
Neuronal Reprograming of Protein Homeostasis by Calcium-Dependent Regulation of the Heat Shock Response
Protein quality control requires constant surveillance to prevent misfolding, aggregation, and loss of cellular function. There is increasing evidence in metazoans that communication between cells has an important role to ensure organismal health and to prevent stressed cells and tissues from compromising lifespan. Here, we show in C. elegans that a moderate increase in physiological cholinergic signaling at the neuromuscular junction (NMJ) induces the calcium (Ca2+)-dependent activation of HSF-1 in post-synaptic muscle cells, resulting in suppression of protein misfolding. This protective effect on muscle cell protein homeostasis was identified in an unbiased genome-wide screening for modifiers of protein aggregation, and is triggered by downregulation of gei-11, a Myb-family factor and proposed regulator of the L-type acetylcholine receptor (AChR). This, in-turn, activates the voltage-gated Ca2+ channel, EGL-19, and the sarcoplasmic reticulum ryanodine receptor in response to acetylcholine signaling. The release of calcium into the cytoplasm of muscle cells activates Ca2+-dependent kinases and induces HSF-1-dependent expression of cytoplasmic chaperones, which suppress misfolding of metastable proteins and stabilize the folding environment of muscle cells. This demonstrates that the heat shock response (HSR) can be activated in muscle cells by neuronal signaling across the NMJ to protect proteome health.
Published in the journal:
Neuronal Reprograming of Protein Homeostasis by Calcium-Dependent Regulation of the Heat Shock Response. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003711
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003711
Summary
Protein quality control requires constant surveillance to prevent misfolding, aggregation, and loss of cellular function. There is increasing evidence in metazoans that communication between cells has an important role to ensure organismal health and to prevent stressed cells and tissues from compromising lifespan. Here, we show in C. elegans that a moderate increase in physiological cholinergic signaling at the neuromuscular junction (NMJ) induces the calcium (Ca2+)-dependent activation of HSF-1 in post-synaptic muscle cells, resulting in suppression of protein misfolding. This protective effect on muscle cell protein homeostasis was identified in an unbiased genome-wide screening for modifiers of protein aggregation, and is triggered by downregulation of gei-11, a Myb-family factor and proposed regulator of the L-type acetylcholine receptor (AChR). This, in-turn, activates the voltage-gated Ca2+ channel, EGL-19, and the sarcoplasmic reticulum ryanodine receptor in response to acetylcholine signaling. The release of calcium into the cytoplasm of muscle cells activates Ca2+-dependent kinases and induces HSF-1-dependent expression of cytoplasmic chaperones, which suppress misfolding of metastable proteins and stabilize the folding environment of muscle cells. This demonstrates that the heat shock response (HSR) can be activated in muscle cells by neuronal signaling across the NMJ to protect proteome health.
Introduction
Cellular health and organismal lifespan are critically dependent upon the fidelity of the proteome and the proteostasis network [1]. What are the molecular events that control proteostasis across tissues to activate protective responses at the cellular level to ensure organismal health? At the cellular level the heat shock response (HSR) and the unfolded protein response (UPR) respond to acute forms of proteotoxic stress with precise and rapid activation to restore homeostasis [2], [3]. In contrast to transient extreme stress, the chronic forms of protein damage and toxicity challenge the quality control machinery by their persistence and amplification effects on cumulative protein damage [4], [5]. How proteostasis monitors and responds to physiological stress is an area of active research [2], [6]–[8]. Yet, we know little about the regulation of stress responses under physiological conditions and at the organismal level. Elucidating these regulatory mechanisms is essential for a better understanding of diseases of altered protein conformation and age-related decline in cellular function [1], [9]–[11].
Much of our understanding of the HSR and the proteostasis network has come from studies using cultured cells and model organisms. The invertebrate animals C. elegans and D. melanogaster have been particularly amenable genetic models for identification of proteostasis components and modifiers of protein misfolding and toxicity [12]–[22]. These modifiers include cell autonomous factors such as molecular chaperones, proteasome subunits, components of the autophagy machinery, and the FOXO and heat shock factor 1 (HSF-1) transcriptomes that promote protein folding and clearance within the cell [20]. At the organismal level, the cell non-autonomous role of neuroendocrine signaling pathways and trans-cellular chaperone signaling has been shown to be important for lifespan, stress resistance, innate immunity and proteostasis [8], [9], . Moreover, tissue-specific regulation of mitochondrial function, including the electron transport chain and the mitochondrial UPR, was shown to affect the rate of aging [27]. Efforts to understand how cell autonomous and non-autonomous processes are integrated and co-regulated at the organismal level offer new genetic and pharmacological strategies to enhance the maintenance of proteostasis and health span.
In this study, we describe a new pathway for the heat shock response involving calcium signaling, in which gei-11 knockdown-dependent upregulation of cholinergic receptor levels at the neuromuscular junction (NMJ) triggers activation of HSF-1. This reveals that under normal physiological conditions, the balance between cholinergic and GABAergic signaling at the NMJ regulates protein homeostasis in body-wall muscle (BWM) cells [28]. In contrast to the situation where complete inhibition of GABAergic signaling leads to overstimulation of muscle cells and dysfunction of post-synaptic cell proteostasis [29], we show here that a moderate increase in ACh receptors (AChR) at the NMJ, attained by down-regulation of gei-11, is beneficial to muscle cells. We demonstrate that there is a critical range of cholinergic activity at the NMJ leading to the Ca2+-dependent activation of HSF-1 and expression of molecular chaperones that results in an enhanced protective state in post-synaptic muscle cells.
Results
Identification of gei-11, a Genetic Modifier of Protein Aggregation and Toxicity
A genome-wide RNA interference (RNAi) screen performed in C. elegans for genetic modifiers of muscle proteostasis identified gei-11 RNAi as a potent suppressor of polyglutamine (polyQ) aggregation in BWM cells (Figure 1A: II, IV, VI) [30]. Knockdown of gei-11 also cross-protected against other aggregation-prone proteins, including polyQ37 that presents an earlier onset and more dramatic foci pattern (Figure S1A: I–IV) and mutant SOD1G93A (Figure S1A: V–VIII). Suppression of Q35 aggregation by gei-11 knockdown was achieved by maintaining the polyQ protein in a diffuse soluble state (Figure 1A–VI), as determined by fluorescence recovery after photobleaching (FRAP) (Figure 1B), and not by reducing the expression of Q35 mRNA or protein (Figure S1:B–D). Moreover, gei-11-mediated suppression of Q35 aggregation also led to the rescue of cellular toxicity as the motility of Q35 animals was restored to 100%, similar to knockdown of the polyQ transgene by yfp-RNAi (Figure 1C), without affecting the motility of wild-type (wt) animals. These results reveal that gei-11 knockdown has potent suppressor activity of polyQ aggregation and toxicity.
