
Dysplastické změny megakaryopoézy podle WHO klasifikace 2008
Dysplastic changes in megakaryopoiesis according to WHO classification 2008
Megakaryocytic dysplasia is frequent finding not only in patients with diagnose of myelodysplastic syndromes but also in wide spectrum of the other clinic situations. In this paper authors summarized the possibilities and also limitations of peripheral blood and bone marrow smears evaluation in the process of diagnose of myelodysplastic syndrome. Usual megakaryopoiesis with respect of morphology point of view is also mentioned during introduce of the paper.
Key words:
megakaryopoiesis – dysplastic changes – myelodysplastic syndrome
Authors:
A. Buliková; J. Kissová; M. Antošová; L. Antošová; I. Trnavská
Authors‘ workplace:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
Published in:
Vnitř Lék 2010; 56(Supplementum 1): 31-38
Category:
16th Parizek's Days, Ostrava-Poruba, March 25th –26th 2010
Overview
Dysplazie v megakaryocytech je častým nálezem nejen u nemocných s diagnózou myelodysplastického syndromu, ale taktéž v širokém spektru jiných klinických situací. V tomto článku autoři shrnuli možnosti a také limitace vyšetření nátěrů periferní krve a kostní dřeně v průběhu diagnostiky myelodysplastického syndromu. V úvodu je rovněž zmíněna normální megakaryopéza z morfologického pohledu.
Klíčová slova:
megakaryopoéza – dysplastické změny – myelodyplastický syndrom
Megakaryopoéza a trombopoéza
Krevní destičky jsou důležitými elementy periferní krve, které se kromě účasti na krevním srážení podílí i na řadě dalších procesů probíhajících v lidském organizmu, jako jsou např. zánět, přirozená imunita, neoangiogeneze a metastazování nádoru [1]. Vznikají v procesu nazývaném megakaryopoéza. Ten začíná v hemopoetických multipotentních kmenových buňkách. Následně dochází k vývoji cestou multipotentního progenitoru, který již má omezenou schopnost sebeobnovy, dále pak myeloidního progenitoru schopného vyzrávat do všech myeloidních linií a dále pak společného progenitoru pro erytroidní - megakaryocytovou řadu. Někteří věří, že tento typ buňky může vznikat i přímo z hemopoetické kmenové buňky [2]. Megakaryocytární progenitor dává vznik koloniím megakaryocytů v tkáňových kulturách. Je charakterizován imunofenotypizačním nálezem CD34, CD31 a CD133. Podle exprese HLA-DR lze odlišit méně zralý progenitor, tj. BFU - MK, který má expresi tohoto znaku nízkou, a dále více zralý, tj. CFU - MK, který má expresi HLA-HR vysokou. První buňkou, která může být za jistých okolností rozeznána cytologicky, je megakaryoblast [3]. Běžně je těchto elementů přítomno okolo 3 % z CD34 pozitivních buněk [1] a v případě normální krvetvorby je od jiných blastů nerozeznáme, neboť jde o malou diploidní buňku s proliferačním potenciálem [3]. Do tohoto stadia prekurzory megakaryocytů podléhají normálním procesům proliferace, které zahrnuje zdvojení obsahu DNA a následné rozdělení buňky na dvě dceřiné. Poté, co je zahájena syntéza destičkových proteinů, nastupuje proces zvaný endomitóza. Již dlouho je známo, že na rozdíl od jiných buněčných linií, u nichž je amplifikace provázená duplikací DNA s následným rozdělením buňky, megakarocyty násobí obsah DNA bez buněčného dělení. Amplifikace tak vede k polypoidizaci buňky, a tím i k jejímu zvětšování, nicméně procesy maturace charakterizované vývojem specifických organel jsou zahájeny až poté, co je amplifikace ukončena [4]. Endomitóza spočívá v odchylném procesu buněčného cyklu, kdy proběhne normální fáze G1, S i G2, resp. i začátek mitózy, tj. profáze a metafáze a nakonec i první část anafáze (tzv. anafáze A), která je charakterizovaná separací sesterských chromatid a pohybem chromozomů k pólům [5]. Následná část anafáze (tzv. anafáze B) probíhá odchylně – nenásleduje po ní normální rozdělení jaderného chromatinu do dvou separátních jader a následně i rozdělení buňky, tzv. cytokineze. Potenciální dceřiné buňky se naopak pohybují k sobě, splývá jaderný chromatin i celá buňka. Na konci endomitózy vzniká megakaryocyt s jedním jádrem a jednou jadernou membránou. Navíc u polyploidních megakaryocytů je dělicí vřeténko taktéž multipolární, přičemž počet těchto pólů koresponduje s úrovní „ploidity“ [1]. Duplikace DNA může být v megakaryocytu ukončena kdykoli mezi 2N (tedy normální diploidní buňka) a 64N, možná i 128N. Většina lidských megakaryocytů (okolo 50 %) má 16N, tedy jádro osmkrát větší než normální diploidní buňka. V průběhu tohoto procesu se navyšuje syntéza proteinů důležitých pro funkci destiček a současně dochází ke zvětšování objemu buňky na hodnotu více než 80násobnou. Současně s narůstáním ploidity dochází k navýšení rozměru buňky – u buněk 2N mají megakaryocyty rozměr 21 ± 4 µm, zatímco u buněk s 64N 56 ± 8 µm, a tím se zvyšují i schopnosti megakaryocytů produkovat trombocyty [6]. Každý lalok jádra odpovídá příslušné oblasti multipolárního dělicího vřeténka a jejich počet odráží stupeň ploidity [5]. Není úplně vyjasněno, jaký význam má vývoj polyploidního jádra v megakaryocytech. Zdá se, že je to cesta ke zvyšování syntézy proteinů a modifikování genové exprese spojené s vyzráváním a polyploidizace je zřejmě integrována do diferenciačního programu megakaryocytů [1]. Stupeň ploidity je v přímé korelaci s obsahem DNA a se stupněm produkce destiček [6]. Spolu s endomitotickým procesem se začíná měnit i cytoplazma, stává se objemnější, navyšuje se počet granulí a dochází k produkci membránových systémů. Zralý megakaryocyt se přesunuje do blízkosti venózních sinusů kostní dřeně. Ve výběžcích jeho cytoplazmy vznikají „protodestičky“, ty se větví a prodlužují a zasahují do vlastního sinusu [7]. Velké protodestičky se pak oddělují od mateřského megakaryocytu a vlastní destičky z nich vznikají až v cirkulaci [1]. Celý proces megakaryopoézy trvá asi 8 – 10 dní [8].
