
Idiopatická plicní fibróza. Umíme ji vždy správně diagnostikovat a léčit?
Idiopathic pulmonary fibrosis. Can we always diagnose and treat it right?
Idiopathic pulmonary fibrosis (IPF) is a severe pulmonary disease characterized by fibrotisation of lung tissue based on pathological healing of alveolar lesions. The disease is initiated in individuals of middle and older age who have a genetical disposition to fibroproliferation. Clinical presentation is not uniform. The patients can have a rapidly progressive disease, eventually severe acute exacerbations, however some of them can have relatively benign course of the disease. Problems in diagnosis are usually caused by atypical radiologic findings and no or nondiagnostic lung biopsy. Causal treatment of this life-threatening disease is available since 2011, nevertheless this treatment is not curative but only slows down declination of lung function and prolongation of survival. Thus it is important to identify the patients with IPF and start treatment early. Section for interstitial lung diseases of the Czech pneumologic and Phtiseologic Society founded the registry of idiopathic pulmonary fibrosis patients (EMPIRE), which has been then adopted by other European and Asian countries and become the biggest registry of this disease in the world. The registry can offer an answer on many questions about this disease in real world and thus is helpful for further knowledge of idiopathic pulmonary fibrosis included the atypical manifestations.
Key words:
atypical clinical and radiologic manifestation – idiopathic pulmonary fibrosis – registry EMPIRE
Authors:
Martina Vašáková
Authors‘ workplace:
Pneumologická klinika 1. LF UK a Thomayerovy nemocnice Praha
Published in:
Vnitř Lék 2017; 63(11): 796-801
Category:
Reviews
Idiopatická plicní fibróza (IPF) je závažné plicní onemocnění charakterizované progresivní fibrotizací plicní tkáně na podkladě patologického vzoru hojení alveolárních lézí. Onemocnění vzniká u jedinců středního a staršího věku, kteří nesou genetickou dispozici k fibroproliferaci. Klinický obraz není jednotný. Pacienti mohou mít rychle progredující onemocnění, případně mohou mít závažné akutní exacerbace, ale mohou mít i relativně benigní průběh nemoci. Problémy v diagnostice činí netypické radiologické obrazy nemoci a nemožnost provést plicní biopsii, případně nediagnostická plicní biopsie. Kauzální léčba této smrtící nemoci je dostupná od roku 2011, nicméně nevede k vyléčení, ale pouze ke zpomalení poklesu plicních funkcí a prodloužení přežití.
Overview
Idiopatická plicní fibróza (IPF) je závažné plicní onemocnění charakterizované progresivní fibrotizací plicní tkáně na podkladě patologického vzoru hojení alveolárních lézí. Onemocnění vzniká u jedinců středního a staršího věku, kteří nesou genetickou dispozici k fibroproliferaci. Klinický obraz není jednotný. Pacienti mohou mít rychle progredující onemocnění, případně mohou mít závažné akutní exacerbace, ale mohou mít i relativně benigní průběh nemoci. Problémy v diagnostice činí netypické radiologické obrazy nemoci a nemožnost provést plicní biopsii, případně nediagnostická plicní biopsie. Kauzální léčba této smrtící nemoci je dostupná od roku 2011, nicméně nevede k vyléčení, ale pouze ke zpomalení poklesu plicních funkcí a prodloužení přežití. Proto je důležité nemocné podchytit včas a včas zahájit léčbu. Sekce intersticiálních plicních procesů České pneumologické a ftizeologické společnosti založila registr nemocných s idiopatickou plicní fibrózou (EMPIRE), který byl posléze přijat i dalšími zeměmi Evropy a Asie a stal se tak největším registrem této nemoci na světě. Registr odkáže odpovědět na řadu otázek o této nemoci v reálném životě a je tak nápomocný v dalším poznání idiopatické plicní fibrózy včetně jejích netypických manifestací.
