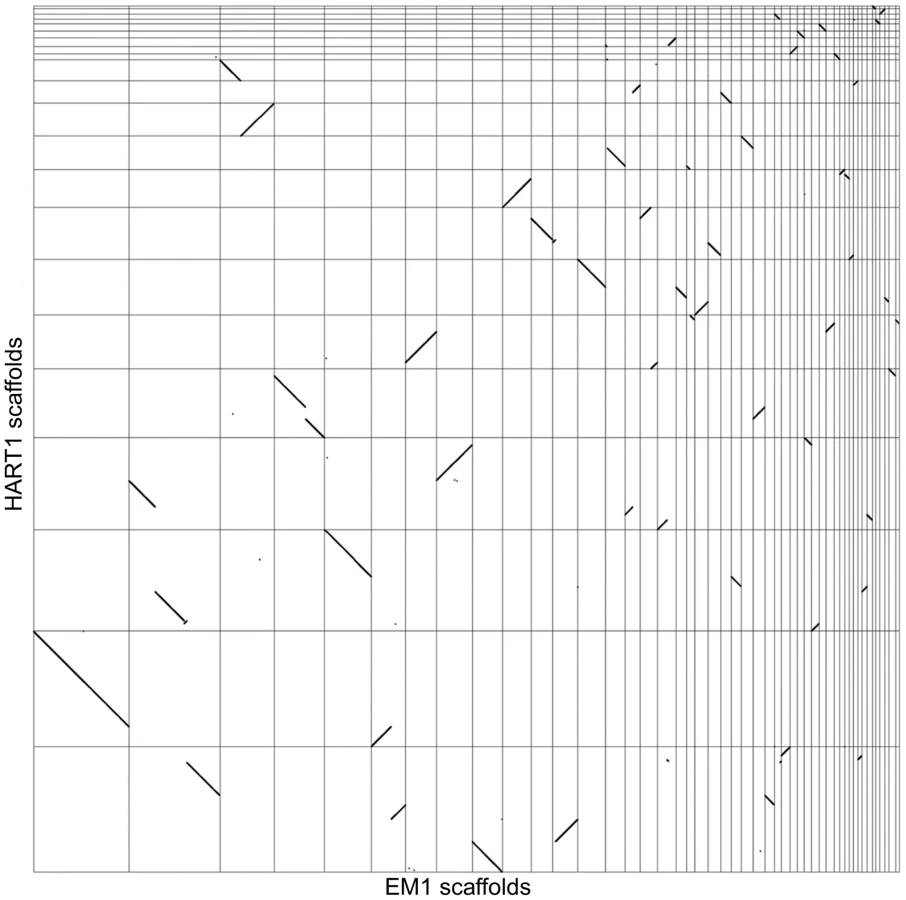
We analyzed synteny between EM1 and HART1 using dot-plots (Figure 4). Synteny was conserved between EM1 and HART1, with most of the synteny breaks corresponding to scaffold boundaries in one of the two isolates. Only five bona fide synteny breaks with HART1 were found in the EM1 assembly, and 10 in the HART1 assembly. The syntenic blocks are large (average of 60 genes, median of 35 genes) and usually include several hypothetical PTUs (average 20 ORFs, median of 10) (Figure S11). There is good conservation between PTUs in EM1 and HART1, with at least one boundary in common between EM1 and HART1 for all PTUs (Figure S12A and Figure S13). Significantly, synteny breaks tend to correspond to the boundaries between putative PTUs (Figure S12B), and intergenic distances are well conserved (Figure S9B). To identify putative insertions in one isolate compared to the other, we searched for gene number differences between successive pairs of BRH in syntenic PTUs (Materials and Methods). After filtering possible annotation artifacts (genes missed, splits/fusions, etc) and genes with strong sequence similarity elsewhere in the genome (Table S10), we retained ten genes in EM1 absent at the syntenic position in HART1, including three already identified as lacking a HART1 ortholog. Furthermore, three genes in HART1 lack a syntenic equivalent in EM1, with two already identified as having no ortholog in EM1 (Table S9). The two strategies did not identify the same sets of genes because of slight differences in the very conservative quality controls applied (see Material and Methods). Significantly, ten and three genes in EM1 and HART1 respectively, displayed weak hits in the syntenic region, suggesting that they have diverged in the other isolate; ten and two genes had no evidence for sequence homology, and could thus correspond to insertions or complete deletions. Combining the two approaches, 20 genes from EM1 were confidently determined to be absent from HART1 and 5 genes from HART1 were found to be absent from EM1 (Table S9). Since we could only compare assembled and annotated genes with confidence, these numbers may be underestimates of the true number of non-conserved genes between both isolates, but they are representative of the overall level of synteny and gene repertoire conservation between these two phylogenetically remotely related Phytomonas isolates (Table S9).
Comparison of Phytomonas with other trypanosomatids
OrthoMCL comparisons [69] were performed between Phytomonas EM1 and HART1, and four other trypanosomatids: L. major [29], T. brucei [28], T. cruzi [30] and Trypanosoma vivax [70] (Materials and Methods). This predicted 22,706 clusters of orthologous genes. Their conservation profiles (i.e. the list of species in which they are found) are shown in Table 1. A core of 2,869 genes was conserved between all six species (Table 1). Indeed, expert examination of this group of genes showed that 80.6% of the identified protein kinases shared by both Phytomonas isolates are also present in T. brucei and L. major. This subgroup contained major regulators, including up to 11 cdc2-related kinases (CRKs), WEE1, aurora kinase AUK1, glycogen synthase kinase 3 (GSK3) and casein kinases CK1 and CK2, expected to be present in all eukaryotes (Table S3C). Putative amino acid transporters conserved in all four mammalian parasites were also identified in these Phytomonas isolates. Interestingly, both isolates contained the same repertoire of amino acid transporters (AAPs), but with differing copy numbers (Table S3E; details in Text S1). Several genes with similarity to calmodulin and genes annotated as calmodulin-like in T. cruzi [71] were also present in both Phytomonas genomes.
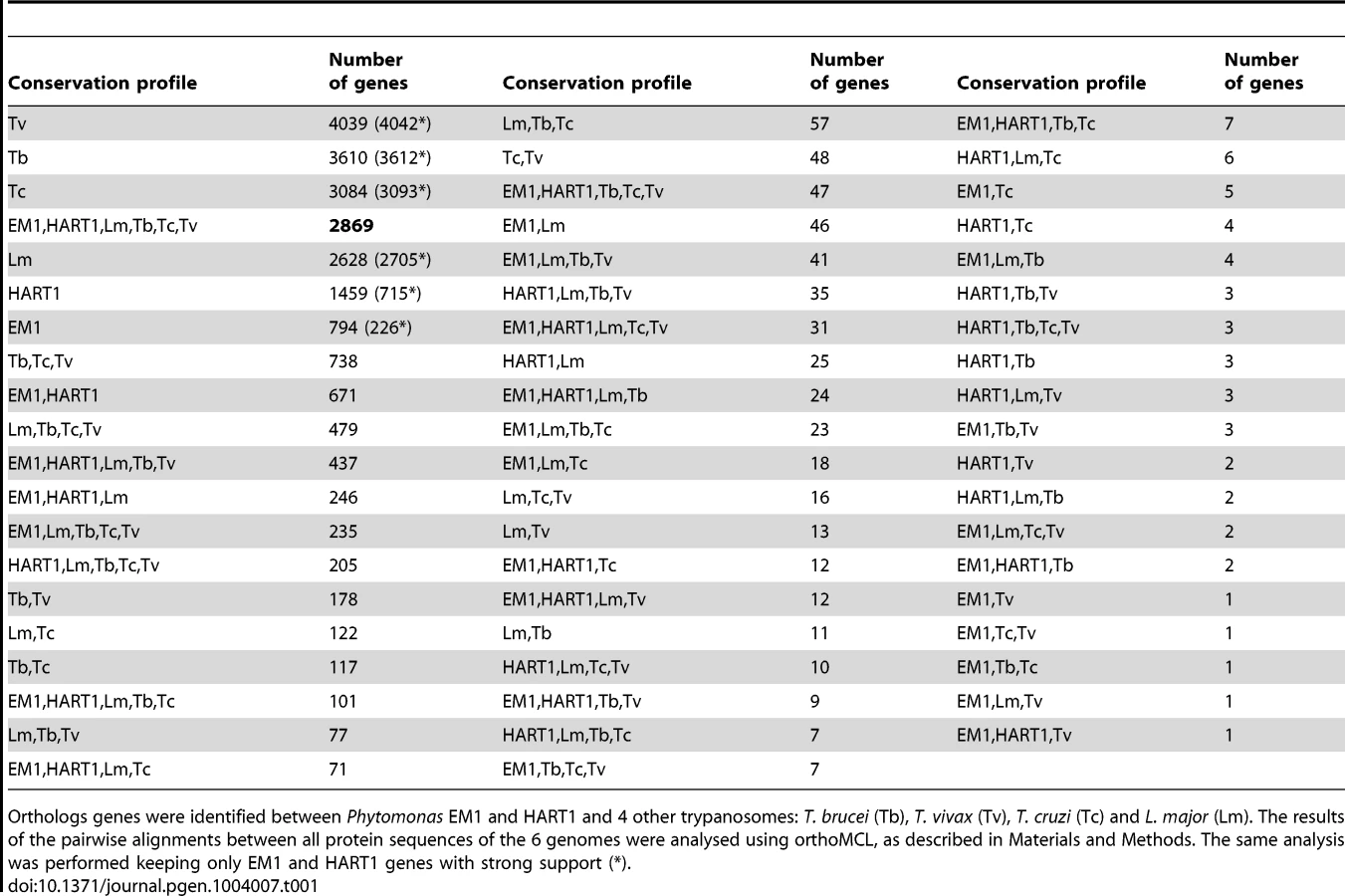
Manual inspection of Phytomonas gene families highlighted many examples of gene conservation within these plant parasites. Four conserved Phytomonas EM1 and HART1 kinases were absent in both T. brucei and L. major: These Phytomonas-specific kinases were one calcium/calmodulin regulated kinase-like, one UNC-51-like kinase, and two unique kinases that do not fall into any defined kinase group (Table S3C; details in Text S1), suggesting that these enzymes could be important for infection of, or survival in, plants. Conservation of the phosphatase complements was also observed in these two isolates; only slight differences were detected between both tyrosine and serine/threonine-specific complements (Table S11; details in Text S1).
The Phytomonas isolates have more genes in common with Leishmania than with the three Trypanosoma spp.: 317 orthoMCL clusters are shared between at least one Phytomonas isolate and L. major but none of the Trypanosoma spp., and only 111 clusters are common to at least one Phytomonas isolate and one Trypanosoma spp. but not L. major. The number of BRH, as well as their percentage of identity, was also significantly higher between Phytomonas and Leishmania than between Phytomonas and trypanosomes (Figure 3). However, the presence of two types of clusters conserved only in Trypanosoma or Leishmania suggests independent secondary losses from an ancestral organism with a substantially larger gene complement.
Significant synteny was observed between Phytomonas and Leishmania (Figure S14), as well as between Phytomonas and trypanosomes (Figure S15, Figure S16 and Figure S17). As expected from the closer phylogenetic relationship of Phytomonas with Leishmania (Figure 1B) [37], [38], [42], more syntenic breaks were observed between the Phytomonas isolates and trypanosomes than Leishmania (Figure S12B). Syntenic blocks usually include several PTUs (Figure S13). We compared the number of synteny breaks that occur at PTU boundaries with what would be expected by chance (Materials and Methods): for all pairs of species, the synteny breaks tended to coincide with PTU boundaries (Figure S12B). The high synteny conservation between trypanosomatids might thus be the result of a selective pressure against intra-PTU rearrangements.
