
Mnohočetná endokrinní neoplazie I (Wermerův syndrom) – formy klinické manifestace: 5 kazuistik
Multiple Endocrine Neoplasia I (Wermer‘s Syndrome), Forms of Clinical Manifestation, 5 Case Studies
Multiple Endocrine Neoplasia (MEN) is a condition in which several endocrine organs of an individual are affected by adenoma, hyperplasia and less often carcinoma, either simultaneously or at different stages of life. Two existing syndromes, MEN1 and MEN2 (2A, 2B), in literature is also mentioned MEN4, are associated also with other non-endocrine disorders. MEN1 (Wermer syndrome) affects the pituitary, parathyroid, and pancreatic area. 95 % of patients show very early manifestation of hyperparathyroidism, often before 40 years of age. Multiple adenomas gradually involve all four parathyroid glands. The first clinical sign of MEN1 includes recurrent nephrolithiasis. The second most frequent manifestation of MEN1 is pancreatic area (pancreas, stomach and duodenum), again multiple malignancies of varying degree which can metastasize. Most often gastrinomas and insulinomas are involved. Pituitary adenomas occur in about one third of MEN1 patients and tend to be larger and less responsive to treatment. Tumors appearing most often are prolactinomas, tumors producing growth hormone, or afunctional adenomas. The other endocrine tumors include carcinoids and adrenal lesions. In the last year we have registered four MEN1 syndrome patients in our center and one patient has been already followed since 2008. In four out of five patients, nephrolithiasis after 30 years of age was the first clinical symptom, but only one of theses cases resulted in MEN1 diagnosis. In all patients, the clinical symptoms intensified and the diagnosis was established between 36 and 40 years of age. A crutial factor is a cooperation with the urology examination of kidney stones formation in young individuals with nephrolithiasis in order to reveal the potential cases of MEN1 syndrome very early on. Consider the MEN1 genetic diagnostics if recurrent primary hyperparathyroidism or recurrent gastroduodenal ulcer disease appear in patients under 40 years of age.
Key words:
carcinoid – gastrinoma – hyperparathyroidism – insulinoma – MEN1 – multiple endocrine neoplasia – nefrolithiasis – neuroendocrine tumor – pancreatic area – pituitary gland
:
Karolína Drbalová 1; Kateřina Herdová 1; Petr Krejčí 1; Monika Nývltová 1; Svatopluk Solař 1; Lenka Vedralová 1; Pavel Záruba 2; David Netuka 3; Petr Bavor 4
:
Interní ambulantní oddělení Interní kliniky 1. LF UK a ÚVN – Vojenská fakultní nemocnice Praha
1; Chirurgická klinika 2. LF UK a ÚVN – Vojenská fakultní nemocnice Praha
2; Neurochirurgická a neuroonkologická klinika 1. LF UK a ÚVN – Vojenská fakultní nemocnice Praha
3; Chirurgická klinika 2. LF UK a FN v Motole, Praha
4
:
Vnitř Lék 2016; 62(Suppl 3): 140-149
:
Case Reports
Mnohočetná endokrinní neoplazie (MEN) je stav, při kterém se u jedince buď současně, nebo v různém pořadí v různém období života vyskytne postižení několika endokrinních orgánů adenomem, hyperplazií, méně často i karcinomem. Rozlišuje se MEN1 a MEN2 (2A a 2B), v literatuře je zmiňován i MEN4, syndromy bývají sdruženy s dalšími neendokrinními poruchami. MEN1 (Wermerův syndrom) postihuje hypofýzu, příštítná tělíska a pankreatickou oblast. Hyperparatyreóza se projevuje velmi časně, ve 40 letech ji má již 95 % pacientů s MEN. Jedná se o mnohočetné adenomy, postupně bývají postižena všechna 4 tělíska. K prvním klinickým příznakům MEN1 patří recidivující nefrolitiáza. Druhou nejčastější manifestací jsou endokrinní neoplazie pankreatické oblasti (slinivka, žaludek a duodenum), které jsou často mnohočetné, mohou být různého stupně malignity a mohou metastazovat. Nejčastěji se jedná gastrinomy a inzulinomy. Hypofyzární adenomy se vyskytují asi u třetiny pacientů s MEN1. Bývají větší a hůře odpovídají na léčbu. Nejčastěji se jedná o prolaktinomy, tumory s produkcí růstového hormonu či afunkční adenomy. K dalším endokrinním tumorům patří navíc karcinoid a léze nadledvin. V posledním roce se v naší ambulanci podařilo zachytit 4 pacienty se syndromem MEN1, jedna pacientka je dispenzarizována již od roku 2008. U 4 z 5 pacientů byla prvním klinickým příznakem nefrolitiáza po 30. roce věku, která však pouze jednou vedla ke stanovení diagnózy MEN1. U všech pacientů klinické obtíže nabyly na intenzitě a diagnóza byla stanovena mezi 36. a 40. rokem věku. Důležité je tedy ve spolupráci s urology nezanedbat vyšetření příčiny tvorby ledvinných kamenů v případě nefrolitiázy u mladých jedinců a pomýšlet na diagnózu MEN1 v případě recidivující primární hyperparatyreózy či recidivující vředové choroby gastroduodenální u pacientů do 40 let věku.
Klíčová slova:
gastrinom – hyperparatyreóza – hypofýza – inzulinom – karcinoid – MEN1 – mnohočetná endokrinní neoplazie – nefrolitiáza – neuroendokrinní tumory – pankreatická oblast
Úvod
Mnohočetná endokrinní neoplazie (MEN) je stav, při němž se u jedince buď současně, nebo v různém pořadí v různém období života vyskytne postižení několika endokrinních orgánů adenomem, hyperplazií, méně často i karcinomem.
Rozlišuje se MEN1 a MEN2 (2A a 2B), v literatuře je zmiňován i MEN4 neboli MENX [1]. Syndromy bývají sdruženy i s dalšími neendokrinními poruchami.
Geneticky se jedná o autozomálně dominantní přenos onemocnění, ale vyskytuje se i sporadicky. Vysoká je penetrace, variabilní je exprese. Liší se mechanizmem vývoje nádoru. MEN1 tumory jsou způsobeny dysfunkcí proteinu meninu, který je kódován MEN1 genem lokalizovaným na 11. chromozomu. Popsáno bylo více než 450 různých mutací. Příčinou MEN2 je mutace aktivující RET protoonkogen. Gen RET je umístěn na 10. chromozomu a jeho mutace vede ke zvýšení transformační aktivity a onkogenezi. Důležitá je rodinná anamnéza příbuzných 1. stupně (rodiče, sourozenci, děti) a jejich dispenzarizace od časného dospívání. MEN4 je způsoben mutací inhibitoru cyklindependentní kinázy.
MEN1 (Wermerův syndrom) postihuje příštítná tělíska (více než 95 %), pankreatickou oblast (75 %) a hypofýzu (50 %).
MEN2A (Sippleův syndrom) je charakterizována přítomností medulárního karcinomu štítné žlázy (90 %), feochromocytomu (50 %) a hyperparatyreózy v důsledku hyperplazie či adenomu příštítného tělíska (10–35 %). MEN2B (Gorlinův syndrom) je charakterizován přítomností medulárního karcinomu, feochromocytomu, mukozálních ganglioneromů a typického marfanoidního habitu. Nutné je genetické vyšetření přímých příbuzných, protože medulární karcinomy se mohou objevit velmi časně. U dětí se zjištěnou RET mutací se totální tyreoidektomie provádí někdy i v předškolním věku.
MEN4 (MENX) je definován postižením příštítných tělísek, nádory hypofýzy, ev. spolu s postižením nadledvin, ledvin a reprodukčních orgánů [1].
Mnohočetná endokrinní neoplazie 1 (Wermerův syndrom)
Syndrom byl poprvé popsán americkým lékařem Paulem Wermerem v roce 1954 [2] (často zaměňováno s německým lékařem C. W. O. Vernerem 1879–1936, zabývajícím se Wernickeovou encefalopatií). Prevalence syndromu je 1–10 případů na 100 000 obyvatel [3]. Penetrance syndromu je více než 95 % ve věku 40 let. Zahrnuje neoplazie postihující příštítná tělíska (více než 95 %), pankreatickou oblast (75 %) a hypofýzu (50 %) [3].
Nejčastější časnou manifestací jsou důsledky hyperparatyreózy, nejčastějším hypofyzárním tumorem je prolaktinom, nejčastějšími tumory pankreatu bývají gastrinomy a inzulinomy.
Hyperparatyreóza se u MEN1 projevuje velmi časně, okolo 20. roku, ve 40 letech ji má již 95 % pacientů. Oproti sporadické formě se jedná o mnohočetné adenomy, postupně bývají postižena všechna 4 tělíska. Ke klinickým příznakům patří nefrolitiáza, osteopenie, hypersekrece žaludečních šťáv až projevy vředové choroby gastroduodenální. V laboratorním nálezu nacházíme vyšší hladiny parathormonu, kalcia, nižší hladiny fosforu, zvýšený je odpad kalcia v moči a zvýšené mohou být hodnoty kostního metabolizmu. Diagnostika bývá snadná, kromě přesvědčivého laboratorního nálezu je většinou sonograficky patrný jeden či více adenomů příštítných tělísek. K potvrzení nálezu pak slouží scintigrafické vyšetření 99mTc-MIBI. U pacientů s MEN1 se s ohledem na prognózu s postupným postižením všech tělísek přistupuje k operačnímu řešení vždy. Pravděpodobná je recidiva onemocnění, během chirurgického výkonu by tedy měla být shlédnuta všechna tělíska a ev. odstraněny i další adenomy. Někdy se přistupuje k exstirpaci všech 4 tělísek a transplantaci jednoho z nich do svalů předloktí či m. sternocleidomastoideus na krku [4,5]. Usnadní to znovuvyhledání tělíska v případě recidivy adenomu. Operace v již předchozí operací změněném terénu je pro chirurga obtížná a pro pacienta riziková.