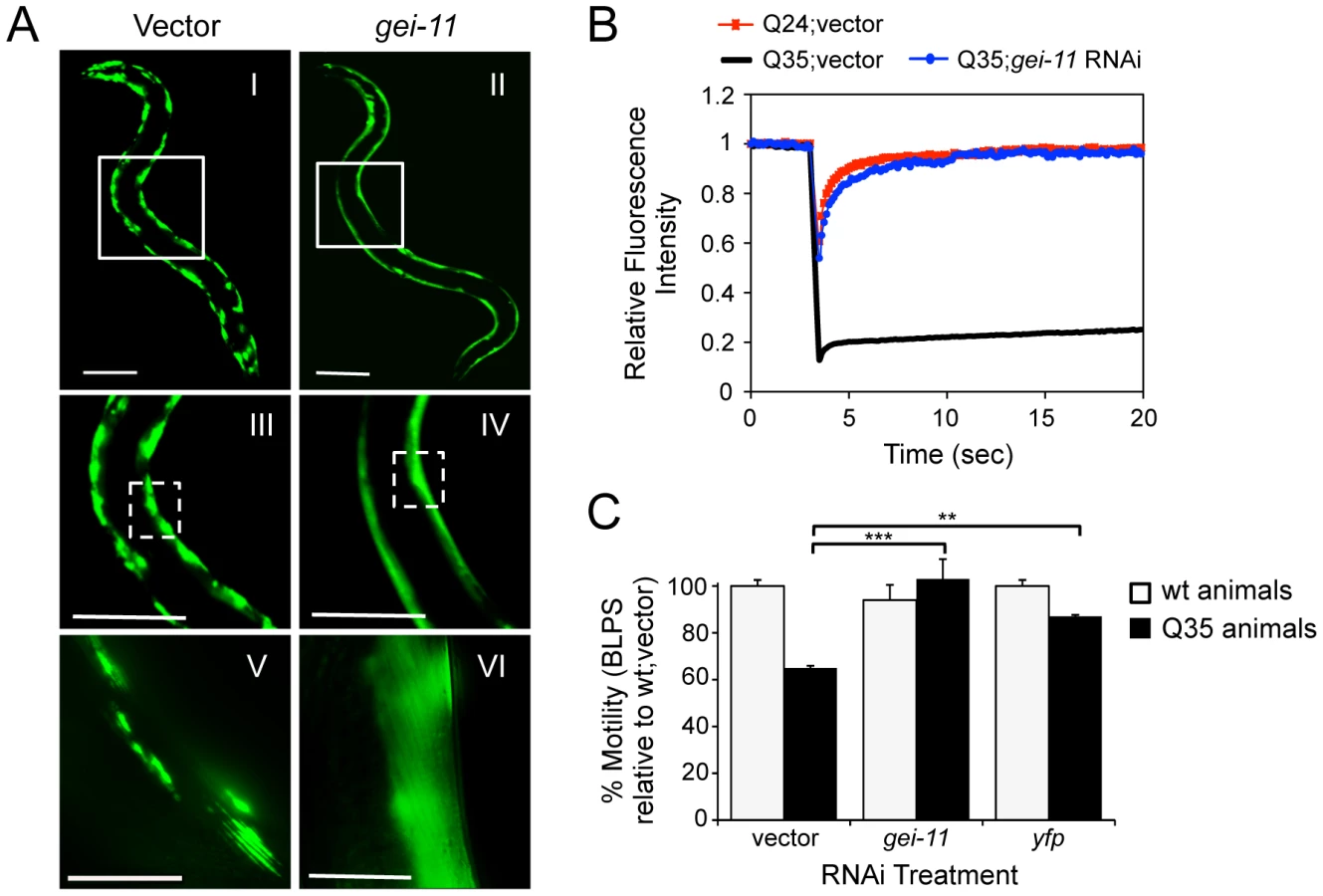
Enhanced Proteostasis Is Due to Increased Expression of L-Type AChR
The gene gei-11 encodes the GEX-3-interacting protein 11 (GEI-11), a member of the Myb superfamily of transcription factors that is homologous to mammalian SNAPC4 (snRNA-activating protein complex subunit 4) [31]. gei-11(tm6548) is the only mutant allele available for this gene, and has a lethal phenotype (National Bioresource Project of Japan). gei-11 has been proposed to have a negative regulatory effect on AChRs in C. elegans BWM cells, and is also expressed in head neurons, germ cells, somatic gonad, and intestine [32], [33].
In C. elegans, two types of ACh receptors, each with distinct subunit composition and pharmacology, are expressed at the NMJ [34]–[36]. To establish the specificity of gei-11 downregulation on the expression of NMJ AChR subtypes, we monitored the expression of the L-type (levamisole-sensitive) AChR subunits unc-29, unc-38, unc-63 and lev-1, and the N-type (nicotine-sensitive) homomeric AChR, acr-16. The effect of gei-11 knockdown increased the expression of only the three essential subunits of the L-AChR (unc-29, unc-38, unc-63) by approximately 3-fold, and had no effect on the expression of N-AChR acr-16 (Figure 2A). Likewise, gei-11 RNAi did not affect the expression of the NMJ GABA receptor (GABAR unc-49, Figure 2A). These results suggest that gei-11 has a highly selective effect on the regulation of the L-type of cholinergic receptors.
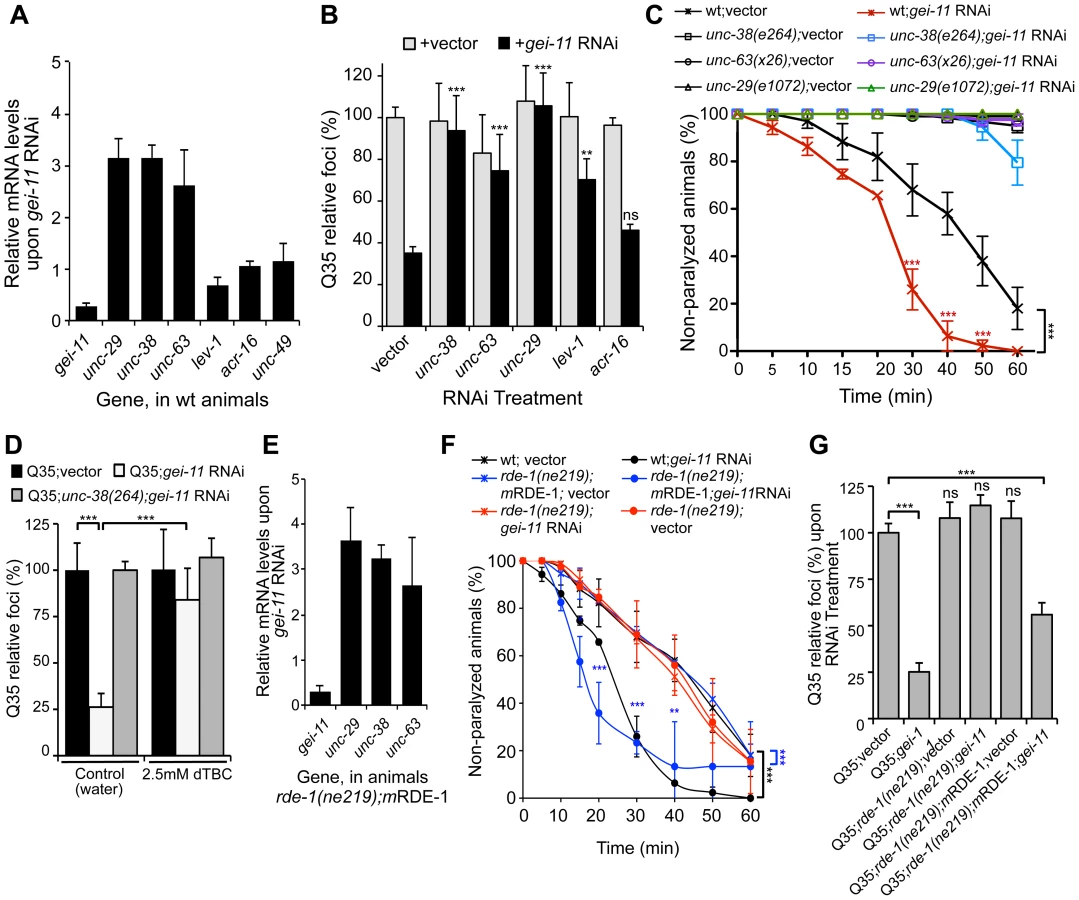
To establish whether gei-11 RNAi-mediated suppression of polyQ aggregation was dependent on elevated expression of the AChR, we performed double RNAi knockdown experiments and downregulated gei-11 together with each of three L-AChR essential subunits (unc-38, unc-63 or unc-29). The results showed that polyQ aggregation was unaffected when an essential L-AChR subunit was co-downregulated with gei-11 (Figure 2B, Table S1). This was further confirmed using an L-AChR mutation (unc-38(e264)), that corroborated the results obtained with RNAi (Figure S2A). Moreover, gei-11 co-downregulation with the non-essential L-AChR subunit lev-1, that only reduces receptor function [37]–[40], only weakened the gei-11 effect on polyQ aggregation (Figure 2B). In contrast, double knockdown of gei-11 with the N-AChR subunit acr-16, still suppressed Q35 aggregation (Figure 2B).
If the elevated expression of three essential L-AChR subunits results in increased L-AChR activity at the NMJ, this would predict increased sensitivity to levamisole, the cholinergic agonist that selectively activates L-AChR and causes hyper-contraction and paralysis [37]. Indeed, gei-11 RNAi-treated wt animals showed a more rapid paralysis on levamisole plates relative to vector RNAi-treated animals (Figure 2C), consistent with the enhanced L-AChR activity at the NMJ. The specificity of levamisole sensitivity to L-AChR activity was confirmed using loss-of-function receptor mutations (unc-38(e264), unc-63(x26), unc-29(e1072)) [28], [35]. These animals were resistant to levamisole upon gei-11 RNAi treatment (Figure 2C), supporting a gei-11 effect dependent on AChR function. Similarly, mutant Q35;unc-38(e264) animals were resistant to levamisole compared to Q35 animals upon gei-11 RNAi (Figure S2B). Consistent with specificity of gei-11 to the L-AChR sub-type, RNAi-treated animals exposed to the agonist nicotine that targets N-AChR [34]–[36], did not show altered sensitivity compared to either wt or N-AChR mutant animals (acr-16(ok789)) (Figure S2C). Additional support for cholinergic-mediated effect on proteostasis was obtained using (+)-Tubocurarine chloride (dTBC), a potent inhibitor of AChR activity [28]. Consistent with our hypothesis, dTBC inhibited gei-11 RNAi suppression of Q35 aggregation in a dose-dependent manner (Figure 2D, ); the specificity of this effect on L-AChR function was consistent with results obtained using the Q35;unc-38(e264) animals (Figure 2D).