Proteins involved in kDNA replication, kRNA editing, modification and translation
The topological complexity of the kDNA network has fascinated replication specialists for decades. The process is not fully understood, but many of the players have been identified. In the model flagellate T. brucei, the machinery is extremely complex, requiring the combined activity of several mitochondrial DNA polymerases, ligases, endonucleases, helicases and topoisomerases [72]. Using a database of 26 genes encoding the kDNA replication machinery of T. brucei, all orthologs have been identified in the Phytomonas EM1 and HART1 isolates.
The transcripts of many maxicircle genes undergo RNA editing in order to be translatable on mitochondrial ribosomes. Editing and processing of these mRNAs require the participation of several dozen proteins. A list of 28 T. brucei orthologs that are confirmed components of the RNA editing core complex or predicted to interact transiently with the complex [73] revealed that both EM1 and HART1 have the same composition, with substantial similarity to T. brucei. With the exception of KREP4, KREP5 and the oligoU-binding protein that have likely been lost or divergent as in L. major, all of the remaining orthologs are present. In both Phytomonas isolates, KREPB7 is duplicated. The available data is compatible with the existence of another complex involved in RNA editing, mitochondrial RNA binding complex 1 (MRB1) being composed of transiently interacting sub-complexes, with up to 32 components [74]. While only recently identified, MRB1 and associated proteins are conserved, as EM1 and HART1 contain all of its known orthologs.
Trypanosomatid flagellates are well known for their uniquely complex kDNA and kRNA. All in all, the gene order, editing patterns, as well as proteins that participate in the metabolism of these organellar nucleic acids, mostly identified in model species T. brucei, L. tarentolae and/or C. fasciculata, are conserved in these Phytomonas isolates.
Phosphorylation, calcium uptake and transporters in Phytomonas spp.: Examples of genome contraction in both EM1 and HART1 isolates
Analysis of the Phytomonas genome sequences provided a global view of the metabolic potential of plant trypanosomatids. Comparison of the gene repertoires from both isolates to other sequenced trypanosomatids revealed a simplified genome, coding for a minimal system with a clear lack of complexity for each isolate. Indeed, both EM1 and HART1 genomes presented diminutive gene sets when compared to T. cruzi, T. brucei and L. major (Table 2, for more details see Table S3), retaining only the most essential functions for the parasite, and often including a considerable fraction of genes that could serve the hosts. Furthermore, both gene repertoires are reduced as a result of both the loss of entire gene families and the reduction of the numbers of paralogs within gene families.

The protein kinase contents of the Phytomonas isolates provide a good example of genome contraction in these plant parasites: eukaryotic protein kinase (ePKs) genes were identified in both isolates (160 and 161 in EM1 and HART1, respectively), but in smaller numbers than in the TriTryp kinomes (Table 2) [31], [75]. Twenty four protein kinases, conserved in T. brucei and L. major, were not present in either of the Phytomonas draft kinomes. (Table S3C). Furthermore, nine T. brucei-only kinases and 24 L. major-only kinases were also absent from both Phytomonas draft kinomes. Even though it is possible that fewer ePKs are required for infection of plants compared to mammals, the similar number of ePKs in the pathogenic isolate HART1 was somewhat unexpected, as it could be considered that additional protein kinases might be required to coordinate virulence factor expression.
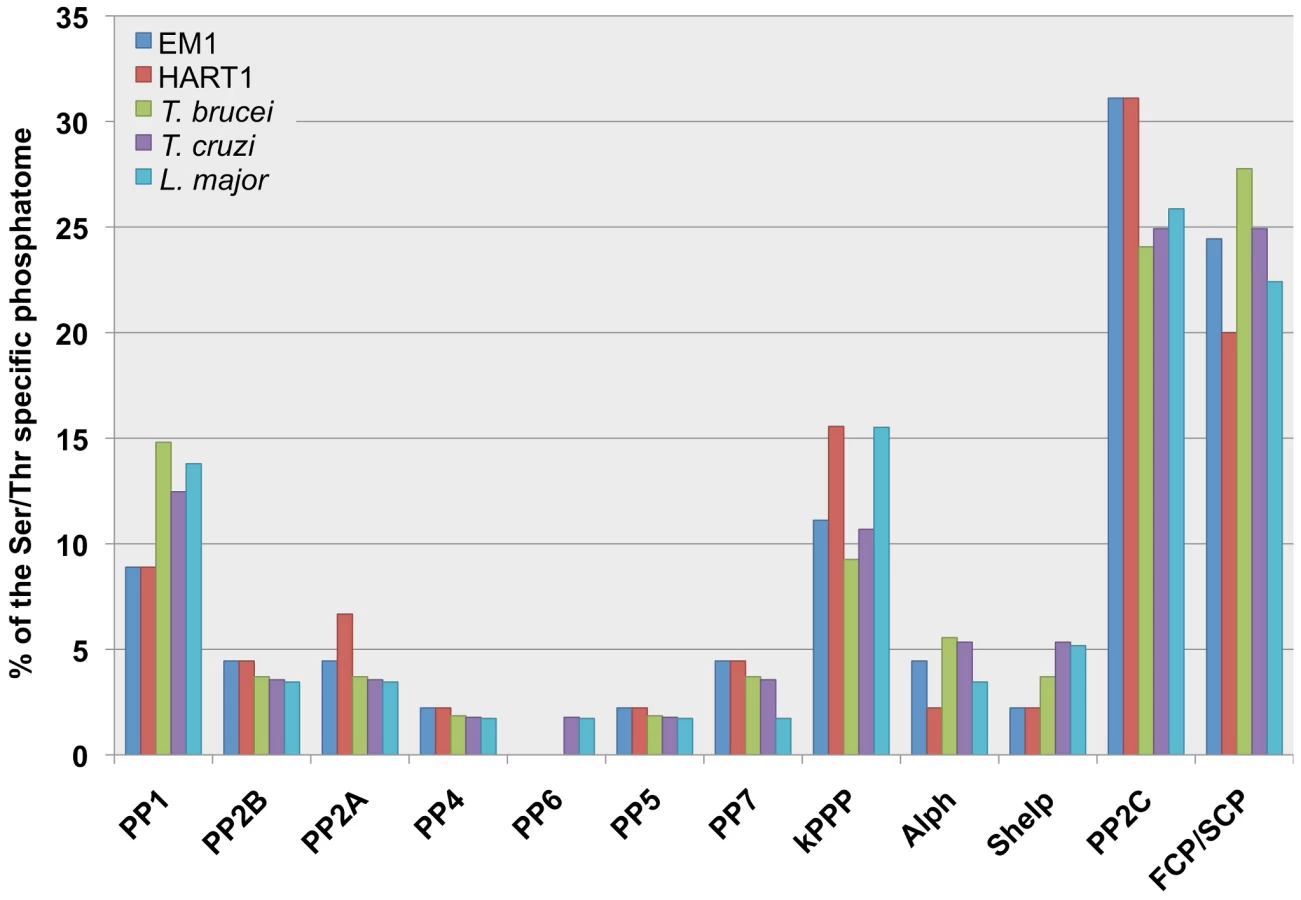
The less investigated partners of the phosphorylation-dephosphorylation regulatory cascades are the protein phosphatases, organized into four major groups, depending on substrate preferences and catalytic signature motifs. Three of these groups corresponds to Ser/Thr specific phosphatases (STP): metallo-dependent protein phosphatases (PPM), phosphoprotein phosphatases (PPP) and aspartate based phosphatases with a DxDxT/V motif. The fourth group corresponds to the protein tyrosine phosphatases (PTP) [76]. The completion of the genome sequences of L. major, T. brucei and T. cruzi [31] has permitted a deeper analysis of the protein phosphatases, showing that the main protein phosphatase groups (Tyr, Ser/Thr and dual specific protein phosphatases) are present in these parasite genomes, as in higher eukaryotes [77].
The Phytomonas phosphatome provides another illustration of the genome reduction observed in these parasites. Comparing the two plant trypanosomes' phosphatomes to the TriTryp phosphatome [78], the main differences were found in the PTP complements: the eukaryotic-like PTPs were absent from both EM1 and HART1 phosphatomes, and no orthologs of PTENs and CDC14s [76] have been identified (Table S11A). PTENs and CDC14s (dual specific phosphatase group) are present in the phosphatomes of all three other kinetoplastids, where they can be grouped into two distinct families, the eukaryotic-like and kinetoplastid-like PTENs, depending on their sequence homology to other eukaryotic PTENs. One kinetoplastid-like PTEN enzyme has been found in the three kinetoplastids T. cruzi, T. brucei and Leishmania [79]. While four eukaryotic-like PTENs have been identified in T. cruzi, only one enzyme was found in L. major. Interestingly, no T. brucei ortholog was identified, thus suggesting a possible role of these enzymes in intracellular parasitism.
When we compared the STP complements of the Phytomonas isolates, we detected a 20% decrease in the total number of phosphatases as compared to the TriTryps, mainly due to the reduced number of type 1 protein phosphatases. The number of PP1s has been augmented in the genomes of T. brucei, T. cruzi and L. major by a gene duplication process (8/7/8) [78]. Still, the functions associated to these apparently higher number of resembling genes have not been characterized. Both in EM1 and HART1, four genes encoding PP1 catalytic subunits were identified, a similar number to those described in other eukaryote PP1 complements. We have also found a two-fold reduction in the number of the bacterial-like phosphatases, Alphs and Shelps [80] in the plant trypanosomatids compared to the TriTryp phosphatomes (Figure 5, Table S11B).
The reduction in the number of members of ABC transporters (Table S12) and amino acid transporter families in these Phytomonas isolates represents another relevant example of genome retrenchment. A unique family of amino acid transporter (AAP) genes from members of the trypanosomatid family (25 in Leishmania, 17 in T. brucei and 19 in T. cruzi) has been identified, based on the existence of amino acid permease pfam domains [81], [82]. This trypanosomatid-specific group of amino acid transporters corresponds to a distinct clade within the amino acid/auxin permease (AAAP) super family [83], [84]. The analysis of these gene families revealed 15 and 16 AAP genes in EM1 and HART1 respectively, fewer than in the mammalian trypanosomatid genomes (Table 2 and Figure S18, details in Table S3E and Text S1).
Eukaryotic cells regulate their cytosolic calcium concentration using numerous channels and transporters located in the mitochondria, the plasma membrane and the endoplasmic reticulum. Additionally, calcium binds to an extensive collection of signaling and regulatory proteins in these eukaryotic cells. In trypanosomatids, acidic organelles known as acidocalcisomes, which have been identified in Phytomonas françai [85], act as the major stock of the intracellular calcium, and are implicated in processes such as calcium homeostasis, osmoregulation and polyphosphate metabolism [71]. Hence, both Phytomonas EM1/HART1 genomes were investigated for the presence of orthologs to trypanosomatid genes known to be involved in calcium and polyphosphate metabolism.