Klíčová slova:
atypické klinické a radiologické manifestace – idiopatická plicní fibróza – registr EMPIRE
Úvod
Dle mezinárodně uznané definice je idiopatická plicní fibróza (IPF – idiopathic pulmonary fibrosis) specifická forma chronického fibrotizujícího intersticiálního plicního procesu dospělých nejasné etiologie s histologickým a radiologickým obrazem obvyklé intersticiální pneumonie (usual interstitial pneumonia – UIP) [1]. Vyskytuje se výhradně u dospělých, a to středního a staršího věku. Prevalence této nemoci je celosvětově odhadována podle výsledků epidemiologických studií na 13–20/100 000 a incidence na 6,8–16,3/100 000 obyvatel. IPF se vyskytuje s o něco vyšší četností u mužů než u žen. IPF se nevyskytuje v mladém věku, jako dolní věková hranice jejího vzniku je uváděno 45 let, s postupujícím věkem pak její výskyt stoupá, takže dvě třetiny pacientů jsou starší 60 let. IPF se obvykle vyskytuje sporadicky, familiární případy jsou vzácné. Pokud se vyskytne familiárně, mívá častěji tendenci k netypické manifestaci a může se vyskytovat ve věku pod 45 let [2].
Disponující faktory
Mezi disponující faktory patří, vedle stárnutí, vlivy zevního prostředí, genetické faktory a mechanické poškození plic, které paradoxně poškozují dýchací cesty v důsledku samotného dýchání. Mezi vlivy zevního prostředí patří kouření tabáku, expozice organickým i anorganickým prachům a infekce, hlavně virové [3]. Na základě těchto zevních faktorů a pravděpodobně i vnitřních faktorů, mezi které patří kupříkladu extraezofageální reflux, vedoucích k drobným alveolárním lézím, vzniká nekontrolovatelné a progredující jizvení. Alveolární makrofágy jsou totiž u pacientů s IPF prostřednictvím Th2 cytokinů pravděpodobně tzv. alternativně aktivovány a zvyšují pak produkci fibronektinu, a tím indukují fibrogenezi ve fibroblastech [4]. Z genetických faktorů hraje zřejmě roli v patogenezi IPF mutace a polymorfizmy genů pro telomerázy, genů pro surfaktantové proteiny a genu pro mucin (MUCB5) [4].
Klinické projevy a vyšetřovací metody
IPF se klinicky se projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxemii i cyanózou. I když je pro IPF obvyklá u většiny pacientů pozvolná progrese nemoci, u některých pacientů se vyskytnou epizody tzv. akutní exacerbace IPF, při nichž dojde k náhlému klinickému zhoršení s poklesem plicních funkcí a radiologickým obrazem tzv. mléčného skla svědčícím pro probíhající floridní alveolitidu. IPF má obvykle nevyhnutelně progredující průběh navzdory jakékoli léčbě a střední přežití pacientů bez léčby není delší než 2,5–3 roky [1,5].
U asi 75 % pacientů se při fyzikálním vyšetření vyskytují fenotypové projevy, jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bázemi [1].
Funkční vyšetření je u pacientů s IPF důležité pro stanovení závažnosti funkčního postižení, které má i prognostický dopad, a pro úhradu nákladné antifibrotické léčby. Základem je spirometrie a vyšetření difuzní kapacity plic. Typickou funkční poruchou u pacientů s IPF je restrikční ventilační porucha (RVP) s poruchou difuzní kapacity a snížením plicní poddajnosti. Reziduální objem (RV – residual volume) je u IPF obvykle zachován a poměr RV/TLC (totální kapacita plic – total lung capacity) je často zvýšen. Funkce dýchacích cest je u intersticiálního plicního procesu (IPP) obvykle dobře zachována. Pouze vzácně se u pacientů s IPF setkáme i s obstrukční ventilační poruchou (OVP), a to u podskupiny nemocných s kombinací plicní rozedmy a plicní fibrózy. Řada pacientů s IPF, pokud je diagnóza stanovena časně, má normální hodnotu VC (vitální kapacita – vital capacity), což může nezkušené lékaře svést ze správné diagnózy nebo je ukolébat představou, že nemoc není ještě závažná. Transfer faktor (TLCO) bývá významně snížen od samého začátku nemoci, a to ve větší míře, než by odpovídalo redukci TLC. Spiroergometrické vyšetření nám pomůže v identifikaci časných fází intersticiálního plicního postižení a je i screeningem pro případnou plicní arteriální hypertenzi. Pro IPF je typické výrazné zhoršení dušnosti již při submaximální zátěži a je snížená vrcholová spotřeba kyslíku a ventilační rezerva.
