
Diagnostika a léčba BCR/ABL-negativních myeloproliferativních onemocnění – principy a východiska doporučení CZEMP
Diagnosis and treatment of BCR/ABL-negative myeloproliferative diseases – principles and rationale of CZEMP recommendations
In 2009, the recommendations of the Czech Collaborative Group for Ph- Myeloproliferative Diseases (CZEMP) for diagnosis and treatment of BCR/ABL-negative myeloproliferative diseases (MPD), i.e. essential thrombocythemia (ET), polycythaemia vera (PV) and primary myelofibrosis (PMF) were updated and extended. The present article gives the rationale of the recommendations in full detail. The CZEMP diagnostic criteria for ET and PMF are based on histopathological (HP) findings, which must unconditionally be in line with the given clinical and laboratory characteristics of ET or of a certain stage of PMF, respectively. The platelet count is not decisive for diagnosis. In cases lacking an adequately taken and read HP finding, the Polycythemia Vera Study Group (PVSG) criteria are recommended. The diagnosis of typical PV is based on demonstration of the V617F mutation of the JAK2 gene along with a significant increase of red cell parameters. If these are close to borderline, the demonstration of increased total red cell mass (RCM) is required. In atypical cases lacking polyglobulia or elevated RCM, the HP picture of PV (in accordance with WHO description) plus JAK2 V617F mutation is satisfactory for diagnosis, or, in cases lacking JAK2 V617F mutation, the HP picture of PV along with polyglobulia (or increased RCM) is sufficient. The treatment principles of ET and other MPDs with thrombocythemia (MPD-T; i.e. the early stages of PMF and PV) are identical. The patients are stratified by their thrombotic risk (preceding thrombosis, another thrombophilic state, JAK2 mutation), presence of disease symptoms (mainly microcirculatory), platelet count and age. Only patients up to 65 years lacking the above mentioned risks with a platelet count < 1 000 × 109/ l are considered as low-risk and do not demand cytoreducing therapy. The others are high-risk ones and have an indication for thromboreduction. In patients older than 65 years, the potentially leukemogenic drug hydroxyurea (HU) may be used. In the younger ones, the choice is between anagrelide (ANG) or interferon-α (IFN). In high-risk patients, the treatment goal is to maintain platelet counts below 400, and in low-risk ones, below 600 × 109/ l. In PV, polycythemia itself is another thrombotic risk factor. The condition is treated by bloodletting or erythrocytaphereses. If hematocrit levels ≤ 45 are not achieved, cytoreductive therapy using HU in patients over 65 years, or IFN in younger individuals is required. All patients with thrombocythemia in PV are high-risk and have an indication for cytoreduction. Acetylsalicylic acid is given to all patients with MPD-T with platelets < 1 000 × 109/ l (at higher counts, hemorrhage is imminent), and to all individuals with PV, unless contraindication is present. In case of platelet count normalization, it may be withdrawn in cases of low-risk ET or PMF, not in JAK2+ PV. The treatment of advanced stages of PMF is symptomatic, with substitution of blood derivatives being the basis. The only curative treatment is allogeneic stem cell transplantation, which should not be indicated too early seeing to its risks, but also not too late – we must not allow transition into acute leukemia, which is heralded by blasts in the blood picture. The indication is the presence of any of the following criteria: values of hemoglobin < 10 g/ dl, WBC < 4 × 109/ l and platelets < 100 × 109/ l, any percentage of blasts or ≥ 10% immature granulocytes in the differential picture, > 1 erythroblast per 100 cells – all at repeated examinations within at least a 2-month interval, and in addition, rapid progression of hepato-/splenomegaly, presence of general symptoms of the disease, portal hypertension and extensive swellings.
Key words:
Ph- myeloproliferative disease – essential thrombocythemia – polycythaemia vera – primary myelofibrosis – diagnosis – risk factors – treatment algorithm – hydroxyurea – anagrelide – interferon-α – acetylsalicylic acid – hematopoietic stem cell transplantation
Autori:
J. Schwarz 1; M. Penka 2; V. Campr 1,3; D. Pospíšilová 4; L. Křen 5; Lucie Nováková 1; C. Bodzásová 6; Y. Brychtová 7; O. Černá 8; P. Ďulíček 9; A. Jonášová 10; J. Kissová 2; Z. Kořístek 7; M. Schutzova 11; I. Vonke 12; L. Walterová 13
Pôsobisko autorov:
Klinický úsek Ústavu hematologie a krevní transfuze Praha, přednosta doc. MUDr. Petr Cetkovský, Ph. D.
1; Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
2; Ústav patologie a molekulární medicíny 2. lékařské fakulty UK a FN Motol Praha, přednosta prof. MUDr. Roman Kodet, CSc.
3; Dětská klinika Lékařské fakulty UP a FN Olomouc, přednosta prof. MUDr. Vladimír Mihál, CSc.
4; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Josef Feit, CSc.
5; Ústav klinické hematologie FN Ostrava, přednosta prim. MUDr. Jaromír Gumulec
6; Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
7; Oddělení klinické hematologie FN Královské Vinohrady Praha, přednosta doc. MUDr. Tomáš Kozák, Ph. D., MBA
8; Oddělení klinické hematologie II. interní kliniky Lékařské fakulty UK a FN Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
9; I. interní klinika 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Marek Trněný, CSc.
10; Hemato-onkologické oddělení FN Plzeň, přednosta prim. MUDr. Vladimír Koza
11; Oddělení klinické hematologie Nemocnice České Budějovice, přednosta prim. MUDr. Ivan Vonke, MBA
12; Oddělení klinické hematologie Krajské nemocnice Liberec, přednostka prim. MUDr. Lenka Walterová
13
Vyšlo v časopise:
Vnitř Lék 2011; 57(2): 189-213
Kategória:
Přehledné referáty
Súhrn
V roce 2009 byla aktualizována a rozšířena doporučení České pracovní skupiny pro Ph negativní (Ph-) myeloproliferativní onemocnění (CZEMP) pro diagnostiku a léčbu BCR/ABL-negativních myeloproliferativních onemocnění (MPO), tj. esenciální trombocytemie (ET), polycythaemia vera (PV) a primární myelofibrózy (PMF). V tomto článku jsou podrobně rozvedena východiska doporučených postupů. Kritéria CZEMP pro diagnostiku ET a PMF se opírají o histopatologický (HP) nález, který však musí být bezpodmínečně v souladu s popsanými klinickými a laboratorními charakteristikami u ET, resp. různých stadií PMF. Počet trombocytů není pro diagnózu rozhodující. U případů, u kterých není k dispozici adekvátně odebraný a odečtený HP nález, doporučujeme užít kritérií Polycythemia Vera Study Group (PVSG). Diagnostika typické PV se opírá o průkaz mutace V617F genu JAK2 za předpokladu významného zvýšení hodnot červeného krevního obrazu. Při jeho hraničních hodnotách je nutný průkaz zvýšené celkové masy erytrocytů (RCM). U atypických případů, není-li přítomna polyglobulie (anebo zvýšená hodnota RCM), postačuje HP obraz PV dle definice WHO plus nález mutace V617F JAK2, anebo není-li přítomna mutace JAK2 V617F, stačí HP obraz PV + průkaz polyglobulie (anebo zvýšeného RCM). Principy léčby ET i ostatních MPO s trombocytemií (MPO-T; tj. raných stadií PMF a PV) jsou identické. Pacienti jsou stratifikováni podle přítomnosti trombotického rizika (předchozí trombózy, dalšího trombofilního stavu, mutace JAK2), přítomnosti symptomů onemocnění (obvykle mikrocirkulačních), počtu trombocytů a věku. Pouze pacienti do 65 let bez uvedených rizik s počtem trombocytů pod 1 000 × 109/ l jsou považováni za nízkorizikové a nevyžadují cytoreduktivní léčbu. Ostatní jsou vysokorizikoví a jsou indikováni k tromboredukci. Od věku nad 65 je možno užít potenciálně leukemogenní hydroxyureu (HU), u mladších volíme mezi anagrelidem (ANG) a interferonem-α (IFN). U vysokorizikových nemocných je cílem udržet trombocyty trvale pod 400, u nízkorizikových pod 600 × 109/ l. Při PV přistupuje jako trombotické riziko i polycytemie samotná. Tu léčíme venepunkcemi nebo erytrocytaferézami, v případě nemožnosti dosáhnout cílových hodnot hematokritu ≤ 45 podáváme cytoreduktivní terapii HU u pacientů nad 65 let, IFN u mladších jedinců. Všichni nemocní s trombocytemií při PV jsou vysokorizikoví a mají indikaci k cytoreduktivní léčbě. Kyselina acetylsalicylová se podává u MPO-T s trombocyty < 1 000 × 109/ l (při vyšších hodnotách hrozí krvácení) i u všech pacientů s PV, nemají-li kontraindikaci. V případě normalizace počtu trombocytů ji lze u nízkorizikové ET nebo PMF vysadit, u JAK2+ pacientů s PV nikoli. Léčba pokročilých stadií PMF s cytopeniemi je symptomatická, základem je substituce transfuzními přípravky. Jedinou kurativní terapií PMF je alogenní transplantace krvetvorných buněk. Tu nesmíme indikovat příliš brzy vzhledem k jejím rizikům, ale zároveň ne příliš pozdě – nesmíme dopustit přechod do akutní leukemie, avizovaný blasty v krevním obrazu. Indikací je přítomnost alespoň jednoho z kritérií: hodnoty hemoglobinu < 100 g/ l, leukocytů < 4 × 109 l a trombocytů < 100 × 109/ l, jakýkoli počet myeloblastů nebo ≥ 10 % nezralých granulocytů v diferenciálním krevním obraze, > 1 erytroblast na 100 buněk – vše při opakovaných vyšetřeních v rozmezí alespoň dvou měsíců, a dále rychlý rozvoj hepato-/splenomegalie, přítomnost celkových symptomů onemocnění, portální hypertenze a rozsáhlých otoků.
Klíčová slova:
Ph- myeloproliferativní onemocnění – esenciální trombocytemie – polycythaemia vera – primární myelofibróza – diagnostika – rizikové faktory – algoritmus léčby – hydroxyurea – anagrelid – interferon-α – kyselina acetylsalicylová – transplantace hematopoetických kmenových buněk
Úvod
Mezi Ph negativní (Ph-; BCR/ABL-) myeloproliferativní onemocnění (MPO) řadí současná WHO klasifikace z roku 2008 [1] řadu různých proliferativních onemocnění myeloidní řady, která nenesou, na rozdíl od chronické myeloidní leukemie, fuzní gen BCR/ABL, produkt tzv. filadelfského chromozomu (Ph). Tato klasifikace je uvedena v tab. 1. Níže uvedená doporučení, vypracovaná Českou pracovní skupinou pro Ph- myeloproliferativní onemocnění (nyní CZEMP) v roce 2009, se týkají pouze 3 nejčastějších onemocnění z této skupiny, a sice esenciální trombocytemie (ET), pravé polycytemie (PV) a primární myelofibrózy (PMF). Tato 3 onemocnění mají mnoho společného, jejich iniciální stadia mají obvykle velmi podobný klinický obraz s výraznou trombocytemií danou zvýšenou proliferací megakaryocytů v kostní dřeni. Společným znakem těchto onemocnění je původ v multipotentní myeloidní kmenové buňce [2–4] (jak již předpokládal Dameshek ve své historické anotaci [5]). Všechna tato onemocnění jsou ve své iniciální fázi charakterizovatelná nejen zvýšenou, autonomní proliferací pluripotentní myeloidní kmenové buňky a megakaryocytárních prekurzorů (a v různé míře i prekurzorů červené a granulocytární řady), ale také plnou schopností těchto buněk vyzrávat do terminálních stadií maturace. Dalším společným rysem těchto onemocnění je možnost výskytu mutace V617F genu tyrozinové kinázy JAK2 [6–8], která dává kmenovým buňkám impuls k proliferaci. Nedávno byl objeven haplotyp genu JAK2 (haplotyp 46/1), který přináší svému nositeli výrazně zvýšenou pravděpodobnost onemocnění MPO, které pak při manifestaci může, ale nemusí nést mutaci JAK2V617F [9–11]. Mutace JAK2 tedy není první událostí v patogenezi MPO [12,13]. Podobně ani nejčastěji detekovaná cytogenetická aberace, del(20)(q), nestojí na počátku vývoje onemocnění [14]. U zhruba 12 % případů MPO, podobně jako i u myeloidních leukemií a myelodysplastického syndromu (MDS), byla zjištěna některá z mutací nebo delecí genu TET2 předcházející ve vývoji MPO mutaci JAK2V617F (ačkoli byly prokázány i u případů bez JAK2 mutace). Význam aberací TET2 genu pro patogenezi MPO však není dosud jasný [15]. Některá Ph- MPO (PV, PMF) mají společnou tu vlastnost, že se mohou transformovat do sekundární akutní myeloidní leukemie (s-AML), tj. onemocnění již charakterizovatelného maturačním blokem.
![Klasifikace myeloidních neoplazií podle WHO 2008 [1]. Uvedena jsou pouze chronická MPO.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d2f8da6f6484f48b26584c1f086b74e1.jpg)
Hlavními riziky ET, PV a PMF jsou v každém okamžiku jejich vývoje možnost krvácení a nebezpečí někdy i fatální trombózy (pro přehled viz [16,17]). Cílem správně vedené terapie je prevence těchto příhod podáváním cytoreduktivní, antiagregační, event. antikoagulační léčby. Přestože jsou uvedená rizika společná pro všechny 3 nosologické jednotky, každá z nich má jinou dlouhodobou prognózu: zatímco ET je onemocněním, které by při správně vedené prevenci trombotických a krvácivých příhod nemělo zkracovat délku života (jelikož nehrozí přechod do s-AML, pakliže jej sami nevyvoláme genotoxickou léčbou cytostatiky), PV ji zkracuje mírně a pokročilá stadia PMF dosti výrazně [16,18]. Jak u PV (především ve stadiu postpolycytemické myelofibrózy), tak ještě více u PMF hrozí spontánní vývoj do terapeuticky velmi svízelně ovlivnitelné s-AML.
V tomto článku budou shrnuty otázky klasifikace Ph- MPO a budou uvedena současná doporučení CZEMP pro diagnostiku a léčbu 3 základních nosologických jednotek: ET, PV a PMF, včetně doporučení pro indikaci alogenní transplantace krvetvorných kmenových buněk (HSCT).
A. Klasifikace MPO
První ucelená koncepce MPO byla publikována Dameshkem v roce 1951 [5], neobsahovala však přesné definice jednotlivých onemocnění. První skutečné definice ET a PV byly vypracovány skupinou PVSG (Polycythemia Vera Study Group), založenou L. Wassermanem na přelomu 60. a 70. let minulého století, s cílem rozběhnout první multiinstitucionální/mezinárodní klinické studie u pacientů s PV a později s dalšími MPO. Starší klasifikace MPO (včetně PVSG kritérií) byly založeny především na různých laboratorních parametrech (hodnotách krevního obrazu, ale i jiných laboratorních a klinických znacích). Modifikovaná PVSG kritéria jsou dodnes často používána [19,20]. Jde v podstatě o non-nosologickou klasifikaci, podle které může jedno onemocnění přecházet v druhé. V definici jednotlivých onemocnění (především ET) chybí pro ně jedinečný rozpoznávací znak, jde tedy o diagnózu per exclusionem [17]. Naopak pozdější diagnostická kritéria, založená (alespoň v případě ET a PMF) především na histopatologické klasifikaci, jsou nosologická v tom smyslu, že pacientova diagnóza se s vývojem onemocnění nemění (mění se pouze stadium v rámci daného onemocnění). Nejde zde o vylučování jiných onemocnění jako u PVSG kritérií, nýbrž o pozitivní identifikaci dané nosologické jednotky [17]. Význam histopatologického nálezu z biopsie kostní dřeně byl rozpoznán patologickými školami v Německu (v Hannoveru a Kolíně nad Rýnem) již koncem 80. let minulého století [18,21]. První klasifikací Ph- MPO založenou na histopatologii byla tzv. „Rotterdamská kritéria“ vypracovaná tzv. Thrombocythemia Vera Study Group (TVSG) v roce 1997. Jejich pozdější úprava z roku 2002 nazvaná ECP, tj. Evropská klinická a patologická kritéria, vyústila v tzv. Evropskou klinickou, molekulární a patologickou klasifikaci (ECMP) v roce 2007 [22–24]. Mezitím v roce 2001 vznikla první WHO klasifikace Ph- MPO, založená na histopatologii [25–27], která byla, podle názoru CZEMP [28], nepříliš šťastně přepracována v roce 2008 [1]. CZEMP ve svých starších doporučeních pro diagnostiku a léčbu Ph- MPO [17,29,30] odkazovala na kritéria WHO 2001, resp. kritéria ECP 2002. Jelikož v současnosti široce používaná WHO 2008 kritéria obsahují řadu nepřesností až chyb kritizovaných nejen CZEMP [28], ale i jinými skupinami z různých úhlů pohledu [31–33], skupina CZEMP se rozhodla pro potřebu ČR vytvořit vlastní diagnostická kritéria, která jsou v principu „nosologická“, a alespoň v případě ET a PMF založená primárně na histopatologickém nálezu. Z výše citovaných kritických prací CZEMP adoptovala princip nutnosti měření celkového erytrocytárního volumu (RCM) u případů s hraničně vysokými hodnotami červeného krevního obrazu [32]. Převratnou novinkou pro diagnostiku Ph- MPO se od doby prvních publikací CZEMP [17,29,30] stal objev mutace JAK2V617F [6–8]. Ta se však v nynějších doporučeních skupiny uplatňuje pouze v diagnostice PV, zatímco u stavů s trombocytemií má především prognostický význam jako významný prediktor trombózy [8,34], který je vždy nutno vyšetřit. Její nález u ET a PMF jinak pouze ověřuje klonální podstatu onemocnění (zatímco její absence nemá žádnou výpovědní hodnotu), a tudíž per se nemá zvláštní diagnostický význam.