Proliferace megakaryocytů a tvorba trombocytů je primárně regulována interakcí mezi trombopoetinem (TPO) a jeho povrchovým buněčným receptorem (MPL). Proces megakaryopoézy je však ovlivňován i řadou transkripčních faktorů, z nichž jedním z nejdůležitějších jsou zejména GATA1 - protein, případně FOG - 1 (friend of GATA - 1). Ty jsou postiženy u X - vázané kongenitální trombocytopenie [9]. GATA - 1 může spolupracovat i s AML-1 (RUNX1), resp. transkripčním komplexem „core binding factor“ (CBF), tedy s faktory, jejichž geny jsou často zasaženy v případech akutní myeloidní leukemie. Na myších modelech vede jejich chybění k dysmegakaryopoéze. Podobně u další vrozené trombocytopenie, tzv. Paris-Trousseauova/Jacobsenova syndromu, je přítomna delece chromozomu 11q23 zahrnující chybění transkripčního faktoru FLI - 1, který spolupracuje s GATA1 a FOG - 1 na expresi genů pro destičkové glykoproteiny. To je jen několik příkladů z mnoha, které by mohly být uvedeny. Mají ilustrovat, jak zásah do těchto regulačních proteinů může vést k vrozené, ale taktéž získané poruše normální megakaryopoézy.
Morfologické nálezy v průběhu normální megakaryopoézy
Z morfologického hlediska rozeznáváme tři stadia zralosti megakaryocytů [3]. Megakaryocyty skupiny I mají silně bazofilní cytoplazmu, vysoký nukleo - cytoplazmatický (N/ C) poměr (obr. 1). Skupina II zahrnuje megakaryocyty s nižším N/ C poměrem, cytoplazma je méně bazofilní a obsahuje azurofilní granula (obr. 2). Megakaryocyty III. skupiny mají objemnou slabě bazofilní cytoplazmu a bohatou azurofilní granulaci. Při okrajích je však cytoplazma agranulární (obr. 3). Tyto poslední megakaryocyty již nesyntetizují DNA, jejich jádra nepodléhají endomitóze, ale jsou schopny produkovat destičky. Ve všech třech skupinách megakaryocytů jsou elementy s jádry 8N (tj. s obsahem chromatinu čtyřnásobným ve srovnání s běžnou diploidní buňkou), 16N a 32N. 4N megakaryocyty jsou obvyklé jen ve skupině I, zatímco 32N megakaryocyty jsou nejčastější ve skupině III. Jádra převážné většiny normálních polyploidních megakaryocytů tvoří nepravidelné laloky spojené chromatinovými vlákny. Poté, co se cestou tvorby protodestiček zasahujících do sinusů kostní dřeně s následnou cytoplazmatickou fragmentací vytvoří krevní destičky, dostávají se megakaryocyty do finálního stadia, kterým je zdánlivě holé jádro, které má ve skutečnosti tenký prstenec cytoplazmy. Velikost megakaryocytů je udávána v širokém rozmezí 30 – 160 µm podle stadia jejich zralosti [3].
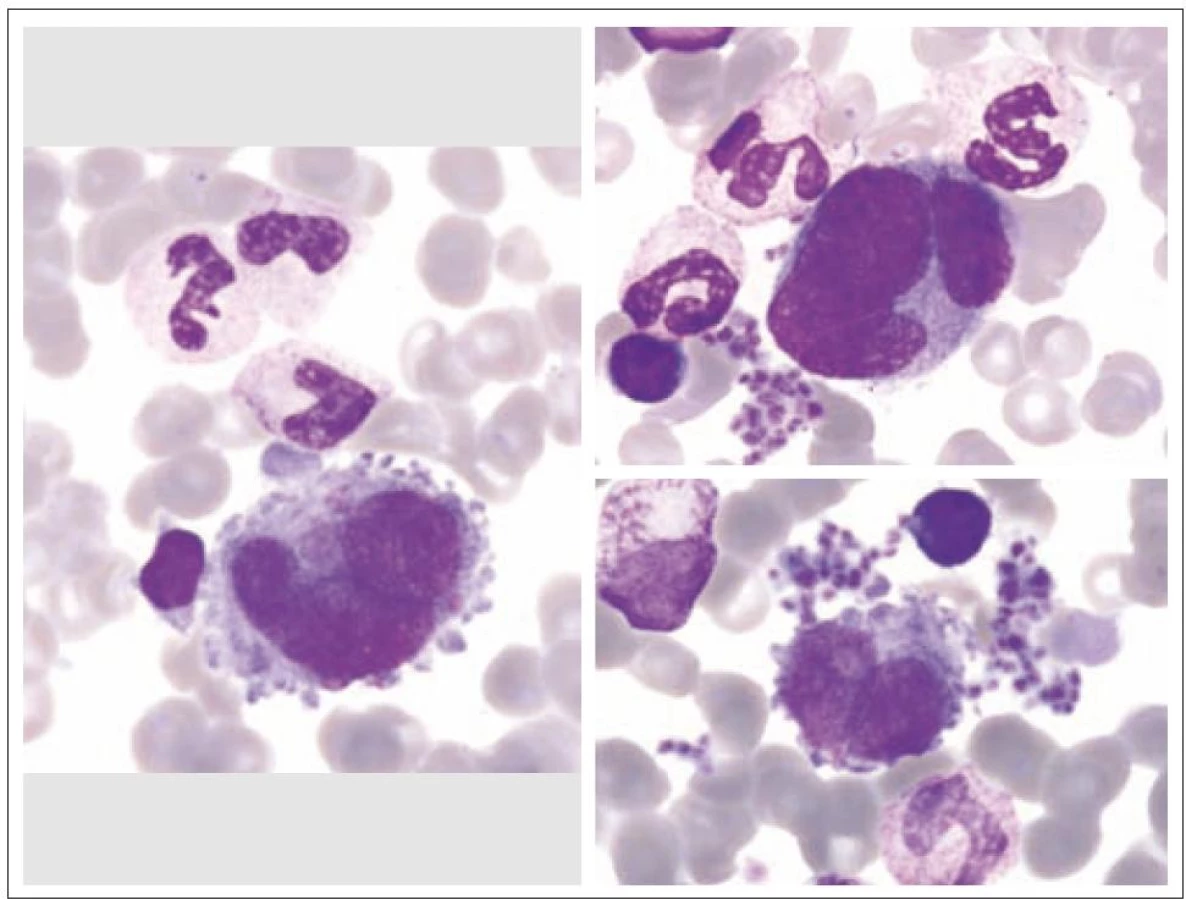
V cytologickém morfologickém nálezu je důležité zhodnocení počtu megakaryocytů. Nicméně toto stanovení je obvykle velmi subjektivní a umožňuje jen říci, zda je megakaryopoéza normální, snížená, či zvýšená [3]. Jindy je udáván počet 2 – 20 megakaryocytů na 1 000 jaderných buněk dřeně [8]. Přesnější určení počtu megakaryocytů umožňuje vyšetření histologické. I zde je však rozdíl, zda je vyšetřeno koagulum z aspirační biopsie či vzorek získaný odběrem kostní dřeně trepanobiopsií. Obvykle je zjišťováno méně megakaryocytů z prvního materiálu, zřejmě proto, že není tak jednoduché velké megakaryocyty aspirovat [3].
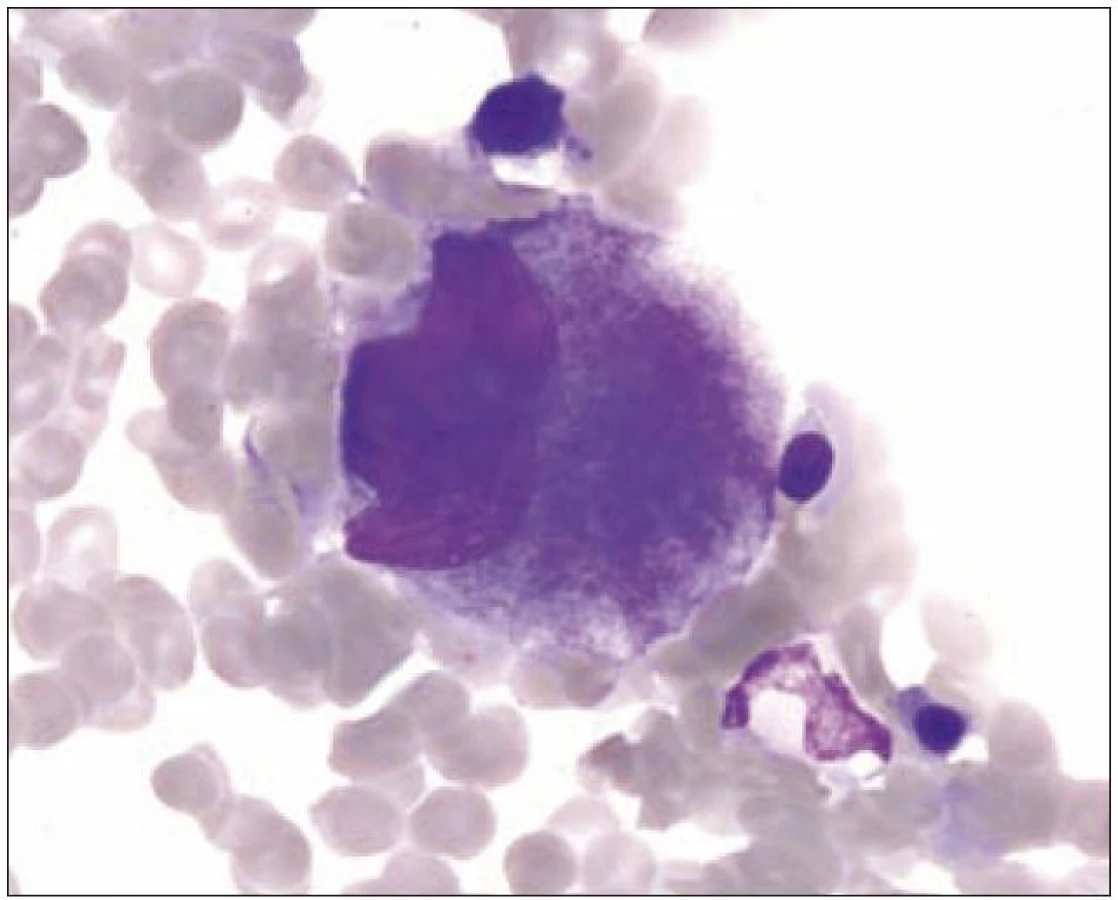
I v normální kostní dřeni lze zachytit proces nazývaný emperipoléza (obr. 3). Jde o zdánlivé pohlcení jiných hematopoetických buněk megakaryocytem. Tento proces se liší od fagocytózy, neboť buňky vstupují do megakaryocytu a cestují v něm dilatovanými dutinami demarkačního membránového systému.
Myelodysplastický syndrom ve WHO klasifikacích
Jako myelodysplastický syndrom (MDS)označujeme skupinu klonálních chorob hemopoetické kmenové buňky. Tato skupina je charakterizována cytopenií či cytopeniemi, dysplazií jedné či více krvetvorných řad, inefektivní hematopoézou a vyšším rizikem vývoje akutní myeloidní leukemie [10]. Vzhledem k tomu, že jde o více typů onemocnění s různou prognózou, byla vždy vyvíjena snaha o nastavení přesných kritérií pro odlišení jednotlivých stavů. Jako první byla řadu let používána klasifikace francouzských, amerických a britských expertů, tj. FAB klasifikace [11]. Od poloviny 90. let minulého století pracovala řada odborníků na WHO klasifikaci nádorů hemopoetických a lymfatických tkání, přičemž tato byla ve finální podobě vydána v roce 2001 a zahrnovala i podmínky diagnostiky a klasifikace MDS [12]. Vzhledem k rozvoji nových metod laboratorní diagnostiky hematologických malignit a současně vzhledem k nárůstu klinických zkušeností s WHO klasifikací z roku 2001 je vyvíjena snaha mezinárodních pracovních skupin pro jednotlivé skupiny diagnóz o průběžné revidování poznatků a jejich praktické uplatnění [13,14]. Tato skutečnost měla za následek vydání nové WHO klasifikace na podzim roku 2008 a zde byla nově revidována i problematika MDS [10]. Nicméně snahy o rychlé a průběžné zapojování nových informací na poli diagnostiky MDS neutuchají ani po vydání revidované klasifikace [15,16].
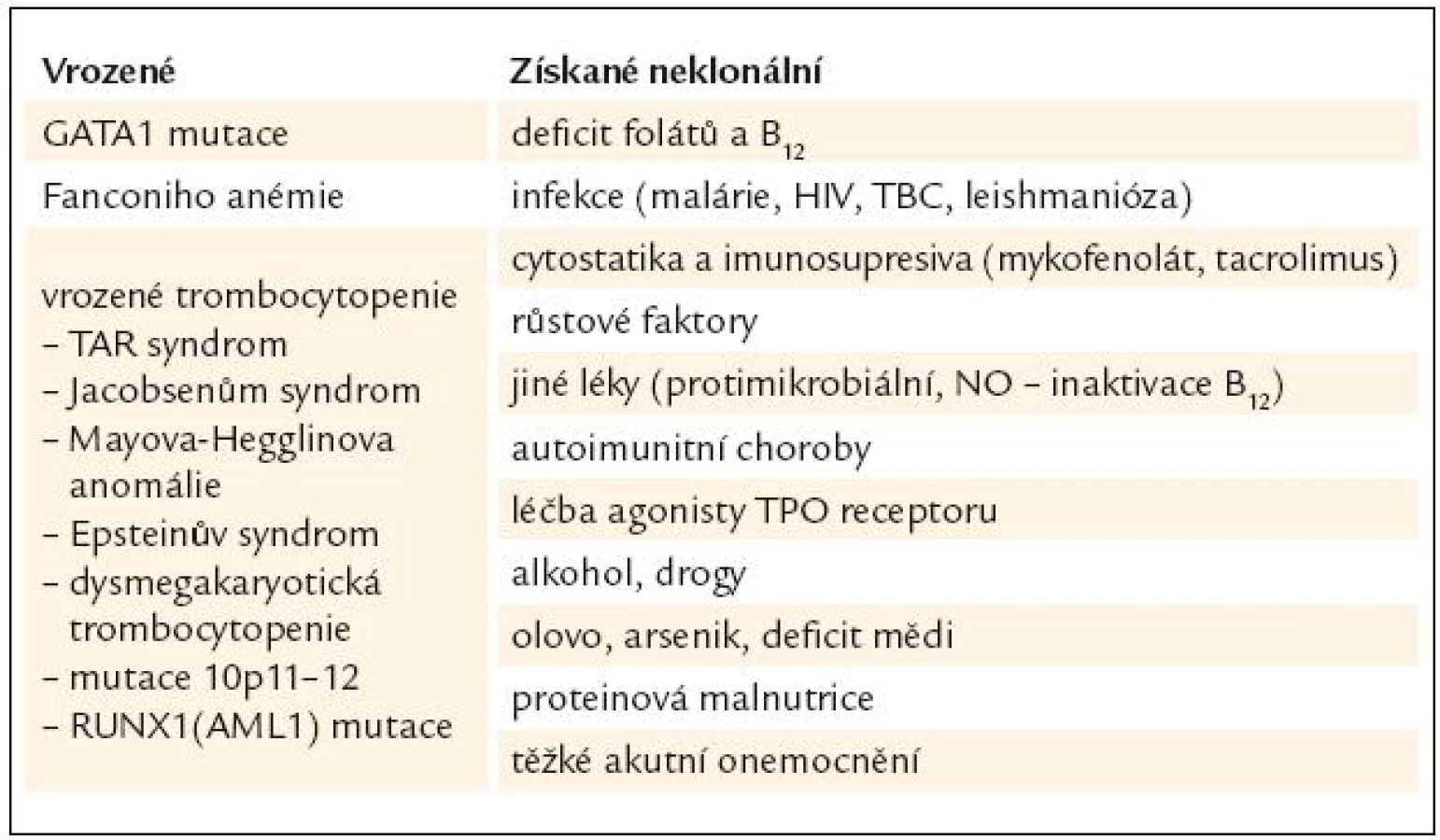
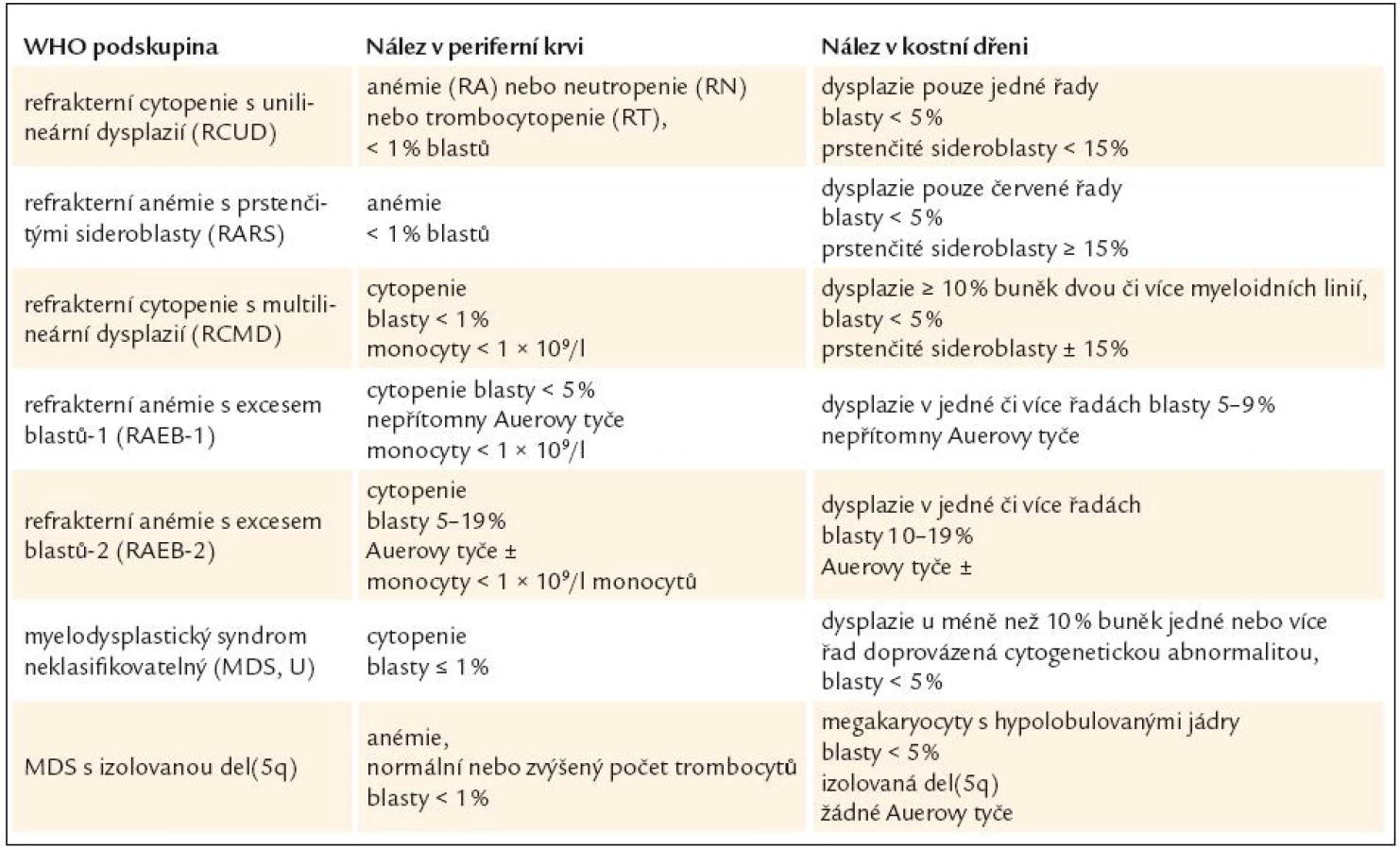
Diagnóza MDS je postavena na klinickém vyšetření, vyhodnocení nálezu krevního obrazu, diferenciálního rozpočtu jaderných buněk periferní krve a kostní dřeně včetně případného cytochemického barvení, vyhodnocení nálezu histologického vyšetření kostní dřeně vzorku získaného trepanobiopsií, event. včetně imunohistochemie, posouzení nálezů z průtokové cytometrie, a to jak u vzorků periferní krve, tak i kostní dřeně, zhodnocení nálezů cytogenetického vyšetření včetně jejich modalit (jako je fluorescenční in situ hybridizace). V diferenciální diagnostice zejména vůči akutním myeloidním leukemiím s nízkým počtem blastů lze s výhodou využít i nálezů molekulární genetiky [3]. Nicméně ani zde práce lékaře, který provádí diagnostiku MDS, zdaleka nekončí. Zejména při výskytu podezření na MDS v dětském věku je zapotřebí vyloučit velkou skupinu vrozených chorob dřeňového selhání, zatímco v pozdějším věku je nutno pomýšlet na řadu klinických situací, které mohou normální krvetvorbu ovlivnit, a to včetně morfologického cytologického nálezu v kostní dřeni. Přehled podává tab. 1.
Nezbytnou podmínkou cytomorfologické diagnostiky MDS je provedení dokonalého nátěru jak z periferní krve, tak z aspirační biopsie kostní dřeně. Tento musí být náležitě nabarven základním barvením (v našich podmínkách nejčastěji dle May - Grünwald - Giemsa, v zahraničí také Wright - Giemsa), resp. pro nátěry zejména z kostní dřeně je běžně využíváno i barvení cytochemické [17]. Rutinně je v případě diagnózy MDS prováděno na obsah železa Perlsovo barvení, které umožní hodnotit zásoby železa, počet sideroblastů, abnormální a zejména prstenčité sideroblasty. Bez tohoto stanovení se klasifikace MDS neobejde. Stejně tak je naprosto nezbytné provést cytochemické barvení na přítomnost myeloperoxidázy v neutrofilních granulocytech. To může odhalit jednak přítomnost Auerových tyčí, jejich nález posouvá kategorizaci MDS do vyššího stadia (tab. 2), jednak snížení myeloperoxidázy ve zralejších stadiích granulopoézy je samostatnou známkou poruchy normálního vývoje těchto buněk. Barvení na obsah glykogenu metodou „Periodic Acid Schiff“ (PAS) může být pozitivní v normoblastech, což je hodnoceno jako nezávislá známka dysplastické erytropoézy, navíc ale může být nápomocno k rozpoznání abnormních megakaryocytů [18].
V základním barvení je proveden rozpočet nejméně na 200 jaderných elementů periferní krve a na 500 jaderných elementů kostní dřeně [10,12]. Normoblasty v periferní krvi a megakaryocyty v periferní krvi i kostní dřeni se do tohoto rozpočtu nezahrnují [17]. Je-li erytropoéza významně zmnožena, pak by měl rozpočet zahrnovat nejméně 100 non-erytroidních elementů. Tento rozpočet slouží zejména ke stanovení počtu myeloblastů, a tím i k přesné kategorizaci typu MDS (tab. 2).