Nedílnou součástí je i vyšetření krevních plynů: u pacientů s IPF je v úvodu onemocnění patrný pokles saturace krve kyslíkem (SaO2) a parciálního tlaku kyslíku (PaO2) v tepenné krvi pouze při zátěži a teprve s progresí onemocnění dochází i ke klidové hypoxemii. Na základě hodnot krevních plynů je pak zvažováno u pacientů s IPF přidělení suplementární léčby kyslíkem, a to buď cestou koncentrátoru kyslíku, nebo kapalného kyslíku. Dobrou informaci o funkčním stavu pacienta nám dá 6minutový test chůzí (6-MWT), který je také nevyhnutelnou součástí vyšetření pro případnou indikaci přidělení kapalného kyslíku [1].
Radiologické vyšetření je jedním ze základních kamenů diagnostiky IPF, a to jmenovitě výpočetní tomografie hrudníku a vysokou rozlišovací schopností (HRCT). IPF má být dle definice charakterizována obrazem obvyklé intersticiální pneumonie (UIP – usual interstitial pneumonia) s typickou subpleurální a bazální retikulací s tlustostěnnými cystami různé velikosti a voštinovitou plíci (tab).

Ukazuje se však, že řada pacientů s IPF obraz voštiny vytvořen nemá, tedy nemá radiologický obraz definitivní UIP, ale pouze možné UIP, což pak činí diagnostické obtíže, a to zvláště u pacientů, kteří nemohou nebo nechtějí podstoupit plicní biopsii. Častým nálezem jsou trakční bronchiektazie, popřípadě i bronchioloektázie (obr. 1) [6–8].

Bronchoalveolární laváž (BAL) by měla být provedena vždy při podezření na intersticiální plicní proces (IPP), tedy i na IPF. V diferenciálním rozpočtu buněk v tekutině získané BAL (BALTe) bývají u pacientů s IPF zmnožené granulocyty, obvykle s malou příměsí eozinofilů, lymfocyty bývají zvýšeny minimálně. Výrazně zvýšené lymfocyty – nad 30 % – již indikují, že by se mohlo jednat o jinou diagnózu, navzdory tomu, že HRCT nález odpovídá UIP. V tomto případě bychom měli vždy pečlivým pátráním vyloučit hlavně exogenní alergickou alveolitidu a systémové nemoci pojiva [1,6].
Plicní biopsie je cestou, jak podpořit diagnózu IPF, a to zvláště pokud radiologický nález není typický, tedy neodpovídá definitivní UIP. Pokud je však klinický obraz kompatibilní s IPF, HRCT nález odpovídá definitivní UIP a vyloučili jsme IPP vyvolaný inhalací organických či anorganických antigenů (exogenní alergickou alveolitidu a pneumokoniózy – zejména azbestózu) a systémové nemoci pojiva, není plicní biopsie požadována. Je třeba si navíc uvědomit, že chirurgická plicní biopsie u pacientů s IPF starších 65 let a s TLCO nižší než 35 % je zatížena signifikantní mortalitou a měli bychom se jí tedy pokud možno vyhnout. Nově přináší bezpečnější alternativu získání vzorku plicní tkáně pro diagnostiku IPF transbronchiální kryobiopsie, která má dobrou výtěžnost a pravděpodobně menší riziko akutní exacerbace spojené s výkonem (obr. 2) [6,9,10].

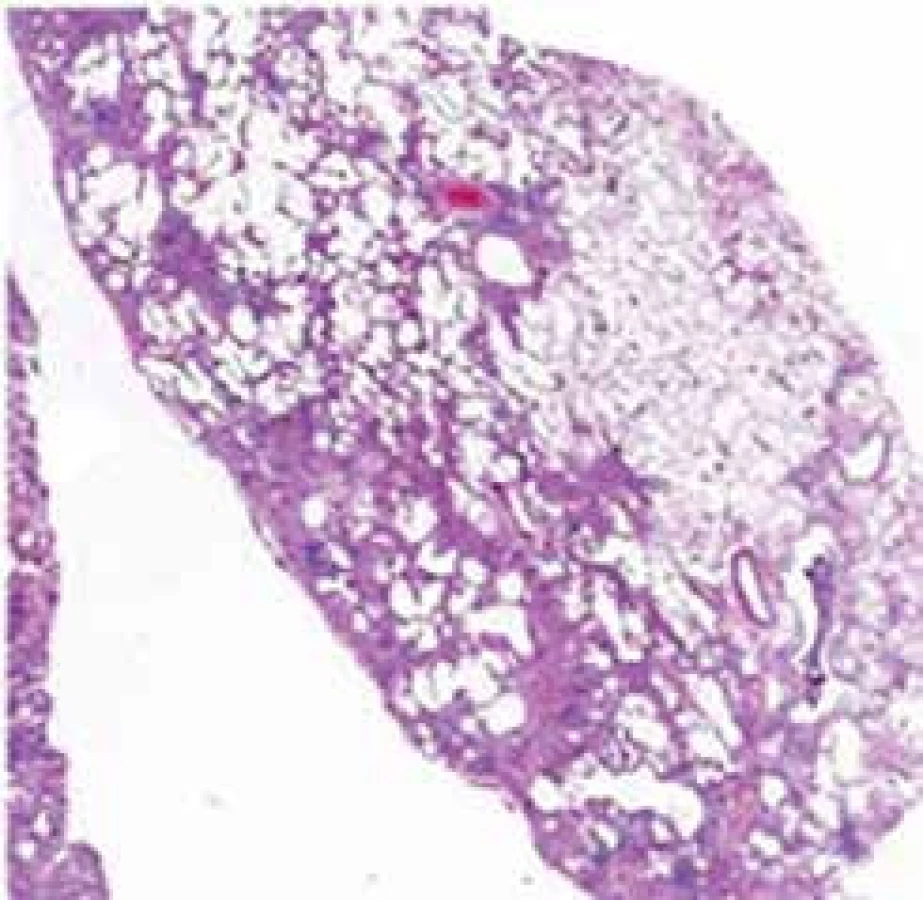
Histopatologickým nálezem u IPF je obvyklá intersticiální pneumonie (UIP) s typickým obrazem fibroticky změněné plicní tkáně s vyzrálým kolagenním vazivem sousedícím s okrsky granulační tkáně s nakupením fibroblastů s myxoidním stromatem. Právě výrazná proměnnost stáří fibrotických změn popisovaná jako „časová heterogenita“ je typická pro obraz UIP. Fibroblastická ložiska reprezentují nejspíše cílová místa opakovaných postižení plicního parenchymu. Když je proces již výrazně pokročilý, dochází ke ztrátě alveolární architektoniky s nápadnou remodelací tkáně, subpleurálně se formují poměrně velké cysty obklopené pruhy husté fibrotizace, pro které se používá historický název „voštinovitá plíce“ (obr. 3) [9].
Laboratorní vyšetření nemá pro podporu diagnózy IPF zásadní přínos, ale laboratorní metody používáme pro screening autoimunitních nemocí (autoprotilátky) a screening protilátek proti organickým antigenům pro vyloučení chronické exogenní alergické alveolitidy [1,6].
Diagnostika
Diagnóza IPF je syntézou klinického, radiologického a případně histopatologického nálezu (tab). Měla by být stanovena na základě multidisciplinárního konsenzu pneumologa, experta v IPP, radiologa a případně pneumopatologa, oba zkušené v problematice IPP [11]. V typických případech, pokud je vyjádřen radiologický obraz definitivní UIP, je diagnóza snadná. Problematické jsou nálezy odpovídající radiologicky možné UIP (tedy bez voštiny), u nichž není možné provést plicní biopsii. Takoví pacienti, pokud jsme vyloučili jiné příčiny IPP, mají tedy možnou IPF. Problém není ani tak v pojmenování nemoci, ale v další léčbě. Tito pacienti mají bohužel shodnou prognózu s těmi, kdo mají definitivní IPF, a proto by měli být léčení jako IPF (graf) [12]. Bohužel není řídkým jevem, že diagnóza správně stanovena není, anebo pojišťovny nemají ochotu platit nákladnou léčbu pacientům s možnou diagnózou IPF.

Léčba
Léčba idiopatické plicní fibrózy musí být komplexní a časná. Základem léčby je farmakologická antifibrotická léčba, která by měla být nabídnuta všem pacientům s IPF, kteří splní kritéria úhrady. Nutné je také již v době diagnózy posoudit, zda bude pacient aktuálně či výhledově kandidátem transplantace plic. Ruku v ruce s farmakologickou léčbou jde léčba symptomatická, podpůrná a paliativní, neboť IPF je bohužel nemocí nevyléčitelnou a cílenou léčbou dokážeme pouze zpomalit průběh nemoci a prodloužit přežití pacientů, případně dovést pacienty k transplantaci [13].
Antifibrotická léčba
Antifibrotická léčba je v současné době reprezentována dvěma dostupnými léky, a to pirfenidonem a nintedanibem.
Pirfenidon kauzálně působí u IPF tím, že snižuje proliferaci fibroblastů a produkci s fibrózou asociovaných proteinů a cytokinů pravděpodobně inhibicí TGFβ1 a destičkového růstového faktoru (platelet-derived growth factor) cestou blokády nukleární translokace proteinu SMAD. Klinická efektivita léčby pirfenidonem byla prokázána v multinárodních studii CAPACITY 004 (PIPF-004) a japonské Shionogi (SP3) a následně i ve studii ASCEND (PIPF-016). Tyto studie prokázaly efekt léčby pirfenidonem na snížení míry poklesu plicních funkcí i na mortalitu na IPF I ze všech příčin ve srovnání s placebem [14]. Pirfenidon je v ČR dostupný od roku 2011 a od 1. 7. 2014 do 30. 6. 2017 byl hrazen z prostředků veřejného zdravotního pojištění v ČR v Centrech pro diagnostiku a léčbu intersticiálních plicních procesů v režimu vysoce inovativního léčebného prostředku (VILP). Podmínkou úhrady byla VC mezi 50–80 % NH a TLCO vyšší než 35 % NH a absence poklesu VC o 10 % a více a TLCO o 15 % a více ve 3 po sobě následujících 6měsíčních intervalech (tzv. stopping rules). Od 1. 7. 2017 je pirfenidon zatím bez úhrady, nicméně mezi Státním ústavem pro kontrolu léčiv, plátci, držitelem rozhodnutí probíhá čilá diskuse, která by měla vést k nastolení definitivní úhrady za zlepšených podmínek pro pacienty. Pirfenidon je podáván v dávce 3krát 3 kapsle po 267 mg/den, tj. 2 403 mg/den. Zvýšené opatrnosti je potřeba při léčbě léky snižujícími aktivitu CYP1A2, při níž může dojít až k významné toxicitě pirfenidonu při současném užívání (např. fluvoxamin, který je při léčbě pirfenidonem kontraindikován, chinolony). Naopak léky zvyšující aktivitu CYP1A2 mohou snižovat účinnost léčby pirfenidonem (rifampicin, omeprazol). Každý pacient musí být při zahájení léčby poučen o nutnosti zanechat kouření tabáku, jinak by byla efektivita léčby ohrožena. Musí být seznámen s fototoxicitou léku a s nutností soustavné ochrany proti slunečnímu záření (celoročně krémy s vysokým ochranným faktorem, tzv. sun-blockery na nechráněná místa kůže, a pokrývka hlavy a ochrana těla oděvem při riziku oslunění). Pacient musí také vědět o gastrointestinálních nežádoucích účincích, hlavně o nevolnosti a nechuti k jídlu a o možnosti snížit dávku při jejich výskytu. Při léčbě pirfenidonem musí být poučen i o možnosti poruchy funkce jater, z toho důvodu se budou v průběhu léčby opakovaně provádět odběry krve.