B. Nosologická diagnostika CZEMP
Nosologický přístup je doporučen přednostně. Jeho základem je histopatologická diagnóza z kostní dřeně, pouze v případě diagnostiky typické PV se lze spolehnout na laboratorní výsledky z periferní krve. Je třeba zdůraznit, že ke směrodatnému histopatologickému nálezu lze dospět pouze u tromboreduktivy neléčeného pacienta, v opačném případě je šance na přesnou nosologickou diagnózu ztracena. Přesné histopatologické obrazy ET, PV a PMF jsou detailně popsány v jiných publikacích – např. v doporučeních WHO z roku 2001 [25–27], doporučeních ECP [24], konsenzus kritériích evropské skupiny pro klasifikaci stupně fibrózy u MPO [35] a dalších [16,36]. Obr. 1 ukazuje základní histopatologické charakteristiky ET, PV a PMF. Dalšími parametry využitými zde uvedenou novou klasifikací a diagnostickými doporučeními CZEMP jsou hodnoty krevní obrazu, měření celkového erytrocytárního a plazmatického volumu a stanovení mutace JAK2V617F [6–8].
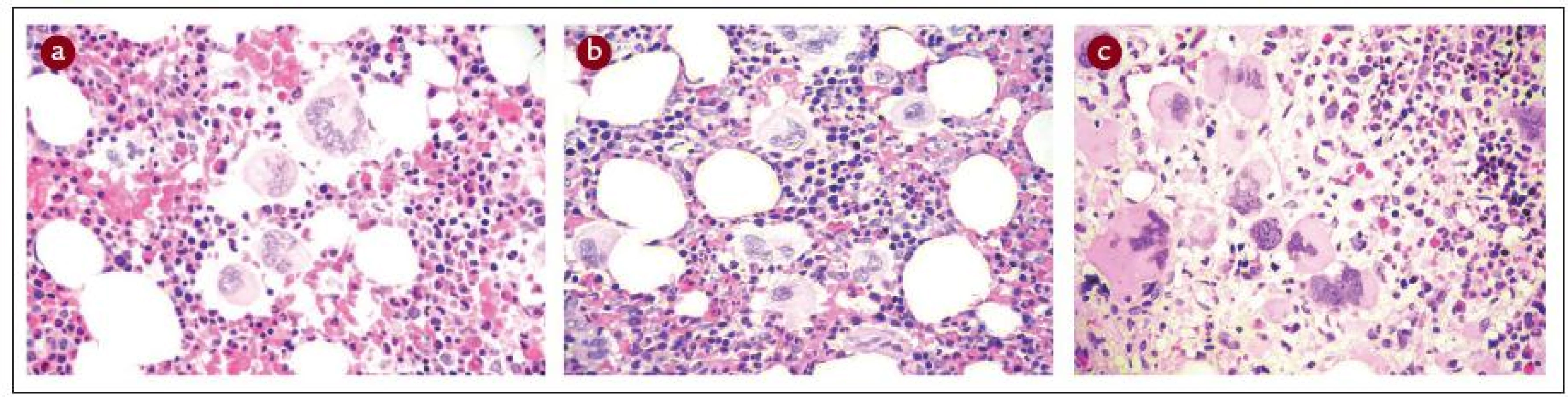
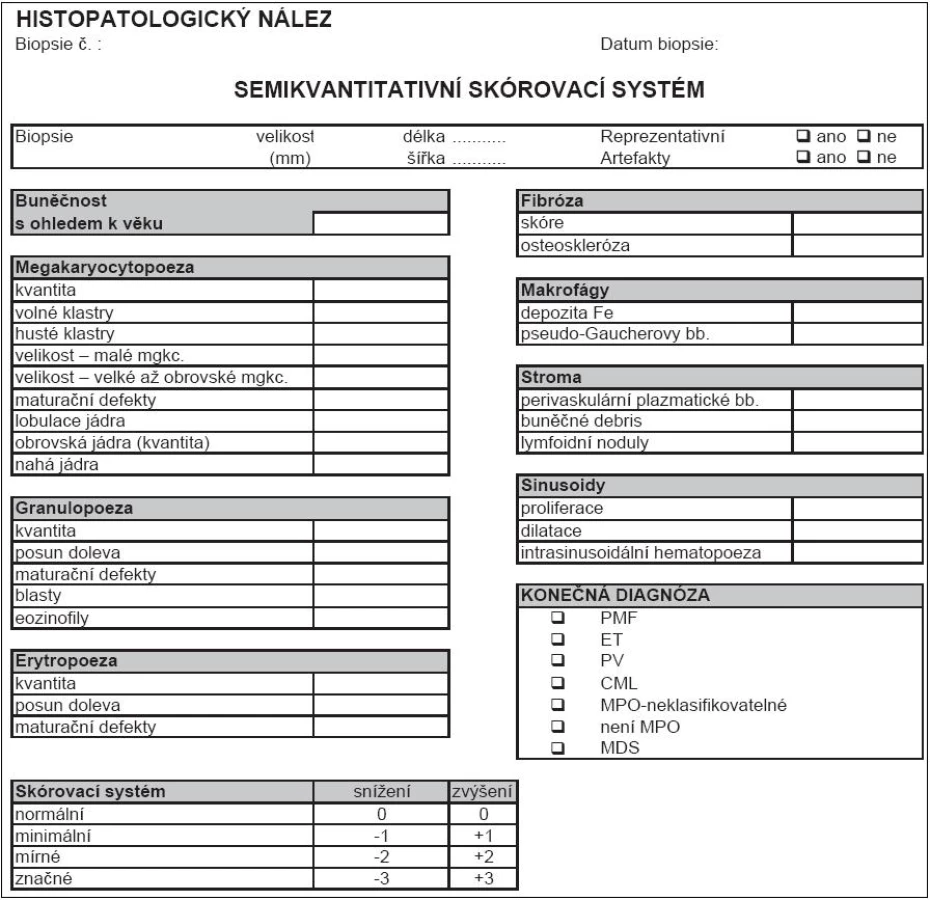
Histopatologická diagnostika Ph- MPO je velmi obtížná, vyžaduje vysokou míru připravenosti patologa po teoretické stránce, a navíc značnou zkušenost. Princip 2. čtení na pracovišti patologa-experta na Ph- MPO by měl být vždy dodržen. Naopak britská skupina nedoporučuje WHO klasifikaci pro její údajně nízkou reprodukovatelnost [37]. Lze se však domnívat, že je to dáno poměrně nízkou zvyklostí britských patologů odečítat biopsie podle principů WHO/ECP. Naopak ve středoevropské studii ANAHYDRET, tj. v zemích, kde je zvykem hodnotit nález prizmatem WHO/ECP klasifikace (Polsko, Srbsko, Česko, Rakousko, Německo), dosahovala míra konkordance diagnostického závěru mezi lokálním patologem a centrálním čtením v Německu (prof. J. Thiele a dr. H. M. Kvasnicka v Kolíně nad Rýnem) 82 % [38]. Skupina prof. Thieleho užívá pro histopatologickou diagnostiku formulář, do kterého musí vyšetřující patolog zanést informace ve 28 polích k přesnému posouzení diagnózy (tab. 2). Je doporučeno takový diagnostický formulář používat v běžné praxi [16]. Uvádíme to v hematologických doporučeních mj. proto, aby měl hematolog představu, co by měl od patologa očekávat. Souhrn diagnostiky 3 hlavních Ph- MPO je uveden v tab. 3 a na obr. 1 (včetně histopatologických charakteristik), v dalších odstavcích budou uvedena diagnostická doporučení podrobně pro každou nosologickou jednotku.

B.1. Diagnóza esenciální trombocytemie
Rozhodujícím kritériem pro diagnózu ET je histopatologický obraz ET v souladu s popisy WHO 2001/ECP 2002 (obr. 1) [24,25]. Tento obraz by měl být v souladu s typickými rysy onemocnění:
- Počet trombocytů je zpravidla > 400 × 109/ l, ale v některých případech může být i nižší, tzn. histopatologický obraz ET může předcházet laboratorní manifestaci ET s trombocytózou > 400 × 109/ l. Většinou jde o vzácné případy trombózy (např. Buddův-Chiariho syndrom), u kterých se nejprve zachytí JAK2 mutace (event. spontánní růst EEC) při počtu trombocytů < 400 x 109/ l, a teprve následné bioptické vyšetření kostní dřeně ukáže na přítomnost MPO včetně ET. Proto někteří autoři již delší dobu připouštějí možnost diagnostikovat ET i u případů s normálním počtem trombocytů [36,39–43]. Z tohoto důvodu (a také proto, že se horní norma počtu trombocytů liší při vyšetření na různých analyzátorech) po mnoha diskuzích upustila CZEMP od pevné definice nejnižšího počtu trombocytů při diagnostice ET, na rozdíl od všech předešlých klasifikací, a připouští diagnózu „pretrombocytemické ET“.
- Bývá normální nebo lehce zvýšený počet leukocytů (zvýšení WBC > 15 × 109/ l je extrémně vzácné, může být spojeno s úrazem, infekcí apod.).
- Hodnoty červeného krevního obrazu anebo RCM nepřekračují normu; JAK2V617F-pozitivní případy mají tendenci k vyšším hodnotám [34] (ale nikoli nad normu – v tom případě by se jednalo o diagnózu PV).
- Nejsou erytroblasty v periferní krvi (PK).
- Nejsou nezralé granulocyty (metamyelocyty a mladší vývojová stadia) v PK.
- Nebývá splenomegalie při palpaci (pakliže není důsledkem např. portální trombózy); mírná splenomegalie při sonografickém vyšetření je však běžná. Spolu s předešlým bodem lze shrnout, že téměř není vyjádřena myeloidní metaplazie.
- Není přítomen Ph chromozom ani fuzní gen BCR/ABL; heterozygotní mutace JAK2V617F bývá zjišťována u asi 50 % nemocných, homozygotní mutace se u ET nepopisuje [6–8,22,44]. Mutace MPLW515K, MPLW515L a event. unikátně popsaná MPLW515S se vyskytují u 1–10 % pacientů převážně, ale nikoli výhradně, JAK2V617F-negativních [45–48]. U individuálních pacientů nesoucích mutaci JAK2 i MPL jde vždy o rozdílné klony buněk [49]. Vzácně se vyskytuje mutace MPLS505N [47], původně popisovaná u familiálních případů trombocytemie [50].
- Cytogenetické aberace jsou vzácné (delece 20q, +8, +9, –Y) [51].
- Hladina sérového erytropoetinu (s-Epo) bývá normální.
- Růst erytropoetin-independentních kolonií (EEC) z KD nebo PK je možný a významně koreluje s mutací JAK2V617F [52].
- CD34+ v PK jsou zvýšené (ale významně menší měrou než u PMF).
- Transformace do myelofibrózy (navzdory nedávné definici „post-ET MF“ [53]) je extrémně vzácná [54].
- V průběhu onemocnění nebývá pozorována transformace do s-AML [54] (není-li způsobena genotoxickou léčbou). Spolu s předešlým bodem lze shrnout, že klinický obraz nekomplikované ET je po dlouhá léta (desetiletí) neměnný [54].
- Rizikovými faktory pro trombózu jsou věk, předchozí trombóza [55,56], počet trombocytů (podle Michielsovy metaanalýzy asi 350–2 000 x 109/ l [16,57,58]). Role (nebo spíše rozhraní) počtu WBC je předmětem diskuze [59–62]. Mutace JAK2V617F je trombogenní [8,34], v českých analýzách jde o nejsilnější prediktivní faktor trombózy [63–65]. Další trombofilní stavy (vedle samotného MPO) rovněž výrazně zvyšují riziko trombózy: jsou to jak dědičné aberace, tak i získané stavy, např. jiné maligní nádory, těhotenství, větší úrazy, imobilizace, hormonální antikoncepce apod. [17,63–75].
- Rizikové faktory pro krvácení jsou počet trombocytů (> 1 000 u starších a > 1 500 × 109/ l u mladších osob), prodloužení časů v globálních koagulačních testech, sekundární von Willebrandova choroba [16,76–78], užívání kyseliny acetylsalicylové (ASA) či jiný typ antiagregační nebo antikoagulační léčby.
- Rizikovým faktorem pro přechod do s-AML je především genotoxická léčba.
B.2. Diagnóza polycythaemia vera
Diagnóza PV je dána splněním vždy 2 kritérií (jde celkem o 3 možné situace):
V typických případech
- 1. polyglobulie (erytrocytóza) nebo zvýšený RCM (nutno jej vyšetřit v případech hraničních hodnot červeného krevního obrazu) + JAK2V617F mutace.
V atypických případech
- 2. není-li přítomna polyglobulie (anebo zvýšená hodnota RCM), histopatologický obraz PV dle definice WHO/ECP + JAK2V617F mutace postačují pro diagnózu;
- 3. není-li přítomna JAK2V617F mutace, histopatologický obraz PV dle definice WHO/ECP + polyglobulie (anebo zvýšený RCM) postačují pro diagnózu.
Diagnóza PV by měla být v souladu se základními klinickými a laboratorními rysy onemocnění:
- Parametry červeného KO jsou zvýšené, nutno však odlišit „nepravou“ či „relativní“ polycytemii u stavů s restrikcí celkového plazmatického objemu (obvykle při dehydrataci) [32]. V každém hraničním či klinicky suspektním případě je doporučeno vyšetřit RCM: vždy při hematokritu 0,48–0,59 u muže a 0,44–0,55 u ženy spolu se stanovením celkového plazmatického objemu. RCM by měl být vztažen nikoli k hmotnosti, nýbrž k tělesnému povrchu pacienta. Za zvýšený RCM arbitrárně (v souladu s doporučeními ICSH a britskými doporučeními pro diagnostiku PV) považujeme hodnoty > 125 % předpokládané výše [32,79]. Výjimečně mohou být hodnoty červeného KO i v rámci normy: 1. u případů s expanzí celkového plazmatického objemu (poměrně běžnou při PV) [32] a 2. v případech zcela iniciálního stadia onemocnění, avšak s již typickým histopatologickým obrazem PV [16], v té chvíli probíhajícím pod obrazem ET definované podle PVSG kritérií – dle Michielse [16] jde o „prepolycytemickou fázi” PV.
- Zvýšený počet leukocytů je obvyklý (ale > 20 × 109/ l vzácně: spíše při poranění, infekci).
- Je normální nebo zvýšený počet trombocytů.
- Erytroblasty/nezralé granulocyty v PK jsou extrémní vzácností, nejedná-li se již o přechod do postpolycytemické myelofibrózy.
- Hodnota s-Epo bývá snížená anebo normální, ale spíše blízko dolní meze.
- Růst EEC je běžný.
- Může být žádná až značná splenomegalie při palpaci (i pod úroveň pupku), při ultrazvukovém vyšetření (USG) je splenomegalie obvyklá.
- Typická, alespoň heterozygotní JAK2V617F mutace (v exonu 14) je přítomna u asi 97–98 % případů při detekci alela-specifickou PCR [6,44]. Homozygozita (tzn. průkaz > 50 % mutovaných alel) je však u pokročilejších fází poměrně častá a je podmíněna ztrátou heterozygozity (tzv. uniparentální disomií) na krátkém raménku chromozomu 9, kde se nachází gen JAK2 [8,80]. Různé mutace (a také delece, ojediněle i s inzercí) v exonu 12 genu JAK2 u V617F-negativních pacientů jsou velmi vzácné, byl však při nich popsán odlišný histopatologický obraz (dle Scottové et al [81] bližší idiopatické erytrocytóze než PV) i poněkud rozdílné hodnoty KO (v průměru vyšší hodnoty parametrů červené řady, ale nižší hodnoty WBC a trombocytů) [81,82]. U PV se nevyskytují mutace genu MPL.
- Cytogenetické aberace jsou nalézány v menšině případů, jsou o málo častější než u ET (–Y, delece 20q, +8, +9) [51].
- Přechod do postpolycytemické myelofibrózy je častý, proces však trvá mnoho let.
- Přímý přechod do s-AML v krátké době je neobvyklý, ale možný (role genotoxické léčby není jasná, je však suspektní). Častěji k s-AML dojde po desetiletích vývoje onemocnění přes stadium postpolycytemické myelofibrózy.
- K rizikovým faktorům pro trombózu počítáme věk, předchozí trombózu, hodnotu hematokritu, zvýšený počet trombocytů [83–85], přídatný trombofilní stav (dědičný nebo získaný), hormonální antikoncepci. Role (resp. spíše mezní hodnota) WBC i úloha nálože JAK2V617F alely jsou předmětem diskuze [59,62].
- Rizikové faktory pro krvácení jsou: léčba ASA, event. jiná antiagregační/antikoagulační léčba, lokální defekty (např. sliznic), v přirozeném vývoji onemocnění počet trombocytů (> 1 000 u starších a > 1 500 × 109/ l u mladších nemocných), nezřídka spojený s prodlouženými časy v globálních koagulačních testech, někdy i se sekundární von Willebrandovou chorobou.
- Rizikové faktory přechodu do myelofibrózy nejsou známy.
- K rizikovým faktorům pro přechod do s-AML nutno počítat genotoxickou léčbu.
B.3. Diagnóza primární myelofibrózy
Hlavním kritériem pro stanovení je histopatologický obraz PMF (včetně prefibrotického stadia) v souladu s definicemi WHO 2001/ECP 2002 [24,27]. Histopatologický nález však musí zhruba zapadat do typického klinického obrazu příslušného stadia PMF. Vývoj onemocnění od raného obrazu (který je v souladu s diagnózou ET podle PVSG kritérií) až do pokročilejších fází s výraznou myeloidní metaplazií trvá obvykle 2–3 desetiletí. Klinické projevy však ne vždy zcela korelují se stupněm fibrózy dřeně (grading fibrózy u MPO je uveden v tab. 4), např. ani ve stadiu osteomyelosklerózy (MF-3) nemusí být palpovatelná splenomegalie. Typické rysy onemocnění od počátečních stadií až k pokročilé PMF jsou shrnuty v tab. 5. Vedle toho uvádíme ještě některé další laboratorní a klinické charakteristiky PMF:
![Stupně fibrózy kostní dřeně podle tzv. evropských konsenzus kritérií [35].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/85a922f38b4e69cf3d0671fcbdee4df9.jpg)
- JAK2V617F mutace může být přítomna (asi 55 % případů při detekci alela-specifickou PCR [6,44]). Homozygozita (> 50 % mutovaných alel) v pokročilejších stadiích je možná. Mutace MPLW515K/L jsou vzácné (asi 5–10 %), patrně však častější než u ET [45–48,86]. I tato mutace se může vyskytnout jako homozygotní.