The expression of GEI-11 is not restricted to muscle cells [33], therefore we determined whether the effect of gei-11 RNAi and increased cholinergic receptor expression on muscle polyQ aggregation was a direct consequence of gei-11 knockdown in muscle cells, rather than a downstream effect from another tissue. For example, gei-11 RNAi did not have an effect on aggregation of polyQ expressed in the intestine (iQ44, Figure S1E). We next examined whether the enhancement of muscle proteostasis was a direct consequence of gei-11 downregulation in muscle by using a C. elegans mutant strain in which the effects of RNAi are restricted to muscle cells: Q35;rde-1(ne219);myo-3p-RDE-1 (here referred to as rde-1(ne219);mRDE-1) [41], [42]. As a negative control, we employed a mutant strain impaired for RNAi in all cells, Q35;rde-1(ne219) (Figure S2E). Knockdown of gei-11 in rde-1(ne219);mRDE-1 animals increased the expression of essential L-AChR subunits (>3-fold, Figure 2E), comparable to organism-wide gei-11 RNAi (compare Figure 2A and C to Figure 2E and F). Likewise, aggregation in Q35;rde-1(ne219);mRDE-1 animals was suppressed by 50% upon gei-11 RNAi relative to vector control, with no effects observed in Q35;rde-1(ne219) animals (Figure 2G). Taken together, these results show that suppression of protein aggregation in muscle cells is due to gei-11 down-regulation in muscle cells and the consequent upregulation of AChR at the NMJ.
Modulatory Effect of Cholinergic Receptors on Muscle Proteostasis
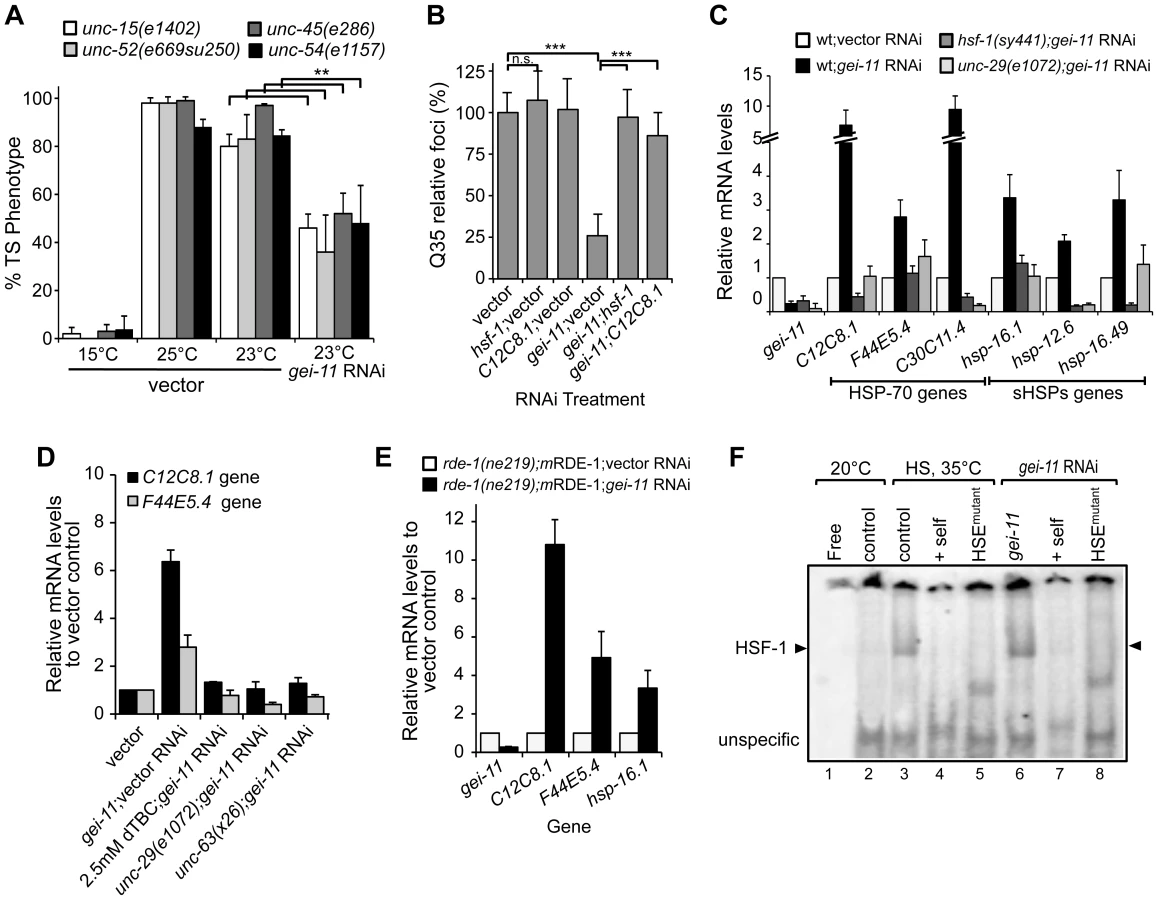
To address whether the effect of gei-11 knockdown-mediated suppression of polyQ aggregation was selective for this class of highly aggregation-prone species or reflected a more general enhancement of proteostasis in the BWM cells, we examined the effects of gei-11 knockdown on the folding stability of four endogenous metastable proteins that function as folding sensors in muscle cells [4]. These metastable proteins exhibit temperature-sensitive (TS) phenotypes and harbor missense mutations in the paramyosin ortholog UNC-15, the basement-membrane protein perlecan UNC-52, the myosin-assembly assisting protein UNC-45, and myosin heavy chain UNC-54 [4], [11]. Thus, in animals held at the permissive temperature (15°C), all four TS-proteins are fully functional, whereas at the restrictive temperatures (23° and 25°C) these sensors misfold and each mutation results in an 80–100% loss of muscle function phenotype (Figure 3A control). However, when gei-11 was downregulated at the restrictive temperature, the loss-of-function phenotype of each TS protein was decreased by 50–60% (Figure 3A). These results establish that downregulation of gei-11 has general protective effects on the stability of multiple muscle cell proteins.
Cholinergic-Mediated Activation of HSF-1 and Expression of Cytoplasmic Chaperones
To establish whether gei-11 RNAi suppression of protein misfolding and aggregation in muscle cells is due to the expression of chaperones, we performed experiments in which gei-11 was knocked-down together with either hsf-1 or hsp-70 (C12C8.1), to reveal that Q35 aggregation was no longer suppressed (Figure 3B). We monitored the expression of cytoplasmic chaperones of the HSP-70 family (C12C8.1, F44E5.4 and C30C11.4) and small heat shock protein family (sHSPs hsp-16.1, hsp-12.6 and hsp-16.49) in wt animals, and show that gei-11 RNAi enhanced the expression of each chaperone gene from 2-to-10-fold (Figure 3C), a level that while substantial, is nevertheless much lower than observed when animals are exposed to acute heat shock treatment (>50-fold, Figure S3A). Moreover, the expression of these chaperones was not induced in the AChR mutant unc-29(e1072), or in HSF-1 mutant hsf-1(sy441) animals (Figure 3C). Therefore, upregulation of the proteostasis machinery by gei-11 RNAi was absolutely dependent upon both cholinergic receptor activity and HSF-1. These results were further corroborated with the cholinergic antagonist dTBC and the L-AChR unc-63(x26) mutation (Figure 3D) that also prevented the upregulation of chaperones upon gei-11 RNAi. The levels of hsps were also upregulated when gei-11 was knocked-down specifically in muscle cells (rde-1(ne219);mRDE-1 animals, Figure 3E), providing additional support that the regulation of cholinergic receptors at the NMJ enhances muscle proteostasis.
To demonstrate directly that knockdown of gei-11 induced the HSR, we monitored the activity of the heat shock transcription factor, HSF-1, using the electrophoresis mobility shift assay (EMSA) [43]. Knockdown of gei-11 induced HSF-1 DNA-binding activity to the heat shock element (HSE) (Figure 3F: lane 6), similar to what was observed in animals exposed to heat shock (Figure 3F: lane 3). The specificity of HSF-1 binding was established using an in vitro competition reaction with excess unlabeled HSE (Figure 3F: lanes 4 and 7) and by using a mutant HSE radiolabeled oligonucleotide incapable of binding by HSF-1 (Figure 3F: lanes 5 and 8). These results demonstrate that gei-11 knockdown activates HSF-1 transcriptional activity.
We further established the downstream regulatory effects of gei-11 knockdown by examining the expression of other components of the proteostasis network, including the expression of the UPR-regulated ER chaperones (hsp-3, hsp-4, dnj-7 and ero-1), metabolic stress FOXO/DAF-16 regulated genes (sod-3 and mtl-1), and oxidative stress regulated genes (hsp-6, gst-4 and gcs-1). As shown in Figure S3B, the expression of none of these other stress responsive genes was induced by gei-11 knockdown in wt animals. Taken together, these results demonstrate that modulation of cholinergic receptors at the NMJ reprograms post-synaptic proteostasis through the activation of HSF-1 and the selective induction of cytoplasmic chaperones.