Nintedanib je dalším lékem, který je dostupný pro léčbu IPF v ČR. Nintedanib je malá molekula, trikinázový inhibitor, cílený na receptor vaskulárního endoteliálního růstového faktoru (VEGFR – vascular endothelial growth factor), růstového faktoru fibroblastů (FGFR – fibroblast growth factor receptors) a destičkového růstového faktoru (PDGFR – platelet-derived growth factor receptors). Nintedanib blokuje tato receptor obsazením vazebních míst pro ATP a tím brání nitrobuněčné signalizaci těchto receptorů. Navíc blokuje též Flt-3 (Fms-like tyrosine protein kinase), LCK (lymphocyte-specific tyrosine-protein kinase), Lyn (tyrosine-protein kinase lyn) a Src (proto-onkogen tyrosine protein-kinase src).
Efektivita a bezpečnost nintedanibu v léčbě IPF byly prokázány ve 2 velkých studiích fáze 3 INPULSIS-1 a INPULSIS-2, do kterých bylo zařazeno 1 066 pacientů s IPF. Nintedanib prokázal v obou těchto studiích efektivitu vyjádřenou významnou redukcí poklesu VC a navíc v INPULSIS-2 i benefit ve smyslu redukce počtu a doby do výskytu akutních exacerbací IPF [15]. Lék je pro perorální užití, ve formě kapslí, užívá se 2krát 1 kapsle po 150 mg denně (přibližně v intervalu 12 hod). Při intoleranci je možné dávku snížit na 2krát 100 mg. Nejčastějšími nežádoucími účinky léčby jsou průjmy a elevace jaterních enzymů. Frekvenci stolic lze snížit podáním loperamidu. Při elevaci transamináz lze při významném vzestupu přechodně přerušit léčbu, častěji však postačí většinou přechodné snížení dávky na 2krát 100 mg denně. Nintedanib je nyní dostupný pro pacienty s IPF v režimu VILP za podmínek VC 50–90 %, TLCO nad 30 % náležitých hodnot, platí pro něj též stopping rules a absolutní výluka kouření.
Další léčba
Dušnost u pacientů v terminálních stadiích IPF obvykle tlumíme opiáty, a to většinou v náplasťových formách s pomalým uvolňováním, obtěžující kašel můžeme zmírnit podáváním malé dávky perorálních kortikosteroidů.
Dlouhodobá domácí oxygenoterapie je doporučena pro všechny pacienty s IPF s klidovou či zátěží indukovanou hypoxemií, kteří splní kritéria ČPFS.
Transplantace plic je pro vhodně vybrané pacienty s IPF rozhodně doporučena. Pětileté přežití po transplantaci plic u pacientů s IPF je odhadováno na 50–56 %.
Umělá plicní ventilace (UPV). Pacienti s IPF by při respiračním selhání způsobeném IPF neměli být paušálně invazivně uměle ventilováni, ale tento způsob léčby může být vhodný pro některé selektované jedince. Mortalita při umělé plicní ventilaci u pacientů s IPF je 96 %. Většinou pouze ti pacienti, kteří mohou být transplantováni z ventilátoru, mají z UPV profit. V praxi to znamená, že by invazivní UPV měla být indikována pro zhoršení IPF s respiračním selháním pouze v případě nemocných, kteří jsou již zařazeni na čekací listině transplantace plic, nebo u kterých existuje šance, že se podaří je urgentně zařadit k transplantaci plic. I tak je pravděpodobnost úspěšné transplantace, a tudíž přežití těchto pacientů velmi malá.
Rehabilitace. Pacienti s IPF by měli být indikováni k plicní rehabilitaci ve většině případů. Je to jedna z možností jak zlepšit kvalitu života pacienta je zlepšení pacientovy funkční výkonnosti a zmírnění dušnosti. Rehabilitace musí být v případě IPF komplexní, zahrnující učení, poradu a behaviorální techniky ke zlepšení sebeobsluhy, redukci symptomů a optimalizaci funkční kapacity [16].
Paliativní péče spočívá v tlumení obtěžujících symptomů. Pro tlumení kašle lze použít systémové kortikosteroidy, a to obvykle v minimální dávce (obvykle do 20 mg prednisonu denně a dávku postupně snižovat), kterou dosáhneme zmírnění úporného kašle. Přesné doporučení pro dávkování kortikosteroidů však není v tomto případě známo. Opioidy indikujeme u těžké dušnosti s kašlem i bez kašle.