- Histologický obraz u případů s mutací MPL zapadá do rámce PMF, ale např. erytropoéza je v průměru chudší [87].
- Cytogenetické aberace jsou relativně časté (pokud je úspěšná aspirace dřeně – krom raných stadií však bývá obtížná), jsou nalézány zhruba u 1/3 případů. Fluorescenční in situ hybridizace (FISH) může být přínosná; interfázickou FISH lze použít i při analýze PK.
- Růst EEC je obvyklý.
- Celková prognóza: k jejímu stanovení doporučujeme Lille skóre [88], Mayo klasifikaci [89], kolínské skóre [90], skóre dle IWG-MFRT (International Working Group for Myelofibrosis Research and Treatment) [91]. Všechna jsou založena pouze na hodnotách krevního obrazu, event. na klinických znacích, a jsou všeobecně aplikovatelná.
Upozornění: k posouzení indikace transplantace hematopoetických buněk je však potřeba vzít v úvahu i další kritéria – viz níže.
B.4. Diagnóza MPO s trombocytemií (MPO-T)
Termínu MPO-T [17,30] lze užít v případě trombocytemie, u které prokazuje histopatologie celkem jednoznačně MPO (anebo je přítomen klonální marker, např. mutace JAK2), avšak její výsledek je v „šedé zóně”, pokud se týká přesnější nosologické diagnózy v rámci Ph- MPO dle WHO/ECP kritérií. Dle PVSG kritérií se jedná obvykle o ET.
C. Klinická – non-nosologická diagnostika
Non-nosologický přístup lze v diagnostice MPO doporučit u případů, u kterých nebyla odečtena histopatologie kostní dřeně podle doporučení WHO/ECP (tj. trepanobiopsie buď nebyla provedena, anebo nebyla odečtena v souladu s těmito WHO/ECP principy, včetně situace, kdy byla provedena až poté, co pacient již absolvoval cytoreduktivní terapii, která může významnou měrou změnit histopatologický obraz). Pro tyto případy je doporučena klasifikace podle PVSG [20]. Podle této klinické klasifikace (ve své podstatě non-nosologické – viz výše – odstavec A) bude většina časnějších stadií PMF klasifikována jako ET. Nicméně diagnostiku PV lze v naprosté většině (JAK2V617F-pozitivních) případů provést podle nosologického principu (viz výše – odstavec B.2.). K odlišení ET od PV podle nosologického principu skupiny CZEMP je však poměrně často potřeba stanovení RCM, které je nutné provést před započetím terapie, jinak je nelze spolehlivě odlišit. PVSG-definovaná ET tedy může být podle nosologických kritérií buď:
- „pravou” ET,
- PMF (stadia MF-0, MF-1, vzácně i MF-2), anebo
- „prepolycytemickou” PV [16].
Diferenciální diagnostika těchto stavů (podle nosologických principů) má zásadní význam především u mladších pacientů, neboť jednotlivá onemocnění se významně liší prognózou [16,18,90].
D. Algoritmy pro vyšetření trombocytózy a polyglobulie
D.1. Algoritmus pro vyšetření trombocytózy
Doporučujeme následující postup:
- Anamnéza včetně rodinné anamnézy (pozitivní rozpoznání familiálního postižení MPO). Vyloučit evidentní sekundární případy (hyposideremie, jiné nádory atd.).
- Vyšetření JAK2V617F mutace a fuze BCR/ABL (k odlišení CML).
- Biopsie (není-li přítomna polyglobulie – v tom případě postupujeme podle příslušného algoritmu pro polyglobulii – viz níže. U případů s hraničními hodnotami parametrů červené řady nutno vyšetřit s-Epo a RCM).
Nutno si uvědomit, že patrně většina případů se zvýšenou hodnotou krevních destiček v praxi internisty či chirurga budou sekundární trombocytózy, zatímco v praxi specializovaných hematologických center se bude častěji jednat o primární MPO-T. Biopsie kostní dřeně odliší s velkou jistotou primární MPO-T od sekundární trombocytózy [30].
D.2. Algoritmus pro vyšetření a diferenciální diagnostiku polyglobulie
- Anamnéza včetně rodinné anamnézy (pozitivní rozpoznání familiálního postižení MPO). Vyloučit evidentní sekundární případy (jiné nádory, ventilační insuficience včetně případů kardiálního selhávání a spánkové apnoe atd.).
- JAK2V617F mutace, saturace hemoglobinu kyslíkem (stačí HbO2 oxymetrie), s-Epo.
- U případů s hraničními hodnotami parametrů červené řady nutno vyšetřit RCM.
- U všech JAK2V617F-negativních případů: provést biopsii dřeně, vyšetřit mutaci JAK2 v exonu 12, vyšetřit růst EEC a patologické hemoglobiny.
- Další mutace (HIF-2α, PHD2, VHL, genu pro Epo-R) podle výsledků s-Epo a EEC.
E. Léčba Ph- MPO
CZEMP doporučuje podávat léčbu podle individuálního rizika daného pacienta, a to jak ve smyslu rizika trombózy a krvácení, tak i biologického vývoje onemocnění, především možného přechodu onemocnění do s-AML. Vždy je třeba vycházet z principu non nocere a každou podanou léčbu vážit z tohoto aspektu s přihlédnutím k odhadu celkového přežití, morbidity a kvality života bez zvažované léčby.
E.1. Léčba MPO s trombocytemií (MPO-T)
Léčebný postup, i nyní doporučovaný CZEMP, byl již skupinou publikován v roce 2005 [17,29]. V těchto publikacích je detailní rozbor literatury, která vedla k formulaci minulé verze doporučení. Doporučení byla založena především na analýze rizikových faktorů pro trombózu a stratifikaci léčby podle přítomnosti těchto rizik. Při formulaci doporučení byl uplatněn princip primum non nocere, tj. vyhnutí se potenciálně leukemogenním látkám při léčbě mladších jedinců pod 60 let. Jako taková byla a nadále jsou naše doporučení založena především na konsenzuálním expertním názoru, neboť v literatuře prakticky chybí kvalitně interpretované studie kategorie A z hlediska tzv. „evidence-based medicine“; většina našich doporučení proto spadá do kategorie B [92].
Od doby první publikace [17,29] doznala doporučení CZEMP pro léčbu MPO-T jen několik málo změn:
- Již v době objevu mutace JAK2V617F v roce 2005 se zdálo pravděpodobné, že je tato mutace spojena s vyšším výskytem trombózy [8,34]. Tento předpoklad byl pak prověřen v reálné praxi v ČR jak na úrovni institucionální, tak i multiinstitucionální v datech registru pacientů léčených anagrelidem [63,64]. JAK2 mutace se zdá být vůbec nejsilnějším protrombotickým faktorem. Proto ji CZEMP zařadil do své rizikové stratifikace pacientů s MPO-T.
- Byla zvýšena hranice rizikovosti pacientů z 60 na 65 let v souladu s rostoucím přežitím české populace. Očekávaná střední délka života mužů, resp. žen ve věku 65 let byla v roce 2008 15,1, resp. 18,4 let, tzn., že medián věku, kterého se dožijí, je 80,1, resp. 83,4 let [93].
Doporučení pro léčbu mají několik základních východisek:
- Analýzu hlavních rizikových
faktorů trombózy, což jsou:
- a) věk [55,56];
- b) předchozí trombóza [55,56];
- c) přídatné trombofilní stavy, a to vrozené, ale i získané (což je podporováno i výsledky z ČR). K těmto trombofilním stavům počítáme především tzv. hereditární trombofilie, tj. dědičný deficit proteinů C nebo S, hetero- i homozygotní „leidenskou“ mutaci genu f. V, mutaci protrombinového genu G→A 20210, deficit antitrombinu. Podobné riziko může představovat i jakýkoli hyperkoagulační stav, např. při nádorových onemocněních, operačních výkonech a v těhotenství [17,64,66–75]. Konečně, zdá se být logické, že v podstatě jakýkoli trombofilní faktor, který se uplatňuje v „normální“ populaci bez MPO-T, může zvýšit riziko trombózy při MPO-T.Může to platit i o zvýšení hladin fibrinogenu (a dysfibrinogenemii), f. VIII, deficitu f. XII, antifosfolipidovém syndromu i pro stavy po chirurgických výkonech a užívání hormonální antikoncepce [17,75,94,95]. Soustavné studie na tato témata však u MPO-T nebyly publikovány;
- d) nově objeveným, avšak možná nejvýznamnějším protrombotickým rizikovým faktorem je mutace JAK2V617F [8,34,63,64]. Názory na to, zda hraje roli i její alelická nálož, se zatím různí [96–99];
- e)
v neposlední řadě také počet
krevních destiček [16,57] a navíc
i doba expozice postiženého jedince trombocytemií jsou
významným faktorem rizika trombózy [55]. Podobně je i riziko
krvácení u MPO-T funkcí počtu trombocytů, jak zachytil
Michiels ve své metaanalýze dat 809 pacientů s ET z celkem
11 studií (obr. 2) [16,58]. U MPO-T dochází k paradoxu,
že v jednom okamžiku je pacient ohrožen jak trombózou, tak
i krvácením – tento stav nazýval Dameshek „double
jeopardy”, tj. dvojí nebezpečí [100]. Michielsova
metaanalýza osvětluje detailně vztah mezi počty trombocytů
a oběma riziky. Zatímco hemoragiemi jsou ohroženi pacienti
s počty trombocytů nad 1 000 × 109/ l
a s přibývajícím počtem destiček lineárně roste
riziko krvácení (obr. 2 – viz klín na obrázku „doutníku
a klínu“), trombotickou komplikací jsou ohroženi pacienti
s počty trombocytů 350–2 200 × 109/ l,
nejvíce pak při počtech kolem 500–1 900 × 109/ l
(viz doutník na obr. 1) [16,58]. Z uvedeného tedy vyplývá,
že obě rizika jsou při počtech asi 1 000–2 200 × 109/ l
aktuální současně. Platnost Michielsových závěrů však není
možno absolutizovat: většina hodnocených trombotických událostí
byla mikrocirkulační povahy (erytromelalgie a akrocyanózy
u asi 30–40 %
pacientů) a arteriálního původu (infarkty myokardu, TIA,
uzávěry periferních tepen u 25–30 %
nemocných). Z dosud nepublikovaných dat registru českých
pacientů léčených anagrelidem naopak vyplývá, že incidence
„velkých“ trombotických událostí arteriálního, resp.
venózního původu byla identická:
53 : 52, tj. 9,7 : 9,5 %.
Na obr. 2 je zachycen Michielsův návrh, jak postupovat právě při
zmíněném dvojím nebezpečí: nejprve cytoreduktivní léčbou
eliminovat riziko krvácení, a dostat tak pacienta do stavu,
kdy je již ohrožen především rizikem trombózy a teprve
tehdy mu nasadit antiagregační léčbu k její prevenci.
Obr. 2. Schéma rizika trombózy a krvácení v závislosti na počtu trombocytů s naznačením léčebných opatření. Podle Michielse et al [16,58]. ![Schéma rizika trombózy a krvácení v závislosti na počtu trombocytů s naznačením léčebných opatření. Podle Michielse et al [16,58].](https://www.prelekara.sk/media/cache/resolve/media_object_image_small/media/image/31355315ed37d7a400b513f3041e10a2.jpg)
- f) při MPO-T se aktivují jak trombocyty, tak i leukocyty, které spolu mohou tvořit komplexy a hrát významnou roli v patogenezi trombózy [101–103]. Potom není překvapivé, že i počet leukocytů se v některých studiích jeví jako prognostický faktor z hlediska rizika trombózy. Jako diskriminující hodnota počtu leukocytů se uvádí hodnota 8,7–15 × 109/ l [59–61]. Nicméně v literatuře se údaje dosti rozcházejí a je třeba dalších studií. Doporučení CZEMP proto zatím neberou zřetel na počet leukocytů. Ošetřující lékař má volnou ruku v tom, zda k nim při volbě terapie přihlédne.
- g) tzv. kardiovaskulární rizika, platná v obecné populaci bez MPO, měla v některých studiích signifikantní dopad na incidenci trombózy při MPO(-T), v jiných však ne. Týká se to např. obezity, zvýšené hladiny cholesterolu a triglyceridů, hypertenze, kouření, přítomnosti diabetes mellitus. Ve výsledcích z českého registru pacientů léčených anagrelidem se žádný z těchto faktorů signifikantně neuplatňuje [104]. Proto CZEMP tato zmíněná rizika nepoužívá pro definici vysokého rizika trombózy u MPO-T, nicméně doporučení CZEMP nebrání ošetřujícímu lékaři brát na tato rizika zřetel při komplexní léčbě pacienta.
- Tromboreduktivní léčba snižuje riziko trombózy u pacientů s vysokým rizikem [105].
- Tromboreduktivní léčba odstraňuje symptomy trombocytemie [106].
- Antiagregační léčba snižuje riziko trombózy u pacientů s nízkým rizikem [107] podobně jako u PV (u druhé z diagnóz důkaz kategorie A [108]). Je významným preventivním faktorem arteriální trombózy u obecné populace [109]. Velmi účinně odstraňuje především mikrovaskulární symptomy [16,110].
- Všechna cytostatika (včetně nejpoužívanější hydroxyurey – HU) jsou prokazatelně, anebo přinejmenším potenciálně leukemogenní. Panuje již konsenzus v tom, že léčba několika cytostatiky po sobě zvyšuje riziko s-AML [17,111–117]. I ve studii s krátkým mediánem sledování byla prokázána leukemogenicita dříve často užívaných typů terapie, a sice alkylátorů (melfalanu, chlorambucilu), radiofosforu, pipobromanu a busulfanu [118]. Nicméně nejsou jednotné názory na to, zda nyní nejčastěji užívané cytostatikum HU je samo o sobě leukemogenní, ačkoli četné in vitro i in vivo studie nasvědčují i tomuto – viz níže odstavec o HU (E.1.2.3.).
![Schéma rizika trombózy a krvácení v závislosti na počtu trombocytů s naznačením léčebných opatření. Podle Michielse et al [16,58].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/31355315ed37d7a400b513f3041e10a2.jpg)
E.1.1. Algoritmus pro léčbu MPO-T
Z výše uvedených východisek vyplývá doporučený terapeutický algoritmus (tab. 6). Uvedený postup byl v červnu roku 2010 přijat i jako standard léčby CEMPO (Central European Myeloproliferative Study Organization) sdružující špičková pracoviště z ČR, Slovenska, Polska, Bavorska, Rakouska, Maďarska, Slovinska, Chorvatska a Rumunska.

Nemocní jsou stratifikováni jednak podle věku (18–65, resp. > 65 let), a jednak podle toho, zda jsou symptomatičtí, zda měli v anamnéze tromboembolickou příhodu, zda mají přídatné trombofilní riziko, anebo zda mají JAK2 mutaci. K tomu, aby byl pacient považován za rizikového, stačí mít alespoň jednu z uvedených charakteristik. Z povahy věci mohou být některé rizikové faktory pouze přechodného charakteru – např. získaný trombofilní stav v podobě dočasné imobilizace, chirurgického výkonu a stavu bezprostředně po něm, těhotenství apod. Dále jsou pacienti stratifikováni podle počtu destiček (event. dle rychlosti jejich nárůstu v kategorii „600–1 000 progresivní” – jde o pacienty se vzestupem počtu trombocytů vyšším než 200 x 109/ l za 2 měsíce). Z uvedeného vyplývá, že nízkorizikoví jsou pouze pacienti s trombocyty < 1 000 × 109/ l, kteří nemají žádnou z uvedených charakteristik vysokého rizika (jde tedy evidentně o menšinu nemocných).
Rizikové pacienty zásadně léčíme tromboreduktivy. Mladší pacienty do 65 let preferenčně anagrelidem (ANG) nebo interferonem-α (IFN), starší pacienty > 65 let hydroxyureou (HU). Byla-li zahájena tromboreduktivní terapie, je třeba v ní pokračovat obvykle kontinuálně (teoreticky s výjimkou odeznělého rizika u jinak nízkorizikových nemocných s počty trombocytů < 1 000 × 109/ l). Rozhodnutí, zda podat u mladého nemocného jako léčbu první linie ANG nebo IFN, je v rukou ošetřujícího hematologa. Vzhledem k tomu, že se o leukocytóze uvažuje jako o možném rizikovém faktoru pro trombózu (viz výše) [59–62], lze nezávazně doporučit spíše použití IFN u pacientů s leukocytózou, tj. počtem WBC nad normu, a ANG u pacientů s normálními počty leukocytů. Arbitrárně stanoveným terapeutickým cílem je dosažení a udržování počtu trombocytů < 400 × 109/ l u vysokorizikových, resp. < 600 × 109/ l u nízkorizikových pacientů. Antiagregační terapie preferenčně kyselinou acetylsalicylovou (ASA) se řídí u MPO-T počtem trombocytů: nepodáváme ji u pacientů s trombocyty > 1 000 × 109/ l, event. u mladších jedinců do 40 let do počtu 1 500 × 109/ l trombocytů, jelikož by silně narůstalo riziko krvácení. Teprve je-li tromboreduktivní léčbou dosaženo stabilní úrovně počtu destiček < 400 × 109/ l, je možno ASA vysadit, nemá-li pacient v anamnéze arteriální trombózu. Překročí-li pacient užívající neleukemogenní léčbu hranici 65 let, není důvod terapii měnit.
E.1.2. Nejdůležitější modality léčby MPO-T
Jak vyplývá z již uvedených terapeutických doporučení, jsou nejdůležitějšími léčivy v terapii MPO-T tromboreduktivní látky ANG, IFN a HU, a antiagregans ASA. Nezastupitelnou roli mají rovněž trombocytaferézy a antikoagulační terapie.