Proteostasis Rescue by a Shift in the Balance of Cholinergic and GABAergic Signaling
While upregulation of AChR at the NMJ induced the heat shock response and suppressed protein misfolding and toxicity in post-synaptic muscle cells, in previous studies we had observed that a null mutation in unc-30, the transcription factor that regulates the GABA operon, had the opposite result leading to enhanced aggregation in BWM cells [29]. This suggested that extreme cholinergic overstimulation was deleterious to muscle cell proteostasis [29]. Taken together with the results presented here for gei-11, we posit that there is a critical balance between the levels and activities of AChR and GABAR (Figure 4A), with a physiological enhancement of AChR activity being proteo-protective and extreme overstimulation having proteotoxic consequences.
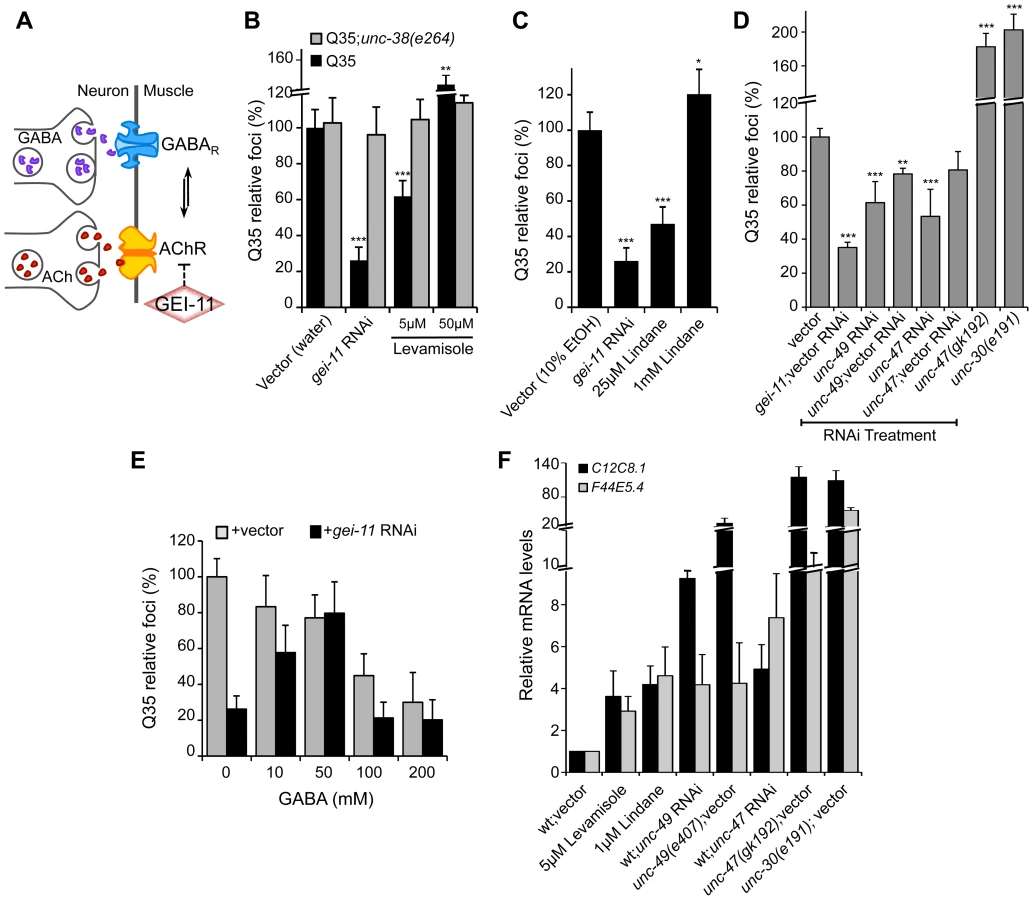
To address this hypothesis, we treated Q35 animals with the L-AChR agonist levamisole over a wide range of concentrations, and monitored aggregation in the post-synaptic muscle cells. At a low concentration of levamisole (5 µM) Q35 aggregation was suppressed by >40% (Figure 4B), whereas at higher levels (50 µM) that caused hyper-contraction, we observed the opposite result of 50–60% enhancement of aggregation (Figure 4B and S4). No effect on aggregation was observed in AChR mutant animals (Q35;unc-38(e264)) (Figure 4B). These results reveal that the beneficial effect on the folding environment is the consequence of a specific physiological range of cholinergic stimulation, and supports our conclusion that overstimulation has deleterious consequences on the folding environment of muscle cells.
We further explored how the imbalance between AChR and GABAR activation at the NMJ affects muscle proteostasis by combining both genetic and small molecule agonists and antagonists probes. Reducing GABAergic activity by exposure to low concentrations (25 µM) of the GABAR antagonist Lindane [29], [44]–[46] led to the suppression of Q35 aggregation (Figure 4C), comparable to a moderate increase of cholinergic signaling. Consistent with the genetic observations after cholinergic overstimulation [29], exposure to the highest concentrations of Lindane (1 mM), that cause BWM cells overstimulation, enhanced Q35 aggregation (Figure 4C). These results provide additional support that shifting the balance at the NMJ can enhance or harm proteostasis in the post-synaptic cell, in a magnitude of signal-dependent manner. To further test this hypothesis, we titrated GABAR expression and GABA release at the synapse by using a combination of RNAi and specific loss-of-function mutations in the GABA pathway: unc-30 (GABA synthesis), unc-47 (GABA transport) and unc-49 (GABAR). Knockdown of unc-47 and unc-49 suppressed Q35 aggregation, and this effect was less penetrant upon dilution of RNAi (1∶1) with vector RNAi (Figure 4D, Table S1). Conversely, eliminating GABA signaling in the unc-30(e191) and unc-47(gk192) mutants had the opposite effect (Figure 4D), consistent with previous results [29]. Finally, we altered the balance between cholinergic and GABAergic signaling by exposing gei-11 RNAi-treated Q35 animals to increasing concentrations of GABA. At low concentrations (≤50 mM), GABA compensated for the gei-11-mediated increase in cholinergic signaling and prevented the suppression of Q35 aggregation, in a dose-dependent manner (Figure 4E). However, at very high GABA concentrations (50 mM–200 mM) this equilibrium shifted to the opposite direction and resulted in suppression of polyQ aggregation by GABAergic signaling (Figure 4E), also consistent with previous results [29]. The highest GABA concentrations tested (>200 mM, not shown) were very toxic and lethal to the animals. These results provide additional support to the importance of the magnitude of cholinergic and GABAergic signaling: an imbalance by either higher AChR or GABAR transmits a signal that is interpreted by the muscle cell to activate a proteo-protective response; however, when this balance is severely disrupted, the consequence is proteotoxic.
Altering the balance between AChR and GABAR, also affected hsp-70 expression. Exposure of wt animals to low levels of the AChR agonist levamisole (5 µM), or to the GABAR antagonist Lindane (25 µM), resulted in elevated expression of the cytoplasmic hsp70 family of chaperone genes (Figure 4F: C12C8.1 and F44E5.4), consistent with enhanced proteostasis. Likewise, and as expected, genetic reduction of GABA signaling using unc-47 or unc-49 RNAi (Table S1), which is equivalent to a moderate increase in cholinergic signaling, also upregulated a low level of hsp-70 (<9-fold, Figure 4F) that restored the folding environment (Figure 4D). Consistent with our previous results, extreme overstimulation in the mutants unc-47(gk192), unc-49(e407) or unc-30(e191) that no longer express GABA or GABAR, led to a massive upregulation of hsp-70 (>50-fold, Figure 4F) that was not proteo-protective and resulted in elevated aggregation (Figure 4D), similar to the deleterious effects of acute heat shock treatment (Figure S3A).
Taken together, these results provide strong support for the importance of the regulation of the equilibrium between cholinergic and GABAergic signaling for optimal proteostasis. Within a critical physiological range, we show that increased AChR activity was beneficial and led to HSF-1-dependent moderate upregulation of cytosolic chaperones in muscle cells to establish a proteo-protective state. In contrast, extreme cholinergic overstimulation, whether obtained by genetics or small molecules, resulted in a dysfunctional proteostasis network that was deleterious.
Upregulation of AChR Leads to Ca2+-Dependent Induction of the HSR
The binding of ACh to receptors in BWM cells initiates a cascade of events that lead to the release of Ca2+ into the cytoplasm for muscle contraction [47] (Figure 5A). We therefore investigated whether the improvement in proteostasis through AChR-mediated HSF-1 activation was dependent on Ca2+ influx. Initially, we utilized the cell permeant Ca2+ chelator BAPTA [48], that alone (15 µM) had no effect on Q35 aggregation, but prevented gei-11 RNAi induction of hsp chaperones and the subsequent suppression of polyQ aggregation (Figure S5A, B).