The trypanosomatid genome projects revealed a vast diversity of Ca2+-binding proteins (as an example for T. cruzi see Table S3A), many of which are not characterized and have little or no homology with non-kinetoplastid proteins. Regulation of cytosolic Ca2+ concentration in Phytomonas isolates EM1 and HART1 appears similar to that of other trypanosomatids. Yet, several differences allow to clearly distinguish these organisms (Table S3A). Though the inositol phosphate/diacylglycerol pathway is present in pathogenic trypanosomatids, no evidence of either a phospholipase C, or a protein kinase C was found in these Phytomonas isolates. However, there are orthologs to the putative InsP3 receptor in both Phytomonas EM1 and HART1 isolates. Another interesting difference is the lack of Phytomonas counterparts to calreticulin, a Ca2+ storage protein located in the endoplasmic reticulum of T. cruzi [86], and the recently characterized polyphosphate kinase (vacuolar transporter chaperone 4) of yeast, pathogenic trypanosomatids, and Apicomplexan.
The membrane trafficking system and the predicted cell surface proteome
To predict both the level of intracellular organellar complexity and the surface composition of Phytomonas, the open reading frame complement of HART1 and EM1 were scanned for around 300 genes involved in membrane trafficking. Both isolates of Phytomonas share essentially identical membrane transport systems, with only one clear example of specialization (Table S3D and Figure S19). Overall, the endomembrane systems are the simplest yet described amongst trypanosomatids; for example the Rab GTPase repertoire, a primary determinant of specificity and organelle identity [87], retains the basic core exocytic and endocytic functions and the trypanosome-specific Rab-like X1 and X2 [88] (Figure 6). However, the system is substantially simpler, with only 12 Rab/Rab-like proteins compared to 16 in T. brucei or 17 in L. major [28], [89]. Given that the losses here are Rab21, 28 and 32, this reduction represents sculpting of the system by secondary loss from the common ancestor and hence is an adaptive streamlining [90]. This simplification is also seen in the secondary loss of the AP4 adaptin sorting complex from both Phytomonas genomes (Table S3D), and in a rather simpler ARF GTPase family compared with other trypanosomatids. Further, these data likely suggest a simplified late endocytic system, to which Rab21, Rab28 and AP4 are all assigned. Overall the view is of a minimal endomembrane system, which conserves the major complexes and pathways, indicating retention of all major organelles, but with an apparent lack of complexity or innovation; adaptation has been via minimization rather than invention.
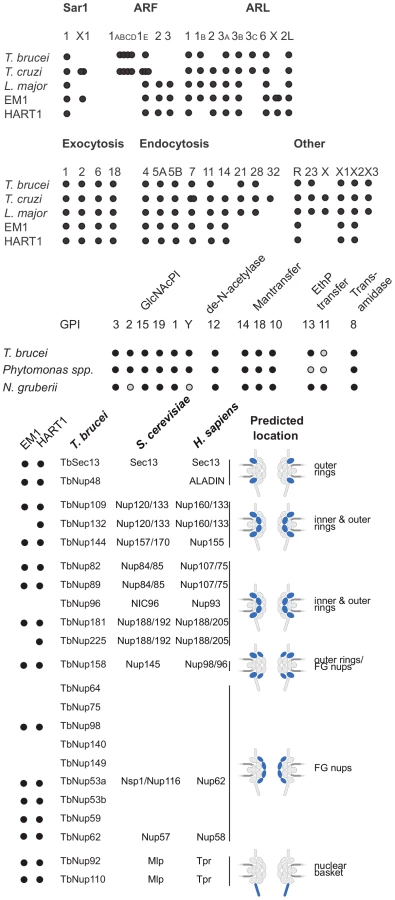
As befits the position of Phytomonas as basal within the trypanosomatid lineage, the surface appears to be rather similar to Leishmania spp., and there is no evidence for mucin-like or variant surface glycoprotein-related protein coding genes, or a dominant, highly expressed, surface antigen as no predicted GPI-anchored protein was encoded by transcripts in the most abundant RNAseq percentiles. The surface system includes full glycosylphosphatidylinositol (GPI) anchor and glycolipid biosynthetic pathways, the enzymatic apparatus for synthesis of a lipophosphoglycan (LPG)-like molecule and evidence for the GPI-anchored gp63 protein (Table S3D, Figure S19).
Metabolism in Phytomonas EM1 and HART1
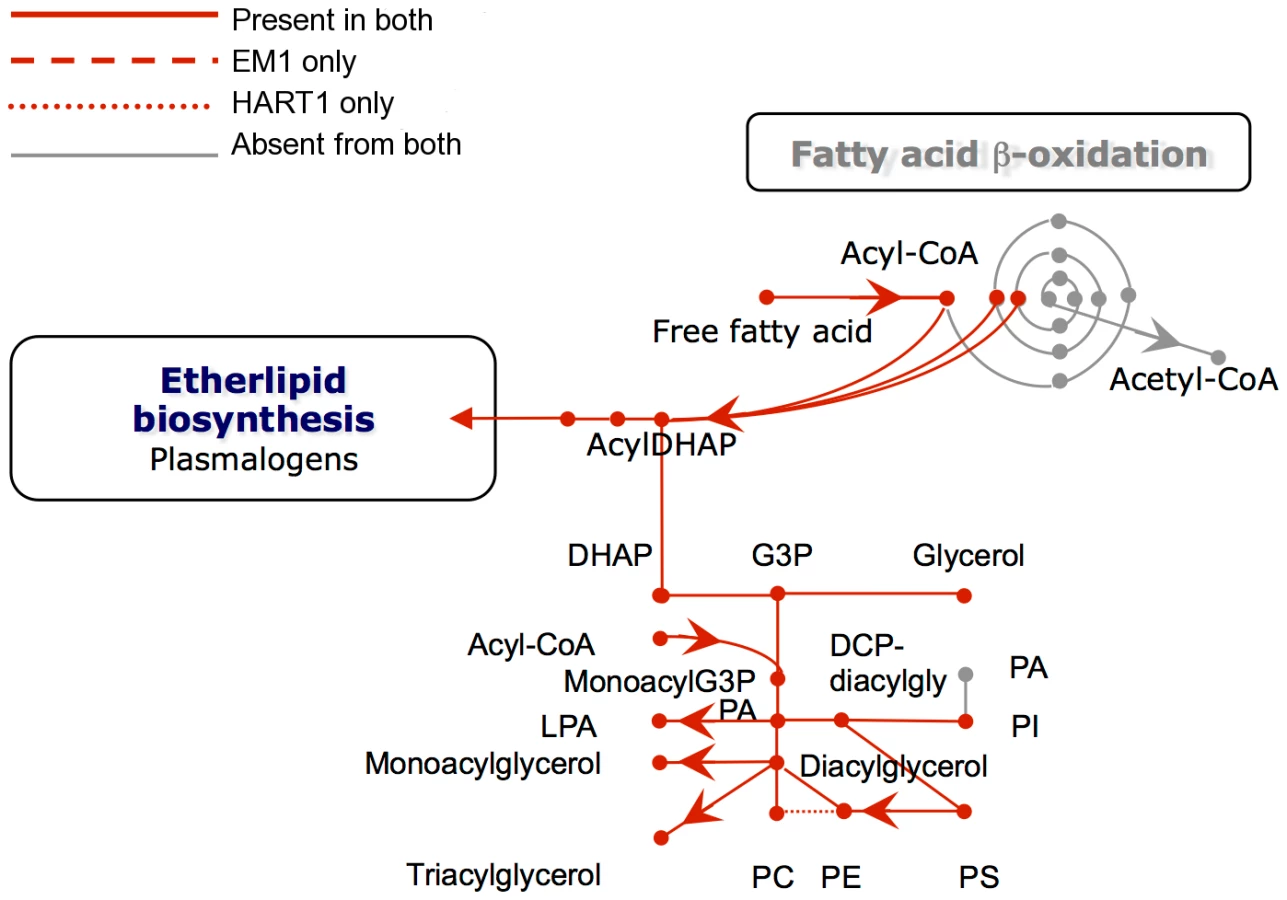
Analysis of the genomes of these two plant trypanosomes provided a global view of the metabolic capacity of Phytomonas. As a consequence of an almost complete absence of tandemly-linked duplicated genes, most of the metabolic genes in Phytomonas were identified as one haploid copy (Figure 7, Figure 8, Figure S20 and Figure S21; for details see Table S3B).
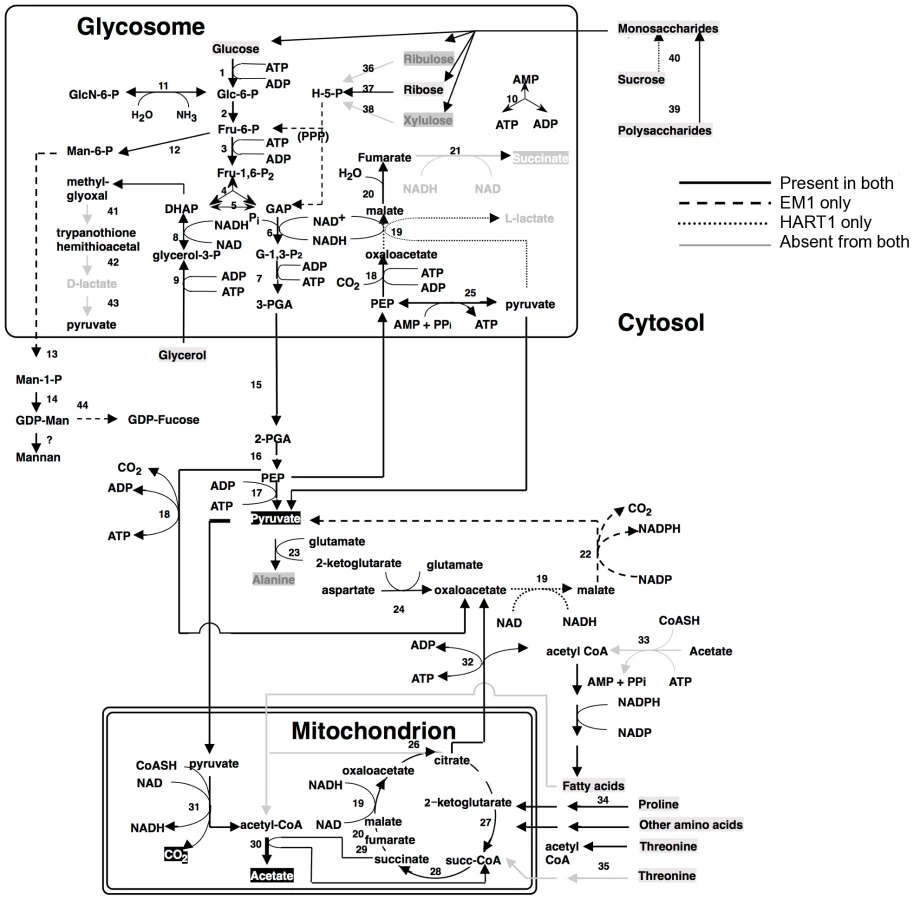
As part of its carbohydrate metabolism (Figure 7; details in Table S3B), Phytomonas not only utilize the plant's sucrose but also its polysaccharide stores as major energy substrates, as confirmed by the identification of genes coding for glucoamylase, alpha-glucosidase and, only in the HART1 isolate, many copies of invertase (beta-fructofuranosidase) homologs (Table S3B).