E.1.2.1. Anagrelid (ANG)
ANG patří do skupiny imidazochinazolinů (strukturálně příbuznými látkami byly např. antiagregans dipyridamol a antihypertenzivum prazosin). ANG, na rozdíl od všech ostatních tromboreduktivních látek, působí selektivně na megakaryocytopoézu (pouze však u člověka, nikoli u zvířat) a neovlivňuje počet leukocytů a erytrocytů. Jde o nemutagenní látku necytostatické povahy: ANG působí až v postmitotické fázi vývoje magakaryocytů, neboť nebrání v proliferaci kmenovým buňkám pro megakaryocytopoézu (CFU-Meg), avšak působí zástavu jejich maturace, snižuje jejich ploidii a velikost (nikoli počet). V důsledku toho neodštěpují tyto prekurzory trombocyty [119–127]. Přesný mechanizmus, jak k tomu dochází, ani význam jednotlivých metabolitů ANG, není zcela vyjasněn. Uvažuje se o možnosti, že ANG blokuje vazbu lidského trombopoetinu (TPO) na jeho receptor C-MPL, který je mezi živočišnými druhy rozdílný [124,127]. Druhově specifická je také míra antiagregačního účinku ANG. U zdravého člověka má sice ANG zřetelný antiagregační účinek, avšak při léčbě MPO není tento efekt ANG v dávkách snižujících trombocytemii (zhruba 10krát nižších) zřejmý [124,127–129].
V současných doporučeních CZEMP (tab. 6) je ANG lékem první volby pro MPO-T u osob do 65 let, pakliže není přítomna výrazná leukocytóza či polycytemie (v tom případě bychom dali spíše přednost IFN). U pacientů starších 60 let je však při jeho podávání nutná vysoká obezřetnost: musíme si být dostatečně jisti, že pacient nemá závažné kardiovaskulární onemocnění. Pozitivně inotropní účinek ANG by teoreticky mohl vést např. k infarktu myokardu [120,122,124,126,127]. Je kontraindikován u nemocných se srdeční slabostí. Také se nesmí užívat v graviditě, neboť jeho molekula je malá (310,5 Da) a prostupuje placentární bariérou (ačkoli i při překročení tohoto doporučení se rodí jinak zdravé děti, které však mohou mít reverzibilní trombocytopenii [130]). Nejčastějším nežádoucím účinkem jsou palpitace a bolesti hlavy spojené s vazodilatačním a mírným pozitivně inotropním účinkem ANG [122,124,131–133], obvykle nevyžadující terapii a spontánně odeznívající. V opačném případě jsou snadno zvládnutelné malou dávkou beta-blokátoru a běžnými analgetiky. U nás registrovaný přípravek Thromboreductin® má na rozdíl od Agrylinu® (nyní Xagridu®, v ČR neobchodovaného) méně nežádoucích účinků, což souvisí s formulací kapslí a jejich farmakokinetikou – u prvního z nich jsou dosahovány nižší špičkové plazmatické hladiny (při ekvivalentní tromboredukci); k nežádoucím účinkům dochází právě při vyšší špičkové hladině léčiva [134]. Proto bylo ve studiích s Agrylinem® dokumentováno mnohem více nežádoucích účinků a nutných vysazení preparátu než ve studiích s Thromboreductinem® [132,135–138]. Při dlouhodobé terapii MPO-T Thromboreductinem® v ČR je léčba přerušena pouze ve 12 % případů, přičemž intolerance léku je minoritním důvodem jeho vysazení [138].
Iniciálně by měla být podávána dávka do 1 mg, maximálně do 2 mg denně (rozděleně po jednotlivých kapslích), podle odhadu potřebné dávky pro kontrolu trombocytemie – vyšší iniciální dávkování může být zatíženo vyšším výskytem nežádoucích účinků [124]. Dávka by měla být individuálně titrována dle odpovědi pacienta k co nejrychlejšímu dosažení terapeutického cíle. Maximální dávka je 5 mg denně. Zpočátku je tedy lépe rozložit dávky po 0,5 mg vícekrát denně, a teprve při toleranci lze zvyšovat jednotlivou dávku do 1,5 mg. ANG zmírňuje mikrovaskulární symptomy paralelně se snižujícím se počtem trombocytů [106]. Na ANG odpovídá zhruba 90–95 % nemocných, v datech českého registru pacientů léčených ANG se dosáhne terapeutického cíle (tj. mnohem „tvrdšího“ kritéria) u 82 % nemocných po 6 měsících léčby [124,127,132,138,139]. Původní zprávy o indukci výrazné anémie při podávání ANG [128,132,140] je nutno brát s rezervou – v uvedených studiích pacientů s ET definovanou podle PVSG kritérií bylo pravděpodobně mnoho pacientů s PMF, u kterých patří anémie k přirozenému vývoji onemocnění. V souladu s tím narůstala ve zmíněných studiích i leukocytóza. Naopak česká data ukazují na to, že poklesy hladin hemoglobinu jsou velmi mírné – asi o 10 g/ l po 4 letech léčby, přičemž hodnoty leukocytů jsou zcela stabilní a dlouhodobá tolerance ANG je velmi příznivá [137–139]. Z povahy selektivního účinku na megakaryocytární řadu vyplývá, že ANG nemůže významněji ovlivnit splenomegalii.
ANG lze s výhodou kombinovat s jinými tromboreduktivy – IFN a HU, jakož i s antiagregancii. Jak jsme již uvedli, ANG byl původně vyvíjen jako antiagregans (je inhibitorem cAMP fosfodiesterázy), avšak u pacientů s MPO v dávkách působících snížení počtu krevních destiček nemá podstatný antiagregační účinek [122,128,129]. Některé klinické studie však naznačují, že může potencovat antiagregační účinek ASA a ojediněle může při této kombinaci docházet ke krvácení [135,141]. Je velmi pravděpodobné, že krvácení může snadno nastat při překročení pravidla, že se ASA nemá podávat pacientům s > 1 000 × 109/ l trombocytů, což (nechtěně) dokumentuje britská studie PT1 [141]. ANG byl srovnán ve 2 randomizovaných studiích s HU [38,141]. První studie, britská PT1 [141] (velmi špatně koncipovaná i interpretovaná – viz naše kritika [17]), svědčila pro větší počet nežádoucích účinků ve větvi s ANG (v kombinaci s ASA) – především se jednalo o zvýšenou incidenci tranzitorních ischemických atak (Petrides však postuloval, že se v této studii mohlo spíše jednat o nerozpoznaná mikrokrvácení do CNS [142], nikoli o arteriální trombózy – jeho logické vysvětlení dle našeho názoru také spíše odpovídá realitě). Naopak druhá studie s názvem ANAHYDRET, provedená u pacientů s WHO-definovanou ET, ukazuje na srovnatelný účinek, pokud se týká kontroly počtu trombocytů i pokud se týká incidence trombotických a krvácivých příhod [38]. Nicméně ze studie PT1 i z dat registru českých pacientů léčených ANG vyplývá, že pacienti léčení ANG jsou v praxi poměrně často poddávkováni, eskalace dávek není dostatečně rychlá, a proto se optimální kontroly počtu trombocytů dosahuje za více než 6 měsíců [137,138,143]. Prodlužuje se tak doba expozice pacienta zvýšenému počtu trombocytů, a tudíž i riziko trombotických komplikací [55]. Hlavní výhodou ANG je pochopitelně jeho neleukemogenicita a absence rizika vyvolání jiné malignity i při dlouhodobém užívání. Preskripce ANG v ČR musí být schvalována „hematologickým centrem“. V budoucnu bude patrně dostupný i retardovaný přípravek ANG.
E.1.2.2. Interferon-α (IFN)
IFN je vedle ANG další neleukemogenní a nekancerogenní tromboreduktivní látkou pro léčbu MPO-T první linie pro pacienty do 65 let, ovšem s naprosto odlišným mechanizmem účinku. Jde o cytokin(y) s imunomodulačním a antiproliferativním účinkem na myeloidní kmenové buňky, může mít i mírně vyjádřené proapoptotické účinky i lehce zkracovat dobu přežití trombocytů [117,144–146]. Tento antiproliferativní efekt je nespecifický z hlediska vývojové řady v rámci myelopoezy, proto je třeba počítat s jeho vedlejšími hematologickými účinky, tj. s anémií a leukopenií. Na druhou stranu je tento poměrně výrazný antiproliferativní účinek spojen i se zřetelným efektem na redukci splenomegalie. V léčbě ET je užíván od poloviny 80. let minulého století, řada studií prokázala jeho schopnost snížit a udržet na nízké hladině hodnoty trombocytů [147–149], na léčbu odpovídá kolem 85 % nemocných [117]. Ve starší metaanalýze léčby IFN u pacientů s ET [145] se uvádělo dosažení takovéto remise až u 12 % pacientů. Tento výsledek je však třeba brát velmi opatrně, neboť v analyzovaných studiích PVSG kritérii definované ET byla jistě převaha pacientů s časnými fázemi PMF. U této diagnózy se s přirozeným vývojem onemocnění z proliferativní do fibrotické fáze postupně snižuje potřeba jakékoli cytoreduktivní terapie. Nicméně i modernější studie naznačují, že IFN patrně nejvýrazněji snižuje nálož mutované alely JAK2 ze všech tromboreduktiv a v některých případech je také schopen navodit i léčbou neudržovanou kompletní remisi a molekulární remisi u původně JAK2V617F-pozitivních pacientů. Zbavuje pacienty symptomů [145,146].
IFN je indikován dle doporučení CZEMP pro případy vysokorizikového MPO-T u pacientů do 65 let věku (tab. 6) jako alternativa léčby ANG. Rozhodnutí mezi těmito dvěma látkami je ponecháno na ošetřujícím hematologovi. IFN je vhodnou alternativou především pro pacienty s leukocytózou a splenomegalií. IFN je lékem volby u těhotných pacientek [117,146,150], potřebují-li tromboredukci. Z italského registru těhotenství při ET vyplývá, že na rozdíl od ASA snižuje IFN významně procento spontánních potratů. V registru je dokumentováno narození živého dítěte v 95 % těhotenství při použití IFN [150].
Původně se léčebně používalo směsi přirozených IFN (dříve Wellferon®, nyní v ČR obtížně dostupný Alfaferone®), v posledních 10 letech se nejčastěji užívají rekombinantní přípravky (v ČR registrované rIFN-α2a Roferon-A® a rIFN-α2b Intron A®). Léčbu de novo pacientů zahajujeme obvykle dávkou 2–3 MIU denně, dávku však nutno (podobně jako u ostatních tromboreduktiv) rychle titrovat k dosažení terapeutického cíle. Při přechodu na IFN z jiné léčby můžeme vystačit např. s dávkou 3 MIU 3krát týdně. Tolerance IFN však může být lepší při užití nižších jednotlivých dávek za cenu častější aplikace. Nepřekračujeme dávku 30 MIU týdně [29] – v případě nedostatečné odpovědi na IFN jej lze kombinovat s ANG nebo HU. Nevýhodou podávání IFN je nutnost injekční s.c. aplikace denně, anebo alespoň několikrát týdně. Léčba IFN je spojena s řadou nežádoucích účinků: zpočátku jsou to především reakce alergické povahy na cizorodou bílkovinu. IFN může vyvolat vysokou horečku, častější však jsou subfebrilie, celková nevolnost charakteru chřipkových příznaků s bolestmi svalů. Obvyklé je i lokální začervenání v místě vpichu. S dobou podávání tyto příznaky většinou mizí, avšak někteří pacienti nemohou pro perzistující horečky a schvácenost pokračovat v terapii. Dlouhodobé podávání může vést k psychickým problémům, anxiózním a častěji depresivním stavům. Někteří pacienti mohou mít anorexii a ztrácet na váze. U řady nemocných lze pozorovat tvorbu antityroidálních protilátek, pouze u menšiny z nich se však projeví hypotyreóza na podkladě autoimunitní thyreoiditis. Vzácněji může být pozorována hepatotoxicita – je proto nutno pravidelně kontrolovat jaterní testy. V různých studiích se udává nutnost vysazení z důvodu intolerance v průměru kolem 20–25 % (v jednotlivých pracích 15–66 %) [117]. V zahraničí se již běžně podávají depotní, pegylované přípravky IFN-α (Pegasys®, Peg-Intron®, navázané na polyethylenglykol – PEG), což vede jednak ke snížení četnosti aplikace (jednou týdně), ale také (alespoň podle některých autorů) i ke snížení nežádoucích účinků [117,146].
E.1.2.3. Hydroxyurea (HU)
HU (hydroxykarbamid; v ČR registrovaný Litalir®) je doporučenou terapií u pacientů starších 65 let. Její molekula byla syntetizována prvně před více než 100 lety. Jde o nealkylační cytostatikum, inhibitor ribonukleotid-reduktázy, a tudíž i DNA syntézy [117,151]. Četné experimentální práce však ukazují na indukci genetické nestability a genotoxicity působením HU [123,151–156]. U experimentálních zvířat je HU teratogenní a u samců brání spermatogenezi [123,157]. Není pak překvapením, že i četné klinické studie ukazují na leukemogenicitu HU významně narůstající po 10 letech léčby HU [158–167]. Navíc HU může indukovat nejen kožní karcinomy, ale působit i další dermatologické problémy: od kvalitu života zhoršující tzv. „hydroxyureové dermopatie” lichenoidní povahy až po špatně se hojící vředy na končetinách [168–172] a afty na sliznicích. U nevelkého procenta nemocných může HU vyvolat horečku jinak nejasné etiologie [173]. Vzácně byla popsána i možná hepatotoxicita [174]. Ačkoli doklady o leukemogenicitě HU sice nejsou z hlediska tzv. „medicíny založené na důkazech“ stále podle řady autorů dostatečné [113,175,176], výše uvedené studie byly naopak dostatečným důvodem, aby CZEMP nedoporučovala dlouhodobou terapii HU (v řádu mnoha let) u osob mladších 65 let.
Na druhé straně je HU v počátku léčby ze všech tromboreduktivních látek nejlépe tolerována, vedlejší účinky (např. nauzea) jsou výjimečné. Navíc odpověď na léčbu lze u HU velmi dobře předvídat a není potřeba zdlouhavé titrace dávek pro dosažení žádaného terapeutického efektu (což může být někdy problém při léčbě ANG i IFN). Proto je HU velmi vhodná v situacích, kdy potřebujeme dosáhnout velmi rychlé tromboredukce. Obvyklá dávka při zahájení terapie se pohybuje kolem 1 g denně, avšak při vysokých hodnotách trombocytů (asi > 2 000 × 109/ l) s hrozícím rizikem krvácení lze krátkodobě podávat i dávku 3 g denně. Proto i CZEMP doporučuje HU na úvod tromboreduktivní terapie i u mladších jedinců s trombocyty > 2 000 × 109/ l, a teprve později (např. při hladině trombocytů < 1 000 × 109/ l, kdy již nehrozí krvácivé komplikace) převést pacienta na léčbu ANG či IFN. HU se také dobře hodí do kombinace s ANG a IFN, což je mimořádně přínosné u pacientů, kteří mají špatně kontrolovatelnou trombocytemii, anebo vedlejší účinky při léčbě ANG nebo IFN – kombinace dovolí podávat nižší dávky všech uvedených látek. Dlouhodobě by dávka HU neměla přesahovat 2 g denně.
Na rozdíl od ANG není HU specifická svým cytoreduktivním účinkem pouze na megakaryocytopoézu. Proto je poměrně časté, že se u pacientů může vyvinout anémie a leukopenie (neutropenie), aniž by bylo dosaženo cílového tromboreduktivního účinku. HU má jistý, ale nepříliš výrazný potenciál redukovat splenomegalii. Jak ukázala randomizovaná studie, snižuje HU prokazatelně svými cytoreduktivními účinky výskyt trombotických komplikací u pacientů s ET, [105]. V obvyklých dávkách mají HU a ANG srovnatelné tromboreduktivní účinky [38,113].
E.1.2.4. Antiagregační léčba
Ačkoli je charakter poruchy trombocytárních funkcí při MPO-T velmi variabilní a agregační testy mohou ukázat jak snížení, tak i zvýšení těchto funkcí, obecně dochází ke zvýšení tvorby destičkově-specifických proteinů, zvýšené tvorbě tromboxanu a zvýšení exprese některých „aktivačních“ epitopů na povrchu trombocytů. Celkem vzato se jedná o stav aktivace trombocytů [177,178]. Podle novějších představ tvoří v krevním řečišti aktivované trombocyty shluky s aktivovanými neutrofily a monocyty [101,102]. Klinicky však je extrémně důležité, že nejen cytoreduktivní, ale i antiagregační léčba může vést k úpravě (nikoli však normalizaci) některých poruch trombocytárních funkcí, zatímco jiné funkce cíleně snižuje [177,179].
Jak vyplývá ze strategii léčby relevantní Michielsovy metaanalýzy (obr. 2) [16,58], antiagregační léčba je možná a zároveň také plně indikovaná u pacientů s trombocyty < 1 000 × 109/ l, event. u mladších jedinců do 40 let do počtu 1 500 × 109/ l trombocytů. Při vyšších počtech krevních destiček již výrazně stoupá riziko krvácení [117,179,180]. Antiagregační léčba snižuje významnou měrou trombotické riziko u MPO [16,107,108,179–181]. Antiagregace je postačující léčbou nemocných s relativně nízkým rizikem trombózy, tj. u osob ≤ 65 let bez dalšího přídatného trombofilního rizika dle doporučení CZEMP (tab. 6) i jiných [112]. U případů s vyšším rizikem trombózy (přídatným trombofilním stavem) je nutno kombinovat antiagregační a cytoreduktivní terapii.
Zdaleka nejčastěji užívaným a zároveň obtížně zastupitelným léčivem je kyselina acetylsalicylová (ASA). Nízká dávka ASA poměrně selektivně snižuje blokem cyklooxygenázy (nověji prostaglandin syntetázy) tvorbu proagregačního tromboxanu A2 v trombocytech, zatímco pouze krátkodobě sníží tvorbu inhibitoru adheze a agregace, prostacyklinu PGI2 v endotelových buňkách [178,182]. Doporučené dávkování u MPO je 50–100 mg denně [16,107,179]. Jelikož jde obvykle o dlouhodobé podávání, volíme formulace co nejméně iritující sliznici GIT. Tyto vlastnosti má ASA ve formulacích s glycinem: s ním je tableta dobře rozpustná ve vodě a lépe a rychleji se vstřebává [183,184]. Glycin je obsažen v přípravku Anopyrin® a ve vyšším množství v tabletě Godasalu®. ASA podáváme zásadně po jídle. ASA lze vysadit při normalizaci počtu trombocytů u nízkorizikových pacientů (to se týká především mladších pacientů). Naopak u starších osob > 65 let ji ponecháme vždy, podobně jako u pacientů se zvýšeným kardiovaskulárním rizikem. ASA je dlouhodobou prevencí recidivy u jedinců s anamnézou arteriální příhody. Podávání ASA je mimořádně efektivní při poruchách mikrocirkulace při MPO-T: např. podání ASA 200 mg po 2–3 dny za sebou dokáže významně zmírnit obtíže při erytromelalgiích [16]. U nemocných s anamnézou krvácení a obzvláště krvácení do GIT nepodáváme ASA raději vůbec, anebo velmi opatrně spolu s inhibitory protonové pumpy (např. omeprazolem). U pozitivních případů by měla předcházet také eradikace Helicobacter pylori. ASA ireverzibilně blokuje funkci trombocytů, je třeba ji vysadit 7–10 dní před plánovanými chirurgickými zákroky (není-li dost času, je třeba indikovat podání trombokoncentrátu) a v případě vysokého trombotického rizika ji nahradit nízkomolekulárním (LMW) heparinem.
Podávání ASA v nízkých dávkách může být podle některých autorů dostatečným opatřením při zabezpečení těhotných pacientek [74]. V těhotenství mají navíc předtím zvýšené hodnoty destiček tendenci klesat. Nicméně gravidita sama o sobě je protrombotickým stavem, tzn., že těhotné pacientky mají v souladu s filozofií doporučení CZEMP vysokorizikové onemocnění, a tudíž zároveň i indikaci k tromboreduktivní terapii (optimálně IFN) s cílem normalizovat počet trombocytů pod 400 × 109/ l, a chránit tak matku. Spontánní potrat, obvykle v důsledku trombotizace placenty, hrozí především v 1. trimestru, obvykle tak končí zhruba 1/3 gravidit [74,150,185–190]. Jak již bylo uvedeno (v odstavci E.1.2.2.), je IFN patrně účinnější prevencí spontánních potratů než ASA [150]. V praxi může být výhodné vysadit ASA před porodem (hrozí riziko zvýšeného krvácení při porodu) a např. od 7. měsíce gravidity raději podávat LMW heparin (matka je ohrožena trombózou).
Někteří pacienti mohou mít na léčbu ASA atypickou vnímavost. Určité procento nemocných (zhruba 20 %) může být na ni rezistentní [191] a naopak někteří výjimečně vnímaví pacienti mohou mít krvácivé komplikace a extrémně sníženou agregaci i při velmi nízké dávce ASA (např. při 50 mg denně i méně). V druhém případě lze zkusit snížit dávku ASA. Primární přecitlivělost ve smyslu Samterova syndromu (trias: astma, nosní polypy a život ohrožující anafylaktoidní reakce) [182] je vzácná, je častější u astmatiků. Bývá spojena s relativně vyššími dávkami ASA. V případech rezistence na ASA, u pacientů s krvácivými komplikacemi a u pacientů s GIT intolerancí ASA (i přes léčbu inhibitorem protonové pumpy) je nutno podávat jiné antiagregans, např. thienopyridiny (tiklopidin, klopidogrel) nebo indobufen v obvyklém dávkování.
E.1.2.5. Trombocytaferéza (TAF)
TAF na separátoru krevních elementů je indikována podle současných i předchozích doporučení u všech pacientů s počtem trombocytů > 3 000 × 109/ l. TAF bychom měli dále indikovat i u symptomatických, krvácejících pacientů při počtech trombocytů > 2 000 × 109/ l. TAF vždy spojíme s cytoreduktivní léčbou, nejlépe HU, podávanou po provedení TAF (tab. 6). Výkon je nutné obvykle opakovat po několik dní. Pokles množství trombocytů je obvykle spojen s úlevou od symptomů a zástavou krvácení, dochází i k obnově agregability destiček [192–199].
E.1.2.6. Antikoagulační léčba
Je-li prokázána čerstvá trombóza stará několik hodin, lze uplatnit trombolytickou léčbu dle běžných standardů (spolu s okamžitou snahou o cytoredukci). Antikoagulační léčba heparinem (nefrakcionovaným nebo nízkomolekulárním) je indikována při zjištění nedávné trombózy arteriální i venózní. V případě venózního tromboembolizmu je pak indikována celoživotní antikoagulační léčba kumarinovým derivátem, obvykle warfarinem. V případě arteriální trombózy se přechází s delším odstupem z heparinu na podávání antiagregační léčby nízkými dávkami ASA, zpravidla 100 mg denně [29].
E.2. Léčba pravé polycytemie (PV)
V souladu s histopatologickým nálezem trilineární myeloproliferace v kostní dřeni („panmyelózy“) často nalézáme v krevním obraze zvýšené hodnoty elementů všech 3 řad. Přitom zvýšení hodnot každé z nich může představovat riziko trombózy. O polycytemii a trombocytemii je to známo již několik desetiletí [16,83,84], zatímco možné, ale nikoli všeobecně přijímané riziko ze strany leukocytózy bylo naznačeno teprve nedávno [62,200]. Dosud nebylo definitivně rozhodnuto, která ze zvýšených řad představuje největší nebezpečí – existuje řada většinou retrospektivních studií ukazujících, že terapeutické snížení jak trombocytemie, tak i polycytemie snižuje riziko trombózy, event. i hemoragie [83,84]. Na významné postavení počtu trombocytů (při MPO navíc aktivovaných) z hlediska trombotického rizika ukazuje fakt, že antiagregační terapie rozumně nízkou dávkou ASA významně snížila incidenci trombotických komplikací v randomizované, placebem kontrolované studii ECLAP [108]. Účelem léčby je především redukce rizik trombózy a krvácení. Žádná léčba však patrně nemůže zpomalit biologický vývoj onemocnění jako takového. PV se vyvíjí z tzv. latentní, prepolycytemické fáze (dle PVSG kritérií se jedná o ET), do manifestní polycytemie, a po mnoha letech (spíše desetiletích) přechází do tzv. vyhořelé fáze („spent phase“), tj. stadia podobného myelofibróze [16,201,202]. PV se může transformovat do s-AML, ať již přímo z polycytemického stadia, tak (častěji) až ze stadia postpolycytemické myelofibrózy. Jedním z úkolů správně vedené léčby u mladších pacientů je nedopustit vývoj do s-AML a zavčas poskytnout jedinou možnou kurativní léčbu, tj. alogenní transplantaci krvetvorných kmenových buněk (HSCT), především ve stadiu postpolycytemické myelofibrózy. Základním kamenem léčby PV jsou venepunkce, antiagregační terapie a při nedostatečnosti předchozích opatření cytoreduktivní terapie. Původní snahy o zlepšení celkového přežití u PV pomocí chemoterapie byly neúspěšné: randomizovaná studie PVSG-01 ukázala zkrácení přežití nemocných léčených chlorambucilem, radiofosforem a venepunkcemi oproti větvi léčené pouze venepunkcemi, především následkem indukce s-AML cytoreduktivní léčbou [203].
E.2.1. Venepunkce a erytrocytaferéza
Před érou terapeutického užití flebotomie přežívali nemocní s PV méně než 2 roky [16,204–207]. Přitom již Galén ve 2. století n. l. užíval venesekce k léčbě pletory [208]. Systematickým užitím venepunkcí s cílovým hematokritem 0,52 se prodloužilo přežití na zhruba 7 let, nicméně průběh onemocnění byl stále zatížen vysokou incidencí trombóz [202,209]. Měřením krevního průtoku mozkem byla prokázána jeho extrémní závislost na hematokritu (resp. viskozitě krve) – proto byl posunut terapeutický cíl na dosažení hematokritu 0,45 [206,207]. V souladu s tím bylo prokázáno, že teprve na této hranici významně klesá incidence trombotických komplikací a medián celkového přežití vzroste na zhruba 13 let [16]. Venepunkce jsou standardní terapií 1. linie u PV [16,79]. Vedlejším efektem tohoto postupu je vývoj nedostatku zásob železa.
Velkoobjemová erytrocytaferéza může nahradit potřebu venepunkcí [210–212]. Někteří ji považují ve srovnání s venepunkcemi za šetrnější způsob snížení hematokritu a trombotického rizika šetřící plazmatické bílkoviny. Podle jiných však nebyly její výhody nikdy spolehlivě doloženy a považují ji pouze za finančně náročnější obdobu venepunkcí. U PV lze erytrocytaferézu doporučit spíše v urgentních situacích s hrozící trombózou, tzn. spíše při velmi vysokých hodnotách hematokritu, dokud se onemocnění nestabilizuje. Pak je možné přejít k opakovaným venepunkcím. Výhodou je možnost spojení s trombaferézou u případů PV s trombocytemií.
E.2.2. Antiagregační terapie
Antiagregační terapie (viz též odstavec E.1.2.4.) pomocí ASA 40–100 mg denně představuje spolu s venepunkcemi standardní terapii PV 1. linie, není-li ASA kontraindikována a není--li přítomna zároveň trombocytemie > 1 000 × 109/ l [16,79]. (Při jakékoli hodnotě trombocytemie je indikována navíc i cytoreduktivní léčba.) U pacientů s PV bez jiné jasné indikace anebo kontraindikace ASA ukázala randomizovaná, placebem kontrolovaná studie ECLAP [108], vysoce signifikantní snížení incidence trombotických příhod za cenu pouze mírného a nesignifikantního zvýšení incidence krvácivých příhod při podávání nízkých dávek ASA. Snížená frekvence byla zaznamenána především u arteriálních, ale zčásti i u venózních trombotických příhod [108].
E.2.3. Cytoreduktivní léčba
Cytoreduktivní léčba je indikována v následujících situacích:
- a) jde-li o vysokorizikového pacienta z hlediska trombózy – jde především o nemocné s předchozí trombotickou událostí, o pacienty se současnou trombocytemií (jakýmkoli počtem trombocytů > 400 × 109/ l), event. s přidruženým trombofilním stavem;
- b) nestačí-li léčba venepunkcemi udržet hodnotu hematokritu pod 0,45;
- c) vyvíjí-li se (hepato-)splenomegalie;
- d) je-li nemocný výrazně symptomatický.
Vzhledem k tomu, že je žádoucí současně snížit počet trombocytů i hematokrit, dáváme obvykle přednost IFN před ANG u pacientů do 65 let, u starších volíme HU. U této druhé skupiny pacientů volíme jednoznačně nejméně leukemogenní cytostatikum – proto dáváme přednost HU před jinými, např. alkylátorem busulfanem. Ten (a v některých zemích také pipobroman) lze použít pouze jako možnost při selhávání léčby HU u velmi starých nemocných s krátkým životním očekáváním jako léčbu 2. linie – u mladších ze skupiny nad 65 let raději volíme IFN, event. kombinaci HU + IFN. Začne-li dominovat při podávání IFN nebo HU trombocytemie, lze do kombinace přidat ANG.
Načasování zahájení cytoreduktivní terapie nemusí být stejné u mladších pacientů indikovaných k léčbě IFN a u starších pacientů s indikací terapie HU, neboť především dlouhodobá toxicita cytostatik ve smyslu možnosti vyvolání sekundární leukemie či jiné malignity velí k opatrnosti při nasazování cytoreduktivní léčby u PV dle principu primum non nocere. Při srovnání výsledků léčby HU a venepunkcí došel Fruchtman [213] k závěru, že HU může prodloužit dobu do vývoje postpolycytemické myelofibrózy, avšak za cenu zvýšené incidence sekundárních leukemií. Obdobné údaje pro IFN v literatuře chybí – lze však předpokládat, že by také mohl oddálit nástup myelofibrózy (tím spíše, že na rozdíl od HU může navodit i remisi onemocnění), aniž by byl tento pozitivní efekt vykoupen leukemogenicitou.
E.2.4. Otázka substituce železa
Po venepunkcích nebo erytrocytaferézách se obvykle vyvíjí stav s výraznou karencí zásobního železa. Železo je potřebné jako katalyzátor enzymatických reakcí a jeho nedostatek může být příčinou značného únavového syndromu. U výrazně proliferativní PV léčené pouze venepunkcemi se však substituce nedoporučuje – obvykle dochází k efektu „přilévání oleje do ohně“. Deplece železa je totiž jedním z mechanizmů pozitivního efektu venepunkcí. Substituci lze s velkou opatrností zkusit u dobře stabilizovaného onemocnění při cytoreduktivní terapii (nemělo by však dojít k eskalaci potřebné dávky léčiva ke kontrole PV), anebo při vyhasínání polycytemické fáze onemocnění při přechodu do „spent phase.“ Jakmile bychom zaznamenali akceleraci polycytemie, musíme železo vysadit.
E.2.5. Antikoagulační léčba
Antikoagulační léčba u posttrombotických stavů při PV se řídí stejnými principy jako u ET (viz odstavec E.1.2.6.).
E.3. Léčba pokročilých stadií primárních MPO s myelofibrózou
Zatímco léčebná strategie u časných, proliferativních stadií PMF je v podstatě identická s postupy popsanými pro Ph- MPO-T (viz kapitola E.1.), pokročilé fáze myelofibrózy vyznačující se cytopeniemi v různých řadách (obvykle nejprve anémií) a rostoucí mírou myeloidní metaplazie (hepato-/splenomegalií a výskytem nezralých buněk neutrofilní řady a erytroblastů v periferní krvi) vyžadují speciální postupy. Není přitom důležité, zda jde o pokročilou fázi PMF, anebo zda jde o postpolycytemickou myelofibrózu. Jediným dostupným kurativním opatřením je provedení alogenní transplantace krvetvorných kmenových buněk (HSCT) – tu musíme zvažovat u všech pacientů mladších 65 let. Cílem je nedopustit, aby onemocnění přešlo do s-AML, která je fatální komplikací, neboť u většiny nemocných se nedaří navodit indukční terapií kompletní remisi, která je předpokladem k provedení HSCT v další fázi. Přirozený přechod do s-AML u Ph- MPO s myelofibrózou je otázkou mnoha let, proto máme relativně dostatek času na organizaci HSCT. Pouze v případech prudkého vývoje s-AML (teoreticky spíše podmíněného předchozí genotoxickou léčbou) nezbývá nic jiného, než transplantovatelného pacienta zaléčit indukční chemoterapií a pak se pokusit o HSCT. Naopak všechna ostatní opatření je třeba vnímat v podstatě jako paliativní, jejich smyslem je poskytnout pacientovi co nejvyšší kvalitu života.
Přechod do stadia pokročilé myelofibrózy, ať již jde o PMF, nebo PV, není nikdy ostrý – obvykle jde o proces trvající léta, spíše desetiletí. U PMF se obvykle vyvíjí anémie ještě ve stadiu, kdy můžeme pozorovat trombocytemii. V této fázi musíme zavedenou tromboreduktivní terapii dávkovat s opatrností, abychom nepřivodili těžkou leukopenii nebo příliš nezhoršili anémii (to se týká jak HU, tak IFN, ale koneckonců i ANG, který svým vazodilatačním účinkem provázeným expanzí plazmatického volumu může prohloubit anémii). Naopak u postpolycytemické myelofibrózy většinou nebývá (alespoň zpočátku) problém s anémií, ale spíše s leukopenií, event. trombocytopenií (většinou bývá v tomto stadiu přítomna výrazná splenomegalie).
E.3.1. Kurativní postup s provedením transplantace hematopoetických kmenových buněk (HSCT)
Jelikož je alogenní HSCT jedinou kurativní metodou léčby, je nutno na ni zavčas pomyslet u každého pacienta s pokročilejší formou PMF nebo postpolycytemické myelofibrózy v transplantabilním věku – v ČR v současnosti do 65 let – a indikovat ji, dokud je čas. Vždy je nutné přihlížet k individuální prognóze pacienta, není vhodné transplantovat pacienty s životním očekáváním delším než 5 let. Podle doporučení ČHS a ČOS je HSCT indikována na základě tzv. „individuální indikace“, tzn. na podkladě zvážení všech rizik daného pacienta [214]. CZEMP již přijala za svá indikační kritéria vypracovaná v ÚHKT Praha [215].
U PMF existuje celá řada prognostických schémat. Tzv. „lilleské“ skóre [88] je založeno pouze na hodnotách krevního obrazu: hladině hemoglobinu a počtu leukocytů (rizika: < 100 g/ l, resp. < 4 nebo > 30 × 109/ l). Medián přežití s jedním a se dvěma z uvedených rizik je pouze 26, resp. 13 měsíců, zatímco při skóre 0 je medián přežití 93 měsíců. Mnohá schémata ještě přibírají další ukazatele: počet trombocytů (< 100, event. < 300 × 109/ l), vyplavování nezralých granulocytů, cirkulující blasty, event. normoblasty, někdy také věk, konstituční symptomy, hepatomegalii, chromozomální aberace, stupeň fibrózy dřeně [89–91,216–223]. Tato kritéria jsou jistě obecně platná, avšak při zvažování HSCT vystupují do popředí i další faktory: 1. včasnou indikací HSCT nesmíme dovolit u pacienta přechod do s-AML, 2. nutno přihlížet k rozsahu choroby – čím větší hepatosplenomegalie (obvykle projev myeloidní metaplazie), tím větší může být riziko potransplantačních komplikací: např. venookluzivní nemoci jater a také relapsu (z tohoto důvodu lze zvažovat splenektomii, event. iradiaci sleziny, optimálně 6–8 týdnů před výkonem). Role (či potřeba) splenektomie před HSCT není sice jasná [224], nicméně z obecně onkologického hlediska lze předpokládat, že se rozsah onemocnění (nálož nádoru) projeví ve výsledcích HSCT (v ÚHKT se splenektomují pacienti se slezinou zhruba pod úroveň pupku). Riziko morbidity i mortality po splenektomii je však u pacientů s PMF velmi vysoké [225] a může se extrémně lišit v závislosti na zkušenosti jednotlivých chirurgických oddělení s tímto výkonem u pacientů s MPO. U splenektomovaných nemocných se zkracuje doba přihojení štěpu [225]. Naopak se stupněm fibrózy dřeně se patrně doba potřebná k přihojení štěpu prodlužuje.
Výsledky HSCT se stále zlepšují, a to za použití myeloablativních [226,227], a ještě významněji za použití nemyeloablativních přípravných režimů (režimů s redukovanou intenzitou) [228,229]. Výsledky s nepříbuznými HLA-identickými dárci jsou srovnatelně dobré jako s příbuznými, ale při HLA-mismatchi bývá přežití horší. Dlouhodobě přežívá v uvedených novějších studiích 60–85 % nemocných [225,230]. Také zkušenost z ÚHKT potvrzuje, že dlouhodobé přežití na transfuzích nezávislých pacientů se blíží 80 % [231]. Načasování HSCT nesmí být ani příliš brzké, ale ani příliš pozdní. HSCT je indikována, splňuje-li pacient kterékoli z kritérií: 1. hodnota hemoglobinu se blíží hranici 100 g/ l (nejde-li o AIHA), trombocyty < 100 a leukocyty < 4 × 109/ l; 2. cirkulující blasty (jakýkoli počet), normoblasty > 1/100 leukocytů, nezralé granulocyty (metamyelocyty až promyelocyty) ≥ 10 % – vše opakovaně v rozmezí alespoň 2 měsíců; 3. progredující hmatná hepato-/splenomegalie; 4. klinické symptomy (poty, ztráta váhy, febrilie bez zjistitelné infekce), rozsáhlé otoky, symptomy portální hypertenze (tab. 7). CZEMP zatím nevydává žádná doporučení ohledně potřeby splenektomie (diskutované výše), ani vhodného přípravného režimu – volba je ponechána na transplantačním centru.

E.3.2. Korekce anémie
Anémie se objevuje u pacientů s myelofibrózou při Ph- MPO celkem pravidelně. Moment, kdy se anémie objeví, je velmi různorodý u jednotlivých pacientů: u některých je relativně pozdním fenoménem (ve stadiu MF-2 a MF-3, a samozřejmě také u postpolycytemické myelofibrózy), u jiných naopak velmi časným, např. ve stadiu MF-1 (nezřídka se anémie objeví po zavedení tromboreduktivní terapie HU, IFN, event. i ANG). U některých pacientů nemusí být přítomna v momentě manifestace anémie ani hmatná splenomegalie – i v tomto ohledu je velká variabilita. Anémie je dána především snížením produkce buněk červené řady ve dřeni. Nicméně může být zhoršována řadou dalších okolností: 1. často hypersplenizmem při splenomegalii a portální hypertenzi, 2. méně často může být přítomna i imunitní hemolýza, 3. mohou se na ní podílet i ztráty krve, např. po krvácení z jícnových varixů, 4. může se objevit makrocytární (megaloblastová) anémie při deficitu folátu.
Základem léčby anémie je substituční léčba transfuzemi erytrocytární masy (analogicky při trombocytopenii indikujeme trombokoncentráty). Anémie se zpravidla vyvíjí pomalu a plíživě, takže se obvykle pacienti na nízké parametry červeného krevního obrazu dobře adaptují. Nemá-li pacient jiné problémy (např. ICHS), netřeba se substitucí spěchat. Indikací k jejímu podávání je v obecné rovině přítomnost klinických anémických symptomů. Při portální hypertenzi a hypoproteinemii se nízký hematokrit může podílet na snížení onkotického tlaku, a tudíž i na rozvoji otoků – proto i tento aspekt nutno zvažovat jako indikaci. U polytransfundovaných pacientů nesmíme zapomenout na potřebu chelatační léčby při přetížení železem.
Vitaminy B řady, především folát, je vhodné podávat u případů myelofibrózy s megaloblastovou (makrocytární) anémií, není-li makrocytóza pouhým důsledkem léčby HU. MPO a obzvláště myelofibróza jsou často sdruženy s deficitem folátu. Podávání kyseliny listové může vést k retikulocytóze, jsou popisovány i případy ztráty závislosti na transfuzích [232]. Ačkoli hyperhomocysteinemie se nezdá být důležitým rizikovým faktorem pro trombózu u Ph- MPO, může být spojena s deficitem kobalaminu nebo folátu a může být korigována jejich substitucí [233].
Kortikoidy se mohou podávat k paliaci konstitučních symptomů u PMF. Pokud je anémie indikací k podávání, mohou být účinné především u případů s výraznou hemolytickou složkou. Efekt lze pozorovat asi u 30 % pacientů s PMF [234,235]. V poslední době se v zahraničí spíše užívají v kombinaci s thalidomidem a jeho deriváty – viz níže.
Androgeny (fluoxymesteron, nandrolon, oxymethalon, testosteron a nejčastěji užívaný danazol) mohou pomoci v léčbě anémie při myelofibróze zhruba u 30–40 % nemocných [235]. Markantnější účinek je pozorován u pacientů bez výraznější splenomegalie. Pro optimální účinek je třeba podávat androgeny dlouhodobě, např. danazol 600 mg denně po dobu 3–6 měsíců [236]. Limitující může být poměrně častá hepatotoxicita.
Erytropoetin (Epo) a darbepoetin mají předpoklad účinnosti zhruba u 50 % nemocných s PMF. Větší šanci na léčebnou odpověď mají pacienti s nižšími hladinami erytropoetinu v séru – pod 125 j/ l [236–238]. Zahajovací dávka Epo může být 10 000 j.3krát týdně, po měsíci při nedostatečné odpovědi lze dávku zdvojit. Podávání Epo může stimulovat progresi onemocnění a splenomegalii [239] – proto je podávání doporučeno pouze na bázi ověřeného snížení hladiny erytropoetinu v séru před zahájením léčby [236], a to ještě s velkou opatrností. Analogicky, se stejným omezením, lze podávat darbepoetin-α jednou týdně 150 μg s.c., lze jej eskalovat na 300 μg týdně [238].
Inhibitory angiogenezy – thalidomid a jeho deriváty lenalidomid a pomalidomid jsou poměrně slibnou možností v léčbě anémie provázející pokročilou myelofibrózu – odpovídá na ně zhruba 40 % pacientů, přičemž lepších výsledků je dosahováno při jejich kombinaci s prednisolonem. Kortikoid jednak zlepšuje toleranci thalidomidu (který má řadu vedlejších účinků), jednak může mít i jistý efekt sám o sobě. Léčba nízkými dávkami thalidomidu a jeho deriváty je často efektivní i v případě trombocytopenie, v menším procentu případů může redukovat i splenomegalii [236,240–243]. V ČR však není žádná z uvedených látek pro léčbu MPO dostupná.
JAK2 inhibitory – výsledky prvních klinických studií byly očekávány s velkými nadějemi, avšak očekávání nebyla zcela naplněna a účinnost dosud zkoušených inhibitorů této tyrozinové kinázy není srovnatelná s účinností inhibitorů tyrozinových kináz u CML. Problémem je především jejich nedostatečná selektivita vůči JAK2 kináze (inhibují obvykle i jiné tyrozinové kinázy), což vede k nežádoucím účinkům, např. k myelosupresi. Na druhou stranu nedostatek selektivity účinku vůči mutované alele JAK2 může být i jistou výhodou – JAK2 inhibitory jsou srovnatelně účinné i u případů bez mutace JAK2 genu [244]. Mohou významně redukovat splenomegalii, zmírňují i konstituční symptomy spojené s MPO (nadměrné pocení, ztráty na váze, únavu, svědivku). Zatím nejvíce prozkoumaným JAK2 inhibitorem u pacientů s PMF je látka označovaná INCB018424 (od firmy Incyte). V ČR zatím nejsou JAK2 inhibitory dostupné.
E.3.3. Splenektomie
Indikací ke splenektomii mohou být případy refrakterní cytopenie (především anémie a trombocytopenie), symptomatická splenomegalie (bolesti z roztahování pouzdra lien, splenický infarkt nezvládnutelný analgetiky) a těžká portální hypertenze [245]. Splenomegalie se navíc může podílet i na kachektizaci pacienta tím, že působí problém v příjmu potravy při útlaku GIT gigantickou slezinou, event. i ascitem. Splenektomie však bývá při velké splenomegalii spojena s významnou morbiditou 9–31 % a mortalitou 9–26 % i na renomovaných pracovištích [245–248] – pooperační mortalita do 3 měsíců i na Mayo Clinic v Rochesteru byla právě zmíněných 26 % [236,248]. Navíc je třeba předpokládat, že výsledky na pracovištích s menšími zkušenostmi mohou být ještě horší. Proto je nutno splenektomii považovat za ultimum refugium v paliativní péči o pacienty s PMF. Častými komplikacemi mohou být krvácení, pooperační trombóza ve splanchniku i jinde, subfrenický absces a jiné infekce a také poměrně značná míra blastické transformace. Záhy po splenektomii může docházet k hepatomegalii a je vždy nutno s jejím vývojem počítat [245–248]. Splenektomie může být indikována jako opatření před HSCT – viz odstavec E.3.1 [224].
E.3.4. Radioterapie (lien a jiné oblasti)
Ozáření lien (obvykle 5 Gy) lze považovat za alternativu splenektomie u pacientů, u kterých je již operativní řešení kontraindikováno. Odpověď je z povahy věci pouze tranzitorní, obvykle trvá zhruba 6 měsíců. Hlavním vedlejším účinkem jsou život ohrožující cytopenie, mortalita při radioterapii může být vysoká až 13 % [245]. Radioterapie je metodou volby u extramedulárních postižení (např. peritonea s ascitem, pleury s tvorbou výpotků, ale i vzácnějších lokalizací: ledviny, centrálního nervového systému, žlučníku a jater). Je-li problémem lymfadenopatie, je možno ozářit uzliny [245].
E.3.5. Opatření při portální hypertenzi
Portální hypertenze může být při PMF způsobena trombózou portálních či hepatických žil, avšak je často přítomna i bez trombotické stenózy. Podle jedné teorie je to dáno zúžením intrahepatálních sinusoidů vlivem myeloidní metaplazie v játrech, podle druhé teorie může být portální hypertenze zapříčiněna zvýšeným průtokem krve v portální oblasti přes zvětšenou slezinu [249]. Krvácení z jícnových varixů u portální hypertenze může být život ohrožující terminální stav (navíc obvykle při současně přítomné trombocytopenii a koagulopatii). Podobně i ascites je typickým symptomem terminálního vývoje PMF. Samozřejmě teoreticky nejúčinnější je zásah vedoucí k potlačení myeloidní metaplazie, tj. např. léčba HU nebo IFN, avšak v praxi obvykle jejich podání nedovolí míra existující cytopenie. Lze zkusit léčbu spironolaktonem (při normo- nebo hypokalemii). Jiná diuretika jsou sice indikována k léčbě ascitu (lze zkusit je podávat s albuminem), avšak nebývají příliš účinná. Ascites lze opakovaně punktovat, avšak doplňuje se velmi rychle. Poměrně slibnou paliací se zdá být zavedení transjugulárního portosystémového shuntu (TIPS), který může zamezit recidivě krvácení z jícnových varixů, snížit míru ascitu, zmírnit splenomegalii i cytopenie i snížit portální hypertenzi u Buddova--Chiariho syndromu [250–254]. TIPS je tedy řešením pro intrahepatální okluzi (v souladu s první teorií). Na druhou stranu může také splenektomie vést ke snížení portální hypertenze (alespoň podle druhé ze zmíněných teorií) [245,253,255]. Avšak splenektomie může být vysoce riziková a doporučujeme ji v této indikaci jen ve zcela výjimečných případech.
Závěr
V přehledu jsme uvedli východiska pro doporučení pro diagnostiku a léčbu 3 hlavních Ph- (BCR/ABL-) MPO. Tato doporučení jsou výsledkem většinového konsenzu skupiny CZEMP (v poměru hlasů 16 : 2, nesouhlasící členové nejsou spoluautory článku). Zkrácená forma doporučení byla předložena výboru České hematologické společnosti České lékařské společnosti J. E. Purkyně ke schválení. Doporučení nejsou dogmatem, které nemůže ošetřující lékař překročit v indikovaných případech, a nejsou z právního hlediska závazná [256]. Jsou pouze návodem, jak lze dle konsenzu odborníků v obvyklých případech postupovat. Základní i klinický výzkum po objevu mutace JAK2 genu akceleruje a lze očekávat, že v následujících letech budou muset být současná doporučení opět aktualizována.
Poděkování
Poděkování
patří Ing. Janě Markové za kritické pročtení rukopisu.
MUDr. Jiří Schwarz, CSc.
www.uhkt.cz
e-mail: jiri.schwarz@uhkt.cz
Doručeno do redakce: 11. 11. 2010
Přijato po recenzi: 22. 12. 2010
Zdroje
1. Tefferi A, Thiele J, Orazi A et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007; 110: 1092–1097.
2. Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood 1978; 51: 189–194.
3. Fialkow PJ, Faguet GB, Jacobson RJ et al. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 1981; 58: 916–919.
4. Adamson JW, Fialkow PJ, Murphy S et al. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med 1976; 295: 913–916.
5. Dameshek W. Some speculation on the myeloproliferative syndromes. Blood 1951; 6: 372–375.
6. Baxter EJ, Scott LM, Campbell PJ et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
7. James C, Ugo V, Le Couédic JP et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
8. Kralovics R, Passamonti F, Buser AS et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
9. Olcaydu D, Harutyunyan A, Jäger R et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet 2009; 41: 450–454.
10. Jones AV, Chase A, Silver RT et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet 2009; 41: 446–449.
11. Pardanani A, Lasho TL, Finke CM et al. The JAK2 46/1 haplotype confers susceptibility to essential thrombocythemia regardless of JAK2V617F mutational status – clinical correlates in a study of 226 consecutive patients. Leukemia 2010; 24: 110–114.
12. Kralovics R, Teo SS, Li S et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006; 108: 1377–1380.
13. Nussenzveig RH, Swierczek SI, Jelinek J et al. Polycythemia vera is not initiated by JAK2-V617F mutation. Exp Hematol 2007; 35: 32–38.
14. Schaub FX, Jäger R, Looser R et al. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood 2009; 113: 2022–2027.
15. Delhommeau F, Dupont S, Della Valle V et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289–2301.
16. Michiels JJ, Kvasnicka HM, Thiele J. Myeloproliferative disorders. Current perspectives on diagnostic criteria, histopathology and treatment in essential therombocythemia, polycythemia rubra vera and chronic idiopathic myelofibrosis. Much (Germany): Verlag ME 2005.
17. Schwarz J, Pytlík R, Doubek M et al. Analysis of risk factors: the rationale of the guidelines of the Czech Hematological Society for diagnosis and treatment of chronic myeloproliferative disorders with thrombocythemia. Semin Thromb Hemost 2006; 32: 231–245.
18. Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann Hematol 2003; 82: 148–152.
19. Berlin NI. Diagnosis and classification of the polycythemias. Semin Hematol 1975; 12: 339–351.
20. Murphy S. Diagnostic criteria and prognosis in polycythemia vera and essential thrombocythemia. Semin Hematol 1999; 36 (1 Suppl 2): 9–13.
21. Georgii A, Buhr T, Buesche G et al. Classification and staging of Ph- negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leuk Lymphoma 1996; 22 (Suppl 1): 15–29.
22. Michiels JJ, De Raeve H, Hebeda K et al. WHO bone marrow features and European clinical, molecular, and pathological (ECMP) criteria for the diagnosis of myeloproliferative disorders. Leuk Res 2007; 31: 1031–1038.
23. Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Semin Thromb Hemost 1997; 23: 339–347.
24. Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera, and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol 2002; 76: 133–145.
25. Imbert M, Pierre R, Thiele J et al. Essential thrombocythemia. In: Jaffe E, Harris N, Stein H et al (eds). WHO Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2001: 39–41.
26. Pierre R, Imbert M, Thiele J et al. Polycythemia vera. In: Jaffe E, Harris N, Stein H et al (eds). WHO Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2001: 32–34.
27. Thiele J, Pierre R, Imbert M et al. Chronic idiopathic myelofibrosis. In: Jaffe E, Harris N, Stein H et al (eds). WHO Classification of Tumours: Pathology & Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2001: 35–38.
28. Schwarz J, Penka M, Indrák K et al. The WHO 2008 classification of Ph-myeloproliferative disorders: statement of the Czech MPD Working Group. Leukemia 2008; 22: 2118–2119.
29. Penka M, Schwarz J, Pytlík R et al. Doporučený postup diagnostiky a terapie esenciální trombocytemie a trombocytemie provázející jiné myeloproliferativní choroby. Vnitř Lék 2005; 51: 741–751.
30. Schwarz J, Penka M. Trombocytózy a trombocytemie. Vnitř Lék 2005; 51: 861–871.
31. Samuelson SJ, Parker CJ, Prchal JT. Revised criteria for the myeloproliferative disorders: too much too soon? Blood 2008; 111: 1741–1742.
32. Spivak JL, Silver RT. The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis: an alternative proposal. Blood 2008; 112: 231–239.
33. Teofili L, Giona F, Martini M et al. The revised WHO diagnostic criteria for Ph-negative myeloproliferative diseases are not appropriate for the diagnostic screening of childhood polycythemia vera and essential thrombocythemia. Blood 2007; 110: 3384–3386.
34. Campbell PJ, Scott LM, Buck G et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005; 366: 1945–1953.
35. Thiele J, Kvasnicka HM, Facchetti F et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005; 90: 1128–1132.
36. Kvasnicka HM, Thiele J. Classification of Ph-negative chronic myeloproliferative disorders – morphology as the yardstick of classification. Pathobiology 2007; 74: 63–71.
37. Wilkins BS, Erber WN, Bareford D et al. Bone marrow pathology in essential thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Blood 2008; 111: 60–70.
38. Gisslinger H, Gotic M, Holowiecki J et al. Final results of the ANAHYDRET-study: non-inferiority of anagrelide compared to hydroxyurea in newly diagnosed WHO-essential thrombocythemia patients. Blood 2008: 112. Abstract 661.
39. De Stefano V, Teofili L, Leone G et al. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Semin Thromb Hemost 1997; 23: 411–418.
40. Lengfelder E, Hochhaus A, Kronawitter U et al. Should a platelet limit of 600 × 10(9)/ l be used as a diagnostic criterion in essential thrombocythaemia? An analysis of the natural course including early stages. Br J Haematol 1998; 100: 15–23.
41. Chait Y, Condat B, Cazals-Hatem D et al. Relevance of the criteria commonly used to diagnose myeloproliferative disorder in patients with splanchnic vein thrombosis. Br J Haematol 2005; 129: 553–560.
42. Boissinot M, Lippert E, Girodon F et al. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood 2006; 108: 3223–3224.
43. Brière JB. Budd-Chiari syndrome and portal vein thrombosis associated with myeloproliferative disorders: diagnosis and management. Semin Thromb Hemost 2006; 32: 208–218.
44. Marková J, Průková D, Volková Z et al. A new allelic discrimination assay using locked nucleic acid-modified nucleotides (LNA) probes for detection of JAK2 V617F mutation. Leuk Lymphoma 2007; 48: 636–639.