Endokrinní neoplazie pankreatické oblasti (slinivka, žaludek a duodenum) jsou druhou nejčastější manifestaci MEN1, s výskytem 75–100 % u pacientů s tímto syndromem, hormonálně aktivní jsou pak u 60 % pacientů. Často jsou mnohočetné a mohou být různého stupně malignity. Nejčastěji se jedná gastrinomy (40 %), které jsou příčinou Zollingerova-Ellisonova syndromu. V klinickém obraze dominují průjmy, peptické ulcerace, které mohou být příčinou až žaludeční perforace. V laboratorním nálezu lze zachytit vysoké hladiny gastrinu. Gastrinomy se v rámci MEN1 objevují až o 10 let dříve než sporadické gastrinomy. Mohou metastazovat do uzlin a jater. Lokalizovány jsou nejčastěji ve slinivce (80–90 %), mohou se ale vyskytovat i duodenu, žaludku, slezině a retroperitoneu. Nejpřesnější diagnostika je pomocí endosonografie (EUS), která posoudí lokalizaci a počet ložisek, umožní též biopsii a následné histologické vyšetření. Mnohočetnost a metastázy přispívají k neúspěchu chirurgické léčby [6]. Stav pacientů může být dlouhodobě stabilizován zavedením účinné antisekreční léčby (H2-antagonisté, inhibitory protonové pumpy) a podáváním dlouhodobě působících analog somatostatinu. Inzulinomy (10 %) jsou druhým nejčastějším pankreatickým neuroendokrinním tumorem u pacientů s MEN1. Často jsou mnohočetné, ve více než 25 % maligní a metastazující. Klinickým projevem jsou hypoglykemie s hypoglykemickými příznaky a rychlým ústupem po podání glukózy. Zvýšené jsou hladiny inzulinu, C-peptidu a proinzulinu. Metodou léčebné volby je chirurgické řešení, a to i u metastatických inzulinomů. Po operaci je recidiva častější než u sporadických případů. Pokles frekvence hypoglykemií lze ovlivnit diazoxidem, jehož vedlejším nežádoucím účinkem je hypertrichóza. Glukagonom je vzácným tumor, u MEN1 se vyskytuje ojediněle. Mezi klinické příznaky patří hyperglykemie s projevy manifestního diabetu, který není inzulindependentní, anémie, trombóza, anorexie a nekrolytický migrující erytém. Bývají maligní a v době diagnózy již s metastázami v játrech. Léčba je chirurgická, dobře odpovídají na analoga somatostatinu. Vipom je ojediněle se vyskytující solitární tumor uvolňující vazoaktivní intestinální polypeptid. Bývá maligní a v době diagnózy již s metastázami v játrech. Klinickým příznakem jsou průjmy, bývá též hyperkalcemie v důsledku zvýšené produkce PTHrP. Vzácný je somatostatinom, projevuje se též diabetem, poruchami resorpce potravy se steatoreou a častým výskytem cholelitiázy. Ostatní tumory enteropankreatické oblasti u MEN1 mohou produkovat GHRH, ACTH, PTHrP a kalcitonin. Asi třetina enteropankreatických tumorů je bez hormonální aktivity. Mohou to být tumory velké, maligní a metastazující. Odhalit je lze zobrazovacími metodami či stanovením hladiny chromograninu [6].
K zobrazení těchto tumorů se používá ultrasonografie, CT, MR, endosonografie a octreoscan.
Léčba inzulinomů, glukagonomů, vipomů a somatostatinomů se neliší od léčby sporadických forem [7,8]. Základem léčby je léčba chirurgická, a to i u metastazujících maligních nádorů [9]. Metastázy (zejména v játrech a uzlinách) lze pak dle počtu řešit buď chirurgicky (v játrech tehdy, lze-li resekovat alespoň 95 % ložisek), analogy somatostatinu (inhibice hormonální sekrece tumoru – léčba je v případě inzulinomů méně účinná než u ostatních neuroendokrinních tumorů), radiofrekvenční ablací, chemoembolizací, chemoterapií, v literatuře je zmiňována i léčba everolimem či transplantace jater. Diskuse je v literatuře vedena nad nutností operace drobných hormonálně neaktivních pankreatických nádorů do 1–2 cm. Přínos tohoto řešení není jednoznačně prokázán [10]. Na rozdíl od sporadických gastrinomů, které bývají solitární, jsou gastrinomy u MEN1 mnohočetné drobné adenomy lokalizované často v oblasti duodena [11]. Operace nebývá plně kurativní metodou a je třeba ji doplnit o antisekreční léčbu (H2-antagonisté, inhibitory protonové pumpy) a ev. podávání dlouhodobě působících analog somatostatinu [11,12].
Hypofyzární adenomy se vyskytují asi u 30–50 % pacientů s MEN1, častěji u žen. Typ nadprodukce hormonů se neliší od sporadických forem. V rámci syndromu MEN1 bývají větší, hůře odpovídají na léčbu, objevují se v životě časněji a jsou provázeny lokálními projevy z útlaku okolních struktur. V 60 % se jedná o prolaktinomy, v 15 % o tumory s produkcí růstového hormonu, v 25 % o afunkční adenomy, ojediněle s nadprodukcí ACTH [13,14]. U prolaktinomů je standardně nejprve léčba konzervativní (kabergolin – Dostinex), která vede k potlačení sekrece, ale i ke zmenšení tumoru. Chirurgické řešení je nutné pouze u objemných adenomů bez dobré reakce na léčbu. Nadbytek růstového hormonu u pacientů s MEN1 je buď důsledkem nadprodukce z hypofyzárního tumoru nebo ektopickou nadprodukcí GHRH tumorem slinivky nebo karcinoidem. Léčba spočívá v chirurgickém odstranění, ev. v podávání dlouhodobě působících antagonistů somatostatinových receptorů. Při špatné odpovědi se využívá radiační terapie Leksellovým gama-nožem.
Mezi další endokrinní tumory v rámci MEN1 patří léze nadledvin a karcinoid. V nadledvinách se jedná buď o adenomy kůry nadledvin bez hormonální aktivity nebo ojediněle o unilaterální, klinicky němý femochromocytom. Karcinoid se vyskytuje u 5–15 % pacientů a na rozdíl od sporadických forem, u nichž se tumory nacházejí hlavně v tenkém střevě a apendixu, se karcinoidy u MEN1 nacházejí v thymu (častěji muži), bronších (častěji ženy) a žaludku. Thymické karcinoidy bývají častěji maligní (70 %) než bronchiální (20 %) a jsou nejčastější příčinou mortality pacientů s MEN1 [15]. Diagnostikují se v pozdějším věku než ostatní tumory (po 45. roce), protože bývají dlouho asymptomatické, klinicky nedominuje karcinoidní syndrom. V žaludku nemívají hormonální nadprodukci, jejich malignita je nízká a bývají obvykle náhodným nálezem [6]. Obecně dobře reagují na léčbu analogy somatostatinu.
Mezi neendokrinní tumory provázející syndrom MEN1 patří mnohočetné obličejové angiofibromy (88 %), kolagenomy (72 %) a lipomatóza kožní i viscerální (17–34 %) [16,17].
Kazuistika 1
37letá pacientka se dostavila na gastroenterologii v prosinci roku 2008 s anamnézou recidivující vředové choroby, již po resekci žaludku (B II roku 2005) se stenózou anastomózy a po eradikaci Helicobacter pylori v roce 2006.
V anamnéze měla operaci krční páteře pro rozsáhlý ependymom (2004, neurochirurgická klinika ÚVN Praha) s pooperační přechodnou kvadruparézou a byla po plastické operaci obou prsů.
Na ultrasonografii (USG) břicha, octeroscanu, endosonografii a CT bylo v lednu roku 2009 zachyceno celkem 5 ložisek, 3 v pahýlu duodena a 2 v hlavě pankreatu (25 mm a 17 mm), histologie z menšího ložiska připouštěla neuroendokrinní povahu nádoru (obr. 1).
V únoru roku 2009 byla provedena distální pankreatektomie se splenektomií a cholecystektomií. Histologicky byly popsány 4 gastrinomy různého stupně malignity (3 s nízkým a 1 s vyšším maligním potenciálem). Pooperační stav byl komplikován v březnu roku 2009 revizí pro perforovaný žaludeční vřed, následnou revizí pro purulentní peritonitidu s evakuací abscesu dutiny břišní, multiorgánovým selháním s kvasinkovou sepsí, hrudní drenáží, oboustrannou pneumonií, nutností umělé plicní ventilace a tracheostomií.