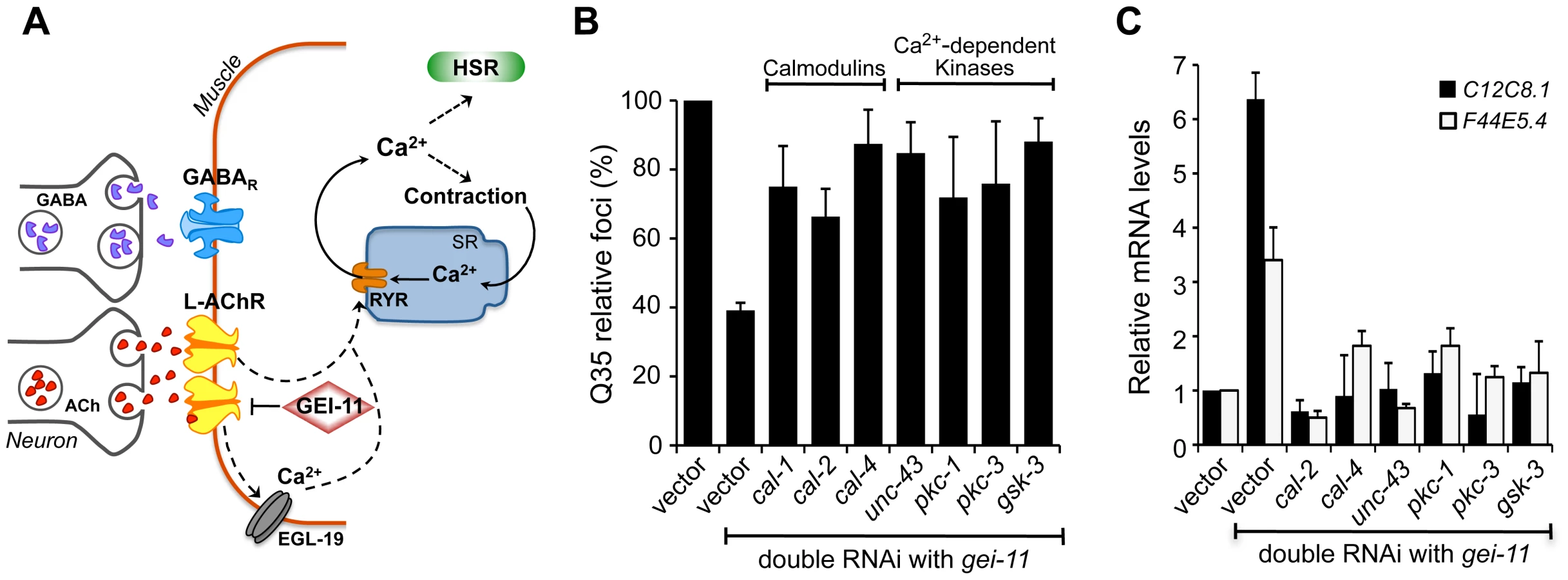
Activation of the HSR by increased levels of cytoplasmic Ca2+ also prompted us to examine the role of Ca2+-dependent kinases, as previous studies from our laboratory and others had identified serine residues of HSF-1 that are stress-inducibly phosphorylated by Ca2+-dependent kinases [49]–[55]. Therefore, we performed a candidate kinase screen to identify the kinases required for the gei-11 RNAi suppression of Q35 aggregation (Figure 5B, Figure S5C) and induction of the HSR in wt animals (Figure 5C). This candidate screen identified calmodulins cal-2 and cal-4; unc-43/CaMKII ortholog; pkc-1, pkc-3; and gsk-3; these genes correspond to the same mammalian kinases previously shown to regulate HSF-1. These results support that AChR upregulation initiates a cascade of Ca2+-signaling events leading to activation of HSF-1.
EGL-19 and Ryanodine Receptors Regulation of Muscle Proteostasis
Cholinergic activity at the NMJ leads to activation of the muscle voltage-gated Ca2+ channel (VGCC), EGL-19, and flux of Ca2+ into the cytoplasm [47] (Figure 5A). We therefore determined the role of EGL-19 activity on muscle proteostasis and polyQ aggregation using a partial (30% reduction) loss-of-function mutant (egl-19(n582)), a weak hypermorphic mutant (egl-19(n582ad952)), and a stronger hypermorphic mutant (egl-19(ad695)) in the background of Q35 [47], [56]. Our results showed that the magnitude of EGL-19 activity, that regulates Ca2+ flux into the muscle, had opposing effects: the strongest hypermorphic mutant ad695 enhanced Q35 aggregation, whereas the weak hypermorphic n582ad952 and weak hypomorphic n582 mutants both suppressed Q35 aggregation (Figure 6A) [29]. Consistent with these effects on aggregation, moderate levels of chaperone upregulation (3-fold) were detected in animals where Ca2+ suppressed aggregation (egl-19(n582) and egl-19(n582ad952); Figure 6B). By comparison, much higher levels of hsp-70 (C12C8.1, F44E5.4; 15-fold) were observed in animals with Ca2+-mediated enhanced aggregation (egl-19(ad695), Figure 6B). These results reveal a consistency between modulation of cholinergic signaling and Ca2+ influx on muscle cell folding environment, reflected by the effect on aggregation. Whereas a mild imbalance in Ca2+ influx, achieved with the weak hypermorph and hypomorph mutants, activated a protective HSR (corresponding to a moderate upregulation of hsp-70), the EGL-19 strong hypermorphic mutation, resulted in a much larger imbalance in signaling and accentuated stress response (corresponding to higher levels of hsp-70). These results provide further support for the importance of a critical physiological stimulation range to establish proteostasis.
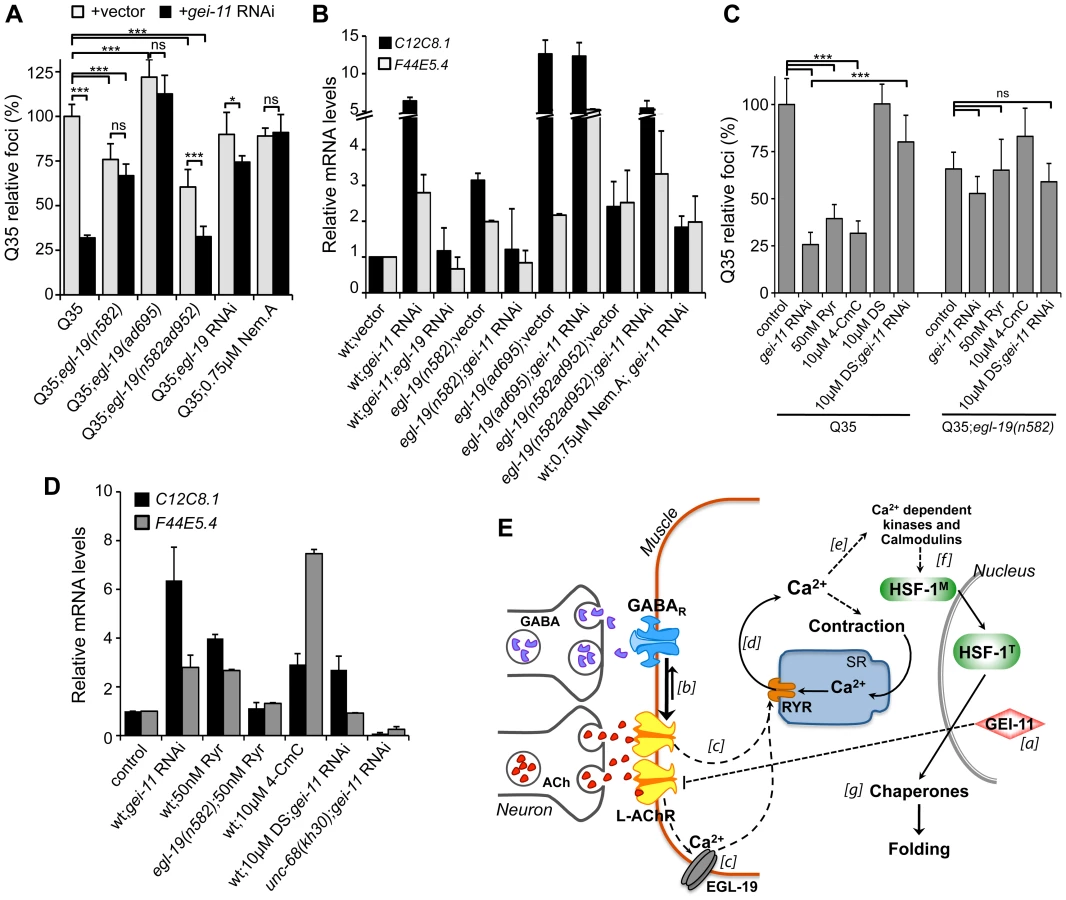
To determine whether the enhanced folding capacity regulated by gei-11 knockdown was dependent on the VGCC, we treated Q35;egl-19 mutant animals with gei-11 RNAi (Figure 6A). Knockdown of gei-11 in hypomorphic egl-19(n582) mutant animals had no effect on Q35 aggregation (Figure 6A) or hsp-70 expression (Figure 6B). This revealed that Ca2+ flux through EGL-19 was required for the beneficial effects of gei-11 knockdown on protein homeostasis. These results were further supported by chemical-genetic approaches using egl-19 RNAi and the EGL-19 inhibitor Nemadipine A [57] that block the gei-11 RNAi effect on muscle proteostasis (Figure 6A, B, S6A, Table S1). Consistent with these observations, the effect of the weak hypermorphic mutant egl-19(n582ad952) was additive to the beneficial effects of gei-11 RNAi on folding (Figure 6A, B), whereas the stronger hypermorphic mutant egl-19(ad695) effect on Ca2+ levels was deleterious (Figure 6A, B). These results establish the role of EGL-19 and Ca2+ influx function downstream of AChR upregulation to the rescue of post-synaptic proteostasis.