Závěr
Idiopatická plicní fibróza je závažnou nemocí s prognózou podobnou maligním nádorům. Je tedy nutné, aby pacienti s IPF byli včas diagnostikováni a aby antifibrotická léčba, která zpomaluje průběh nemoci, byla nasazena co nejdříve. Jedině tak můžeme dát pacientům s IPF šanci na delší přežití.
prof. MUDr. Martina Vašáková, Ph.D.
martina.vasakova@ftn.cz
Pneumologická klinika 1. LF UK a Thomayerovy nemocnice v Praze
www.ftn.cz
Doručeno do redakce 21. 8. 2017
Přijato po recenzi 26. 9. 2017
Sources
1. Kolek V, Kašák V, Vašáková M. Pneumologie. 2. vyd. Maxdorf: Praha 2014 : 261–264. ISBN 978–80–7345–387–9.
2. Nalysnyk L, Cid-Ruzafa J, Rotella P et al. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126): 355–361. Dostupné z DOI: <http://dx.doi.org/10.1183/09059180.00002512>.
3. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 2014; 189(10): 1161–1172. Dostupné z DOI: <http://dx.doi.org/10.1164/rccm.201312–2221PP>.
4. Borensztajn K, Crestani B, Kolb M. Idiopathic pulmonary fibrosis: from epithelial injury to biomarkers-insights from the bench side. Respiration 2013; 86(6): 441–452. Dostupné z DOI: <http://dx.doi.org/10.1159/000357598>.
5. Raghu G, Collard HR, Egan JJ et al. On behalf of the ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med 2011; 183(6): 788–824. Dostupné z DOI: <http://dx.doi.org/10.1164/rccm.2009–040GL>.
6. Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev 2013; 22(128): 158–162. Dostupné z DOI: <http://dx.doi.org/10.1183/09059180.00001213>.
7. Gotway MB, Freemer MM, King TE Jr. Challenges in pulmonary fibrosis. The use of high resolution CT scanning of the lung for the evaluation of patients with idiopathic interstitial pneumonias. Thorax 2007; 62(6): 546–553.
8. Lynch DA, Godwin DJ, Safrin S et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med 2005; 172(4): 488–493.
9. Smith M, Daluzro M, Panse P et al. Usual interstitial pneumonia-pattern fibrosis in surgical lung biopsies. Clinical, radiological and histopathological clues to aetiology. J Clin Pathol 2013; 66(10): 896–903. Dostupné z DOI: <http://dx.doi.org/10.1136/jclinpath-2013–201442>.
10. Fishbein MC. Diagnosis: to biopsy or not to biopsy: assessing the role of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Chest 2005; 128(5 Suppl 1): 520S-525S.
11. Flaherty KR, King TE, Raghu G et al. Idiopathic interstitial pneumonia. What is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8): 904–910.
12. Mura M, Porretta MA, Bargagli E et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40(1): 101–109. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.00106011>.
13. Vašáková M et al. Moderní farmakoterapie v pneumologii. 2. ed. Maxdorf: Praha 2016 : 236–243. ISBN 978–80–7345–506–4.
14. King TE Jr, Bradforf WB, Catsro-Bernardini S et al. [ASCEND Study Group]. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2083–2092. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1402582>.
15. Richeldi L, du Bois RM, Raghu et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2071–2082. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1402584>.
16. Swigris JJ, Brown KK, Make BJ et al. Pulmonary rehabilitation in idiopathic pulmonary fibrosis: A call for continued investigation. Resp Med 2008; 102(12): 1675–1680. Dostupné z DOI: <http://dx.doi.org/10.1016/j.rmed.2008.08.014>.
Labels
Diabetology Endocrinology Internal medicine Pneumology and ftiseology General practitioner for adults RadiodiagnosticsArticle was published in
Internal Medicine

2017 Issue 11
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
Most read in this issue
- Spirometrie – základní vyšetření funkce plic
- Neinvazivní ventilace
- Pneumonie u imunokompromitovaných
- Malobuněčný karcinom plic: epidemiologie, diagnostika a léčba