V tuto chvíli proběhlo první endokrinologické vyšetření a bylo vysloveno podezření na MEN1, a to vzhledem k zaznamenané hyperkalcemii, vyšším hodnotám parathormonu (PTH) 10,13 pmol/l (norma do 6,9 pmol/l) a hyperprolaktinemii 605 mU/l (norma do 496 mIU/l). Hyperparatyreóza byla potvrzena, PTH adenomy byly patrné při dolním pólu obou laloků štítné žlázy.
Kontrolní octreoscan z května roku 2009 popsal znovu 5 ložisek se zvýšenou akumulací radiofarmaka ve střední třetině epigastria a zbytku slinivky.
Zahájena byla terapie somatostatinem.
V září roku 2009 proběhla subtotální paratyreoidektomie, prolaktinom na MRI hypofýzy nebyl popsán, mírně zvýšená hladina prolaktinu byla patrně pouze důsledkem aktuální farmakoterapie a stresu.
Porucha sacharidového metabolizmu nejprve vedla směrem k sekundárnímu pankreatogennímu diabetu, přechodně byla léčena sitagliptinem, postupně byl sitagliptin vysazen a úprava diety vedla k uspokojivému stavu.
Geneticky byla verifikována mutace v genu MEN1.
Od zahájení léčby somatostatinem je pacientka bez obtíží, bez recidivy vředové choroby, bez recidivy ependymomu dle kontrolní MRI v červnu roku 2016, pacientka je normokalcemická. Dispenzarizována je na endokrinologii, diabetologii a neurochirurgii.
Prvním klinickým projevem byly obtíže spojené s rozsáhlým ependymomem krční páteře, následovaly projevy vředové choroby gastroduodenální.
Kazuistika 2
40letý pacient se poprvé dostavil na endokrinologii ÚVN Praha v srpnu roku 2015.
V anamnéze měl 2 ataky nefrolitiázy se spontánním odchodem konkrementů, léky chronicky neužíval žádné. V posledním roce si stěžoval na zhoršené rozostřené vidění, pro které v červenci roku 2015 navštívil očního lékaře. Tím byl na základě změn na očním pozadí odeslán na MRI mozku.
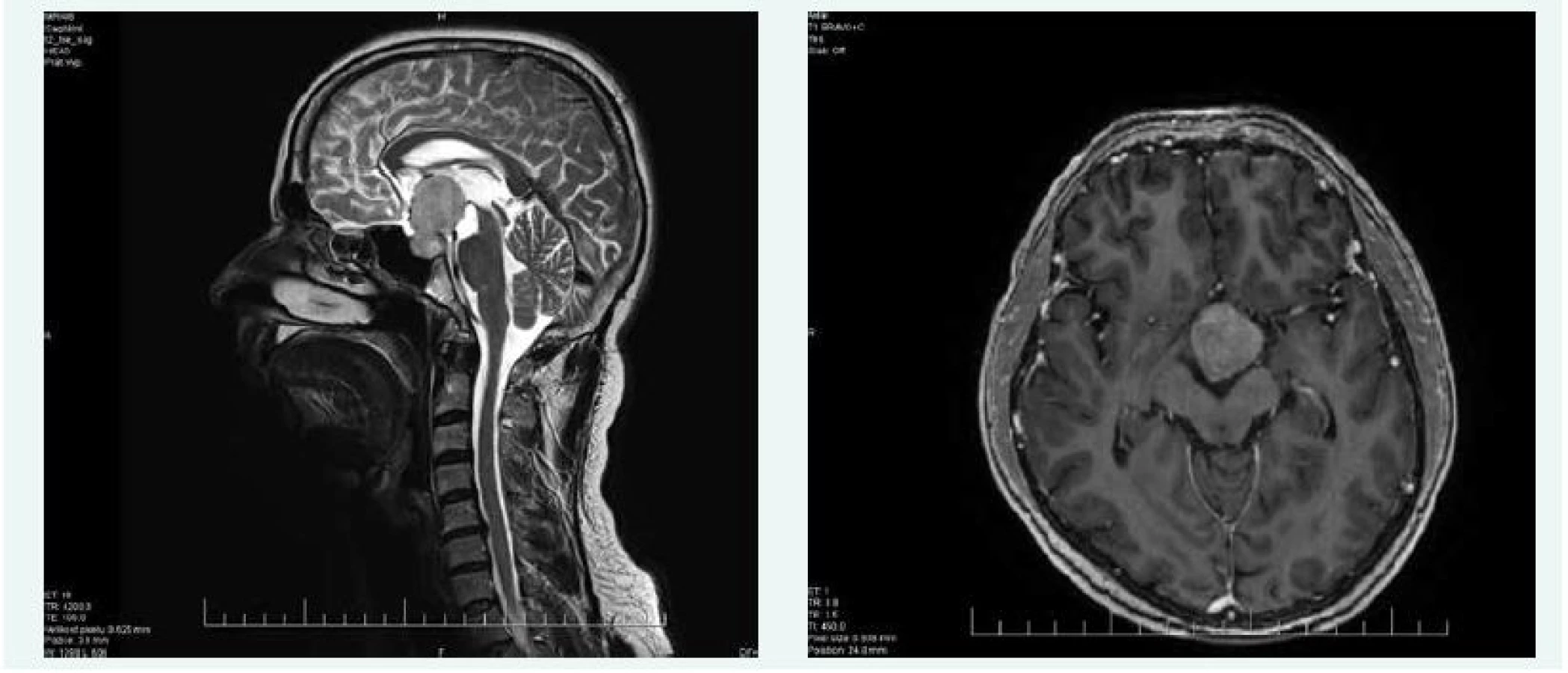
Na MRI mozku byl popsán objemný makroadenom hypofýzy s útlakem okolních struktur, útlakem chiazmatu, III. komory, kaudálně expandující sellu, velikosti 39 × 27 × 24 mm.
Z prvního odběru v rámci hormonálního spektra byla patrná hyperparatyreóza s hodnotou PTH 15,660 pmol/l (norma do 6,9 pmol/l ), hyperkalcemie 3,05 mmol/l (norma do 2,65 mmol/l) a hyperprolaktinemie 827,600 mIU/l (norma do 324 mIU/l).
Ultrasonograficky (srpen roku 2015) byla popsána normální štítná žláza a podezření na 2 PTH adenomy pod oběma póly laloků (obr. 2). Scintigraficky (srpen roku 2015) byl pak nález potvrzen.
Po zjištění 2 PTH adenomů již bylo vysloveno podezření na MEN1, pacient byl odeslán k exstirpaci PTH adenomů, objednáno bylo CT břicha, klinické příznaky hormonální aktivity neuroendokrinních nádorů pankreatické oblasti nebyly patrné.
Ve snaze vrátit se co nejdříve do běžného života si pacient domluvil časný termín exstirpace PTH adenomů na chirurgické klinice FN v Motole, kde při předoperačním lačnění a čekání na výkon upadl do bezvědomí. Zjištěna byla hypoglykemie 1 mmol/l, operační výkon byl odložen a pacient byl přeložen na interní kliniku ÚVN Praha (gastroenterologie) k dovyšetření pro možný inzulinom.
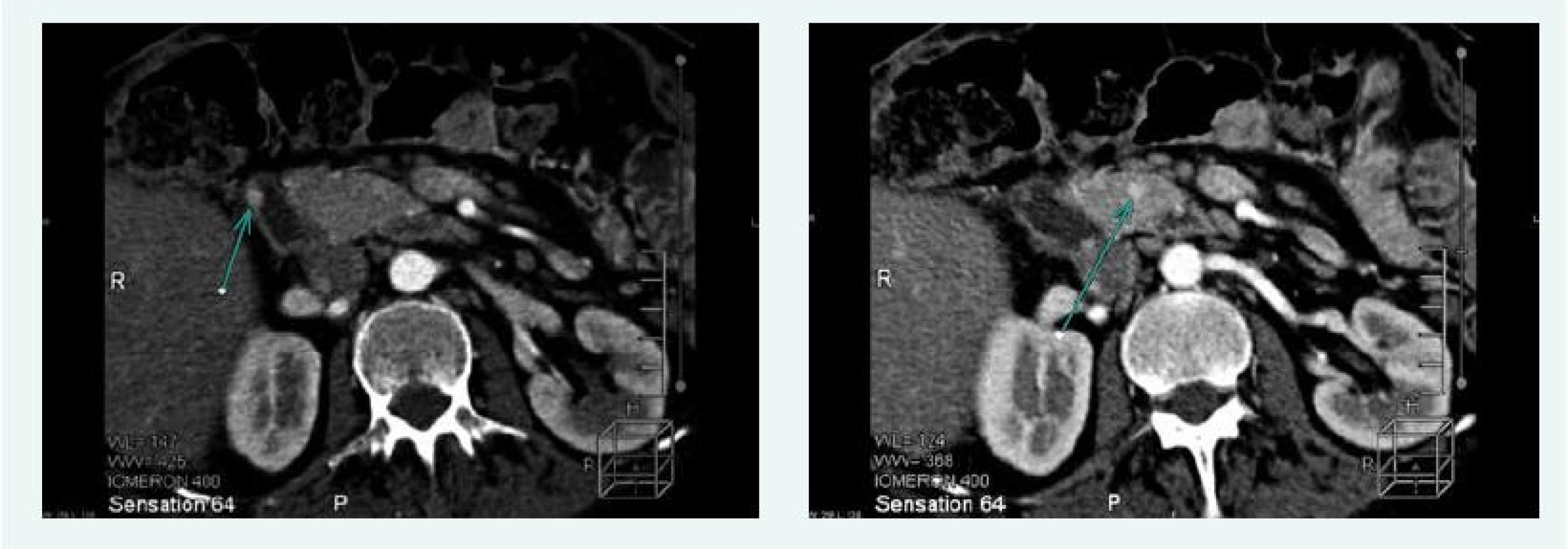
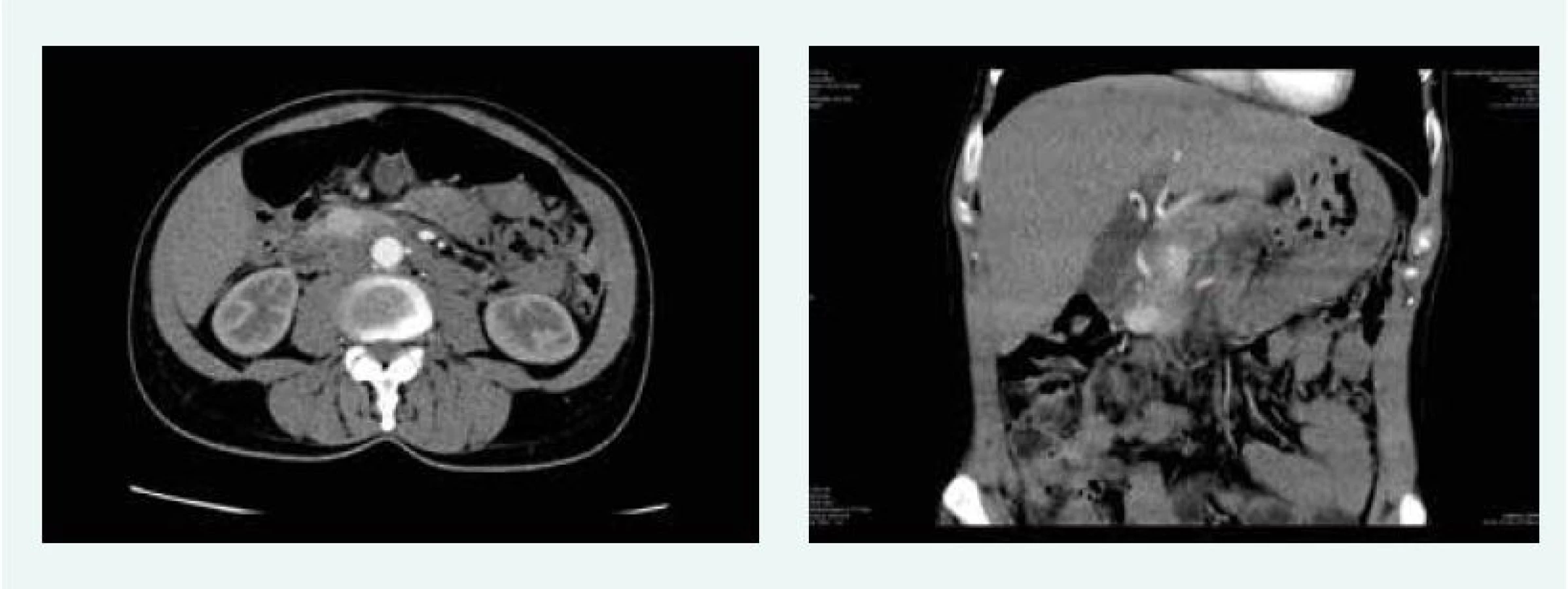
Na CT vyšetření břicha v září roku 2015 byla popsána 3 ložiska ve slinivce a adenom pravé nadledviny 13 mm (obr. 3).
Endosonograficky pak bylo popsáno mnohočetné ložiskové postižení slinivky (3 ložiska v hlavě, 1 v krčku, 1 v těle a 1 v ocasu), navíc pak vřed bulbu duodena v terénu bulbitidy a mírná ezofagitida.
Koncem září roku 2015 pacient podstoupil totální pankreatektomii se splenektomií a cholecystektomií pro mnohočetný inzulinom na chirurgické klinice ÚVN Praha, pooperační průběh byl komplikován revizí dutiny břišní pro infikované hematomy.
Pro sekundární inzulindependentní diabetes byla nastavena inzulinoterapie.
Oční obtíže se zcela upravily, nebyly tedy způsobeny útlakem chiazmatu makroadenomem hypofýzy, jak se původně předpokládalo, jednalo se o projev hypoglykemie.
Při kontrolní MRI hypofýzy v říjnu roku 2015 byla popsána progrese makroadenomu hypofýzy na 42 × 25 × 28 mm (obr. 4), perimetr v říjnu roku 2015 popisoval též zhoršení nálezu na obou stranách.
Časně po pankratektomii bylo počátkem prosince roku 2015 rozhodnuto o nutnosti operace makroadenomu hypofýzy na neurochirurgické klinice ÚVN – VFN Praha, po které následovala plastika sfenoidu a revize pro likvoreu. Důsledkem operace byla sekundární parciální centrální hypotyreóza, sekundární parciální diabetes insipidus a sekundární parciální hypokortikalizmus.
V březnu roku 2016 proběhla již zcela bez komplikací exstirpace 2 PTH adenomů, po které došlo k normalizaci kalcemie i parathormonu.
Byla nastavena substituční léčba glukokortikoidy, mineralokortikoidy, levotyroxinem, adiuretinem, inzulinem, pankreatickými enzymy, minimálními dávkami kabergolinu, testosteronem, pacient je navíc léčen pro hypertenzi. V plánu je testování nutnosti substituce růstovým hormonem, hodnoty IGF1 jsou po operaci hypofýzy podnormální.
Hormonální aktivita adenomu pravé nadledviny 13 mm nebyla prokázána, chirurgické řešení není plánováno.
Geneticky byla verifikována mutace v genu MEN1, a to varianta, která nebyla doposud popsána.
U staršího syna (17 let) byla zachycena vyšší kalcemie s normální hodnotou parathormonu, bude geneticky vyšetřen a nadále sledován.
Pacient je dispenzarizován na endokrinologii, diabetologii, neurochirurgii, chirurgii a přechodně i v ambulanci pro poruchy výživy.
Prvním klinickým příznakem byla v tomto případě nefrolitiáza, která vzhledem ke spontánnímu odchodu konkrementů nebyla nikdy dovyšetřována. K lékaři pacienta přivedlo až zhoršené rozostřené vidění, které bylo jediným projevem hypoglykemických stavů.
Kazuistika 3
39letá pacientka, matka 3 dětí, byla poprvé na endokrinologii ÚVN – VFN Praha vyšetřena v říjnu roku 2015 na žádost rodiny, doposud byla sledována na jiném pracovišti.
V anamnéze měla již opakované ataky nefrolitiázy v letech 2011 a 2012, exstirpaci dvou PTH adenomů ve dvou dobách květnu a září roku 2015, byla po totální tyreoidektomii ve dvou dobách: v září roku 2015 byla provedena levostranná tyreoidektomie s histologicky popsanou folikulární variantou papilárního karcinomu 2 mm, pravostranná tyreoidektomie pak byla doplněna v říjnu roku 2015 s již benigní histologií.
Klinicky pacientku nejvíce obtěžovaly průjmy, pro které odmítala i dostatečnou substituci tyreoidálními hormony.
Vzhledem k postižení dvou příštítných tělísek u mladé osoby bylo pomýšleno na MEN1. V aktuálních laboratorních výsledcích byla patrná pooperační hypotyreóza – TSH 6,550 mU/l (norma do 4,2 mU/l), fT4 4,92 pmol/l (norma od 11 pmol/l), normokalcemie s lehce vyšší hodnotou parathormonu PTH 9,480 pmol/l (norma do 6,9 pmol/l), hyperprolaktinemie 984,600 mIU/l (norma do 496 mIU/l) a zvýšené hodnoty IGF1 – 527,19 μg/l (norma do 324 μg/l). Klinicky byla zmiňována sekundární amenorea a průjmy. Pacientce byly zvýšeny substituční dávky levotyroxinu, nasazen byl kabergolin a odeslána byla na CT břicha a MRI hypofýzy.
V listopadu roku 2015 bylo na CT popsáno ložisko v hlavě pankreatu 21 mm. Doplněna byla endosonografie, která potvrdila ložisko, ale v krčku slinivky, 19 mm, histologicky suspektní neuroendokrinní tumor. Při indikační vizitě před operací se dospělo k závěru, že se jedná o 2 různá ložiska – 21 mm v hlavě a 19 mm v krčku pankreatu (obr. 5).
V lednu roku 2016 byla doplněna MRI hypofýzy s nálezem makroadenomu hypofýzy 22 × 24 × 24 mm, s útlakem chiazmatu, infiltrací levého kavernózního splavu a přerůstáním do sfenoidální dutiny (obr. 6).