45. Pardanani AD, Levine RL, Lasho T et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108: 3472–3476.
46. Schnittger S, Haferlach C, Beelen DW et al. Detection of three different MPLW515 mutations in 10.1% of all JAK2 V617 unmutated ET and 9.3% of all JAK2 V617F unmutated OMF: a study of 387 patients. Blood 2007: 110. Abstr. 2546.
47. Beer PA, Campbell PJ, Scott LM et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008; 112: 141–149.
48. Hussein K, Bock O, Theophile K et al. JAK2V(V617F) allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp Hematol 2009; 37: 1186–1193.
49. Beer PA, Jones AV, Bench AJ et al. Clonal diversity in the myeloproliferative neoplasms: independent origins of genetically distinct clones. Br J Haematol 2009; 144: 904–908.
50. Ding J, Komatsu H, Wakita A et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood 2004; 103: 4198–4200.
51. Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol 2009; 6: 627–637.
52. Goerttler PS, Steimle C, März E et al. The Jak2V617F mutation, PRV-1 overexpression and EEC formation define a similar cohort of MPD patients. Blood 2005; 106: 2862–2864.
53. Barosi G, Mesa RA, Thiele J et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008; 22: 437–438.
54. Thiele J, Kvasnicka HM, Orazi A. Bone marrow histopathology in myeloproliferative disorders-current diagnostic approach. Semin Hematol 2005; 42: 184–195.
55. Cortelazzo S, Viero P, Finazzi G et al. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol 1990; 8: 556–562.
56. Besses C, Cervantes F, Pereira A et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999; 13: 150–154.
57. Michiels JJ, Kutti J, Stark P et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and myelofibrosis. Neth J Med 1999; 54: 46–62.
58. Michiels JJ, Berneman ZN, Schroyens W et al. Pathophysiology and treatment of platelet-mediated microvascular disturbances, major thrombosis and bleeding complications in essential thrombocythaemia and polycythaemia vera. Platelets 2004; 15: 67–84.
59. Wolanskyj AP, Schwager SM, McClure RF et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006; 81: 159–166.
60. Carobbio A, Finazzi G, Guerini V et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and Jak2 mutation status. Blood 2007; 109: 2310–2313.
61. Carobbio A, Antonioli E, Guglielmelli P et al. Leukocytosis and risk stratification assessment in essential thrombocythemia. J Clin Oncol 2008; 26: 2732–2736.
62. Tefferi A. Leukocytosis as a risk factor for thrombosis in myeloproliferative neoplasms-biologically plausible but clinically uncertain. Am J Hematol 2010; 85: 93–94.
63. Schwarz J, Markova J, Volkova Z et al. Chronic myeloproliferative disorders with thrombocythemia: thrombosis associated with inherited thrombophilia or JAK2 V617F gene mutation. Haematologica 2007, 92 (Suppl 1): 385–386. Abstr. 1044.
64. Schwarz J, Penka M, Doubek M et al. JAK2 mutation and additional thrombophilic state are major prothrombotic risk factors in myeloproliferations with thrombocythemia – data from a registry of anagrelide-treated patients. Haematologica 2008; 93 (Suppl 1): 57–58. Abstr. 0144.
65. Penka M, Schwarz J, Pavlík T et al. Jak léčíme nemocné s esenciální trombocytémií a dalšími myeloproliferacemi provázenými trombocytemií a co může být prediktivní známkou rizika trombózy u těchto nemocných – zpráva z registru pacientů léčených Thromboreductinem. Vnitř Lék 2008; 54: 775–782.
66. Conlan MG, Haire WD. Low protein S in essential thrombocythemia with thrombosis. Am J Hematol 1989; 32: 88–93.
67. Bucalossi A, Marotta G, Bigazzi C et al. Reduction of antithrombin III, protein C, and protein S levels and activated protein C resistance in polycythemia vera and essential thrombocythemia patients with thrombosis. Am J Hematol 1996; 52: 14–20.
68. Ruggeri M, Gisslinger H, Tosetto A et al. Factor V Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Am J Hematol 2002; 71: 1–6.
69. Amitrano L, Guardascione MA, Ames PR et al. Thrombophilic genotypes, natural anticoagulants, and plasma homocysteine in myeloproliferative disorders: relationship with splanchnic vein thrombosis and arterial disease. Am J Hematol 2003; 72: 75–81.
70. Kessler CM. Propensity for hemorrhage and thrombosis in chronic myeloproliferative disorders. Semin Hematol 2004; 41 (2 Suppl 3): 10–14.
71. Schwarz J, Hrachovinova I, Vorlova Z et al. Thromboembolism in thrombocythemia patients with an additional thrombophilic state. Hematol J 2004; 5 (Suppl 2): S321. Abstr. 974.
72. Gisslinger H, Müllner M, Pabinger I et al. Mutation of the prothrombin gene and thrombotic events in patients with polycythemia vera or essential thrombocythemia: a cohort study. Haematologica 2005; 90: 408–410.
73. Falanga A. Thrombosis and malignancy: an underestimated problem. Haematologica 2003; 88: 607–610.
74. Griesshammer M, Grünewald M, Michiels JJ. Acquired thrombophilia in pregnancy: essential thrombocythemia. Semin Thromb Hemost 2003; 29: 205–212.
75. Schafer AI, Levine MN, Konkle BA et al. Thrombotic disorders: diagnosis and treatment. Hematology (Am Soc Hematol Educ Program) 2003: 520–539.
76. Tatewaki W, Shibata A. Acquired von Willebrand disease in patients with chronic myeloproliferative disorders. Leuk Lymphoma 1989; 1: 51–57.
77. van Genderen PJJ, Michiels JJ, van der Poel-van de Luytgaarde SCPAM et al. Acquired von Willebrand disease as a cause of recurrent mucocutaneous bleeding in primary thrombocythemia: relationship with platelet count. Ann Hematol 1994; 69: 81–84.
78. van Genderen PJJ, Budde U, Michiels JJ et al. The reduction of large von Willebrand factor multimers in plasma in essential thrombocythaemia is related to the platelet count. Br J Haematol 1996; 93: 962–965.
79. McMullin MF, Bareford D, Campbell P et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol 2005; 130: 174–195.
80. Jones AV, Kreil S, Zoi K et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005; 106: 2162–2168.
81. Scott LM, Tong W, Levine RL et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356: 459–468.
82. Schnittger S, Haferlach C, Geer T et al. Detection of JAK2 exon 12 mutations in 10 patients (10.1%) of V617F negative PV: a study of 211 cases with PV or suspected PV. Blood 2007: 110. Abstr. 2541.
83. Dawson AA, Ogston D. The influence of the platelet count on the incidence of thrombotic and haemorrhagic complications in polycythaemia vera. Postgrad Med J 1970; 46: 76–78.
84. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet 1978; 2: 1219–1222.
85. Berk PD, Goldberg JD, Donovan PB et al. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol 1986; 23: 132–143.
86. Pikman Y, Lee BH, Mercher T et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
87. Vannucchi AM, Antonioli E, Guglielmelli P et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood 2008; 112: 844–847.
88. Dupriez B, Morel P, Demory JL et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood 1996; 88: 1013–1018.
89. Dingli D, Schwager SM, Mesa RA et al. Prognosis in transplant-eligible patients with agnogenic myeloid metaplasia: a simple CBC-based scoring system. Cancer 2006; 106: 623–630.
90. Kvasnicka HM, Thiele J, Werden C et al. Prognostic factors in idiopathic (primary) osteomyelofibrosis. Cancer 1997; 80: 708–719.
91. Cervantes F, Dupriez B, Pereira A et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895–2901.
92. U.S. Preventive Services Task Force Ratings: grade definitions. Guide to clinical preventive services, third edition: Periodic updates, 2000-2003. Available from: http://www.ahrq.gov/clinic/3rduspstf/ratings.htm.
93. Střední délka života ve věku 65 let – muži. Střední délka života ve věku 65 let – ženy. Praha: ÚZIS (Ústav zdravotnických informací a statistiky ČR) 2009. Dostupné z: http://www.uzis.cz/cz/dps/data/frame.html?T0=r2008+&T1=UKZ+0054+0064+&T2=REG+CZE+.
94. Nowak-Göttl U, Kosch A, Schlegel N et al. Thromboembolism in children. Curr Opin Hematol 2002; 9: 448–453.
95. Tripodi A. Levels of coagulation factors and venous thromboembolism. Haematologica 2003; 88: 705–711.
96. Antonioli E, Guglielmelli P, Poli G et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 2008; 93: 41–48.
97. Larsen TS, Pallisgaard N, Møller MB et al. High prevalence of arterial thrombosis in JAK2 mutated essential thrombocythaemia: independence of the V617F allele burden. Hematology 2008; 13: 71–76.
98. Pemmaraju N, Moliterno AR, Williams DM et al. The quantitative JAK2 V617F neutrophil allele burden does not correlate with thrombotic risk in essential thrombocytosis. Leukemia 2007; 21: 2210–2212.
99. Kittur J, Knudson RA, Lasho TL et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer 2007; 109: 2279–2284.
100. Mitus AJ, Schafer AI. Thrombocytosis and thrombocythemia. Hematol Oncol Clin North Am 1990; 4: 157–178.
101. Falanga A, Marchetti M, Barbui T et al. Pathogenesis of thrombosis in essential thrombocythemia and polycythemia vera: the role of neutrophils. Semin Hematol 2005; 42: 239–247.
102. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC et al. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 2006; 91: 169–175.
103. Maugeri N, Giordano G, Petrilli MP et al. Inhibition of tissue factor expression by hydroxyurea in polymorphonuclear leukocytes from patients with myeloproliferative disorders: a new effect for an old drug? J Thromb Haemost 2006; 4: 2593–2598.
104. Schwarz J, Penka M. The Czech Collaborative Group for Ph- MPD. Thromboreductin patient registry, a Czech success story. Turracher Höhe (Rakousko): AOP Orphan 2008.
105. Cortelazzo S, Finazzi G, Ruggeri M et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995; 332: 1132–1136.
106. Laguna MS, Kornblihtt LI, Marta RF et al. Effectiveness of anagrelide in the treatment of symptomatic patients with essential thrombocythemia. Clin Appl Thromb Hemost 2000; 6: 157–161.
107. Michiels JJ. Aspirin and platelet-lowering agents for the prevention of vascular complications in essential thrombocythemia. Clin Appl Thromb Hemost 1999; 5: 247–251.
108. Landolfi R, Marchioli R, Kutti J et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004; 350: 114–124.
109. Colwell JA. Aspirin for the primary prevention of cardiovascular events. Timely Top Med Cardiovasc Dis 2006; 10: E25.
110. Michiels JJ. Platelet-mediated microvascular inflammation and thrombosis in thrombocythemia vera: a distinct aspirin-responsive arterial thrombophilia, which transforms into a bleeding diathesis at increasing platelet counts. Pathol Biol (Paris) 2003; 51: 167–175.
111. Finazzi G, Barbui T. Efficacy and safety of hydroxyurea in patients with essential thrombocythemia. Pathol Biol (Paris) 2001; 49: 167–169.
112. Barbui T, Barosi G, Grossi A et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89: 215–232.
113. Harrison CN. Essential thrombocythaemia: challenges and evidence-based management. Br J Haematol 2005; 130: 153–165.
114. Barbui T. The leukemia controversy in myeloproliferative disorders: is it a natural progression of disease, a secondary sequela of therapy, or a combination of both? Semin Hematol 2004; 41 (2 Suppl 3): 15–17.
115. Briere J, Guilmin F. Management of patients with essential thrombocythemia: current concepts and perspectives. Pathol Biol (Paris) 2001; 49: 178–183.
116. Fruchtman SM. Treatment paradigms in the management of myeloproliferative disorders. Semin Hematol 2004; 41 (2 Suppl 3): 18–22.
117. Birgegård G. Long-term management of thrombocytosis in essential thrombocythaemia. Ann Hematol 2009; 88: 1–10.
118. Finazzi G, Caruso V, Marchioli R et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005; 105: 2664–2670.
119. Mazur EM, Rosmarin AG, Sohl PA et al. Analysis of the mechanism of anagrelide-induced thrombocytopenia in humans. Blood 1992; 79: 1931–1937.
120. Spencer CM, Brogden RN. Anagrelide. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the treatment of thrombocythaemia. Drugs 1994; 47: 809–822.
121. Solberg LA Jr, Tefferi A, Oles KJ et al. The effects of anagrelide on human megakaryocytopoiesis. Br J Haematol 1997; 99: 174–180.
122. Pescatore SL, Lindley C. Anagrelide: a novel agent for the treatment of myeloproliferative disorders. Expert Opin Pharmacother 2000; 1: 537–546.
123. Hong Y, Erusalimsky JD. Comparison of the pharmacological mechanisms involved in the platelet lowering actions of anagrelide and hydroxyurea: a review. Platelets 2002; 13: 381–386.
124. Petrides PE. Anagrelide: a decade of clinical experience with its use for the treatment of primary thrombocythaemia. Expert Opin Pharmacother 2004; 5: 1781–1798.
125. Pytlík R, Cmunt E, Kleibl Z et al. Úloha anagrelidu v léčbě esenciální trombocytemie. Transf Hematol Dnes 2004; 10: 154–160.
126. Dingli D, Tefferi A. A critical review of anagrelide therapy in essential thrombocythemia and related disorders. Leuk Lymphoma 2005; 46: 641–650.
127. Emadi A, Spivak JL. Anagrelide: 20 years later. Expert Rev Anticancer Ther 2009; 9: 37–50.
128. Balduini CL, Bertolino G, Noris P et al. Effect of anagrelide on platelet count and function in patients with thrombocytosis and myeloproliferative disorders. Haematologica 1992; 77: 40–43.
129. Balduini CL. Primary thrombocythemia: new drugs for an evolving disease. Haematologica 1992; 77: 297–301.
130. Doubek M, Brychtova Y, Doubek R et al. Anagrelide therapy in pregnancy: report of a case of essential thrombocythemia. Ann Hematol 2004; 83: 726–727.
131. Silverstein MN, Petitt RM, Solberg LA Jr et al. Anagrelide: a new drug for treating thrombocytosis. N Engl J Med 1988; 318: 1292–1294.
132. Anagrelide Study Group. Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Am J Med 1992; 92: 69–76.
133. Petrides PE, Beykirch MK, Trapp OM. Anagrelide, a novel platelet lowering option in essential thrombocythaemia: treatment experience in 48 patients in Germany. Eur J Haematol 1998; 61: 71–76.
134. Petrides PE, Gisslinger H, Steurer M et al. Pharmacokinetics, bioequivalence, tolerability, and effects on platelet counts of two formulations of anagrelide in healthy volunteers and patients with thrombocythemia associated with chronic myeloproliferation. Clin Ther 2009; 31: 386–398.
135. Steurer M, Gastl G, Jedrzejczak WW et al. Anagrelide for thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer 2004; 101: 2239–2246.
136. Birgegård G, Björkholm M, Kutti J et al. Adverse effects and benefits of two years of anagrelide treatment for thrombocythemia in chronic myeloproliferative disorders. Haematologica 2004; 89: 520–527.
137. Penka M, Schwarz J, Pavlík T et al. Esenciální trombocytemie a další myeloproliferace s trombocytemií v údajích registru pacientů léčených Thromboreductinem® do konce roku 2006. Vnitř Lék 2007; 53: 653–661.
138. Penka M, Schwarz J, Pavlík T et al. Výsledky léčby nemocných s esenciální trombocytemií a dalšími myeloproliferacemi provázenými trombocytemií – zpráva z registru pacientů léčených Thromboreductinem®. Vnitř Lék 2009; 55: I–XII.
139. Schwarz J, Penka M. The Czech Ph- MPD study group (CZEMP). Patient registry in the Czech Republic – updated results. Turracher Höhe (Rakousko): AOP Orphan 2010.
140. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001; 97: 863–866.
141. Harrison CN, Campbell PJ, Buck G et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005; 353: 33–45.
142. Petrides PE. Anagrelide: what was new in 2004 and 2005? Semin Thromb Hemost 2006; 32: 399–408.
143. Penka M, Doubek M, Schwarz J et al. Anagrelid v léčbě esenciální trombocytemie a dalších myeloproliferací s trombocytemií sledovaných v registru pacientů v ČR. Vnitř Lék 2006; 52: 498–503.
144. Wadenvik H, Kutti J, Ridell B et al. The effect of alpha-interferon on bone marrow megakaryocytes and platelet production rate in essential thrombocythemia. Blood 1991; 77: 2103–2108.
145. Lengfelder E, Griesshammer M, Hehlmann R. Interferon-alpha in the treatment of essential thrombocythemia. Leuk Lymphoma 1996; 22 (Suppl 1): 135–142.
146. Kiladjian JJ, Chomienne C, Fenaux P. Interferon-alpha therapy in bcr-ABL-negative myeloproliferative neoplasms. Leukemia 2008; 22: 1990–1998.
147. Velu T, Delwiche F, Gangji D et al. Therapeutic effect of human recombinant interferon-alpha-2C in essential thrombocythaemia. Oncology 1985; 42 (Suppl 1): 10–14.
148. Giles FJ, Singer CR, Gray AG et al. Alpha-interferon therapy for essential thrombocythaemia. Lancet 1988; 2: 70–72.
149. Bellucci S, Harousseau JL, Brice P et al. Treatment of essential thrombocythaemia by alpha 2a interferon. Lancet 1988; 2: 960–961.
150. Melillo L, Tieghi A, Candoni A et al. Outcome of 122 pregnancies in essential thrombocythemia patients: a report from the Italian registry. Am J Hematol 2009; 84: 636–640.
151. Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol 1992; 19 (3 Suppl 9): 1–10.
152. Seino Y, Nagao M, Yahagi T et al. Mutagenicity of several classes of antitumor agents to Salmonella typhimurium TA98, TA100, and TA92. Cancer Res 1978; 38: 2148–2156.
153. Mehnert K, Vogel W, Benz R et al. Different effects of mutagens on sister chromatid exchange induction in three Chinese hamster cell lines. Environ Mutagen 1984; 6: 573–583.
154. Schroeder AL. Chromosome instability in mutagen sensitive mutants of Neurospora. Curr Genet 1986; 10: 381–387.
155. Marsteinstredet U, Brunborg G, Bjørås M et al. DNA damage induced by 3-chloro-4-(dichloromethyl)-5-hydroxy-2[5H]-furanone (MX) in HL-60 cells and purified DNA in vitro. Mutat Res 1997; 390: 171–178.