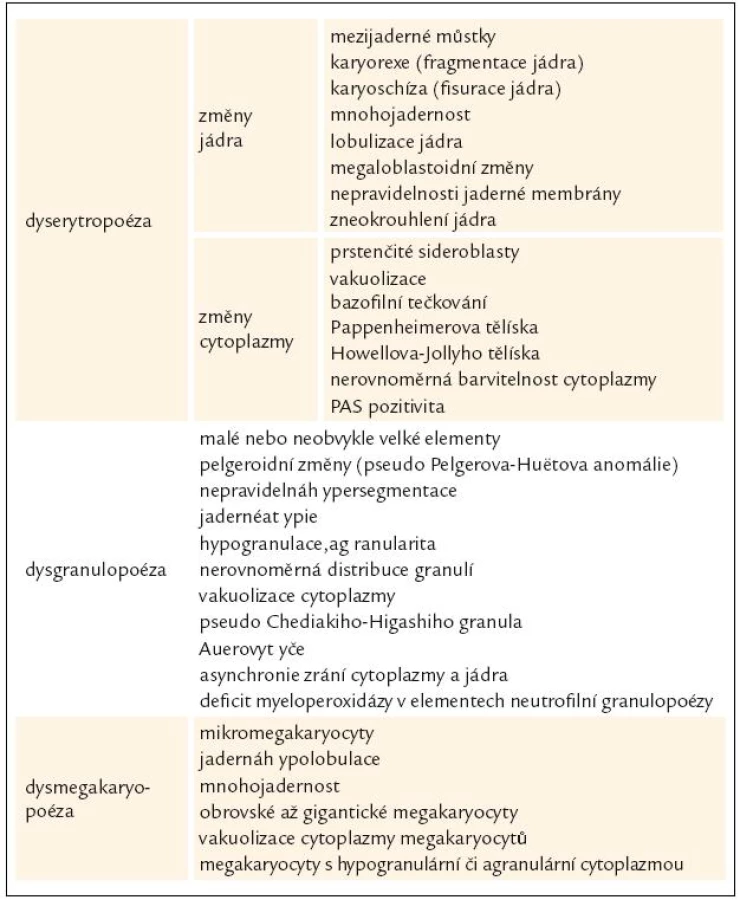
WHO klasifikace navíc stanovuje pro morfologickou diagnózu myelo-dysplazie, že pro určení dysplastických změn pro potřeby diagnózy MDS musí být přesně určenými morfologickými odchylkami postiženo nejméně 10 % buněk každé vývojové řady [10,12]. Přehled těchto odchylek v jednotlivých buněčných liniích shrnuje tab. 3 [3,10,19]. WHO klasifikace 2008 upřesňuje, že procento zastoupených dysplastických změn musí být určeno na 200 buňkách granulopoézy, 200 buňkách erytropoézy a nejméně 30 megakaryocytech [10].
Známky dysplazie v megakaryocytární řadě
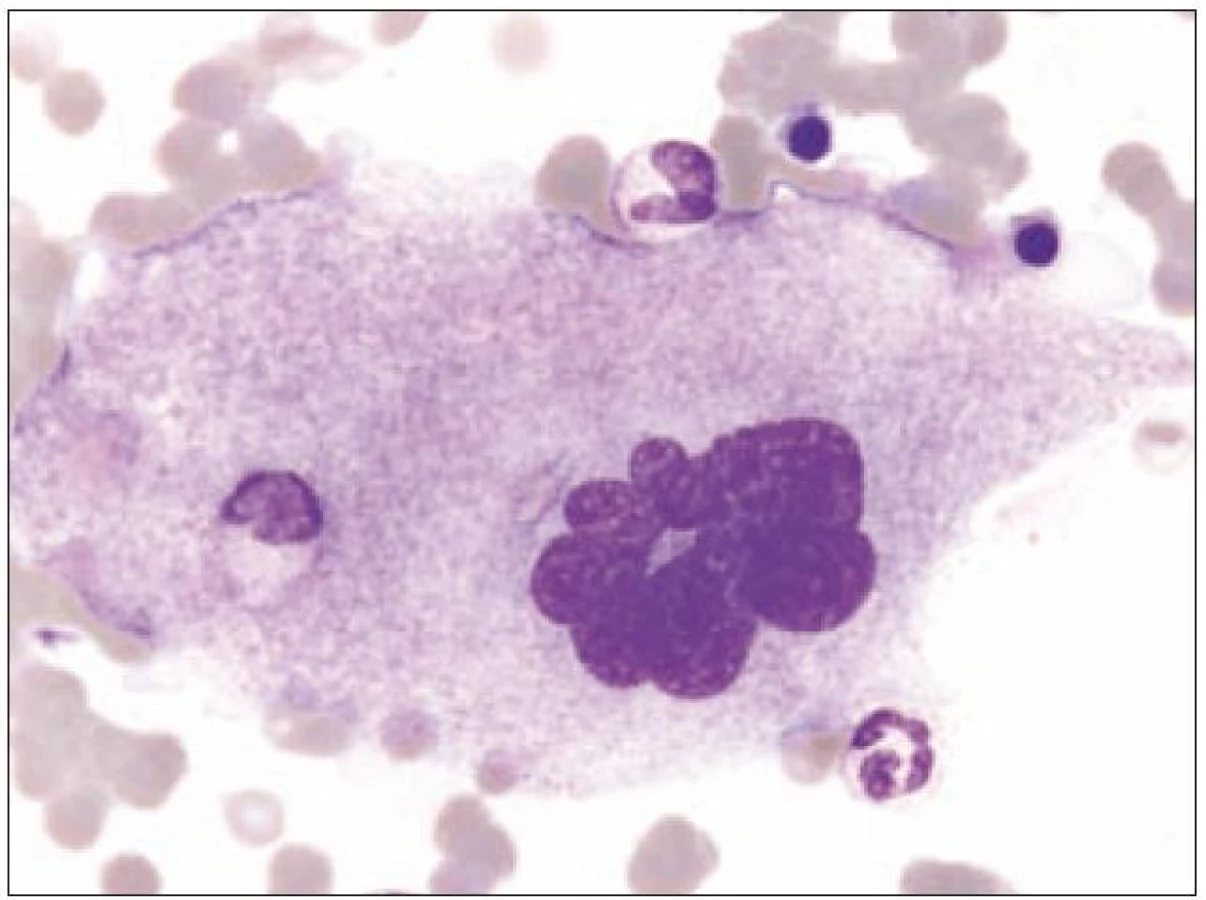
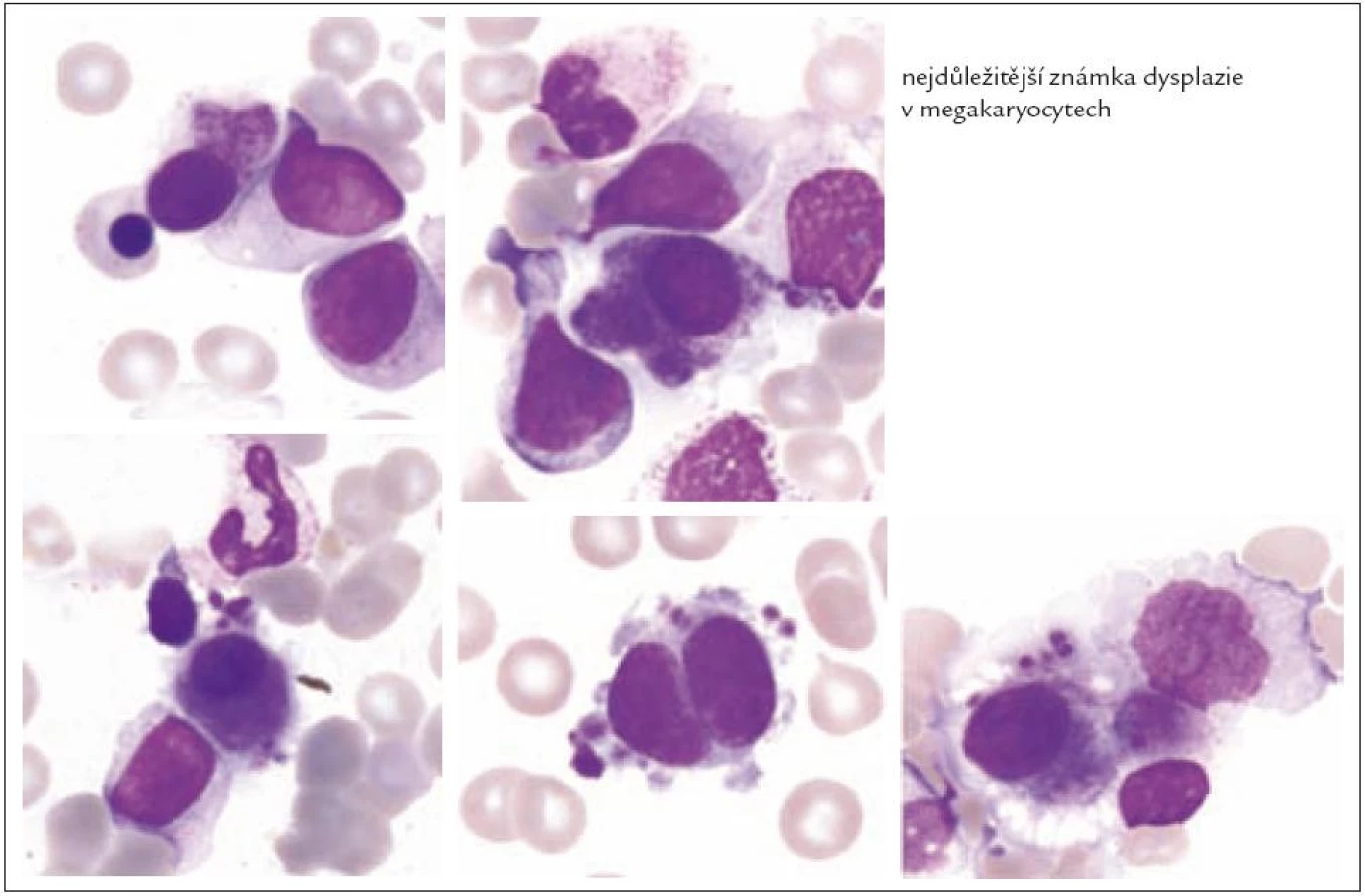
Z tohoto pohledu je jedním z nejdůležitějších nálezů průkaz velmi malých forem megakaryocytů, tzv. „mikromegakaryocyty“ (obr. 4). Není arbitrárně stanoveno, jak „malý“ má být mikromegakaryocyt. Diskutuje se o rozměrech menších než 15 – 30 µm. Praktické se zdá doporučení Bainové z roku 2010, že jde o buňku přibližně velikosti myeloblastu s jedním či dvěma malými kulatými jádry [18]. Tyto buňky nebyly nikdy nalezeny v nátěrech z aspirační biopsie zdravých jedinců. Již o něco méně významný je průkaz nesegmentovaných jader zralých megakaryocytů, případně i multinuklearita, tj. nález více než jednoho zcela separovaného jádra v jednom megakaryocytu (obr. 5). Tyto formy mohou být event. v normální dřeni v nízkém procentu zastiženy [3] a je důležité v těchto případech provést skórování, zda nález morfologických abnormit překročí hranici 10 % [10,12]. Nález megakaryocytů s nelobulizovanými jádry je typický pro MDS spojený s izolovanou delecí krátkého raménka chromozomu 5 [20] (obr. 6), relativně často je v tomto případě i hypoplazie erytropoézy. Nicméně výlučně podle morfologického vzhledu megakaryocytů nelze s jistotou stanovit ani přesnou kategorii MDS ani jasné podezření na tu či onu cytogenitickou abnormitu. Ostatní odchylky považované za dysplastické změny jsou v tab. 3 (obr. 7).