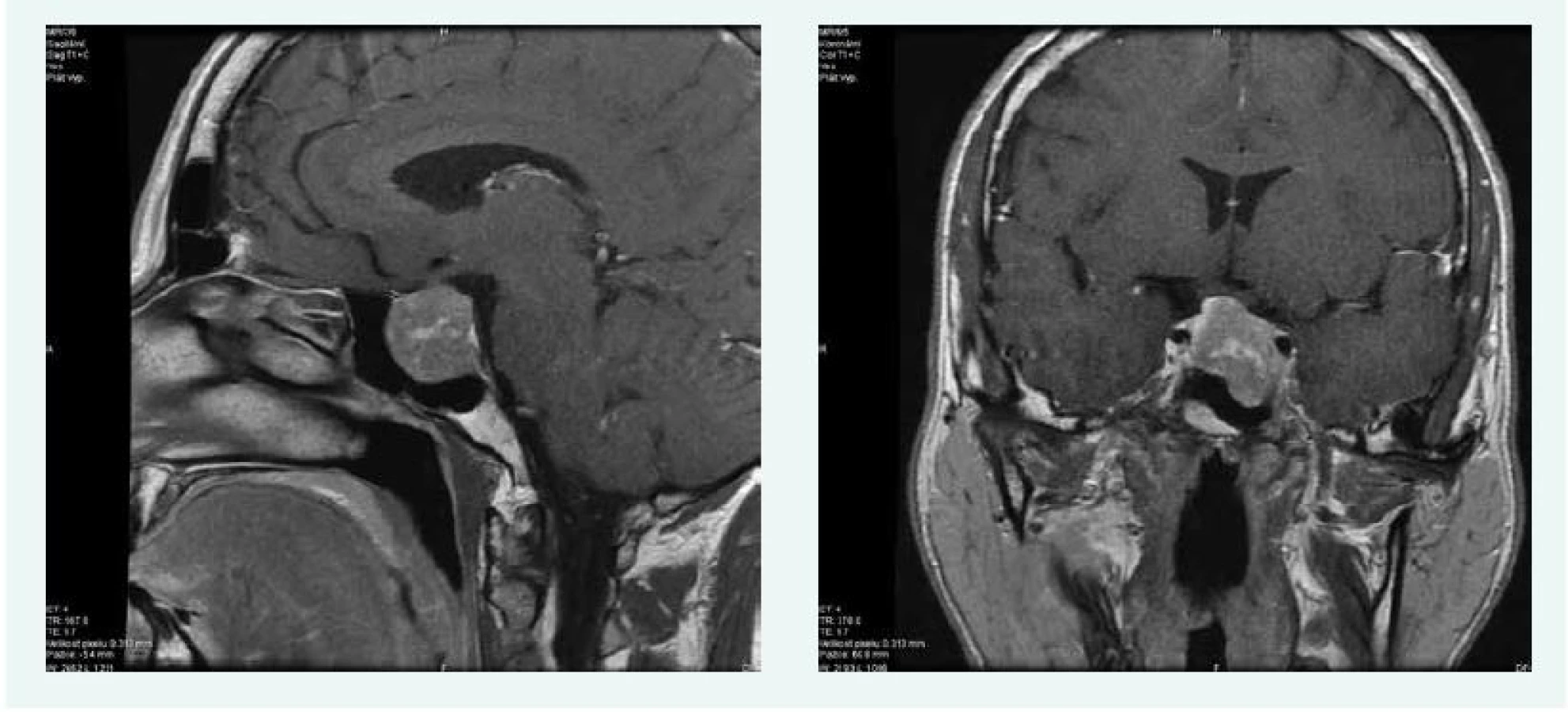
Oční vyšetření včetně perimetru bylo normální.
V lednu roku 2016 byla provedena hemipankreatodudenektomie s cholecystektomií na chirurgické klinice ÚVN Praha, histologicky byl potvrzen NET G1. Pooperačně přetrvávala bolest v oblasti epigastria, jejíž intenzita se zvyšovala. Provedena byla tedy gastroskopie s průkazem ulcerace duodena, která dobře reagovala na konzervativní terapii omeprazolem. S myšlenkou na ev. další ložiska gastrinomu pankreatické oblasti mimo slinivku bylo provedeno vyšetření octreoscanem s negativním nálezem.
V květnu roku 2016 byla provedena operace makroadenomu hypofýzy na neurochirurgii ÚVN Praha, pooperačně bez patrného rezidua tkáně. Prolaktin klesl do podnormálních hodnot, důsledkem operace je parciální centrální hypokortikalizmus, IGF1 přetrvává mírně zvýšené, tyreoidální hormony jsou plně substituovány již od doby totální tyreoidektomie.
Pacientka je dispenzarizována na endokrinologii, chirurgii a neurochirurgii.
Genetické vyšetření zatím neproběhlo.
Prvním klinickým příznakem byla u pacientky nefrolitiáza v důsledku hyperparatyreózy, poté sekundární amenorea v důsledku hyperprolaktinemie a chronické průjmy jako projev hormonálně aktivních gastrinomů.
Kazuistika 4
39letá pacientka, matka 3 dětí, byla přijata do ÚVN Praha v listopadu roku 2015 k endosonografickému vyšetření s biopsií pro již dříve zjištěné ložisko ve slinivce.
V anamnéze měla ataku idiopatické proktokolitidy v roce 2013 a nefrolitiázu, která byla opakovaně řešena litotrypsí rázovou vlnou. Chronicky byla léčena mesalazinem.
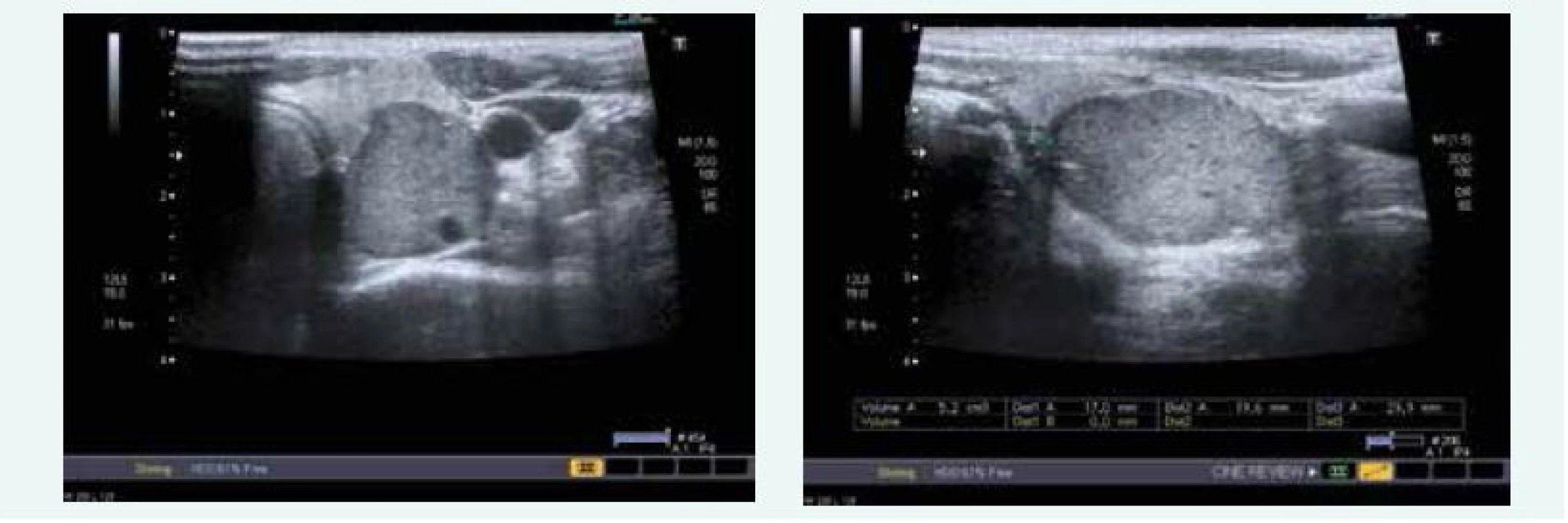
Endosonograficky bylo popsáno několik ložisek do 10 mm v hlavě a těle pankreatu, navíc pak solidně cystické ložisko v procesus uncinatus 16 mm (obr. 7), histologicky byly potvrzeny neuroendokrinní nádory G1. Vzhledem k zachyceným asymptomatickým hypoglykemiím až 1,7 mmol/l se předpokládalo, že se jedná o inzulinomy. Hospitalizace byla komplikována postbioptickou akutní pankreatitidou.
Během hospitalizace byla zjištěna hyperkalcemie 2,71 mmol/l (norma do 2,65 mmol/l) v důsledku primární hyperparatyreózy, PTH 19,89 nmol/l (norma do 6,9 pmol/l) a sonograficky byl patrný objemný adenom levého dolního příštítného tělíska (obr. 8).
Vysloveno bylo podezření na MEN1.
Na CT břicha byl popsán adenom levé nadledviny 6 mm, hladiny nadledvinových hormonů byly v normě.
V prosinci roku 2015 byla pacientce provedena totální pankreatoduodenektomie + splenektomie, pro sekundární inzulindependentní diabetes byla nastavena inzulinoterapie.
V lednu roku 2016 byla doplněna MRI hypofýzy s nálezem mikroadenomu hypofýzy 3 × 3,5 mm nepůsobící útlak chiazmatu, jehož hormonální aktivita nebyla prokázána.
Hodnoty kalcemie se zvýšily až na 3,6 mmol/l, v červenci roku 2016 podstoupila pacientka odstranění adenomu levého příštítného tělíska na ORL ve FN v Motole, který byl histologicky potvrzen. Těsně pooperačně však byla znovu zaznamenaná hyperkalcemie 2,88 mmol/l a hyparparatyreóza – PTH 15,76 nmol/l. Sonograficky další adenom nebyl patrný, scintigraficky byl popsán možný adenom příštítného tělíska za pravým lalokem. Pacientku čeká další operace.