Activation of the VGCC, and the flux of Ca2+ into the cytoplasm of muscle cells, triggers the opening of the ryanodine receptor (RYR) at the sarcoplasmic reticulum (SR), releasing additional Ca2+ into the cytosol (Figure 5A) [58], [59]. We examined the contribution of RYR to induction of the HSR by stimulating RYR activity to mimic the effect of enhanced cholinergic signaling at the NMJ. Ryanodine (Ryr), a plant alkaloid with high affinity to the RYR, is a pharmacological agent widely used to study intracellular Ca2+ signaling in muscle cells [58], [59]. At low (nM) concentrations, Ryr acts as an agonist and sensitizes RYR channels to activation by Ca2+ [60], [61]. Treatment of Q35 animals with Ryr (50 nM) caused suppression of aggregation (Figure 6C) and upregulation of hsp-70 (Figure 6D), and this effect was reduced in the background of the hypomorphic mutant egl-19(n582) (Figure 6C), supporting the model where enhancing both Ca2+ channels has beneficial effects on proteostasis. As observed for the natural agonist ryanodine, the clinically-used small molecule RYR activator 4-Chloro-m-cresol (4-CmC) [62] also up-regulated hsp-70 levels and significantly reduced Q35 aggregation by more than 60% (Figure 6C, D, S6B). At high concentrations of 4-CmC (>1 mM), we only observed toxicity and no effect on aggregation (Figure S6B). These results establish that Ca2+ release by the RYR is involved in the enhancement of folding in muscle cells.
Recognizing that Ca2+ regulates many signaling cascades, we examined the specificity of gei-11 RNAi-dependent Ca2+ release by the RYR on induction of hsp-70 and suppression of polyQ aggregation, by testing a RYR-specific antagonist and a RYR mutant. We employed dantrolene sodium (DS), a clinically relevant muscle relaxant that selectively targets RYR and blocks Ca2+ release from the SR during muscle contraction [62], [63]. This antagonist prevented induction of hsp-70 and suppression of Q35 aggregation by gei-11 RNAi (Figure 6C, D, S6C). Similar results were obtained with the RYR mutant unc-68(kh30) (Figure 6D) [58]. From these observations, we conclude that Ca2+ flux from the RYR-SR is an important component of this new signaling pathway regulating muscle HSR. Finally, treatment of egl-19 hypomorphic mutant animals (Q35;egl-19(n582)) with the RYR modulators had no significant effect on aggregation (Figure 6C) supporting the epistatic relationship of the two Ca2+ channels (Figure 6E). Taken together, these results demonstrate that the downstream events of AChR upregulation involve Ca2+-dependent activation of the HSR, and establish a new proteo-protective state in BWM cells with enhanced folding capacity (Figure 6E).
Discussion
We demonstrate that cholinergic-dependent calcium signaling across the synaptic junction induces an atypical heat shock response that promotes protein homeostasis and suppresses misfolding and aggregation in post-synaptic muscle cells. These molecular events are dependent upon HSF1, but are distinct from the classical HSR regulated by transient exposures to acute heat shock stress. The key feature of this novel neuromuscular stress signaling mechanism, centers around the balance between cholinergic and GABAergic signaling at the NMJ. The importance of balanced signaling is highlighted by observations that muscle overexcitation caused by the complete absence of GABA is deleterious to the folding environment and results in a proteotoxic cellular environment [29]. Our present studies provide additional support for the importance of neuronal signaling in the control of somatic cell protein homeostasis, demonstrating that signaling balance at the NMJ can be perturbed to have either beneficial or detrimental consequences on the HSR-dependent proteome stability.
The identification of gei-11 as a new genetic modifier of protein folding reveals a new strategy by which metazoans ensure proteostasis across tissues. Our biological observations suggest that the regulation of receptor expression in muscle cells can initiate a protective mechanism against stress and degeneration, such as age-dependent sarcopenia [11], [64], [65]. These results also suggest that neuronal signaling control of post-synaptic receptor function can achieve the same outcome. This may be highly relevant for complex pathologies, including neurodegenerative diseases and other neuromuscular disorders, where scenarios of protein misfolding initiated at one tissue have both autonomous (cell or tissue specific) and non-autonomous (inter-cellular) consequences on cellular function and organismal health. For example, neurodegeneration leads to muscle weakness and paralysis in motor neuron disorders such as ALS, hereditary spastic paraplegia, and spinal muscular atrophy [66]. Consequently, an understanding of the regulatory signaling cascades that trigger protective responses across tissues is of fundamental importance to delay or prevent the organismal collapse of proteostasis [22]. Modulation of signaling events at the NMJ to rescue muscle function, as described here upon knockdown of the gene gei-11, could suggest novel therapeutic targets for proteostasis maintenance with possible benefit for the patients suffering from somatic wasting diseases. Overall, it emphasizes the importance of dissecting neuronal signaling pathways that affect organismal stress responses and cellular function.
We propose that induction of the HSR by physiological regulation of cholinergic receptors reveals a new class of regulatory pathways of HSF-1 and chaperone networks that is distinct from the classical activation of the HSR. gei-11 was identified from a genetic screen for proteostasis regulators that enhanced the cellular folding environment [30], and found to modulate AChR levels. The levels of ACh and GABA that activate the HSR are in contrast with the extreme imbalance and overstimulation of muscle cells caused by the absence of GABA, leading to proteotoxicity in the muscle cells [29]. An intermediate increase in cholinergic signaling at the NMJ, whether by genetic or small molecule upregulation of AChR activity or downregulation of GABAergic signaling, led to selective activation of the HSR and rescued folding capacity in muscle cells. As for cholinergic signaling, the folding rescue effect of Ca2+ influx in muscle cells homeostasis also occurs at a critical range. The activation of both EGL-19 and RYR channels by cholinergic signaling (Figure 6E) led to increased levels of cytoplasmic Ca2+ and activation of HSF-1 and chaperones that were physiologically beneficial, and well below the deregulated levels of signaling of these pathways that cause proteotoxicity. These results shift the emphasis from extreme environmental forms of stress to a new view on the roles of physiologically relevant in vivo stress signaling pathways regulation.
Aging and chronic stress challenge the cellular quality control systems by the accumulation of misfolded toxic proteins. Our findings strongly suggest that control of HSR and proteostasis, at the cellular level and at cell-non-autonomous level through neuronal signaling, are critical mechanisms in the cellular challenge to activate proteo-protective pathways and maintain homeostasis at the level of the organism. “Fine-tuning” of post-synaptic receptor expression, and therefore regulation of neuronal cholinergic signaling within physiologically relevant levels, may provide a potential strategy to enhance the functional properties of the proteostasis network. Our results contribute to the growing understanding of the properties of stress response networks, as an integrated organismal response to diverse challenges to the health and lifespan of the organism.
Materials and Methods
C. elegans Strains
Animals were maintained according to standard methods, at 20°C on nematode growth media (NGM) with OP50 E. coli [67]. The strains utilized in this work were previously described: wild-type (wt) Bristol strain N2; polyQ strains Q24 AM138 (rmIs130[Punc-54::q24::yfp]II), Q35 AM140 (rmIs132[Punc-54::q35::yfp]I), Q37 AM470 (rmIs225[Punc-54::q37::yfp]II) [16], [17]; human SOD1G93A AM265 (rmIs177[Punc-54::sod1G93A::yfp]) [5]; intestinal Q44 GF80 dgEx80[pAMS66 Pvha-6::q44::yfp + rol-6(su1006) +pBluescript II] [68]; temperature sensitive (TS) mutant strains CB1402 [unc-15(e1402)], CB1157 [unc-54(e1157)], HE250 [unc-52(e669su250)] and CB286 [unc-45(e286)] [4]; WM27 [rde-1(ne219)] and WM118 [rde-1(ne219);neIs9[myo-3::HA::RDE-1+pRF4(rol-6(su1006))]] [41], [42]; CB1072 [unc-29(e1072)], CB904 [unc-38(e264)], ZZ26 [unc-63(x26)], RB918 [acr-16(ok789)], VC311 [unc-47(gk192)], CB845 [unc-30(e191)], CB407 [unc-49(e407)] [29], MT1212 [egl-19(n582)], DA952 [egl-19(n582ad952)], DA695 [egl-19(ad695)] [47], [56], HK30 [unc-68(kh30)], PS3551 [hsf-1(sy441)]. Where indicated, genetic crosses between mutant animals and Q35 animals were generated.