The presence of an alpha, alpha-trehalose phosphorylase in both isolates suggested that Phytomonas is also capable of using the abundant plant disaccharide trehalose for its carbohydrate needs. The presence of this bacterial-type enzyme illustrates that the adaptation of the plant parasite to their sojourn in their specific hosts may have been facilitated by HGT events. In agreement with previous studies on the carbohydrate metabolism of Phytomonas [91], [92], genome analysis revealed the presence of a complete set of glycolytic enzymes, the majority of which seem to be sequestered inside glycosomes, similar to other trypanosomatids. The existence of glycosomes in Phytomonas, previously demonstrated, was now confirmed by the presence of peroxisomal targeting signals at either the C- or N-termini of the encoded glycolytic enzymes as well as by the identification of a number of genes for peroxisome biogenesis proteins or so-called peroxins.
Besides the horizontal alpha,alpha-trehalose phosphorylase transfer event described here, other HGT events were previously described for other Phytomonas isolates. A zinc-containing alcohol dehydrogenase from a trypanosomatid isolated from the lactiferous plant Euphorbia characias, previously identified as an isopropanol dehydrogenase of bacterial origin, was also acquired by an event of lateral gene transfer from a strictly aerobic bacterium to an ancestral trypanosomatid [93]. The addition of this gene could explain a selective advantage for a plant colonizing-flagellate living in the phloemic or lactiferous tubes of infected plants, supported by the fact that this enzyme was only identified in all plant trypanosomes analyzed thus far, while absent from the rest of the trypanosomatid family. This zinc-containing alcohol dehydrogenase, together with a glycosomal malate dehydrogenase (Table S3), allowed us to assume that EM1 and HART1 would be able to produce small amounts of lactate, as observed for other Phytomonas isolates [94].
Almost nothing is known about the amino acid metabolism in Phytomonas. Amino acid metabolism of Phytomonas resembles that of the other trypanosomatids. The so-called non-essential amino acids can either be degraded and utilized as energy sources, or be formed from other metabolites. However, Phytomonas lacks the capacity to oxidize aromatic amino acids and is predicted to require an external supply of most of the essential amino acids. The absence of a fatty acid beta-oxidation pathway and of ETF predicts that Phytomonas is unable to oxidize both long chain and side chain amino acids (Results in Figure S21, for details see Table S3).
An arginine kinase was detected as a single copy gene in both isolates. This enzyme may have been acquired by horizontal gene transfer from the arthropod vector during evolution, as previously shown for Phytomonas Jma [95]. The genomes revealed that overall the interconversion and breakdown of amino acids is very similar to what has been described for the other trypanosomatids. However, while amino acids serve as the most important source of energy for the other trypanosomatids inside their insect vector, this cannot be the case in Phytomonas because of its limited mitochondrial capabilities [91]. Owing to the fact that their insect vector(s) feed exclusively on plant juices that are rich in carbohydrates, the switch from plant to insect host would probably not require a metabolic switch from carbohydrate to amino acid metabolism as occurs in the mammalian trypanosomes. The absence of such a switch may have allowed the irreversible loss of a number of mitochondrial functions such as a respiratory chain required for beta oxidation of fatty acids and the complete oxidation of amino acids. Indeed, no genes coding for any of the mitochondrial cytochromes could be found.
The enzymes of the hexose monophosphate pathway, as well as the ones involved in gluconeogenesis are present in Phytomonas, even though no evidence for the synthesis of glycogen has been detected. Few genes were found for the formation of storage polysaccharides. However, several mannosyl transferases, possibly involved in the synthesis of mannan polysaccharides, were detected, suggesting that mannans rather than glycogen may serve as a polysaccharide store.
Protein glycosylation differs in the two Phytomonas isolates (Figure S20, Table S3B). The genes required for the incorporation of glucose, mannose, galactose, N-acetylglucosamine, glucuronic acid, xylose and fucose into glycoproteins, but not for sialic acid, were identified in the genome of the EM1 isolate. The HART1 isolate seems to lack the genes necessary for the incorporation of N-acetylglucosamine and fucose.
With respect to lipid metabolism, fatty acyl dehydrogenase or, oxidase, multifunctional enzyme and thiolase were absent in both parasite isolates, indicating that Phytomonas is not capable of oxidizing any fatty acids via the beta oxidation pathway. On the other hand, Phytomonas should be capable of fatty acid biosynthesis, since the genes coding for the responsible enzymes have been identified in both parasite genomes (Type II fatty acid synthesis in mitochondrion, and Type I fatty acid synthesis absent but synthesis taking place by a set of elongases) (Figure 8).
Oxidant stress protection in trypanosomatids is based on trypanothione, an adduct of one spermidine and two molecules of glutathione [96]. Thus the Phytomonas proteome was searched for the presence of enzymes involved in this metabolism. Phytomonas has a trypanothione reductase as well as a homolog of glutathionylspermidine synthase, or trypanothione synthase, as well as the enzymes thioredoxin (tryparedoxin), several thioredoxin (tryparedoxin) peroxidases, peroxiredoxin, and trypanothione peroxidase. Several mitochondrial and cytosolic superoxide dismutases and an iron/ascorbate oxidoreductase, but no catalase, were identified. The reducing equivalents in the form of NADPH are provided by the enzymes NADP-dependent isocitrate dehydrogenase in the mitochondrion and by the hexose-monophosphate pathway enzymes glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase. A plant-like ascorbate peroxidase, as described for T. cruzi and Leishmania, was not detected (Table S3B).
Phytomonas lacks the capacity for RNAi, since the argonaut AGO1 (Tb10.406.0020) and the two dicer proteins DCL1 (Tb927.8.2370) and DCL2 (Tb927.3.1230) present in both T. brucei and in L. brasiliensis but not in T. cruzi and L. major, two organisms that lack RNAi, were also absent in both EM1 and HART1 genomes (see Table S3B). In fact, the lack of these gene products agrees with the presence of a double stranded RNA virus reported in the phloem-restricted isolates [97] that could serve as an indication for the absence of defense mechanisms against invasion by foreign RNA. Similar viruses have been reported for Leishmania spp. as well [98], [99].
Analysis of the Phytomonas HART1 and EM1 secretome
Virtually no information is available about the existence of effectors of pathogenicity in Phytomonas spp. and their possible role in the interaction with the host. We investigated the secretome of Phytomonas EM1 and HART1 isolates for potential virulence factors, by selecting those sequences having a secretion signal peptide, no transmembrane domains and no glycophosphotidylinositol (GPI) anchors. We detected 282 putative secreted proteins in both HART1 and EM1 (Table S13). Among these proteins, only 43 proteins in HART1 and 44 in EM1 had a PFAM domain annotation. The secretome was classified into molecular function and biological process using the Gene Ontology annotation (Figure S22). However, we noted the presence of numerous false positives in the set of predicted secreted proteins. This is due to the high divergence between the trypanosomatid sequences and the one used by SignalP for learning, mostly from fungi, animals, plants or bacteria origin.
In the set of putative secreted proteins, we looked for proteins involved in plant carbohydrate degradation. One protein having a glycoside hydrolase family 31 domain was present in both HART1 and EM1 isolates, but the EST data did not show any expression of the two corresponding genes. We also found a secreted protein in HART1 (GSHART1T00001406001) coding for glycosyl hydrolase family 32 that corresponded to one of the beta-fructofuranosidases (see Metabolism of HART1 and EM1 section); other beta-fructofuranosidases harbored a signal peptide and GPI anchor. We did not identify any secreted proteins that were supported by expression data and likely to be involved in plant cell wall degradation. This finding is consistent with the fact that Phytomonas is directly injected in the host phloem by an insect vector, thus it does not need to degrade the plant cell wall to penetrate into the host and gain access to the phloem sap.
We screened for secreted proteins having a proteolytic activity that may lead to degradation of host proteins. Three genes were found in EM1 coding for an S24 serine peptidase, an M3A metallo-peptidase and an A1 aspartyl protease (AP); one AP was also found in the secretome of HART1. Cathepsin D-like A1 family AP genes have not been found in other known trypanosomatid genomes such as Leishmania and Trypanosoma. However, APs are known to be secreted and involved in the virulence of several pathogenic fungi. In the case of the fungal animal pathogen, Candida albicans, ten APs that contribute to the dissemination of the pathogen in mice are present [100]. Fourteen APs are also present in the genome of the ascomycete plant pathogen Botritys cinerea, including BCap8, which was found to constitute up to 23% of the total secreted proteins [101].
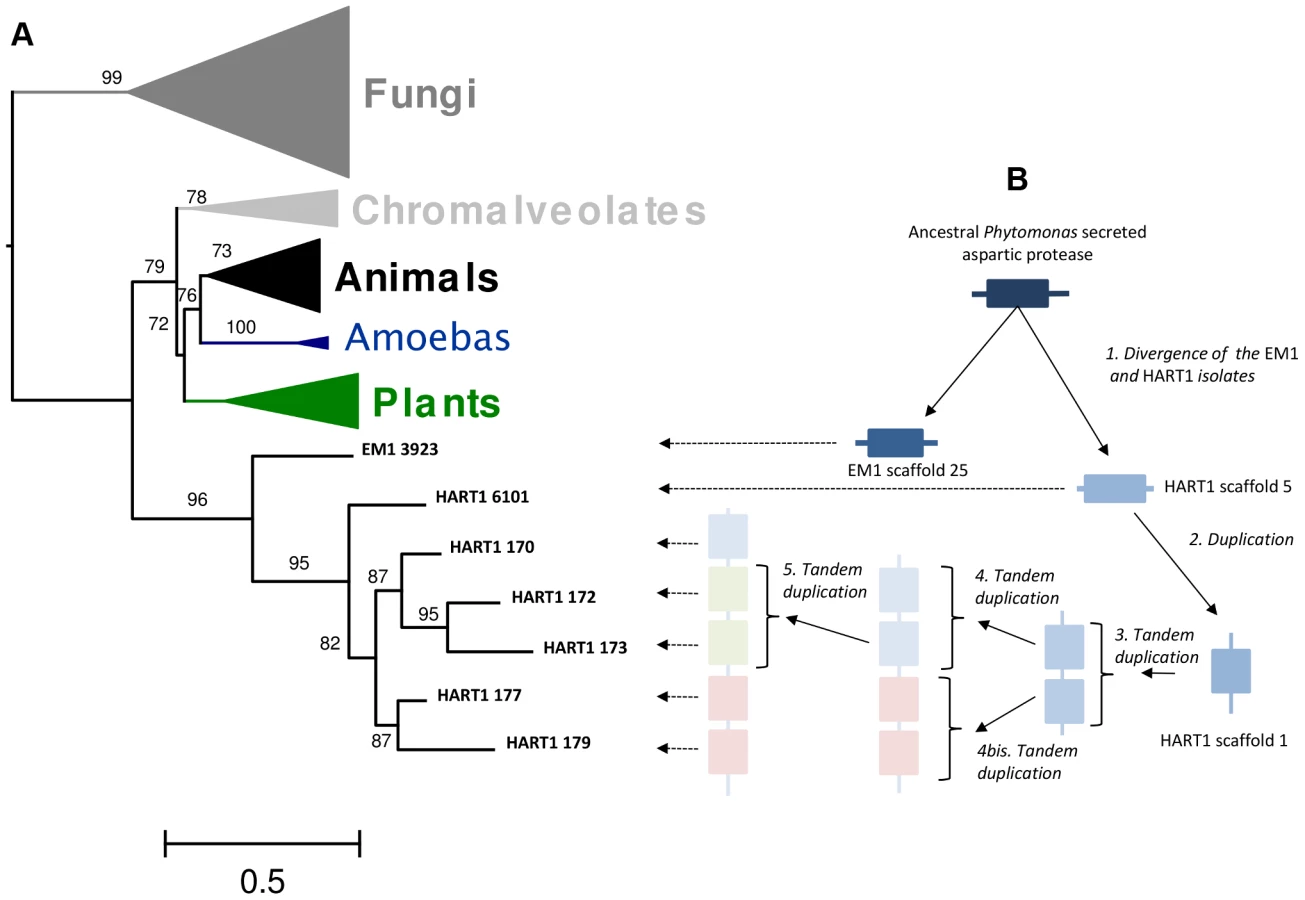
Since secreted Leishmania proteins with proteolytic activities may contribute to pathogenesis [102], [103], we looked for other AP coding genes in the HART1 and EM1 genome. EM1 did not have any extra APs, while HART1 harbors a cluster of five APs located in scaffold 1 (Table S3). These five tandem genes, absent in the syntenic region of EM1, were not detected by the “synteny” approach because of the stringency of filtering (see Material and Methods). The “true” first methionine of each protein of the cluster was located in intercontig gaps. When extending the N-terminal region of each of these proteins, a signal peptide could only be detected for the most extended gene (GSHART1T00000177001). For the four remaining APs, the N-terminal extension was not long enough to detect a probable signal peptide, and none of the five APs harbored a GPI anchor.