Je dispenzarizována na endokrinologii, diabetologii, gastroenterologii a chirurgii.
Genetické vyšetření nepotvrdilo mutaci MEN1 genu, ale třetina MEN1 syndromů je bez mutace v genu nebo se u nich jedná o větší poškození MEN1 genu, které se přímou sekvenací neodhalí.
Prvním klinickým příznakem byla v tomto případě nefrolitiáza, která byla sice opakovaně řešena litotrypsí, ale její etiologie nebyla vyšetřována. Dále pak dominovaly nespecifické zažívací obtíže, které vedly k sonografickému vyšetření břicha s nálezem ložiska ve slinivce.
Kazuistika 5
37letá pacientka byla poprvé vyšetřena na endokrinologii ÚVN Praha v červenci roku 2015, doporučena byla z urologie, protože v letech 2008–2015 byla 7krát operována pro ureteronefrolitiázu vlevo.
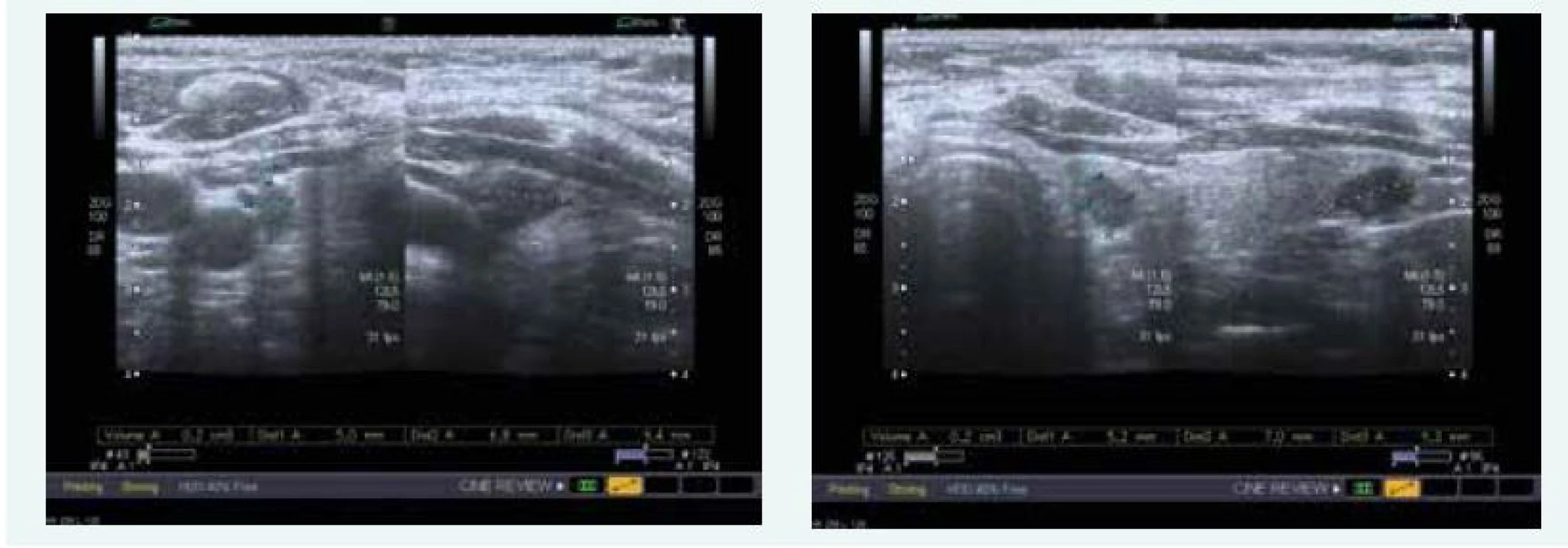
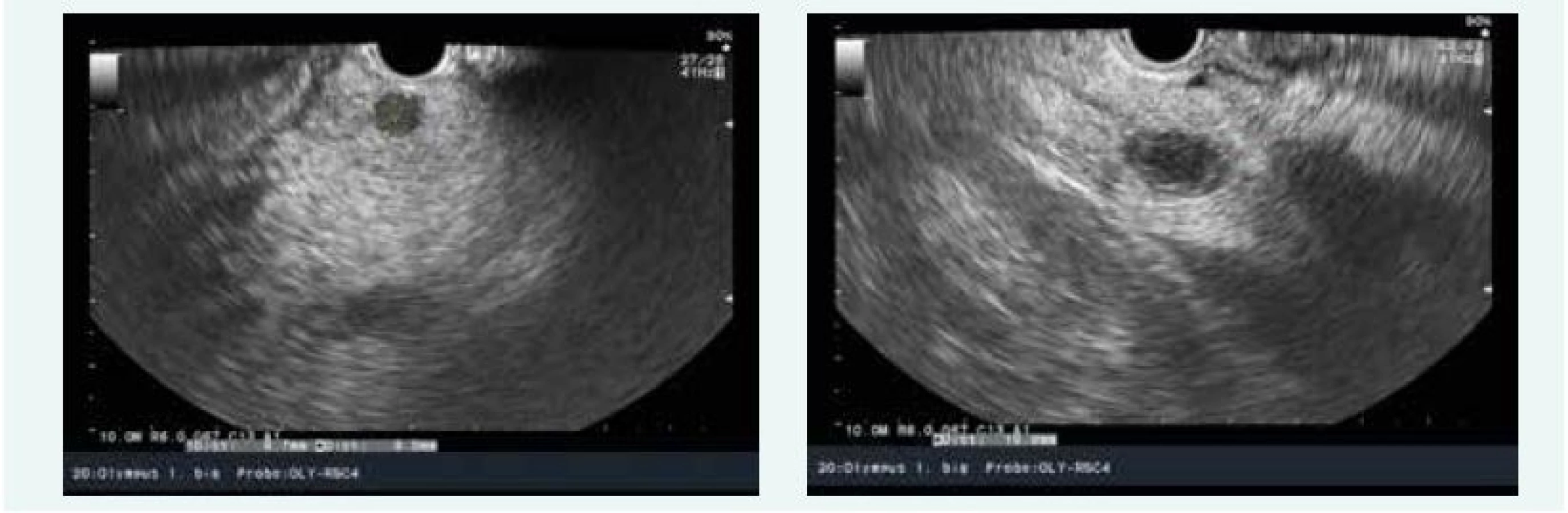
Zjištěna byla primární hyperparatyreóza s hodnotou PTH 13,61 nmol/l (norma do 6,9 pmol/l) a hyperkalcemie s hodnotou Ca 3,12 mmol/l (norma do 2,65 mmol/l), sonograficky byl přesvědčivý PTH adenom za dolním pólem levého laloku (obr. 9), drobné ložisko pak ještě pod dolním pólem pravého laloku (obr. 9). Scintigraficky byl v červenci roku 2015 potvrzen PTH adenom za levým lalokem.
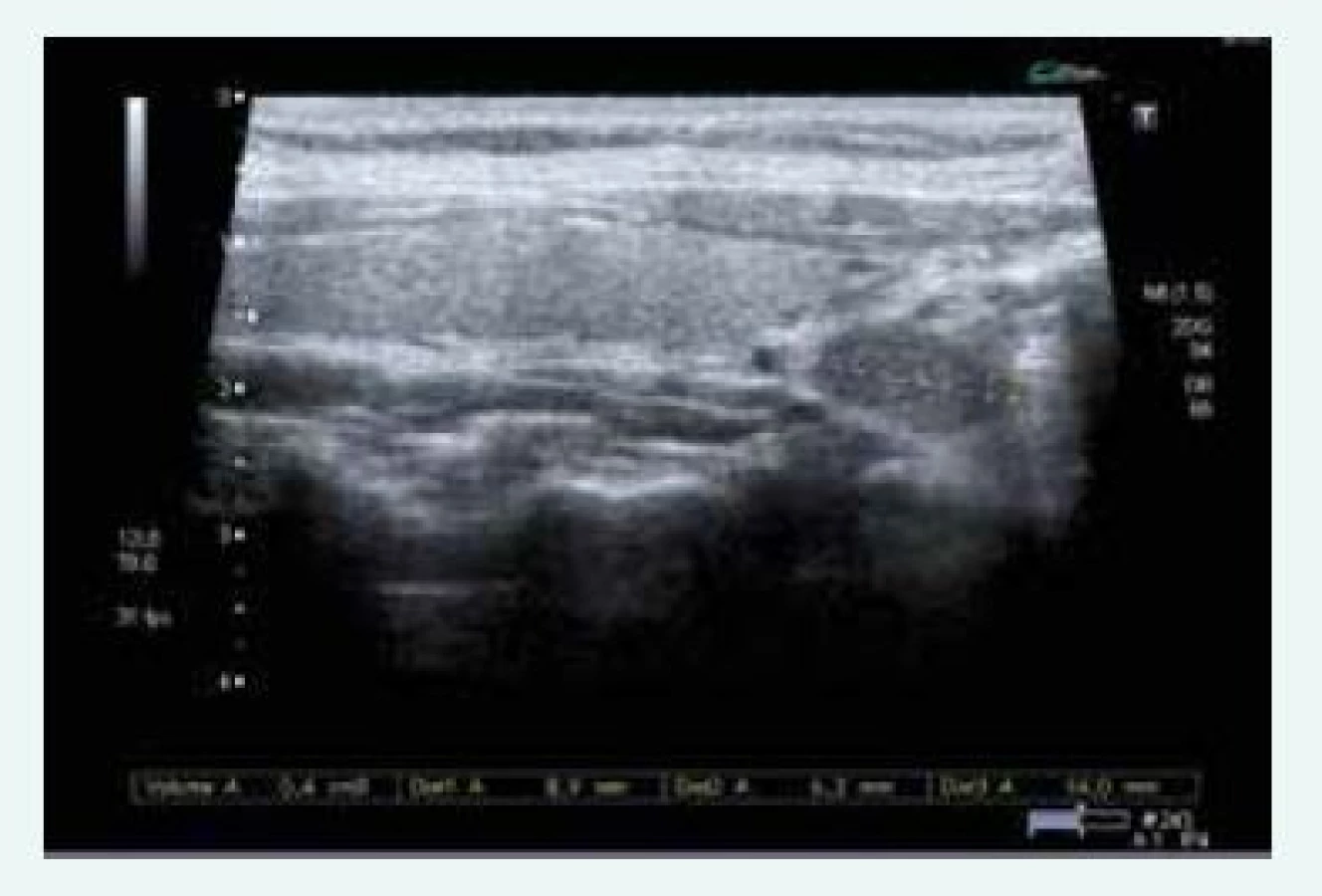
Exstirpace PTH adenomu proběhla na Chirurgii FN Motol Praha v říjnu roku 2015, histologicky byl adenom příštítného tělíska potvrzen. Při kontrole v listopadu roku 2015 byl zaznamenán pokles kalcemie i parathormonu, ne však jejich normalizace, sonograficky byl patrný PTH adenom za levým lalokem ve stejné lokalitě jako před operací (obr. 10)!