RNAi Assays
RNAi gene knockdown in C. elegans was performed as described previously, using the commercial RNAi library (GeneService, USA) [30], [69]. Briefly, animals were added to RNAi bacteria (in liquid or RNAi-seeded NGM plates) at the L1 stage (first larval, day 1), incubated at 20°C for 5 days and scored for number of aggregates at 6 days old (which corresponds to 3 days after the onset of Q35 aggregation) [30], using the stereomicroscope Leica MZ16FA (Leica Microsystems, Switzerland). Q35 aggregation was scored in at least 50 animals, for each condition (n≥3). As a negative control, animals were fed bacteria carrying the L4440 empty vector (vector). Liquid RNAi treatment was performed in 96-well plates, with a total volume of 60 µl per well, consisting of 15–20 worms and RNAi bacteria. Bacteria was grown overnight (∼16 h), induced with isopropyl β-D-thiogalatoside (IPTG Sigma, 1 mM for 3 h at 37°C), pelleted and resuspended in S-medium complete (S-Basal supplemented with 3 mM MgSO4, 3 mM CaCl2, 10 mM Potassium Citrate, 100 mg/ml Ampicillin and 1 mM IPTG) so that the final OD595 nm was 0.9 in the well. RNAi assays on plates were performed as described previously [30], and for double knockdown experiments, equal volumes of each RNAi bacteria were mixed (1∶1 ratio) prior to plate seeding. Fluorescent microscopy images were taken using an Axiovert 200 microscope with a Hamamatsu digital camera C4742-98 (Carl Zeiss, Germany). All RNAi plasmids were sequenced to confirm correct and specific gene-target identity. Gene knockdown by RNAi was confirmed by PCR analysis.
FRAP Analysis
Animals were mounted on a 3% (w/v) agar pad on a glass slide, immobilized with 2 mM levamisole (Sigma), and subjected to FRAP analysis using the Zeiss LSM510 confocal microscope (Carl Zeiss, Germany) as previously described [30], [70].
Motility Assays
The movement of 6 day old animals grown on RNAi-seeded NGM plates (>75 animals per experiment, n≥3) were digitally recorded using a Leica M205 FA microscope with a Hamamatsu digital camera C10600-10B (Orca-R2, Leica Microsystems, Switzerland), and the Hamamatsu Simple PCI Imaging software. Animals were tracked using a custom ImageJ plugin wrMTrck [30]. The average speed of each animal was calculated by dividing the distance of each track, corrected for body length, by the duration (in seconds) of the track (body length per second BLPS) (n≥3). Results are shown relative to wt animals' speed on L4440-RNAi vector control plates (100%).
Real-Time qPCR Analysis
RNA from ∼50 animals was extracted with Trizol (Invitrogen), followed by DNase treatment (Applied Biosystems #AM1906). mRNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad #170-8891). cDNA real-time PCR amplification was done using the iQ-SYBR Green Supermix (Bio-Rad #170-8880) and the iCycler system (Bio-Rad) (see Protocol S1). Expression levels of each gene were determined using the Comparative CT Method (Real-Time PCR Applications Guide, Bio-Rad), normalized to actin (act-1) in the same sample, and relative to the non-treated or vector control sample. Measurements were performed for ≥3 biological samples.
Paralysis Assays
Five day old animals grown on RNAi-seeded NGM plates at 20°C (≤40) were transferred onto NGM plates, equilibrated at 20°C, containing 1 mM Levamisole (Sigma), 30 mM Nicotine (Sigma) or the solvent (water or ethanol, respectively). Sensitivity to the drugs was followed by visual inspection every 2 to 5 min and defined as paralysis, or lack of movement in response to prodding on the nose and tail of the animal (n = 3). Compound stock solutions: 800 mM levamisole (Sigma) in water, 300 mM nicotine (Sigma) in ethanol.
Compound Assays
Compound assays were performed in liquid culture as described previously [71], with 60 µl of final volume per well, 15–20 animals (in S-Basal complete), compound at the appropriate concentration and bacteria (OP50 or RNAi) at a final OD595 nm of 0.9 (resuspended in S-Basal complete). Replicates of each condition were included in each assay/plate. Animals were incubated with each drug, at the concentrations indicated in the respective Figures, from L1 stage (levamisole, GABA), L2 stage (Lindane, Ryanodine, Nemadipine A, Dantrolene Sodium, 4-CmC) or L4 stage (dTBC, BAPTA), until day 6 of age, at 20°C (n≥3) (see Protocol S1). At this time animals were transferred from liquid culture onto NGM plates for aggregate quantification (Leica MZ16FA) and collected for real-time qPCR analysis.
Assay for TS Phenotypes
Temperature sensitive (TS) mutant animals were age-synchronized to L1 stage, grown on RNAi-seeded NGM plates (∼20 animals per plate) from day 1 at a sensitized temperature of 23°C (to maintain the RNAi suppressor effect on aggregation), or at the control restrictive (25°C) and permissive (15°C) temperatures, and scored for phenotypes on day 5, as previously described [30]. >50 animals were scored for each TS phenotype, per assay: slow movement/paralysis assay for unc-15(e1402) and unc-54(e1157), stiff paralysis for unc-52(e669su250), and egg-laying phenotype for unc-45(e286) (partially paralyzed animals with a large belly of accumulated eggs) (n = 3) [4], [29], [30].
Gel Mobility Shift Analysis
Native nuclear protein extracts were prepared from 200 µl of pelleted worms (grown on NGM RNAi-seeded plates), with the commercial Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit (# 78835), as described previously [72]. Electrophoretic mobility shift analysis (EMSA) was performed as before [43] using a [32P]-labeled probe containing the proximal heat shock element (HSE) from the C. elegans hsp-70 (C12C8.1) gene promoter. Nuclear extracts (40 µg) were incubated with the [32P]-labeled probe (HSE or mutant) for 20 min at room temperature. For heat shock treatment (HS) the samples were pre-incubated at 35°C for 30 min. For competition experiments, a 100-fold molar excess of the same unlabeled oligonucleotide was added to the mixture. The samples were analyzed by electrophoresis on a 4% (w/v) polyacrylamide native gel that was dried and scanned using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). Oligonucleotide probes: HSE-F: taaattgtagaaggttctagaagatgccaga; HSE-R: tctggcatcttctagaaccttctacaattta; HSEmut-F: taaattgtaaaaggaaataaaagatgccaga; HSEmut-R: tctggcatcttttatttccttttacaattta.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BalchWE, MorimotoRI, DillinA, KellyJW (2008) Adapting proteostasis for disease intervention. Science 319: 916–919.
2. MorimotoRI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22: 1427–1438.
3. Morimoto RI (2012) The Heat Shock Response: Systems Biology of Proteotoxic Stress in Aging and Disease. In: Press CSH, editor. Cold Spring Harbor Symposia on Quantitative Biology: Metabolism and Disease. pp. 91–99.
4. GidalevitzT, Ben-ZviA, HoKH, BrignullHR, MorimotoRI (2006) Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311: 1471–1474.
5. GidalevitzT, KrupinskiT, GarciaS, MorimotoRI (2009) Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS genetics 5: e1000399.
6. VoisineC, PedersenJS, MorimotoRI (2010) Chaperone networks: tipping the balance in protein folding diseases. Neurobiology of disease 40: 12–20.
7. GidalevitzT, PrahladV, MorimotoRI (2011) The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harbor perspectives in biology 3: pii: a009704.
8. PrahladV, CorneliusT, MorimotoRI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320: 811–814.
9. PrahladV, MorimotoRI (2009) Integrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol 19: 52–61.
10. SotoC, EstradaLD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65: 184–189.
11. Ben-ZviA, MillerEA, MorimotoRI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A 106: 14914–14919.
12. BrignullHR, MorleyJF, MorimotoRI (2007) The stress of misfolded proteins: C. elegans models for neurodegenerative disease and aging. Adv Exp Med Biol 594: 167–189.
13. DriscollM, GerstbreinB (2003) Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet 4: 181–194.
14. MarshJL, ThompsonLM (2006) Drosophila in the study of neurodegenerative disease. Neuron 52: 169–178.
15. van HamTJ, BreitlingR, SwertzMA, NollenEA (2009) Neurodegenerative diseases: Lessons from genome-wide screens in small model organisms. EMBO Mol Med 1: 360–370.
16. MorleyJF, BrignullHR, WeyersJJ, MorimotoRI (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99: 10417–10422.
17. NollenEA, GarciaSM, van HaaftenG, KimS, ChavezA, et al. (2004) Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A 101: 6403–6408.
18. CohenE, BieschkeJ, PerciavalleRM, KellyJW, DillinA (2006) Opposing activities protect against age-onset proteotoxicity. Science 313: 1604–1610.