The phylogenetic analysis (Figure 9A) revealed that these Phytomonas APs evolved from a common gene that branched deeply in the tree with high aLRT support (aLRT support = 96). This result suggested the existence of an ancestral AP gene in the trypanosomatid lineages that may have been lost in Leishmania and Trypanosoma. The integration of the APs genomic positions on scaffold 1 and the topology of the HART1 clade allowed the reconstruction of the events that led to the creation of a pathogenicity gene cluster in HART1 (Figure 9B). HART1 and EM1 had initially one copy of the gene coding for a secreted AP. Then, the HART1 gene duplicated once from scaffold 5 to scaffold 1. The cluster of five genes was created in the scaffold 1 of HART1 by four successive tandem duplications. The presence of a signal peptide in the AP from EM1, the AP in scaffold 5 and one AP in the cluster of scaffold 1 let us speculate about the presence of a signal peptide in the other four APs, but their sequences were too short to detect it. The scaffold gaps between the five AP genes may correspond to repeated elements that may have mediated the AP tandem gene duplication. The EST data provided evidence for the expression of the five AP genes which comprise the AP cluster in the Phytomonas HART1 isolate (Figure S23), suggesting that, similarly to the function of the AP family in the fungi Candida and Bothrytis [101], [104], the Phytomonas HART1 AP gene cluster could be involved in virulence, an example of convergent evolution between distant organisms.
Pathogen versus non-symptomatic genomes: Examples and possible biological implications
The genus Phytomonas encompasses flagellates that differ substantially in their pathogenic potential. Despite most genes being shared between EM1 and HART1 isolates with respect to both gene count and content, several differences are still present (Table S3, Table S9). Among the members of the Phytomonas EM1 and HART1 kinase repertoires, only two specific genes were identified: a CMGC/DYRK EM1-specific kinase, absent from the HART1 genome, and an AGC/RSK only present in the pathogenic HART1 isolate. Both specific kinases have no orthologs in T. brucei or L. major. Since the function of these kinases has not been studied in any trypanosomatid, and little is known about protein kinase signaling pathways in the TriTryps, the biological implications are not clear at present. While two CDC25 phosphatases were also identified in EM1, no orthologs were found in HART1, as in the case of T. brucei. The CDC25/CDD25-like phosphatases were identified in Leishmania spp. and T. cruzi, suggesting distinct roles for the protein phosphatases only present in the two intracellular trypanosomatids.
Phytomonas is adapted specifically to infect and live in plants, where an abundant and diverse supply of carbohydrates is available for the parasite. Surprisingly, genomes of both EM1 and HART1 isolates contained only one sugar transporter, an ORF encoding a GT2 homolog (Table S3E). The presence of only a single sugar transporter is intriguing. It would suggest that both EM1GT2 and HART1GT2 have a broader substrate specificity than the mammalian trypanosome GT2, and would be in agreement with a much more simplified metabolic life cycle.
The pathogenic HART1 isolate seems to be specialized in metabolizing sucrose, as is suggested by the presence of many copies of an invertase (fructofuranosidase) homolog only detected in this strain. The main difference between the pathogenic and asymptomatic Phytomonas isolates resides in their specific location inside the host: EM1 multiplies in latex tubes, while HART1 colonizes the phloem sap inside the sieve tubes. It is not yet clear whether this difference in habitat is related to the presence, or not, of multiple invertase genes. The presence of an alpha, alpha-trehalose phosphorylase in Phytomonas may be the explanation of why this plant parasite can survive in the insect hemolymph by using trehalose, a disaccharide of glucose, as an energy substrate, rather than amino acids, as is the case in midgut-dwelling trypanosomatids of hematophagous insects. Trehalose, originally regarded as a sugar characteristic of certain lower plants, is also a major blood sugar of insects [105]. Since the phosphorylase is present in all trypanosomatids for which the genome has been sequenced so far, it is unlikely that the enzyme would be involved in the parasite's energy metabolism when dwelling in the plant host. It is more likely that it fulfills a major role in the passage of trypanosomatids through their insect vector rather than to their survival in the widely different types of mammalian and plant hosts, where only some plants have high concentrations of trehalose.
Both isolates seem to differ in the make-up of their surface glycoproteins (examples in the Text S1 and Table S3D). Most significantly, HART1 and EM1 have radically distinct gp63 repertoires, with only two genes detected in EM1 but over 20 in HART1. These are clearly derived from a single common precursor, with multiple expansions in HART1, and suggesting a more complex surface for HART1 than EM1 that potentially facilitated adaptation to a greater range of conditions, host species or tissue spaces.
Perhaps the pathogenic effects of HART1 are primarily due to their location in the sap, containing the products of photosynthesis and essential for plant growth. The death of palms, coffee trees and Alpinia may be the result of competition for essential metabolites that are more efficiently scavenged by Phytomonas. Biological inoculation experiments using the isolate EM1 in palms would address this hypothesis. The specific relationships among Phytomonas, its vector, and the host make this experiment hard to endeavour. The intraphloemic trypanosomatids associated with wilts form a very distinct group, especially for their cultivation [15]. Parasites could not be isolated without the help of feeder cells in the cultures, while the cultures of latex isolates like EM1 or fruit isolates were obtained in an axenic medium. Further comparative analysis of the two Phytomonas genomes may reveal the source of these differences.
Phytomonas genome: A minimized gene repertoire as a hint for survival in the plant host
The Phytomonas genomes consist essentially of single copy genes for the bulk of their metabolic enzymes, whereas Leishmania and Trypanosoma possess numerous duplicated genes or large gene families. While such gene duplications may have helped some trypanosomatids to adapt to multiple, widely different hosts, i.e. poikilothermic insects and warm-blooded mammals, their absence in the two Phytomonas genomes analyzed here suggests that plant trypanosomatids have been confronted much less with strikingly different metabolic environments and temperatures, and have hence lost or never needed these additional paralogs.
The unlimited availability of sugars in both plant and insect hosts is a situation that normally leads to suppression of mitochondrial activities, such as cyanide-sensitive respiration and oxidative phosphorylation. Eventually this may result in an irreversible loss of the genes coding for all of the above functions. The irreversible suppression observed in Phytomonas resembles the adaptation of some African trypanosomes to a permanent stay in the bloodstream of their mammalian hosts, without the possibility for cyclic transmission via insects. This also leads to the loss of mitochondrial genes and results in the appearance of dyskinetoplastic or akinetoplastic trypanosomes, unable to survive in the tsetse fly. Interestingly, Phytomonas spp. possess orthologs to the mitochondrial calcium uniporter recently described [106] suggesting that, as bloodstream forms of T. brucei, they also utilize the mitochondrial ATPase in reverse to maintain a membrane potential that drives Ca2+ uptake through the uniporter. Some Phytomonas genes likely gained via HGT may have permitted increased flexibility of genome expression, enabling the successful adaptation of Phytomonas spp.
Significant genome reduction has been identified in microbial lineages living in selective environments. Selection plays a key role during the initial phases of such adaptation removing “accessory” genes [107]. High gene density bear witness of genome contraction in several obligate intracellular parasites. In the case of microsporidia, genome-size variation resulted from varying frequencies of repeat elements without affecting gene density. Furthermore, Phytomonas shows important host dependency pictured by considerable gene losses. These adaptations combined with genome compaction led to gene size reduction and simplification of certain cellular processes [108].
Also phytoplasmas, specialized bacteria living as obligate parasites of plant phloem tissue and transmitting insects [109], have suffered extreme genome shrinkage, which resulted in a gene repertoire that is specific for survival in plant hosts [110]. In the case of phytoplasmas, this adaptation was made possible thanks to the presence of repeated DNAs, which allowed survival in different environments. Here also adaptation is particularly important, as their host environments, including phloem tissues of plants, and guts, salivary glands, and other organs and tissues of the insect host, are extremely variable [110].
Phytomonas spp. are highly specialized trypanosomes, with central differences in life history and infection strategy compared to eukaryotic plant pathogens like fungi and oomycetes. Leaf, fruit and stem are some of the surfaces colonized by plant pathogens. Wind-blown rain, fog and any plant visitor are some of the mechanisms by which phytopathogens like filamentous fungi and oomycetes are disseminated to the host plants. Still, these phytopathogens should penetrate by themselves in order to colonize and circulate inside the host. In this process, several biological mechanisms are triggered to colonize and propagate into the host, by the use of enzymes (cutinase, cellulase, pectinase), hormones, toxins and frequently by the interaction with metabolites produced by the plant in response to the invasion [111].
Contrary to these phytopathogens, plant trypanosomatids do not need to degrade cell walls to settle inside the plant since they are deposited into very specialized tissues or cells in the host thanks to insects that acts as their natural carriers. Yet, the discovery of a Phytomonas HART1 AP gene cluster, known to be secreted and involved in the virulence of several pathogenic fungi [100], [101] but missing in animal parasitic trypanosomids and the non pathogenous EM1 isolate, could be described as a good example of convergent evolution between these distant phytopathogen organisms.
The genome completion is the first step towards development of effective chemical control agents against Phytomonas spp., which is not only of economic interest, but may have relevance for other Trypanosomatidae pathogenic to humans and animals, since they share similar metabolic routes and many other biological mechanisms [112], [113]. Comparative studies between plant, human and animal pathogenic trypanosomatids as well as free living species will assist in the identification of gene cohorts specifically linked to various pathogenesis mechanisms. These comparisons will also contribute towards better and safer control methods for trypanosomatid diseases of animals, plants and humans and provide better insights into the evolution of parasitic and pathogenic mechanisms.
Materials and Methods
Genome sequencing and assembly
The sequencing strategy used for both Phytomonas genomes corresponds to a mix of three technologies: 454/Roche for most coverage; Solexa/Illumina for automatic corrections of low-quality regions (especially around homopolymers); and classical Sanger sequencing at low coverage with large-insert clones (10 kb-insert containing plasmids and fosmids) to organize the contigs into scaffolds. The assembly method is described in detail in Text S1.