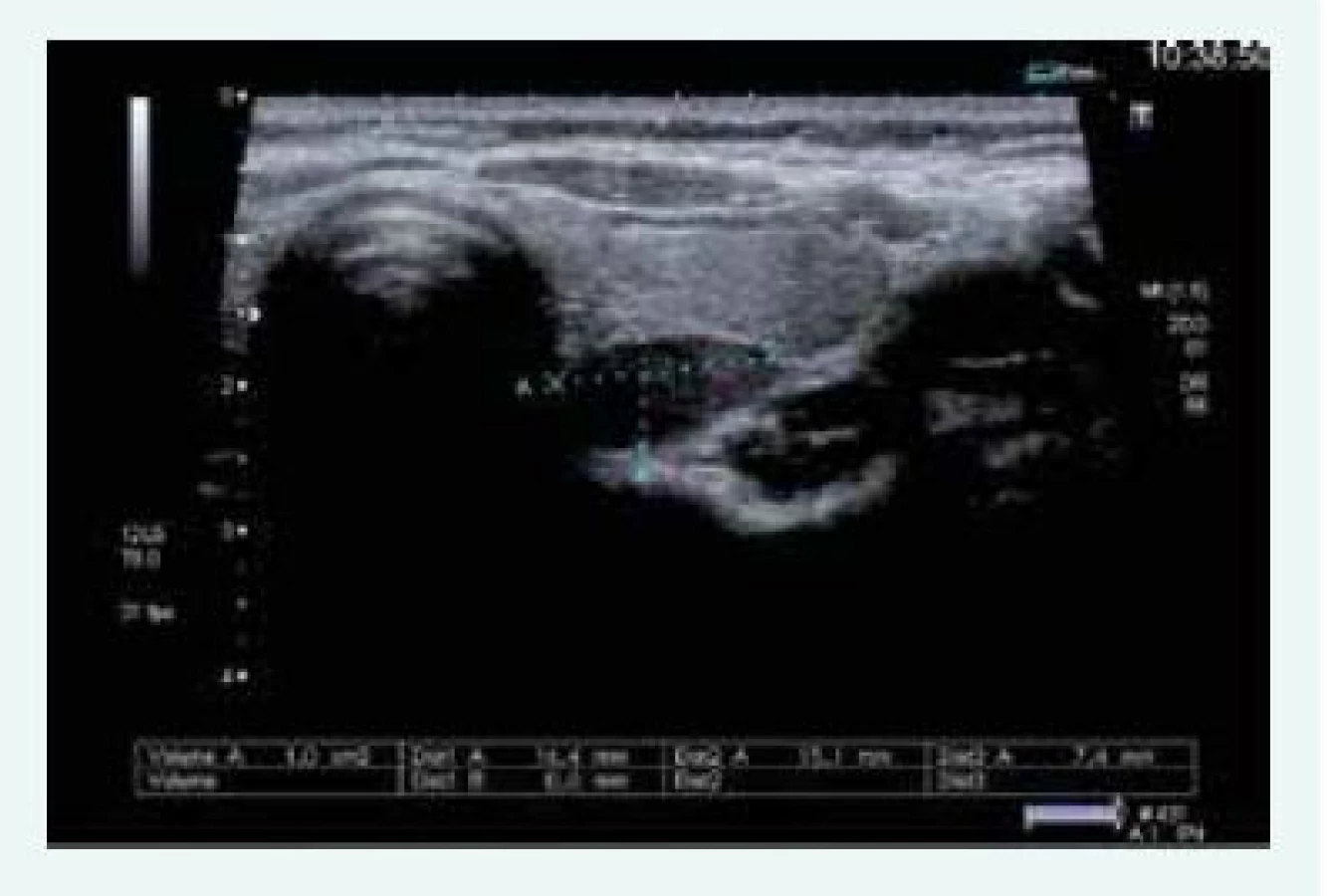
Kontrolní scintigrafie MIBI z března roku 2016 popisuje 2 PTH adenomy, a to za dolním pólem pravého laloku a za horním pólem levého laloku. Pacientka byla znovu odeslána k exstirpaci výše popsaných adenomů a již s myšlenkou na MEN1 byla objednána MRI hypofýzy a CT břicha k vyloučení dalších neuroendokrinních nádorů. Hormonální spektrum hypofyzárních a nadledvinových hormonů včetně plazmatických metanefrinů a chromograninu bylo v normě.
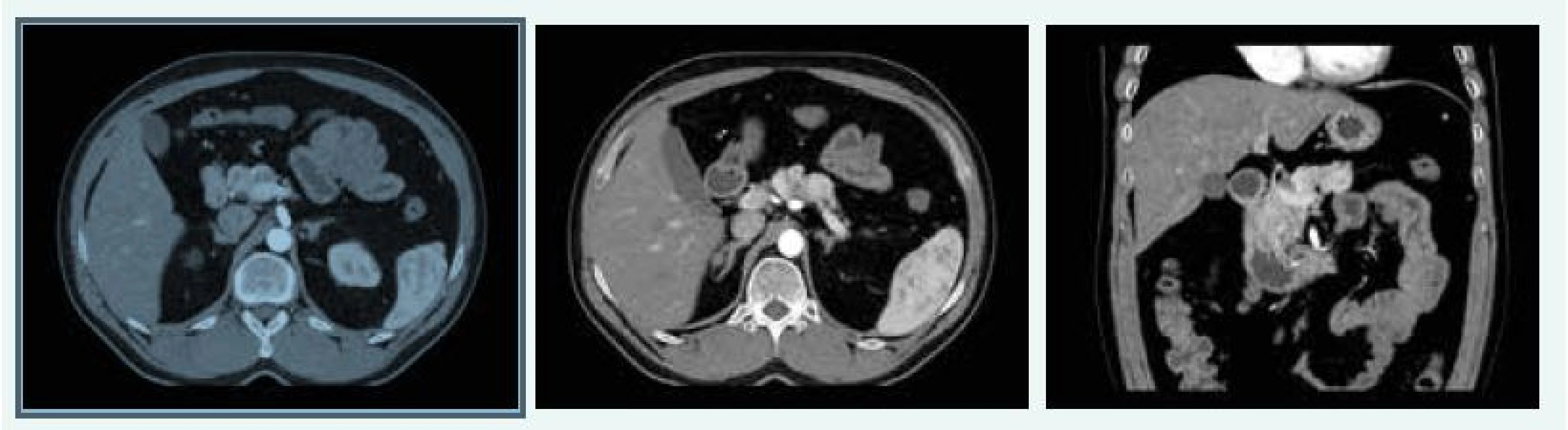
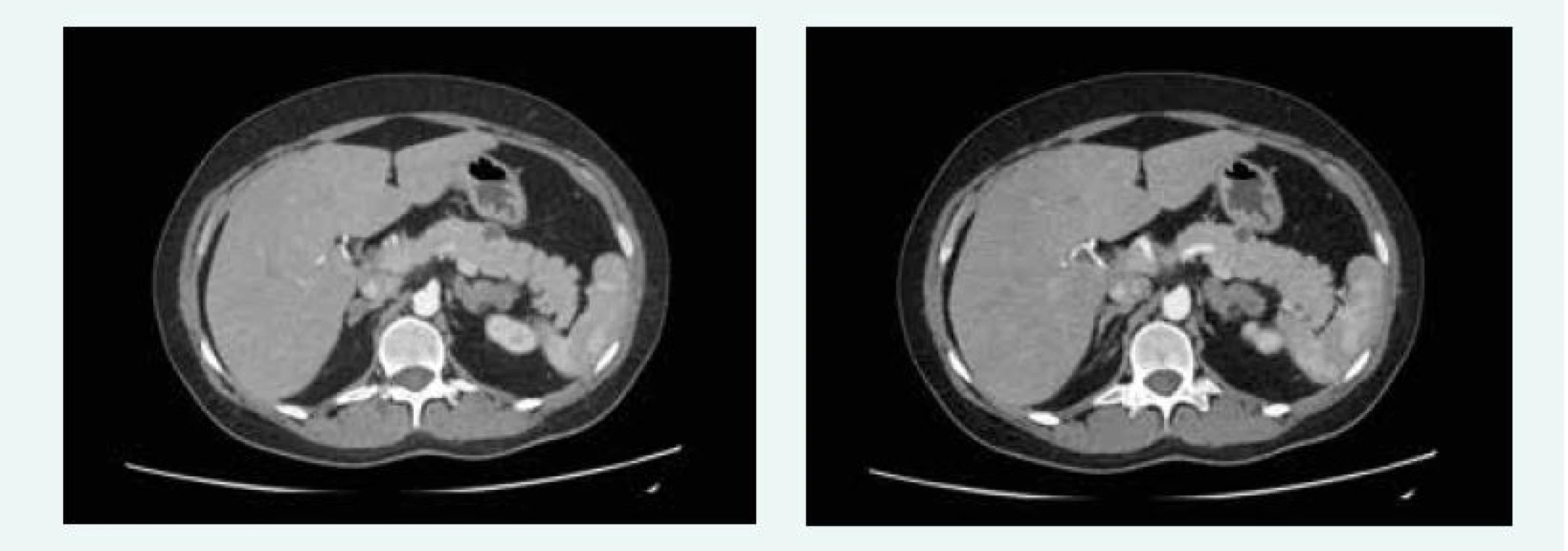
V červnu roku 2016 byla MRI hypofýzy negativní, popsáno bylo pouze nehomogenní sycení v levé dorzální části hypofýzy, na CT břicha byla popsána cholecystolitiáza 13 mm, ložiskové změny obou nadledvin difuzně, největší ložisko pak 28 × 29 mm vlevo, ložiskový proces je mnohočetný, ve zjevné velikostní progresi od nativního CT z roku 2012, nález připouští i možnost tumorózního procesu primárního charakteru. V kaudě pankreatu jsou ložiska 15 mm a 13 mm (obr. 11), bez růstové progrese od roku 2012.
V červnu roku 2016 proběhla druhá exstirpace PTH adenomů za pravým i levým lalokem na chirurgické klinice FN v Motole s následnou normalizací kalcemie i hladiny parathormonu.
Endosonograficky byla v červenci roku 2016 popsána ve shodě s CT 2 ložiska ve slinivce – v těle solidně cystický tumor 18 × 12 × 13 mm, v kaudě 9 × 7 mm. Indikována je distální pankreatektomie spolu s operací levé nadledviny na chirurgické klinice ÚVN Praha.
Pacientka je dispenzarizována na endokrinologii, urologii a chirurgii, genetické vyšetření mutace genu MEN1 nebylo zatím provedeno.
Prvním klinickým příznakem byla v tomto případě recidivující nefrolitiáza, pro kterou byla pacientka z urologie odeslána na endokrinologii po 7 letech a 7 operacích. CT nález z roku 2012 s popisem mnohočetných adenomů nadledvin a ložisek ve slinivce byl u tehdy 33leté pacientky ponechán bez povšimnutí.
Závěr
U 4 z 5 výše uvedených pacientů byla prvním klinickým příznakem nefrolitiáza po 30. roce věku, která však pouze jednou vedla ke stanovení diagnózy MEN1. Nebyla totiž vyšetřována etiologie tvorby ledvinných kamenů, a to ať už docházelo ke spontánnímu odchodu konkrementů nebo bylo třeba onemocnění opakovaně řešit operačně. U jedné pacientky byl první klinickou známkou rozsáhlý ependymom krční páteře, který byl neurochirurgicky řešen ve 33 letech věku.
Diagnostický proces, který vedl ke stanovení diagnózy MEN 1, rozpoutala v prvním z případů recidivující vředová choroba gastroduodenální, ve druhém případě pak rozostřené vidění a nález na očním pozadí, který vedl očního lékaře k vyšetření mozku, ve třetí kazuistice recidiva hyperparatyreózy s nutností opakované exstirpace PTH adenomu v krátkém časovém intervalu spolu s chronickými průjmy, ve čtvrté kazuistice pak bolesti břicha a neurčité zažívací obtíže, které vedly k vyšetření a diagnostice ložiska ve slinivce, v páté kazuistice hyperparatyreóza zjištěná při vyšetření recidivující nefrolitiázy na urologii, ale až po 7 letech dispenzarizace a 7 urologických výkonech.
U všech pacientů klinické obtíže nabyly na intenzitě a diagnóza byla stanovena mezi 36. a 40. rokem věku.
Důležité je tedy ve spolupráci s urology nezanedbat vyšetření příčiny tvorby ledvinných kamenů v případě nefrolitiázy u mladých jedinců a pomýšlet na diagnózu MEN1 v případě recidivující primární hyperparatyreózy či recidivující vředové choroby gastroduodenální u pacientů do 40 let věku.
MUDr. Karolína Drbalová
karolina.drbalova@uvn.cz
Ústřední vojenská nemocnice – Vojenská fakultní nemocnice Praha
www.uvn.cz
Doručeno do redakce 17. 8. 2016
Přijato po recenzi 2. 9. 2016
Sources
1. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 2014; 386(1–2): 2–15. Dostupné z DOI: <http://dx.doi.org/10.1016/j.mce.2013.08.002>.
2. Wermer P. Genetic aspect of adenomatosis of endocrine glands. Am J Med 1954; 16(3): 363–371.
3. Carroll W. Multiple endocrine neoplasia type 1 (MEN1). Asia Pac J Clin Oncol 2013; 9(4): 297–309. Dostupné z DOI: <http://dx.doi.org/10.1111/ajco.12046>.
4. Waldmann J, Lopez CL, Langer P et al. Surgery for multiple endocrine neoplasia type 1 – associated primary hyperparathyroidism. Br J Surg 2010; 97(10): 1528–1534. Dostupné z DOI: <http://dx.doi.org/10.1002/bjs.7154>.
5. Montenegro FLM, Lourenco DM Jr, Tavares MR. Total parathyroidectomy in a large cohort of cases with hyperparathyroidism associated with multiple endocrine neoplasia type 1: experience from a single academic center. Clinics 2012; 67(Suppl 1): S131-S139.
6. Brunová J, Bruna J. Mnohočetná endokrinní neoplazie. In: Brunová J, Bruna J. Klinická endokrinologie a zobrazovací diagnostika endokrinopatií. Maxdorf Jessenius: Praha 2009: 321–333. ISBN 978–80–7345–190–5.
7. Tonelli F, Giudici F, Giusti F et al. Gastroenteropancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1. Cancers 2012; 4(2): 504–522. Dostupné z DOI: <http://dx.doi.org/10.3390/cancers4020504>.
8. Cryer PE, Axelrod L, Grossman AB et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2009; 94(3): 709–728. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2008–1410>.
9. Hanazaki K, Sakurai A, Munekage M et al. Surgery for a gastropancreatic neuroendocrine tumor (GEPNET) in multiple endocrine neoplasia type 1. Surg Today Mar 2013; 43(3): 229–236. Dostupné z DOI: <http://dx.doi.org/10.1007/s00595–012–0376–5>.
10. Falconi M, Plöckinger U, Kwekkeboom DJ et al. Well-differentiated pancreatic nonfunctioning tumors/carcinoma. Neuroendocrinology 2006; 84(3): 196–211.
11. Jensen RT, Niederle B, Mitry E et al. Gastrinoma (duodenal and pancreatic). Neuroendocrinology 2006; 84(3): 173–182.
12. Thakker RV, Newey PJ, Walls GV et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012; 97(9): 2990–3011. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2012–1230>.
13. Vergès B, Boureille F, Goudet P et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 Multicenter Study. J Clin Endocrinol Metab 2002; 87(2): 457–465.
14. Burgess JR, Shepherd JJ, Parameswaran V et al. Spectrum of pituitary disease in multiple endocrine neoplasia type 1 (MEN 1): clinical, biochemical, and radiological features of pituitary disease in a large MEN 1 kindred. J Clin Endocrinol Metab 1996; 81(7): 2642–2646.
15. Christakis I, Qiu W, Silva Fiqueroa AM et al. Clinical Features, Treatments, and Outcomes of Patients with Thymic Carcinoids and Multiple Endocrine Neoplasia Type 1 Syndrome at MD Anderson Cancer Center. Horm Cancer Aug 2016; 7(4): 279–287. Dostupné z DOI: <http://dx.doi.org/10.1007/s12672–016–0269-y>.
16. Asgharian B, Turner ML, Gibril F et al. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab 2004; 89(11): 5328–5336.
17. Vidal A, Iglesias MJ, Fernández B et al. Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. J Eur Acad Dermatol Venereol 2008; 22(7): 835–838. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1468–3083.2008.02578.x>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue Suppl 3
Most read in this issue
- Congenital Adrenal Hyperplasia in Adults
- Multiple Endocrine Neoplasia I (Wermer‘s Syndrome), Forms of Clinical Manifestation, 5 Case Studies
- Vitamin D and autoimmune thyroid diseases
- Central Thyroid Disorders