V této souvislosti je nutno zmínit skutečnost, že megakaryocyty jsou nejčastěji postiženy v případech neklonální myelodysplazie navozené širokou paletou získaných klinických stavů (tab. 1), zatímco změny v erytropoéze a granulocytech nastávají zřetelně méně často. Z tohoto pohledu je zapotřebí hodnotit morfologické odchylky v megakaryocytech mnohem více obezřetně, než tomu je v jiných hemopetických řadách. Budeme-li respektovat tento přístup, pak je jasné, že diagnóza refrakterní cytopenie charakteru refrakterní trombocytopenie v rámci MDS typu refrakterní cytopenie s unilineární dyplázií (RCUD), resp. její diferenciální diagnóza vůči jiným, zejména periferním trombocytopeniím, je na základě morfologické diagnostiky nejméně nelehká a v řadě případů až nemožná [21]. Nález dysplastických změn v jiné hematopoetické linii než jen v megakaryocytech činí diagnózu MDS vždy více pravděpodobnou, a to bez ohledu na povinnost komplexního klinického a laboratorního vyšetření u všech nemocných s podezřením na MDS.
Závěr
I když jsou morfologické abnormity megakryocytů častou známkou MDS a mohou v mnoha případech být prvním signálem ke správnému stanovení této diagnózy, není v tomto ohledu postavení cytologické morfologické diagnostiky nikterak výlučné. I dle současných doporučení je nezbytné využít komplexní klinický a laboratorní pohled na potvrzení diagnózy MDS u suspektních nemocných a morfologické nálezy v periferní krvi či kostní dřeni chápat jako jeden z dílů mozaiky takovéhoto multidisciplinárního přístupu [3,10].
MUDr. Alena Buliková, Ph.D.
www.fnbrno.cz
e-mail: abulik@fnbrno.cz
Doručeno do redakce: 24. 3. 2010
Sources
1. Chang Y, Bluteau D, Debili N et al. From hematopoietic stem cells to platelets. J Throm Haemost 2007; 5 (Suppl 1): 318 – 327.
2. Fosberg EC, Serwold T, Kogan S et al. New evidence supporting megakarocyte - erythrocyte potential of flk2/flt3+ multipotent hematopoietic progenitors. Cell 2006; 126 : 415 – 426.
3. Bain BJ, Clark DM, Wilkins BS. Bone marrow pathology. 4th ed. Wiley - Blackwell 2010 : 20 – 26.
4. Bessis M. Blood smears reinterpreted (transl. Brechner G). Heidelberg Springer - Verlag Berlin 1977 : 129 – 144.
5. Roy L, Coullin P, Virat N et al. Asymmetrical segregation of chromosomes with a normal metaphase/ anaphase checkpoint in polyploid megakaryocytes. Blood 2001; 97 : 2238 – 2247.
6. Mattia G, Vulcano F, Milazzo L et al. Different ploidity levels of megakaryocytes generated from peripheral or cord blood CD34+ cells are correlated with different levels of platelet release. Blood 2002; 99 : 888 – 897.
7. Italiano JE, Patel - Hett S, Hartwig JH. Mechanics of proplatelet elaboration. J Thromb Haemost 2007; 5 (Suppl 1): 18 – 23.
8. Pecka M, Malý J, Fátorová I et al. Nové pohledy na vývoj megakaryopoézy a trombopoézy. Trans Hemat dnes 2008; 4 (Suppl 1): 21 – 22.
9. Bolton - Maggs PHB, Chalmers EA, Collins PW et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol 2006; 135 : 603 – 633.
10. Brunning RD, Orazi A, Germing U et al. Myelodysplastic syndromes/ neoplasms, overwiew. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO classification of tumours of haematopoietic and lymphoid tissues. Geneva: WHO Press 2008 : 88 – 93.
11. Bennett JM, Catovsky D, Daniel MT et al. Proposal for the classification of the acute leukaemias (FAB cooperative group). Br J Haematol 1976; 33 : 451 – 458.
12. Brunning RD, Bennett JM, Flandrin G et al. Myelodysplastic syndromes: Introduction. In: Jaffe ES, Harris NL, Stein H et al (eds). World Health Organization classification of tumours: Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: ICAC Press 2001 : 63 – 67.
13. van de Loosdrecht AA, Westers TM, Westra AH et al. Identification of district progrostic subgroups in low - and intermediate - 1 - risk myelodysplastic syndromes by flow cytometry. Blood 2008; 111 : 1067 – 1077.
14. Mufti GJ, Bennett JM, Goasguent J et al. Diagnosis and classification of myelodysplastic syndrome: International working group in morphology of myelodysplastic syndrome (IWGM - MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008; 93 : 1712 – 1717.
15. van de Loosdrecht AA, Alhan C, Béne MCh et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndrome. Haematologica 2009; 94 : 1124 – 1134.
16. Steensma DP. The changing classification of myelodysplastic syndromes: what’s in a name? Hematology (Am Soc Hematol Educ Program) 2009 : 645 – 655.
17. Verdiman JW, Brunning RD, Arber DA et al. Introduction and overview of the classification of the myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL et al. (Eds). WHO classification of tumours of haematopoietic and lymphoid tissues. Geneva: WHO Press 2008 : 18 – 30.
18. Bain BJ, Clark DM, Wilkins BS. Bone marrow pathology. 4th ed. Wiley - Blackwell 2010 : 208 – 228.
19. Kačírková P, Campr V. Hematoonkologický atlas krve a kostní dřeně. Praha: Grada Publishing 2007 : 55 – 88.
20. Hasserrjian RP, Le Beau MM, List AF et al. Myelodysplastic syndrome with isolated del (5q). In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO classification of tumours of haematopoietic and lymphoid tissues. Geneva: WHO Press 2008 : 102 – 103.
21. Brunning RD, Hasserjian RP, Porwizt Aet al. Refractory cytopenia with unilineage dysplasia. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO classification of tumours of haematopoietic and lymphoid tissues. Geneva: WHO Press 2008 : 94 – 97.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue Supplementum 1
Most read in this issue
- Trombocytopenie a koagulopatie u hepatopatie: úvod do problematiky
- Monitorace parametrů koagulace a možnosti jejich ovlivnění u pacientů s jaterní cirhózou před invazivními výkony
- Diferenciální diagnostika trombocytopenie v těhotenství
- Využití parametru IPF (Immature platelet fraction) v laboratorní diagnostice