Chromosome read depth analysis
Illumina reads were mapped to the corresponding Phytomonas genome using SOAP version 1.10 [114], under the guidance of a custom perl script. The number of bases mapping to each position in each scaffold was recorded, and used to determine the total number of read bases mapping to each scaffold and the median read depth for each scaffold. Observing that a majority of the scaffolds displayed similar median read depths, and interpreting this as a nominal ‘ploidy’, a within-genome normalisation was performed by setting the average of the read depth of the four longest ‘euploidic’ scaffolds to 2. The read depth for each scaffold was subsequently normalized to this value. Results of the scaffolds “somy” are shown in Table S1.
Genome annotation
Protein-coding genes are predicted by combining ab initio gene model predictions (already trained on manually annotated genes) and homology searches, using collections of expressed sequences - full length cDNAs, ESTs or massive-scale mRNA sequences from the same or closely related organisms – proteins or other genomic sequences. Details on the pipeline are given in the Text S1. Moreover, tRNA-Scan [115] was used to detect tRNAs in both Phytomonas assembled sequences.
After a final integration of all gene evidence using GAZE [116], the final proteome was delivered with computed annotation data, such as ortholog and paralog associations, functional domains and ontology relationships.
Identification of candidate horizontal gene transfers
Phytomonas proteins were used against the protein nr database (blastx, [117]), with the parameters “-f 100 -X 100 -e 0.00001 -E 2 -W 5”, and the best hits were retained using the following criteria: only BLAST scores greater than 90% of the best score outside kinetoplastids (so that horizontal gene transfers shared between kinetoplastids could be detected) and above 100 were retained. Then, the proteins with all their best hits in bacteria or archaea were retained as candidates to have arisen from bacterial/archeal horizontal gene transfers. All the candidates were then manually inspected, which provided 87 final candidate HGT genes, 80 of which have orthologs in other trypanosomatids, and 8 have no orthologs in Leishmania spp. nor Trypanosoma spp. and might thus be specific of Phytomonas (Table S4).
Detection of orthologs between EM1, HART1 and other trypanosomatids
We identified orthologous genes between Phytomonas EM1 and HART1, and 4 other trypanosomatids: L. major [29], T. brucei [28], T. cruzi [30] and T. vivax [70] (Tritryp release 2.1). Each pair of annotated genes was aligned with the Smith-Waterman algorithm, and alignments with a score higher than 300 (BLOSUM62, gapo = 10, gape = 1) were retained. Orthologs were defined as best reciprocal hits (BRH), i.e. two genes, A from genome GA and B from genome GB, were considered orthologs if B is the best match for gene A in GB and A is the best match for B in GA. Indeed, 5006 gene pairs (representing 77.6% for HART1 and 78.4% for EM1 genes), similar in gene size and intergenic length, were detected using this approach (Figure S9A and B). The number of BRH for each comparison and their average and median %id are displayed in Figure 3; the distribution of %id for these BRH between different pairs of species designated both Phytomonas isolates as being much closer to each other than to other trypanosomes (average %id of 70% for EM1 and HART1; 56 to 57.6% between the different pairs of trypanosomes).
The results of the pairwise alignments between all protein sequences of the 6 genomes were then inputted to the orthoMCL software V1.4 [69], in order to assemble clusters of orthologous genes between both Phytomonas EM1 and HART1, and other trypanosomatids. This approach was complementary to what was observed by the BRH strategy, since it permitted us to ascertain orthologs for multigenic families, not seen by the BRH strategy alone. This procedure provided 7,694 clusters of orthologs genes, gathering 5,188 EM1 and 4,643 HART1 genes in clusters containing genes from both isolates (regardless of the presence or absence of genes from other trypanosomes) (Table 1).
We also ran orthoMCL on the subset of genes from EM1 and HART1 that have strong support (i.e that are overlapping uniprot genewise hits, or cDNA reads as well as ab initio predictions): EM1 contains 5,237 such genes, and HART1 5,247 genes (Table 1).
Identification of genes absent from one isolate compared to the other
1/“homology” approach
After combining BRH and orthoMCL approaches, 1,171 EM1 genes remained with no ortholog detected in HART1, and 1,343 HART1 genes remained with no ortholog detected in EM1. But the fact that no orthologous gene could be detected was likely due to detection issues rather than differences between the isolates. In order to filter out those detection issues, we first aligned the protein sequences against the genome, with the same pipeline as the one used for aligning uniprot proteins, i.e. using BLAT [118] followed by genewise [119]; 504 genes from EM1 and 602 genes from HART1 displayed a genomic match in the other isolate, which correspond to missed genes or wrong annotations (Figure S10). The genes that displayed no match on the genome were subsequently aligned against the reads using tblastn [120] with an e-value cutoff of 10e−04. Genes matching on at least 5 reads, and covered on > = 25% of their length with an average %id > = 35% were retained: 247 genes from EM1 and 359 genes from HART1 are present in the reads but not in the assembly. Among the remaining genes, a substantial proportion was suspected to correspond to annotation artefacts. They were filtered out by retaining only genes overlapping uniprot genewise hits or cDNA reads and ab initio predictions, or genes sharing homology with other trypanosomatids (i.e. present in orthoMCL clusters). 82 genes remained in EM1 and 83 genes in HART1. We then investigated the syntenic regions of those genes and discarded the genes for which we could not find a syntenic region (genes upstream and downstream were not in the same operon), and for which there were intercontig gaps in the intergenic region; such genes were likely not assembled but possibly present in the genome (and not detected by the mapping on the reads because of the stringency of the criteria). Finally, manual curation of the remaining candidates allowed elimination of doubtful candidates (with doubtful structures or some homology with the syntenic region). This procedure provided 13 EM1 genes with no ortholog in HART1 and 4 HART1 genes with no ortholog in EM1 (Figure S10).
2/“synteny” approach
We also investigated genes from each isolate that had no counterpart in the syntenic region of the other isolate. We first identified pairs of successive BRH between EM1 and HART1 that were in the same operon and harbored different numbers of genes between them. They were filtered to eliminate structural differences of annotation (splits/fusions) between the two isolates: if the two orthologous genes of at least one BRH pair surrounding the region differed in length by more than 500 bp, the region was discarded. Discrepancies likely due to missed genes in the annotation were also filtered out: we only retained syntenic regions with lengths differing by more than 1000 bp between the two Phytomonas isolates. Regions containing intercontig gaps were also discarded. Finally, all candidates were inspected manually, and aligned using blastn [121] to the genome of the other isolate in order to sort out cases where the gene was absent from the whole genome and cases where the gene was absent in synteny but present somewhere else in the genome. The approach was sensitive enough to detect gene order changes (gene inversions) as well as translocations: the 7 EM1 genes and 5 HART1 genes that were detected in another position on the genome of the other isolate are described in Table S10. We detected 10 genes in EM1 that are neither present in the syntenic position in HART1 nor anywhere else (3 of which were already identified as having no ortholog), and 3 genes in HART1 that are not present in the syntenic position in EM1 nor anywhere else (2 of which were already identified as having no ortholog): Table S9. The “synteny” approach retained genes with weak homology with other genes anywhere on the genome (because they share a common domain for instance) that had been discarded by the alignment on the genome/reads in the “orthology” approach. On the other hand, some of the genes detected by the “orthology” approach were not detected by the “synteny” approach because the very stringent gene structure filter from the “synteny” approach discarded some genes identified by the “orthology” approach.
Combining the two complementary approaches, 20 genes from EM1 and 13 genes from HART1 are identified. Since the two approaches are not capturing the same genes, we suspect that more genes specific of each isolate have been missed in the automatic detection process, but our aim was to be conservative and keep only cases with strong support. Refining the selection would require extensive manual curation.
Coincidence of synteny breaks with operon boundaries
An in-house perl script was used to draw the dot plots and build syntenic blocks between species. The clustering was performed by single linkage clustering using the euclidian distance between genes. Those distances were calculated with the gene index in each scaffold rather than the genomic position. The minimal distance between two orthologous genes was set to 10 on both counterparts and we only retained clusters that were composed of at least 5 pairs of paralogous genes. The boundaries of the syntenic clusters were then filtered in order to eliminate those occuring at the end of scaffolds and corresponding to “assembly breaks” rather than synteny breaks. As a consequence, for genomes with a more fragmented assembly, the number of synteny breaks detected is lower because some real syntenic breaks occur at scaffolds boundaries and are discarded. This is the case for T. cruzi (41 scaffolds) that appears to have less synteny breaks with Phytomonas compared to T. vivax and T. brucei (11 chromosomes). We performed a simulation to distribute randomly the same number of synteny breaks as observed for each scaffold (1000 iterations) and counted the number of randomly distributed synteny breaks that coincided with operon boundaries. In all cases, the observed number of synteny breaks at operon boundaries was significantly higher than expected randomly (Figure S12B).
Search for specific gene families in Phytomonas
Both genomic EM1 and HART1 assemblies were queried using sequence probes encompassing selected Interpro domains, by a series of reciprocal sequence comparisons using the BLAST server, accessed through the SeqTryplant Genome Browser or directly on a secure web site. Likewise, the reads not included in the assembly as well as the contigs smaller than 5 kb and so excluded from the assembled sequence, were scanned with the same probes. The results obtained were subsequently examined by the experts of the Phytomonas consortia in order to validate the gene models. Details on the probes and procedure used for each gene family can be found in the Text S1 file.
Comparison of trypanosome gene repertoires
The same gene probes used to search for gene families in both Phytomonas genomes were later employed to query the TriTrypDB 4.0 Released, in order to obtain the corresponding genes in the T. brucei, T. cruzi and L. major genome annotations. Later on, these sequences were applied to query the OrthoMCL DB (version 5), and copy number, as automatically defined by the OrthoMCL approach was reported. Moreover, T. brucei, T. cruzi and/or Leishmania spp. gene copy number for members of certain families (e.g. kinases and transporters) was obtained from the literature or human expertise when available (Table 2 and Table S3).
Analysis of the Phytomonas secretome
Proteins with a signal peptide were detected with SignalP version 3.0 [122], transmembrane domains were detected with TMHMM 2.0 [123] and GPI anchors with KOHGPI version 1.5 (http://gpi.unibe.ch/). Proteins harboring a signal peptide, not containing transmembrane domains nor GPI anchors were considered to be secreted by Phytomonas, and their annotation was performed using BLASTp against the non-redundant NCBI database, Interproscan and Gene Ontology. We retrieved aspartic proteases from others clades (amoebae, plants, chromalveolates, fungi and animals) using the Phytomonas aspartic proteases amino acid sequences as queries with BLASTp on the NCBI nr database [121]. Phylogenetic analysis was executed on the Phylogeny.fr platform [124] as described in [125], with the parameters “minimum length of a block after gap cleaning: 5, no gap positions were allowed in the final alignment, all segments with contiguous non conserved positions bigger than 8 were rejected, minimum number of sequences for a flank position: 85%” for Gblocks v0.91b [126].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DanielsJP, GullK, WicksteadB (2010) Cell biology of the trypanosome genome. Microbiol Mol Biol Rev 74: 552–569.
2. MalvyD, ChappuisF (2011) Sleeping sickness. Clin Microbiol Infect 17: 986–995.
3. CouraJR, Borges-PereiraJ (2010) Chagas disease: 100 years after its discovery. A systemic review. Acta Trop 115: 5–13.
4. den BoerM, ArgawD, JanninJ, AlvarJ (2011) Leishmaniasis impact and treatment access. Clin Microbiol Infect 17: 1471–1477.
5. PennisiE (2010) Armed and dangerous. Science 327: 804–805.
6. RaffaeleS, KamounS (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol 10: 417–430.
7. KamperJ, KahmannR, BolkerM, MaLJ, BrefortT, et al. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444: 97–101.
8. KemenE, GardinerA, Schultz-LarsenT, KemenAC, BalmuthAL, et al. (2011) Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol 9: e1001094.
9. AmselemJ, CuomoCA, van KanJA, ViaudM, BenitoEP, et al. (2011) Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 7: e1002230.
10. DolletM (1984) Plant diseases caused by flagellate protozoa (Phytomonas). Annual Review Phytopathology 22: 115–125.
11. CamargoEP (1999) Phytomonas and other trypanosomatid parasites of plants and fruit. Adv Parasitol 42: 29–112.
12. Wallace FG, Roitman I, Camargo EP (1992) Trypanosomatids of plants. In: Kreir JP, Baker JR, editors. Parasitic protozoa. 2nd ed. New York: Academic Press. pp. 55–84.
13. Camargo EP, Wallace G (1994) Vectors of plant parasites of the genus Phytomonas (Protozoa, Zoomastigophorea, Kinetoplastida); Harris KF, editor. New York: Springer-Verlag.
14. Desmier De ChenonR (1984) Research on the genus Lincus Stal, Hemiptera Pentatomidae Discocephalinae, and its possible role in the transmission of Marchitez of oil palm and hartrot of coconut. Oléagineux 39: 1–6.
15. Dollet M., Alvanil F., Diaz A., Louvet C., Gargani D., et al.. (1993) Les Pentatomides vecteurs des Trypanosomes associés au Hartrot du cocotier et Marchitez du palmier. In: Plantes ANdlPd, editor. 3ème Conférence Internationale sur les Ravageurs en Agriculture. Montpellier, France. pp. 1321–1328.
16. LouiseC, DolletM, MariauD (1986) Research into Hartrot of the Coconut, a Disease Caused by Phytomonas (Trypanosomatidae), and into Its Vector Lincus Sp (Pentatomidae) in Guiana. Oléagineux 41: 437–446.
17. DonovanC (1909) Kala-Azar in Madras, especially with regard to its connexion with the dog and the bug (Conorrhinus). Lancet 177: 1495–1496.
18. MullerE, GarganiD, BanulsAL, TibayrencM, DolletM (1997) Classification of plant trypanosomatids (Phytomonas spp.): parity between random-primer DNA typing and multilocus enzyme electrophoresis. Parasitology 115 (Pt 4) 403–409.
19. DolletM (2001) Phloem-restricted trypanosomatids form a clearly characterised monophyletic group among trypanosomatids isolated from plants. Int J Parasitol 31: 459–467.
20. DolletM, SturmNR, CampbellDA (2001) The spliced leader RNA gene array in phloem-restricted plant trypanosomatids (Phytomonas) partitions into two major groupings: epidemiological implications. Parasitology 122: 289–297.
21. SturmNR, DolletM, LukesJ, CampbellDA (2007) Rational sub-division of plant trypanosomes (Phytomonas spp.) based on minicircle conserved region analysis. Infect Genet Evol 7: 570–576.
22. DolletM, SturmNR, CampbellDA (2012) The internal transcribed spacer of ribosomal RNA genes in plant trypanosomes (Phytomonas spp.) resolves 10 groups. Infect Genet Evol 12: 299–308.
23. CamargoEP, KasteleinP, RoitmanI (1990) Trypanosomatid parasites of plants (phytomonas). Parasitol Today 6: 22–25.
24. StahelG (1931) Zur Kenntnis der Siebröhrenkrankheit (Phloëmnekrose) des Kaffeebaumes in Surinam. Mikroskopische Untersuchungen und Infektionsversuche 4: 65–82.
25. VermeulenH (1968) Investigations into the cause of the phloem necrosis disease of Coffea liberica in Surinam, South America. Netherlands Journal of Plant Pathology 74: 202–218.
26. LopezG, GentyP, OllagnierM (1975) Control preventivo de la “Marchitez sorpresiva” del Elaeis guineensis en America Latina. Oleagineux 30: 243–250.
27. ParthasarathyMV, WGVANS, SoudantC (1976) Trypanosomatid flagellate in the Phloem of diseased coconut palms. Science 192: 1346–1348.
28. BerrimanM, GhedinE, Hertz-FowlerC, BlandinG, RenauldH, et al. (2005) The genome of the African trypanosome Trypanosoma brucei. Science 309: 416–422.
29. IvensAC, PeacockCS, WortheyEA, MurphyL, AggarwalG, et al. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science 309: 436–442.
30. El-SayedNM, MylerPJ, BartholomeuDC, NilssonD, AggarwalG, et al. (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309: 409–415.
31. El-SayedNM, MylerPJ, BlandinG, BerrimanM, CrabtreeJ, et al. (2005) Comparative genomics of trypanosomatid parasitic protozoa. Science 309: 404–409.
32. PeacockCS, SeegerK, HarrisD, MurphyL, RuizJC, et al. (2007) Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet 39: 839–847.
33. AslettM, AurrecoecheaC, BerrimanM, BrestelliJ, BrunkBP, et al. (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38: D457–462.
34. RaymondF, BoisvertS, RoyG, RittJF, LegareD, et al. (2012) Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res 40: 1131–1147.
35. JacksonAP, BerryA, AslettM, AllisonHC, BurtonP, et al. (2012) Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proc Natl Acad Sci U S A 109: 3416–3421.
36. KorenyL, SobotkaR, KovarovaJ, GnipovaA, FlegontovP, et al. (2012) Aerobic kinetoplastid flagellate Phytomonas does not require heme for viability. Proc Natl Acad Sci U S A 109: 3808–3813.
37. MarinC, AlbergeB, DolletM, PagesM, BastienP (2008) First complete chromosomal organization of a protozoan plant parasite (Phytomonas spp.). Genomics 91: 88–93.
38. MarinC, DolletM, PagesM, BastienP (2009) Large differences in the genome organization of different plant Trypanosomatid parasites (Phytomonas spp.) reveal wide evolutionary divergences between taxa. Infect Genet Evol 9: 235–240.
39. AuryJM, CruaudC, BarbeV, RogierO, MangenotS, et al. (2008) High quality draft sequences for prokaryotic genomes using a mix of new sequencing technologies. BMC Genomics 9: 603.
40. RogersMB, HilleyJD, DickensNJ, WilkesJ, BatesPA, et al. (2011) Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res 21: 2129–2142.
41. AkopyantsNS, KimblinN, SecundinoN, PatrickR, PetersN, et al. (2009) Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science 324: 265–268.
42. MarcheS, RothC, PhilippeH, DolletM, BaltzT (1995) Characterization and detection of plant trypanosomatids by sequence analysis of the small subunit ribosomal RNA gene. Mol Biochem Parasitol 71: 15–26.
43. ArnerE, KindlundE, NilssonD, FarzanaF, FerellaM, et al. (2007) Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants. BMC Genomics 8: 391.
44. AnderssonB (2011) The Trypanosoma cruzi genome; conserved core genes and extremely variable surface molecule families. Res Microbiol 162: 619–625.
45. MauricioIL, GauntMW, StothardJR, MilesMA (2007) Glycoprotein 63 (gp63) genes show gene conversion and reveal the evolution of Old World Leishmania. Int J Parasitol 37: 565–576.
46. d'Avila-LevyCM, SantosLO, MarinhoFA, DiasFA, LopesAH, et al. (2006) Gp63-like molecules in Phytomonas serpens: possible role in the insect interaction. Curr Microbiol 52: 439–444.
47. SantosAL, d'Avila-LevyCM, DiasFA, RibeiroRO, PereiraFM, et al. (2006) Phytomonas serpens: cysteine peptidase inhibitors interfere with growth, ultrastructure and host adhesion. Int J Parasitol 36: 47–56.
48. EliasCG, ChagasMG, Souza-GoncalvesAL, PascarelliBM, d'Avila-LevyCM, et al. (2012) Differential expression of cruzipain- and gp63-like molecules in the phytoflagellate trypanosomatid Phytomonas serpens induced by exogenous proteins. Exp Parasitol 130: 13–21.
49. SiegelTN, HekstraDR, KempLE, FigueiredoLM, LowellJE, et al. (2009) Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev 23: 1063–1076.
50. Martinez-CalvilloS, Vizuet-de-RuedaJC, Florencio-MartinezLE, Manning-CelaRG, Figueroa-AnguloEE (2010) Gene expression in trypanosomatid parasites. J Biomed Biotechnol 2010: 525241.
51. MairG, ShiH, LiH, DjikengA, AvilesHO, et al. (2000) A new twist in trypanosome RNA metabolism: cis-splicing of pre-mRNA. RNA 6: 163–169.
52. OpperdoesFR, MichelsPA (2007) Horizontal gene transfer in trypanosomatids. Trends Parasitol 23: 470–476.
53. HannaertV, SaavedraE, DuffieuxF, SzikoraJP, RigdenDJ, et al. (2003) Plant-like traits associated with metabolism of Trypanosoma parasites. Proc Natl Acad Sci U S A 100: 1067–1071.
54. NawatheanP, MaslovDA (2000) The absence of genes for cytochrome c oxidase and reductase subunits in maxicircle kinetoplast DNA of the respiration-deficient plant trypanosomatid Phytomonas serpens. Curr Genet 38: 95–103.
55. LukesJ, HashimiH, ZikovaA (2005) Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet 48: 277–299.
56. SimpsonL, MaslovDA (1999) Evolution of the U-insertion/deletion RNA editing in mitochondria of kinetoplastid protozoa. Ann N Y Acad Sci 870: 190–205.
57. VotypkaJ, KlepetkovaH, JirkuM, KmentP, LukesJ (2012) Phylogenetic relationships of trypanosomatids parasitising true bugs (Insecta: Heteroptera) in sub-Saharan Africa. Int J Parasitol 42: 489–500.
58. MaslovDA, VotypkaJ, YurchenkoV, LukesJ (2013) Diversity and phylogeny of insect trypanosomatids: all that is hidden shall be revealed. Trends Parasitol 29: 43–52.
59. MaslovDA, HollarL, HaghighatP, NawatheanP (1998) Demonstration of mRNA editing and localization of guide RNA genes in kinetoplast-mitochondria of the plant trypanosomatid Phytomonas serpens. Mol Biochem Parasitol 93: 225–236.
60. SimpsonL (1997) The genomic organization of guide RNA genes in kinetoplastid protozoa: several conundrums and their solutions. Mol Biochem Parasitol 86: 133–141.
61. DolletM, SturmNR, AhomadegbeJC, CampbellDA (2001) Kinetoplast DNA minicircles of phloem-restricted Phytomonas associated with wilt diseases of coconut and oil palms have a two-domain structure. FEMS Microbiol Lett 205: 65–69.
62. BringaudF, BiteauN, ZuiderwijkE, BerrimanM, El-SayedNM, et al. (2004) The ingi and RIME non-LTR retrotransposons are not randomly distributed in the genome of Trypanosoma brucei. Mol Biol Evol 21: 520–528.
63. BringaudF, BartholomeuDC, BlandinG, DelcherA, BaltzT, et al. (2006) The Trypanosoma cruzi L1Tc and NARTc non-LTR retrotransposons show relative site specificity for insertion. Mol Biol Evol 23: 411–420.
64. BringaudF, GhedinE, BlandinG, BartholomeuDC, CalerE, et al. (2006) Evolution of non-LTR retrotransposons in the trypanosomatid genomes: Leishmania major has lost the active elements. Mol Biochem Parasitol 145: 158–170.
65. BringaudF, MullerM, CerqueiraGC, SmithM, RochetteA, et al. (2007) Members of a large retroposon family are determinants of post-transcriptional gene expression in Leishmania. PLoS Pathog 3: 1291–1307.
66. BringaudF, BerrimanM, Hertz-FowlerC (2009) Trypanosomatid genomes contain several subfamilies of ingi-related retroposons. Eukaryot Cell 8: 1532–1542.
67. SmithM, BringaudF, PapadopoulouB (2009) Organization and evolution of two SIDER retroposon subfamilies and their impact on the Leishmania genome. BMC Genomics 10: 240.
68. BringaudF, Garcia-PerezJL, HerasSR, GhedinE, El-SayedNM, et al. (2002) Identification of non-autonomous non-LTR retrotransposons in the genome of Trypanosoma cruzi. Mol Biochem Parasitol 124: 73–78.
69. LiL, StoeckertCJJr, RoosDS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189.
70. GuerreiroLT, SouzaSS, WagnerG, De SouzaEA, MendesPN, et al. (2005) Exploring the genome of Trypanosoma vivax through GSS and in silico comparative analysis. OMICS 9: 116–128.
71. Ulrich P, Cintron R, Docampo R (2010) Calcium homeostasis and acidocalcisomes in Trypanosoma cruzi. In: de Souza W, editor. Structures and Organelles in Pathogenic Protists. Berlin: Springer-Verlag. pp. 299–318.
72. LiuB, LiuY, MotykaSA, AgboEE, EnglundPT (2005) Fellowship of the rings: the replication of kinetoplast DNA. Trends Parasitol 21: 363–369.
73. StuartKD, SchnauferA, ErnstNL, PanigrahiAK (2005) Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105.
74. AmmermanML, DowneyKM, HashimiH, FiskJC, TomaselloDL, et al. (2012) Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res 40: 5637–5650.
75. ParsonsM, WortheyEA, WardPN, MottramJC (2005) Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6: 127.
76. AlonsoA, SasinJ, BottiniN, FriedbergI, FriedbergI, et al. (2004) Protein tyrosine phosphatases in the human genome. Cell 117: 699–711.
77. SzoorB (2010) Trypanosomatid protein phosphatases. Mol Biochem Parasitol 173: 53–63.
78. BrenchleyR, TariqH, McElhinneyH, SzoorB, Huxley-JonesJ, et al. (2007) The TriTryp phosphatome: analysis of the protein phosphatase catalytic domains. BMC Genomics 8: 434.
79. StegmeierF, AmonA (2004) Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet 38: 203–232.
80. AndreevaAV, KutuzovMA (2004) Widespread presence of “bacterial-like” PPP phosphatases in eukaryotes. BMC Evol Biol 4: 47.
81. AkermanM, Shaked-MishanP, MazarebS, VolpinH, ZilbersteinD (2004) Novel motifs in amino acid permease genes from Leishmania. Biochem Biophys Res Commun 325: 353–366.
82. JacksonAP (2007) Origins of amino acid transporter loci in trypanosomatid parasites. BMC Evol Biol 7: 26.
83. SaierMHJr (1999) A functional-phylogenetic system for the classification of transport proteins. J Cell Biochem Suppl 32–33: 84–94.
84. BuschW, SaierMHJr (2003) The IUBMB-endorsed transporter classification system. Methods Mol Biol 227: 21–36.
85. MirandaK, RodriguesCO, HentchelJ, VercesiA, PlattnerH, et al. (2004) Acidocalcisomes of Phytomonas francai possess distinct morphological characteristics and contain iron. Microsc Microanal 10: 647–655.
86. FuruyaT, OkuraM, RuizFA, ScottDA, DocampoR (2001) TcSCA complements yeast mutants defective in Ca2+ pumps and encodes a Ca2+-ATPase that localizes to the endoplasmic reticulum of Trypanosoma cruzi. J Biol Chem 276: 32437–32445.
87. BrighouseA, DacksJB, FieldMC (2010) Rab protein evolution and the history of the eukaryotic endomembrane system. Cell Mol Life Sci 67: 3449–3465.
88. NatesanSK, PeacockL, LeungKF, MatthewsKR, GibsonW, et al. (2009) The trypanosome Rab-related proteins RabX1 and RabX2 play no role in intracellular trafficking but may be involved in fly infectivity. PLoS One 4: e7217.
89. AckersJP, DhirV, FieldMC (2005) A bioinformatic analysis of the RAB genes of Trypanosoma brucei. Mol Biochem Parasitol 141: 89–97.
90. EliasM, BrighouseA, Gabernet-CastelloC, FieldMC, DacksJB (2012) Sculpting the endomembrane system in deep time: high resolution phylogenetics of Rab GTPases. J Cell Sci 125: 2500–2508.
91. Sanchez-MorenoM, LasztityD, CoppensI, OpperdoesFR (1992) Characterization of carbohydrate metabolism and demonstration of glycosomes in a Phytomonas sp. isolated from Euphorbia characias. Mol Biochem Parasitol 54: 185–199.
92. ChaumontF, SchanckAN, BlumJJ, OpperdoesFR (1994) Aerobic and anaerobic glucose metabolism of Phytomonas sp. isolated from Euphorbia characias. Mol Biochem Parasitol 67: 321–331.
93. MolinasSM, AltabeSG, OpperdoesFR, RiderMH, MichelsPA, et al. (2003) The multifunctional isopropyl alcohol dehydrogenase of Phytomonas sp. could be the result of a horizontal gene transfer from a bacterium to the trypanosomatid lineage. J Biol Chem 278: 36169–36175.
94. UttaroAD, OpperdoesFR (1997) Characterisation of the two malate dehydrogenases from Phytomonas sp. Purification of the glycosomal isoenzyme. Mol Biochem Parasitol 89: 51–59.
95. CanepaGE, CarrilloC, MirandaMR, SayeM, PereiraCA (2011) Arginine kinase in Phytomonas, a trypanosomatid parasite of plants. Comp Biochem Physiol B Biochem Mol Biol 160: 40–43.
96. FairlambAH, CeramiA (1992) Metabolism and functions of trypanothione in the Kinetoplastida. Annu Rev Microbiol 46: 695–729.
97. MarcheS, RothC, ManoharSK, DolletM, BaltzT (1993) RNA virus-like particles in pathogenic plant trypanosomatids. Mol Biochem Parasitol 57: 261–267.
98. WidmerG, ComeauAM, FurlongDB, WirthDF, PattersonJL (1989) Characterization of a RNA virus from the parasite Leishmania. Proc Natl Acad Sci U S A 86: 5979–5982.
99. WeeksR, AlineRFJr, MylerPJ, StuartK (1992) LRV1 viral particles in Leishmania guyanensis contain double-stranded or single-stranded RNA. J Virol 66: 1389–1393.
100. NaglikJR, ChallacombeSJ, HubeB (2003) Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 67: 400–428 table of contents.
101. ten HaveA, EspinoJJ, DekkersE, Van SluyterSC, BritoN, et al. (2010) The Botrytis cinerea aspartic proteinase family. Fungal Genet Biol 47: 53–65.
102. SilvermanJM, ChanSK, RobinsonDP, DwyerDM, NandanD, et al. (2008) Proteomic analysis of the secretome of Leishmania donovani. Genome Biol 9: R35.
103. CorralesRM, SerenoD, Mathieu-DaudeF (2010) Deciphering the Leishmania exoproteome: what we know and what we can learn. FEMS Immunol Med Microbiol 58: 27–38.
104. HorvathP, NosanchukJD, HamariZ, VagvolgyiC, GacserA (2012) The identification of gene duplication and the role of secreted aspartyl proteinase 1 in Candida parapsilosis virulence. J Infect Dis 205: 923–933.
105. WyattGR, KaleGF (1957) The chemistry of insect hemolymph. II. Trehalose and other carbohydrates. J Gen Physiol 40: 833–847.
106. DocampoR, LukesJ (2012) Trypanosomes and the solution to a 50-year mitochondrial calcium mystery. Trends Parasitol 28: 31–37.
107. LeeMC, MarxCJ (2012) Repeated, selection-driven genome reduction of accessory genes in experimental populations. PLoS Genet 8: e1002651.
108. PeyretailladeE, El AlaouiH, DiogonM, PolonaisV, ParisotN, et al. (2011) Extreme reduction and compaction of microsporidian genomes. Res Microbiol 162: 598–606.
109. KubeM, MitrovicJ, DudukB, RabusR, SeemullerE (2012) Current view on phytoplasma genomes and encoded metabolism. ScientificWorldJournal 2012: 185942.
110. BaiX, ZhangJ, EwingA, MillerSA, Jancso RadekA, et al. (2006) Living with genome instability: the adaptation of phytoplasmas to diverse environments of their insect and plant hosts. J Bacteriol 188: 3682–3696.
111. Tyler BM, Rouxel T (2013) Effectors of fungi and oomycetes: their virulence and avirulence functions and translocation from pathogen to host cells. In: Sessa G, editor. Molecular Plant Immunity: Wiley-Blackwell. pp. 123–154.
112. UttaroAD, MirkinN, RiderMH, MichelsM, OpperdoesFR (1999) Phytomonas sp. A model of trypanosomatid metabolism and drug. Mem Inst Oswaldo Cruz 94.
113. MaganR, MarinC, SalasJM, Barrera-PerezM, RosalesMJ, et al. (2004) Cytotoxicity of three new triazolo-pyrimidine derivatives against the plant trypanosomatid: Phytomonas sp. isolated from Euphorbia characias. Mem Inst Oswaldo Cruz 99: 651–656.
114. LiR, LiY, KristiansenK, WangJ (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24: 713–714.
115. LoweTM, EddySR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964.
116. HoweKL, ChothiaT, DurbinR (2002) GAZE: a generic framework for the integration of gene-prediction data by dynamic programming. Genome Res 12: 1418–1427.
117. AltschulSF, MaddenTL, SchafferAA, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
118. KentWJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12: 656–664.
119. BirneyE, DurbinR (2000) Using GeneWise in the Drosophila annotation experiment. Genome Res 10: 547–548.
120. GertzEM, YuYK, AgarwalaR, SchafferAA, AltschulSF (2006) Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol 4: 41.
121. AltschulSF, GishW, MillerW, MyersEW, LipmanDJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410.
122. BendtsenJD, NielsenH, von HeijneG, BrunakS (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795.
123. KroghA, LarssonB, von HeijneG, SonnhammerEL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305: 567–580.
124. DereeperA, GuignonV, BlancG, AudicS, BuffetS, et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36: W465–469.
125. BinetR, MaurelliAT (2009) The chlamydial functional homolog of KsgA confers kasugamycin sensitivity to Chlamydia trachomatis and impacts bacterial fitness. BMC Microbiol 9: 279.
126. CastresanaJ (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552.
127. MannaPT, KellyS, FieldMC (2013) Adaptin evolution in kinetoplastids and emergence of the variant surface glycoprotein coat in African trypanosomatids. Mol Phylogenet Evol 67: 123–128.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2014 Číslo 2
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Genome-Wide Association Study of Metabolic Traits Reveals Novel Gene-Metabolite-Disease Links
- A Cohesin-Independent Role for NIPBL at Promoters Provides Insights in CdLS
- Classic Selective Sweeps Revealed by Massive Sequencing in Cattle
- Arf4 Is Required for Mammalian Development but Dispensable for Ciliary Assembly