156. Andersson M, Agurell E, Vaghef H et al. Extended-term cultures of human T-lymphocytes and the comet assay: a useful combination when testing for genotoxicity in vitro? Mutat Res 2003; 540: 43–55.
157. Ficsor G, Ginsberg LC. The effect of hydroxyurea and mitomycin C on sperm motility in mice. Mutat Res 1980; 70: 383–387.
158. Löfvenberg E, Nordenson I, Wahlin A. Cytogenetic abnormalities and leukemic transformation in hydroxyurea-treated patients with Philadelphia chromosome negative chronic myeloproliferative disease. Cancer Genet Cytogenet 1990; 49: 57–67.
159. Nand S, Messmore H, Fisher SG et al. Leukemic transformation in polycythemia vera: analysis of risk factors. Am J Hematol 1990; 34: 32–36.
160. Nand S, Stock W, Godwin J et al. Leukemogenic risk of hydroxyurea therapy in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Am J Hematol 1996; 52: 42–46.
161. Nielsen I, Hasselbalch HC. Acute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphan. Am J Hematol 2003; 74: 26–31.
162. Weinfeld A, Swolin B, Westin J. Acute leukaemia after hydroxyurea therapy in polycythaemia vera and allied disorders: prospective study of efficacy and leukaemogenicity with therapeutic implications. Eur J Haematol 1994; 52: 134–139.
163. Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea and pipobroman in 292 patients under the age of 65 years. Blood 1997; 90: 3370–3377.
164. Kiladjian JJ, Rain JD, Bernard JF et al. Long-term incidence of hematological evolution in three French prospective studies of hydroxyurea and pipobroman in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost 2006; 32: 417–421.
165. Sterkers Y, Preudhomme C, Laï JL et al. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood 1998; 91: 616–622.
166. Tóthová E, Fričová M, Štecová N et al. Leukemic transformation of polycythemia vera after treatment with hydroxyurea with abnormalities of chromosome 17. Neoplasma 2001; 48: 389–392.
167. Kiladjian JJ, Rain JD, Bernard JF et al. Long-term incidence of hematological evolution in three French prospective studies of hydroxyurea and pipobroman in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost 2006; 32: 417–421.
168. Papi M, Didona B, DePità O et al. Multiple skin tumors on light-exposed areas during long-term treatment with hydroxyurea. J Am Acad Dermatol 1993; 28: 485–486.
169. Best PJ, Petitt RM. Multiple skin cancers associated with hydroxyurea therapy. Mayo Clin Proc 1998; 73: 961–963.
170. Young HS, Khan AS, Kendra JR et al. The cutaneous side-effects of hydroxyurea. Clin Lab Haematol 2000; 22: 229–232.
171. Ravandi-Kashani F, Cortes J, Cohen P et al. Cutaneous ulcers associated with hydroxyurea therapy in myeloproliferative disorders. Leuk Lymphoma 1999; 35: 109–118.
172. Daoud MS, Gibson LE, Pittelkow MR. Hydroxyurea dermopathy: a unique lichenoid eruption complicating long-term therapy with hydroxyurea. J Am Acad Dermatol 1997; 36: 178–182.
173. Lannemyr O, Kutti J. Hydroxyurea as a cause of drug fever in essential thrombocythaemia. Eur J Haematol 1999; 62: 354–355.
174. Hallam MJ, Kolesar JM. Hydroxyurea induced acute elevations in liver function tests. J Oncol Pharm Pract 2008; 14: 61–63.
175. Finazzi G, Barbui T. Risk-adapted therapy in essential thrombocythemia and polycythemia vera. Blood Rev 2005; 19: 243–252.
176. Randi ML, Ruzzon E, Luzzatto G et al. Safety profile of hydroxyurea in the treatment of patients with Philadelphia-negative chronic myeloproliferative disorders. Haematologica 2005; 90: 261–262.
177. Wehmeier A, Südhoff T, Meierkord F. Relation of platelet abnormalities to thrombosis and hemorrhage in chronic myeloproliferative disorders. Semin Thromb Hemost 1997; 23: 391–402.
178. Rao AK. Molecular and biochemical basis for the platelet dysfunction in myeloproliferative disorders. Semin Hematol 2004; 41 (2 Suppl 3): 6–9.
179. van Genderen PJ, Mulder PG, Waleboer M et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997; 97: 179–184.
180. Griesshammer M, Bangerter M, van Vliet HH et al. Aspirin in essential thrombocythemia: status quo and quo vadis. Semin Thromb Hemost 1997; 23: 371–377.
181. Michiels JJ. Normal life expectancy and thrombosis-free survival in aspirin treated essential thrombocythemia. Clin Appl Thromb Hemost 1999; 5: 30–36.
182. Dzúrik R, Dzúriková V. Kyselina acetylsalicylová v antiagregačnej terapii. Slovakofarma Rev 1992; 2: 31–36.
183. Müller P, Dammann HG, Bergdolt H et al. Verbesserung der gastroduodenalen Verträglichkeit von Azetylsalizylsäure durch Ranitidin. Eine endoskopische kontrollierte Doppelblindstudie an gesunden Probanden. Arzneimittelforschung 1991; 41: 638–639.
184. Schnell O, Lang E, Bertholdt H. Untersuchungen zur thrombozytenaggregationshemmenden und analgetischen Wirkung der Acetylsalicylsäure. Arzneimittelforschung 1980; 30: 1917–1922.
185. Randi ML, Rossi C, Fabris F et al. Essential thrombocythemia in young adults: treatment and outcome of 16 pregnancies. J Intern Med 1999; 246: 517–518.
186. Ruggeri Z, Cannata ML, Stella NC et al. Pregnancy in essential thrombocythemia during aspirin treatment. Arch Gynecol Obstet 2003; 268: 209–210.
187. Niittyvuopio R, Juvonen E, Kaaja R et al. Pregnancy in essential thrombocythaemia: experience with 40 pregnancies. Eur J Haematol 2004; 73: 431–436.
188. Harrison C. Pregnancy and its management in the Philadelphia negative myeloproliferative diseases. Br J Haematol 2005; 129: 293–306.
189. Tefferi A, Passamonti F. Essential thrombocythemia and pregnancy: Observations from recent studies and management recommendations. Am J Hematol 2009; 84: 629–630.
190. Gangat N, Wolanskyj AP, Schwager S et al. Predictors of pregnancy outcome in essential thrombocythemia: a single institution study of 63 pregnancies. Eur J Haematol 2009; 82: 350–353.
191. Patrono C, Rocca B. Aspirin, 110 years later. J Thromb Haemost 2009; 7 (Suppl 1): 258–261.
192. Taft EG, Babcock RB, Scharfman WB et al. Plateletpheresis in the management of thrombocytosis. Blood 1977; 50: 927–933.
193. Boughton BJ. Chronic myeloproliferative disorders: improved platelet aggregation following venesection. Br J Haematol 1978; 39: 589–598.
194. Panlilio AL, Reiss RF. Therapeutic plateletpheresis in thombocythemia. Transfusion 1979; 19: 147–152.
195. Beard ME, Blacklock HA, Varcoe AR. Control of thrombocytosis by plateletpheresis using a cell separator. N Z Med J 1980; 91: 136–138.
196. Orlin JB, Berkman EM. Improvement of platelet function following plateletpheresis in patients with myeloproliferative diseases. Transfusion 1980; 20: 540–545.
197. Fabris F, Belloni M, Casonato A et al. Improvement of platelet aggregation abnormalities in thrombocytosis after thrombocytopheresis. Folia Haematol Int Mag Klin Morphol Blutforsch 1981; 108: 853–862.
198. Baron BW, Mick R, Baron JM. Combined plateletpheresis and cytotoxic chemotherapy for symptomatic thrombocytosis in myeloproliferative disorders. Cancer 1993; 72: 1209–1218.
199. Greist A. The role of blood component removal in essential and reactive thrombocytosis. Ther Apher 2002; 6: 36–44.
200. Landolfi R, Di Gennaro L, Barbui T et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007; 109: 2446–2452.
201. Dameshek W. Physiopathology and course of polycythemia vera as related to therapy. J Am Med Assoc 1950; 142: 790–797.
202. Wasserman LR. Polycythemia vera: its course and treatment; relation to myeloid metaplasia and leukemia. Bull N Y Acad Med 1954; 30: 343–375.
203. Berk PD, Goldberg JD, Silverstein MN et al. Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N Engl J Med 1981; 304: 441–447.
204. Rosenthal N, Bassen F. Course of polycythaemia. Arch Intern Med 1938; 62: 903–907.
205. Stephens D.J, Kaltreider N. The therapeutic use of venesection in polycythemia. Ann Intern Med 1937; 10: 1565–1581.
206. Thomas DJ, du Boulay GH, Marshall J et al. Cerebral blood-flow in polycythaemia. Lancet 1977; 2: 161–163.
207. Thomas DJ, Marshall J, Russell RW et al. Effect of haematocrit on cerebral blood-flow in man. Lancet 1977; 2: 941–943.
208. Steensma DP. The chronic myeloproliferative disorders: an historical perspective. Curr Hematol Rep 2003; 2: 221–230.
209. Videbaek A. Polycythaemia vera. Course and prognosis. Acta Med Scand 1950; 138: 179–187.
210. Kaboth U, Rumpf KW, Liersch T et al. Advantages of isovolemic large-volume erythrocytapheresis as a rapidly effective and long-lasting treatment modality for red blood cell depletion in patients with polycythemia vera. Ther Apher 1997; 1: 131–134.
211. Kaboth U, Rumpf KW, Lipp T et al. Treatment of polycythemia vera by isovolemic large-volume erythrocytapheresis. Klin Wochenschr 1990; 68: 18–25.
212. Valbonesi M, Bruni R. Clinical application of therapeutic erythrocytapheresis (TEA). Transfus Sci 2000; 22: 183–194.
213. Fruchtman SM, Mack K, Kaplan ME et al. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997; 34: 17–23.
214. Koza V, Cetkovský P, Faber E et al. Indikace k alogenním a autologním transplantacím krvetvorných buněk. Doporučení České hematologické společnosti ČLS JEP a České onkologické společnosti ČLS JEP. Klin Onkol 2006; 19: 310–316.
215. Schwarz J, Marková M, Vítek A et al. Indikace k transplantaci hematopoetických kmenových buněk u Ph- myeloproliferativních onemocnění (MPO). (Abstr. H-O-032). Transf Hematol Dnes 2008; 14 (Suppl 2): 52–53.
216. Njoku OS, Lewis SM, Catovsky D et al. Anaemia in myelofibrosis: its value in prognosis. Br J Haematol 1983; 54: 79–89.
217. Barosi G, Berzuini C, Liberato LN et al. A prognostic classification of myelofibrosis with myeloid metaplasia. Br J Haematol 1988; 70: 397–401.
218. Visani G, Finelli C, Castelli U et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br J Haematol 1990; 75: 4–9.
219. Hasselbalch H, Jensen BA. Prognostic factors in idiopathic myelofibrosis: a simple scoring system with prognostic significance. Eur J Haematol 1990; 44: 172–178.
220. Cervantes F, Pereira A, Esteve J et al. Identification of “short-lived” and “long-lived” patients at presentation of idiopathic myelofibrosis. Br J Haematol 1997; 97: 635–640.
221. Reilly JT, Snowden JA, Spearing RL et al. Cytogenetic abnormalities and their prognostic significance in idiopathic myelofibrosis: a study of 106 cases. Br J Haematol 1997; 98: 96–102.
222. Kreft A, Weiss M, Wiese B et al. Chronic idiopathic myelofibrosis: prognostic impact of myelofibrosis and clinical parameters on event-free survival in 122 patients who presented in prefibrotic and fibrotic stages. A retrospective study identifying subgroups of different prognoses by using the RECPAM method. Ann Hematol 2003; 82: 605–611.
223. Strasser-Weippl K, Steurer M, Kees M et al. Age and hemoglobin level emerge as most important clinical prognostic parameters in patients with osteomyelofibrosis: introduction of a simplified prognostic score. Leuk Lymphoma 2006; 47: 441–450.
224. Li Z, Deeg HJ. Pros and cons of splenectomy in patients with myelofibrosis undergoing stem cell transplantation. Leukemia 2001; 15: 465–467.
225. Hoffman R, Prchal JT, Samuelson S et al. Philadelphia chromosome-negative myeloproliferative disorders: biology and treatment. Biol Blood Marrow Transplant 2007; 13 (1 Suppl 1): 64–72.
226. Guardiola P, Anderson JE, Bandini G et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Société Française de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood 1999; 93: 2831–2838.
227. Deeg HJ, Gooley TA, Flowers ME et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003; 102: 3912–3918.
228. Kröger N, Zabelina T, Schieder H et al. Pilot study of reduced-intensity conditioning followed by allogeneic stem cell transplantation from related and unrelated donors in patients with myelofibrosis. Br J Haematol 2005; 128: 690–697.
229. Rondelli D, Barosi G, Bacigalupo A et al. Allogeneic hematopoietic stem-cell transplantation with reduced-intensity conditioning in intermediate- or high-risk patients with myelofibrosis with myeloid metaplasia. Blood 2005; 105: 4115–4119.
230. Kröger N, Holler E, Kobbe G et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009; 114: 5264–5270.
231. Markova M, Schwarz J, Vitek A et al. Allogeneic transplantation for myelofibrosis in myeloproliferative disease-a single center experience. Haematologica 2008; 93 (Suppl 1): 134. Abstr. 0328.
232. Chanarin I. Folate deficiency in the myeloproliferative disorders. Am J Clin Nutr 1970; 23: 855–860.
233. Faurschou M, Nielsen OJ, Jensen MK et al. High prevalence of hyperhomocysteinemia due to marginal deficiency of cobalamin or folate in chronic myeloproliferative disorders. Am J Hematol 2000; 65: 136-140.
234. Jack FR, Smith SR, Saunders PW. Idiopathic myelofibrosis: anaemia may respond to low-dose dexamethasone. Br J Haematol 1994; 87: 877–878.
235. Arana-Yi C, Quintás-Cardama A, Giles F et al. Advances in the therapy of chronic idiopathic myelofibrosis. Oncologist 2006; 11: 929–943.
236. Cervantes F. Modern management of myelofibrosis. Br J Haematol 2005; 128: 583–592.
237. Cervantes F, Alvarez-Larrán A, Hernández-Boluda JC et al. Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature. Br J Haematol 2004; 127: 399–403.
238. Cervantes F, Alvarez-Larrán A, Hernández-Boluda JC et al. Darbepoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol 2006; 134: 184–186.
239. Iki S, Yagisawa M, Ohbayashi Y et al. Adverse effect of erythropoietin in myeloproliferative disorders. Lancet 1991; 337: 187–188.
240. Berrebi A, Feldberg E, Spivak I et al. Mini-dose of thalidomide for treatment of primary myelofibrosis. Report of a case with complete reversal of bone marrow fibrosis and splenomegaly. Haematologica 2007; 92: e15–e16.
241. Tefferi A, Verstovsek S, Barosi G et al. Pomalidomide is active in the treatment of anémia associated with myelofibrosis. J Clin Oncol 2009; 27: 4563–4569.
242. Quintás-Cardama A, Kantarjian HM, Manshouri T et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 2009; 27: 4760–4766.
243. Mesa RA, Steensma DP, Pardanani A et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003; 101: 2534–2541.
244. Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program 2009: 636–642.
245. Reilly JT. Idiopathic myelofibrosis: pathogenesis to treatment. Hematol Oncol 2006; 24: 56–63.
246. Barosi G, Ambrosetti A, Buratti A et al. Splenectomy for patients with myelofibrosis with myeloid metaplasia: pretreatment variables and outcome prediction. Leukemia 1993; 7: 200–206.
247. Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood 2009; 113: 5394–5400.
248. Tefferi A, Mesa RA, Nagorney DM et al. Splenectomy in myelofibrosis with myeloid metaplasia: a single-institution experience with 223 patients. Blood 2000; 95: 2226–2233.
249. Abu-Hilal M, Tawaker J. Portal hypertension secondary to myelofibrosis with myeloid metaplasia: a study of 13 cases. World J Gastroenterol 2009; 15: 3128–3133.
250. Bělohlávek J, Schwarz J, Jirásek A et al. Idiopathic myelofibrosis complicated by portal hypertension treated with a transjugular intrahepatic portosystemic shunt (TIPS). Wien Klin Wochenschr 2001; 113: 208–211.
251. Doki N, Irisawa H, Takada S et al. Transjugular intrahepatic portosystemic shunt for the treatment of portal hypertension due to idiopathic myelofibrosis. Intern Med 2007; 46: 187–190.
252. Mishchenko E, Tefferi A. Treatment options for hydroxyurea-refractory disease complications in myeloproliferative neoplasms: JAK2 inhibitors, radiotherapy, splenectomy and transjugular intrahepatic portosystemic shunt. Eur J Haematol 2010; 85: 192–199.
253. Tefferi A, Barrett SM, Silverstein MN et al. Outcome of portal-systemic shunt surgery for portal hypertension associated with intrahepatic obstruction in patients with agnogenic myeloid metaplasia. Am J Hematol 1994; 46: 325–328.
254. Dulíček P, Malý J. Budd-Chiari syn-drom – úloha hematologa v multidisciplinárním přístupu. Vnitř Lék 2008; 54: 842–845.
255. Silverstein MN, ReMine WH. Splenectomy in myeloid metaplasia. Blood 1979; 53: 515–518.
256. Mach J. Profesní standardy, doporučené postupy a závazná stanoviska. Tempus medicorum 2010; 19: 6–8.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2011 Číslo 2
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Statiny indukovaná myopatie: Jak na diferenciální diagnostiku?
- Nech brouka žít… Ať žije astma!
- Co dělat při intoleranci statinů?
Najčítanejšie v tomto čísle
- Bikuspidální aortální chlopeň – etiopatogeneze a přirozený vývoj
- Alkoholová kardiomyopatia – diagnóza stále aktuálna
- Diagnostika a léčba BCR/ABL-negativních myeloproliferativních onemocnění – principy a východiska doporučení CZEMP
- Hodnotenie kvality života a funkčného stavu u pacientov s reumatoidnou artritídou