19. MorleyJF, MorimotoRI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Molecular biology of the cell 15: 657–664.
20. GuisbertE, CzyzDM, RichterK, McMullenPD, MorimotoRI (2013) Identification of a tissue-selective heat shock response regulatory network. PLoS Genet 9: e1003466.
21. Kirstein-MilesJ, SciorA, DeuerlingE, MorimotoRI (2013) The nascent polypeptide-associated complex is a key regulator of proteostasis. EMBO J 32: 1451–1468.
22. van Oosten-HawleP, PorterRS, MorimotoRI (2013) Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153: 1366–1378.
23. RussellSJ, KahnCR (2007) Endocrine regulation of ageing. Nature reviews Molecular cell biology 8: 681–691.
24. StyerKL, SinghV, MacoskoE, SteeleSE, BargmannCI, et al. (2008) Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322: 460–464.
25. SternbergEM (2006) Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6: 318–328.
26. SunJ, SinghV, Kajino-SakamotoR, AballayA (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332: 729–732.
27. DurieuxJ, WolffS, DillinA (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144: 79–91.
28. RichmondJE, JorgensenEM (1999) One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2: 791–797.
29. GarciaSM, CasanuevaMO, SilvaMC, AmaralMD, MorimotoRI (2007) Neuronal signaling modulates protein homeostasis in Caenorhabditis elegans post-synaptic muscle cells. Genes Dev 21: 3006–3016.
30. SilvaMC, FoxS, BeamM, ThakkarH, AmaralMD, et al. (2011) A Genetic Screening Strategy Identifies Novel Regulators of the Proteostasis Network. PLoS Genet 7: e1002438.
31. TsuboiD, QadotaH, KasuyaK, AmanoM, KaibuchiK (2002) Isolation of the interacting molecules with GEX-3 by a novel functional screening. Biochem Biophys Res Commun 292: 697–701.
32. GottschalkA, AlmedomRB, SchedletzkyT, AndersonSD, YatesJR (2005) Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis elegans. EMBO J 24: 2566–2578.
33. NiuW, LuZJ, ZhongM, SarovM, MurrayJI, et al. (2011) Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome research 21: 245–254.
34. BoulinT, GielenM, RichmondJE, WilliamsDC, PaolettiP, et al. (2008) Eight genes are required for functional reconstitution of the Caenorhabditis elegans levamisole-sensitive acetylcholine receptor. Proc Natl Acad Sci U S A 105: 18590–18595.
35. RayesD, FlaminiM, HernandoG, BouzatC (2007) Activation of single nicotinic receptor channels from Caenorhabditis elegans muscle. Mol Pharmacol 71: 1407–1415.
36. TouroutineD, FoxRM, Von StetinaSE, BurdinaA, MillerDM (2005) acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J Biol Chem 280: 27013–27021.
37. CulettoE, BaylisHA, RichmondJE, JonesAK, FlemingJT, et al. (2004) The Caenorhabditis elegans unc-63 gene encodes a levamisole-sensitive nicotinic acetylcholine receptor alpha subunit. The Journal of biological chemistry 279: 42476–42483.
38. FlemingJT, SquireMD, BarnesTM, TornoeC, MatsudaK, et al. (1997) Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits. J Neurosci 17: 5843–5857.
39. LewisJA, WuCH, BergH, LevineJH (1980) The genetics of levamisole resistance in the nematode Caenorhabditis elegans. Genetics 95: 905–928.
40. RandJB (2007) Acetylcholine. WormBook 1–21.
41. BolzDD, TenorJL, AballayA (2010) A conserved PMK-1/p38 MAPK is required in caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. The Journal of biological chemistry 285: 10832–10840.
42. QadotaH, InoueM, HikitaT, KoppenM, HardinJD, et al. (2007) Establishment of a tissue-specific RNAi system in C. elegans. Gene 400: 166–173.
43. MosserDD, TheodorakisNG, MorimotoRI (1988) Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Molecular and cellular biology 8: 4736–4744.
44. BamberBA, RichmondJE, OttoJF, JorgensenEM (2005) The composition of the GABA receptor at the Caenorhabditis elegans neuromuscular junction. Br J Pharmacol 144: 502–509.
45. WaffordKA, LummisSC, SattelleDB (1989) Block of an insect central nervous system GABA receptor by cyclodiene and cyclohexane insecticides. Proc R Soc Lond B Biol Sci 237: 53–61.
46. ZhangHG, ffrench-ConstantRH, JacksonMB (1994) A unique amino acid of the Drosophila GABA receptor with influence on drug sensitivity by two mechanisms. J Physiol 479 ((Pt 1)) 65–75.
47. JospinM, JacquemondV, MariolMC, SegalatL, AllardB (2002) The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J Cell Biol 159: 337–348.
48. Ghosh-RoyA, WuZ, GoncharovA, JinY, ChisholmAD (2010) Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. The Journal of neuroscience : the official journal of the Society for Neuroscience 30: 3175–3183.
49. JorqueraG, JureticN, JaimovichE, RiverosN (2009) Membrane depolarization induces calcium-dependent upregulation of Hsp70 and Hmox-1 in skeletal muscle cells. Am J Physiol Cell Physiol 297: C581–590.
50. MosserDD, KotzbauerPT, SargeKD, MorimotoRI (1990) In vitro activation of heat shock transcription factor DNA-binding by calcium and biochemical conditions that affect protein conformation. Proceedings of the National Academy of Sciences of the United States of America 87: 3748–3752.
51. PriceBD, CalderwoodSK (1991) Ca2+ is essential for multistep activation of the heat shock factor in permeabilized cells. Mol Cell Biol 11: 3365–3368.
52. AkerfeltM, MorimotoRI, SistonenL (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545–555.
53. HolmbergCI, HietakangasV, MikhailovA, RantanenJO, KallioM, et al. (2001) Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J 20: 3800–3810.
54. HolmbergCI, RoosPM, LordJM, ErikssonJE, SistonenL (1998) Conventional and novel PKC isoenzymes modify the heat-induced stress response but are not activated by heat shock. Journal of cell science 111 ((Pt 22)) 3357–3365.
55. WhitesellL, LindquistS (2009) Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets 13: 469–478.
56. LeeRY, LobelL, HengartnerM, HorvitzHR, AveryL (1997) Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. The EMBO journal 16: 6066–6076.
57. KwokTC, RickerN, FraserR, ChanAW, BurnsA, et al. (2006) A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature 441: 91–95.
58. AdachiR, KagawaH (2003) Genetic analysis of ryanodine receptor function in Caenorhabditis elegans based on unc-68 revertants. Mol Genet Genomics 269: 797–806.
59. MaryonEB, SaariB, AndersonP (1998) Muscle-specific functions of ryanodine receptor channels in Caenorhabditis elegans. J Cell Sci 111 ((Pt 19)) 2885–2895.
60. LaporteR, HuiA, LaherI (2004) Pharmacological modulation of sarcoplasmic reticulum function in smooth muscle. Pharmacol Rev 56: 439–513.
61. RobertsonAP, ClarkCL, MartinRJ (2010) Levamisole and ryanodine receptors. I: A contraction study in Ascaris suum. Mol Biochem Parasitol 171: 1–7.
62. WesterbladH, AndradeFH, IslamMS (1998) Effects of ryanodine receptor agonist 4-chloro-m-cresol on myoplasmic free Ca2+ concentration and force of contraction in mouse skeletal muscle. Cell calcium 24: 105–115.
63. FruenBR, MickelsonJR, LouisCF (1997) Dantrolene inhibition of sarcoplasmic reticulum Ca2+ release by direct and specific action at skeletal muscle ryanodine receptors. The Journal of biological chemistry 272: 26965–26971.
64. HerndonLA, SchmeissnerPJ, DudaronekJM, BrownPA, ListnerKM, et al. (2002) Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419: 808–814.
65. RueggMA, GlassDJ (2011) Molecular mechanisms and treatment options for muscle wasting diseases. Annual review of pharmacology and toxicology 51: 373–395.
66. DionPA, DaoudH, RouleauGA (2009) Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nature reviews Genetics 10: 769–782.
67. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94.
68. PrahladV, MorimotoRI (2011) Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A 108: 14204–14209.
69. KamathRS, FraserAG, DongY, PoulinG, DurbinR, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237.
70. PhairRD, MisteliT (2000) High mobility of proteins in the mammalian cell nucleus. Nature 404: 604–609.
71. VoisineC, VarmaH, WalkerN, BatesEA, StockwellBR, et al. (2007) Identification of potential therapeutic drugs for huntington's disease using Caenorhabditis elegans. PLoS One 2: e504.
72. MahAK, ArmstrongKR, ChewDS, ChuJS, TuDK, et al. (2007) Transcriptional regulation of AQP-8, a Caenorhabditis elegans aquaporin exclusively expressed in the excretory system, by the POU homeobox transcription factor CEH-6. The Journal of biological chemistry 282: 28074–28086.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2013 Číslo 8
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription