
Species and Population Level Molecular Profiling Reveals Cryptic Recombination and Emergent Asymmetry in the Dimorphic Mating Locus of
Heteromorphic sex-determining regions or mating-type loci can contain large regions of non-recombining sequence where selection operates under different constraints than in freely recombining autosomal regions. Detailed studies of these non-recombining regions can provide insights into how genes are gained and lost, and how genetic isolation is maintained between mating haplotypes or sex chromosomes. The Chlamydomonas reinhardtii mating-type locus (MT) is a complex polygenic region characterized by sequence rearrangements and suppressed recombination between its two haplotypes, MT+ and MT−. We used new sequence information to redefine the genetic contents of MT and found repeated translocations from autosomes as well as sexually controlled expression patterns for several newly identified genes. We examined sequence diversity of MT genes from wild isolates of C. reinhardtii to investigate the impacts of recombination suppression. Our population data revealed two previously unreported types of genetic exchange in Chlamydomonas MT—gene conversion in the rearranged domains, and crossover exchanges in flanking domains—both of which contribute to maintenance of genetic homogeneity between haplotypes. To investigate the cause of blocked recombination in MT we assessed recombination rates in crosses where the parents were homozygous at MT. While normal recombination was restored in MT+×MT+ crosses, it was still suppressed in MT−×MT− crosses. These data revealed an underlying asymmetry in the two MT haplotypes and suggest that sequence rearrangements are insufficient to fully account for recombination suppression. Together our findings reveal new evolutionary dynamics for mating loci and have implications for the evolution of heteromorphic sex chromosomes and other non-recombining genomic regions.
Published in the journal:
Species and Population Level Molecular Profiling Reveals Cryptic Recombination and Emergent Asymmetry in the Dimorphic Mating Locus of. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003724
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003724
Summary
Heteromorphic sex-determining regions or mating-type loci can contain large regions of non-recombining sequence where selection operates under different constraints than in freely recombining autosomal regions. Detailed studies of these non-recombining regions can provide insights into how genes are gained and lost, and how genetic isolation is maintained between mating haplotypes or sex chromosomes. The Chlamydomonas reinhardtii mating-type locus (MT) is a complex polygenic region characterized by sequence rearrangements and suppressed recombination between its two haplotypes, MT+ and MT−. We used new sequence information to redefine the genetic contents of MT and found repeated translocations from autosomes as well as sexually controlled expression patterns for several newly identified genes. We examined sequence diversity of MT genes from wild isolates of C. reinhardtii to investigate the impacts of recombination suppression. Our population data revealed two previously unreported types of genetic exchange in Chlamydomonas MT—gene conversion in the rearranged domains, and crossover exchanges in flanking domains—both of which contribute to maintenance of genetic homogeneity between haplotypes. To investigate the cause of blocked recombination in MT we assessed recombination rates in crosses where the parents were homozygous at MT. While normal recombination was restored in MT+×MT+ crosses, it was still suppressed in MT−×MT− crosses. These data revealed an underlying asymmetry in the two MT haplotypes and suggest that sequence rearrangements are insufficient to fully account for recombination suppression. Together our findings reveal new evolutionary dynamics for mating loci and have implications for the evolution of heteromorphic sex chromosomes and other non-recombining genomic regions.
Introduction
Heteromorphic sex chromosomes and mating-type loci can be dynamic genomic regions with large non-recombining blocks of rearranged sequences, high transposon and repeat density, low-protein coding gene density, and high rates of sequence evolution compared to autosomes [1]–[3]. Sex chromosomes undergo decay and gene loss [4], but have also been found to be sources of genetic innovation [5]. Sex determining or mating-type regions in haploid species are diverse and can be controlled by small mating-type loci with one or two genes, as in the case of yeasts [6], by complex heteromorphic mating-type loci such as those found and algae and some fungi [7]–[11], or by sex chromosomes in bryophytes [12], [13].
Volvocine algae are an emerging model for investigating the evolution of sex chromosomes and mating-type loci [14]. These haploid green algae form a coherent phylogenetic group that encompasses unicellular species such as Chlamydomonas reinhardtii and multicellular species such as Volvox carteri. Volvocine algae show convergent evolution with other multicellular clades in their sexual cycles: isogamy (equal-sized gametes) is predominant in small colonial genera and unicellular species such as Chlamydomonas, while anisogamy (large and small gametes) or oogamy (eggs and sperm) are predominant in larger colonial genera such as Volvox, Pleodorina and Eudorina. Homothallic and heterothallic mating systems also evolved within different Volvocine algal sub-lineages making them a highly diverse group [15]–[17].
Chlamydomonas reinhardtii is a heterothallic species with two mating types, plus (MT+) and minus (MT−), which are defined by alleles at its mating locus (MT) located near one telomere of Chromosome 6. Haploid cells of either mating type can propagate mitotically when supplied with sufficient light and nutrients, but differentiate into mating-competent gametes in the absence of nitrogen. Gametes of opposite mating type recognize each other and fuse to form dormant diploid zygospores. When returned to light and nutrients zygospores undergo meiosis to produce two MT+ and two MT− progeny that reenter the vegetative mitotic reproductive cycle (Figure S1).
While MT segregates as a single Mendelian trait, it is a genetically complex region encompassing around 200–400 kb of sequence that is rearranged between the two mating-type haplotypes. This rearranged region (R-domain) is flanked by telomere proximal (T) and centromere-proximal (C) domains that are collinear between the mating types, but where recombination is also suppressed [18].
Within the R-domain of MT are sex-limited genes (present in only one of the two mating haplotypes, MT+ or MT−) that are involved in sex determination and other aspects of the sexual cycle. However most of the genes in the R domain are shared genes with alleles present in both MT+ and MT− that are arranged in different relative order and/or orientation between the two haplotypes.
A previous study of the MT genes and their expression patterns was done before either haplotype was sequenced. Restriction fragment probe hybridization to Northern blots revealed both sex-regulated and constitutively-expressed genes within MT, but was limited to finding well-expressed genes with favorable hybridization characteristics [19]. More recent sequencing of the full genome of a MT+ strain, and of the MT− haplotype allowed a more comprehensive identification and prediction of Chlamydomonas MT genes [11], [20]. However, gene model validation and expression patterns for many of these genes have not been previously reported.
Although sex-linked polymorphisms are evident between MT+ and MT− alleles of genes in the R, C and T domains, the degree of haplotype differentiation in Chlamydomonas is unexpectedly low when compared to the male and female MT haplotypes of Volvox carteri that are physically much larger (>1 Mb), but derived from a region of Volvox linkage group I that is syntenic with Chlamydomonas MT and chromosome 6 [11]. Assuming no recombination occurred within MT for either species, it is expected that the MT+ and MT− alleles for genes in Chlamydomonas would be at least as diverged as those from Volvox female and male MT haplotypes [14], [21], but this is not the case: Volvox MT neutral divergence levels are about 100-fold higher than those for Chlamydomonas MT genes. This divergence paradox might be explained if rare recombination or genetic exchange occurred between MT+ and MT− genes of Chlamydomonas [14].
A second unexplained difference between MT of Chlamydomonas and MT of Volvox is the rates of recombination observed in their C and T domains that are the collinear regions immediately proximal to either side of the R domain of MT. In Chlamydomonas the C/T regions show suppressed recombination over several hundred kb, whereas in Volvox, crossovers were observed <30 kb from the R-domain [11]. Thus, proximity to a large rearranged region appears to be insufficient to explain suppressed recombination in nearby flanking collinear regions. The causes of different recombination behaviors between the Chlamydomonas and Volvox MT regions have not been previously investigated.
Here we examined gene content and expression of Chlamydomonas MT genes in greater detail than previously possible. Our investigations revealed new R-domain sequences caused by translocations into the MT+ locus bringing the total of such events to three and substantially increasing the size of the MT+ R-domain. We validated expression for 29 MT gene models and found sex-regulated expression patterns for a subset of uncharacterized MT genes. In addition we used population genetic data for Chlamydomonas MT genes to reassess their history of genetic exchange and potential for recombination. These experiments revealed a history of gene conversion in the R-domain as well as genetic exchange in the C and T domains. Finally, we examined the recombination potential of MT genes by performing crosses where each of the parents contained the same MT haplotype (MT+×MT+ or MT−×MT). These crosses revealed an underlying asymmetry between MT+ and MT− and suggest the presence of sequences in MT− that repress recombination in MT even when a collinear partner is available for meiotic pairing.
Results
Our results are divided into four sections. First, we describe new structural features of the C. reinhardtii mating locus revealed from sequencing both haplotypes. Second, we describe sexually controlled expression patterns of newly-described mating locus genes. Third, we use population genetics to identify rare genetic exchange events between MT haplotypes. Finally, we examine the potential for recombination in MT in crosses engineered so that both MT haplotypes are identical and collinear.
Revised description of structure and genetic content for the C. reinhardtii MT locus
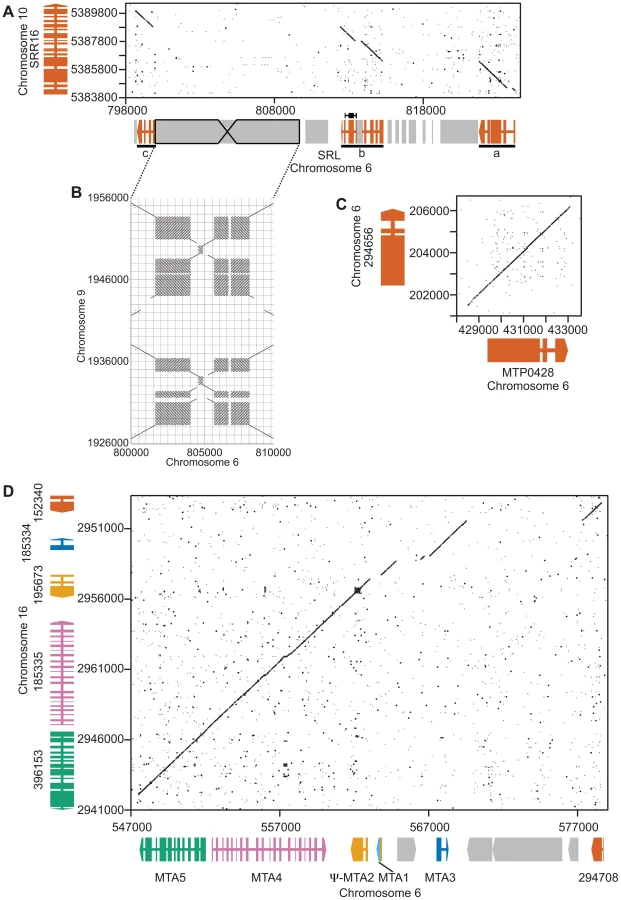
Structural data on the Chlamydomonas reinhardtii mating locus (hereafter referred to as Chlamydomonas MT) was previously based on a restriction-enzyme-mapped phage walk through both mating types [22]. In addition, the published V3 genome sequence contains portions of the plus haplotype (MT+) but its assembly was not contiguous through the mating locus [20]. An updated assembly of Chromosome 6 available through Phytozome [23] is contiguous through the MT+ region, though there are still some repeats whose copy number has not been accurately determined. We recently cloned and sequenced the minus haplotype (MT−) that allowed direct comparisons between nearly complete sequences of both mating types from Chlamydomonas (Figure 1 and [11]). Below we describe new and updated analyses of Chlamydomonas MT including two regions of the MT+ haplotype that derive from autosomal insertions, a redefined border for the R-domain, and a revised description of the 16 kb repeat region.

Autosomal insertions into MT
SRL region. Similarity searches done with MT sequences queried against autosomes revealed a domain of MT+ that we termed the SRL region whose discovery extends the R-domain by ∼30 kb (Figure 1). SRL arose through duplication-insertion of a ∼5.7 kb segment of SRR16 from Chromosome 10 into the MT+ locus (Figure 2A, Table S1). The full-length SRR16 gene encompasses ∼60 kb and encodes a predicted transmembrane scavenger receptor protein of 797 kDa with two scavenger receptor (SR) like domains followed by a glycosyl hydrolase (GH) domain and fourteen C-type lectin (CTL) domains [24], none of which are present in the translocated SRL region. Further analyses of the SRL region showed that additional rearrangements and secondary insertions took place after it moved into the mating locus (Figure 2A). These secondary insertions divided the SRR16-homologous region into three large blocks that are designated SRLa, SRLb and SRLc. The largest secondary insertion into SRL comes from Chromosome 9 and derives from an uncharacterized segmental repeat that has undergone at least two cycles of duplication-inversion (Figure 2B).
MTP0428 gene:
The telomere proximal border of the MT+ R-domain (previously described as region b in [19]), contains an uncharacterized gene, MTP0428, that has a full length duplicate copy and two partial copies on autosomal portions of Chromosome 6 (Figures 1, 2C, and Table S1). The predicted MTP0428 protein has no identifiable domains and no identifiable homologs outside of Chlamydomonas.
MTA region:
The mating-type a region (MTA) found in the MT+ haplotype was previously described and found to be derived from an autosomal translocation [19]. Here we identify its source as a ∼25 kb contiguous portion of Chromosome 16 that inserted between the MT+ genes 522875 and PKY1 (Figure 1). The MTA region contains three full-length genes from Chromosome 16—MTA2, MTA3 and MTA4—and two partially duplicated genes—294708 and MTA5—whose autosomal homologs straddle the translocation breakpoints (Figure 2D, Table S1). MTA2 was subsequently modified by insertion of sequences from Chromosome 7 into its first exon to generate a chimeric gene, MTA1, while the downstream exons of MTA2 became a pseudogene [19]. MTA4 acquired a premature stop codon mutation about half way through its coding region relative to its autosomal counterpart. All of the MTA/Chromosome 16 genes in the translocated region encode proteins that are lineage specific: MTA2/195673 is a putative hydroxyproline-rich glycoprotein (HRGP) with no homologs outside of Chlamydomonas, while the remaining encoded proteins have autosomal homologs in Volvox carteri but nowhere outside of Volvocine algae (data not shown).
Divergence of autosomally-derived MT genes
We expected that the MTP0428, MTA and SRL regions might behave as “strata” [25], [26] with neutral divergence correlated with the timing of each separate insertion/duplication event as has been proposed for mating-type chromosomes in other systems [8], [27]–[29]. Intron divergence was used as a metric for neutral rates of evolution, but we also examined intergenic regions in MTA and silent substitutions in coding regions for all three duplicated segments (Figure 3, Table S2). The neutral divergence patterns of the MTA region were highly variable. On one end of the MTA region is 294708 that has a relatively low intronic divergence value of 0.0186 (98% alignment identity), while on the other end is MTA5 with an intronic divergence value of 0.0844 (89% alignment identity). MTA4 shows a similar pattern as MTA5 while MTA2 and MTA3 are in between. dS values for coding regions followed a similar pattern as intronic divergence (Figure 3B) while intergenic divergence was less variable (∼0.5–0.8) (Table S2). The divergence data for MTP0428 and the SRL region are not sufficiently different from the MTA region that we can assign a relative time to their insertions into MT+ (Figure 3, Table S2).

The ratio of synonymous (dS) and non-synonymous (dN) substitution rates within coding sequences provide a measure of the strength of selection on one or both duplicate copies of a gene. Low dN/dS ratios imply strong purifying selection on both copies as is seen for MTA4/185335 with a value of 0.059 (Figure 3B, Table S2). Other duplicate genes such as MTP0428/294656 and SRLc/SRR16 have higher dN/dS ratios of 0.682 and 0.723 respectively. Our data cannot determine whether one or both genes in the duplicate pair are under positive selection or are evolving neutrally as the dN/dS ratios indicate. Codon adaptation indices (CAI) [30] can provide an indirect measure of differing selection on homologs [31], but we found no significant differences in CAI or codon mutational bias for MT+ versus autosomal paralogs in this study (Table S3 and data not shown).
16 kb repeat region
A ∼160 kb region of MT+ Chromosome 6 consists of around nine or ten copies of a ∼17 kb (17,217 bp) tandem repeat termed the “16 kb repeats” in [19]. At least three genes are found within the 16 kb repeats: EZY2 encodes a predicted chloroplast protein with no recognizable domains or similarity, and its mRNA is zygote specific [19] (Figure 4D). There are at least six copies of EZY2 in the 16 kb repeat region (Figure 1, Table S4) designated EZY2a-EZY2f and a single EZY2 pseudogene in the MT− locus (Figure 1). Based on its presence in MT+, its zygotic expression pattern, and predicted chloroplast localization EZY2 was proposed to be involved in uniparental chloroplast DNA inheritance [18], [19]. OTU2 encodes a putative otubain-related protease [18]. The three copies of OTU2 that could be distinguished based on polymorphisms are designated OTU2a-OTU2c. A single copy of OTU2a that resides in the MT− R-domain (Figure 1) was not previously described. INT1 encodes a putative retroviral-related integrase that is present in some of the 16 kb repeats but nowhere else in the Chlamydomonas genome (Figure 1 and Table S4). The open reading frame of the INT1 gene contains a frame-shift mutation that would prevent production of a full-length polypeptide in the absence translational frame shifting; however, we were unable to detect any mRNA corresponding to INT1 (Figure 1 and data not shown).
Shared genes in MT
The MT locus contains sex-limited genes (e.g. MID in MT− and FUS1 in MT+), as well as shared genes that have an allele in both mating types (Figure 1). A few shared genes in the rearranged domain of MT encode enzymes involved in primary metabolism such as PDK1 (pyruvate dehydrogenase kinase), GCSH (glycine decarboxylase subunit H), LEU1S (isopropylmalate dehydratase, small subunit) and DLA3 (dihydrolipoamide acetyltransferase). There are also a pair of convergently transcribed genes with overlapping 3′ untranslated regions, PR46a and PR46b, whose configuration and putative protein products are conserved in diverse eukaryotes, including humans, but whose function is not known ([18], [19] and data not shown). Additional shared R-domain genes in Chlamydomonas encode conserved proteins of unknown function (NMDA1, CGL70), possible signaling proteins including a kinase (PKY1), GTP binding protein (DRG1), and ubiquitin hydrolase (UBCH1), a MADS box transcription factor (MADS2), a putative cell wall protein (HRGP1), a splicing factor (SPL2), and nucleolar protein (UTP1). Many of the shared genes in Chlamydomonas MT have homologs in or near Volvox MT [11], but several do not, including DLA3, as well as four genes that encode putative proteins of unknown function, 155027, 522875, MT0796 and MT0828 (Figure 1 and Table S4).
Expression patterns of mating locus genes
We determined the expression patterns of selected MT genes from vegetative, gametic, and early zygotic RNA samples in order to identify those with possible roles in the sexual cycle. Results of our expression studies and summaries of previous such studies are presented in Figures 1, 4, S4, S5, and Table S5.
Sex-limited genes
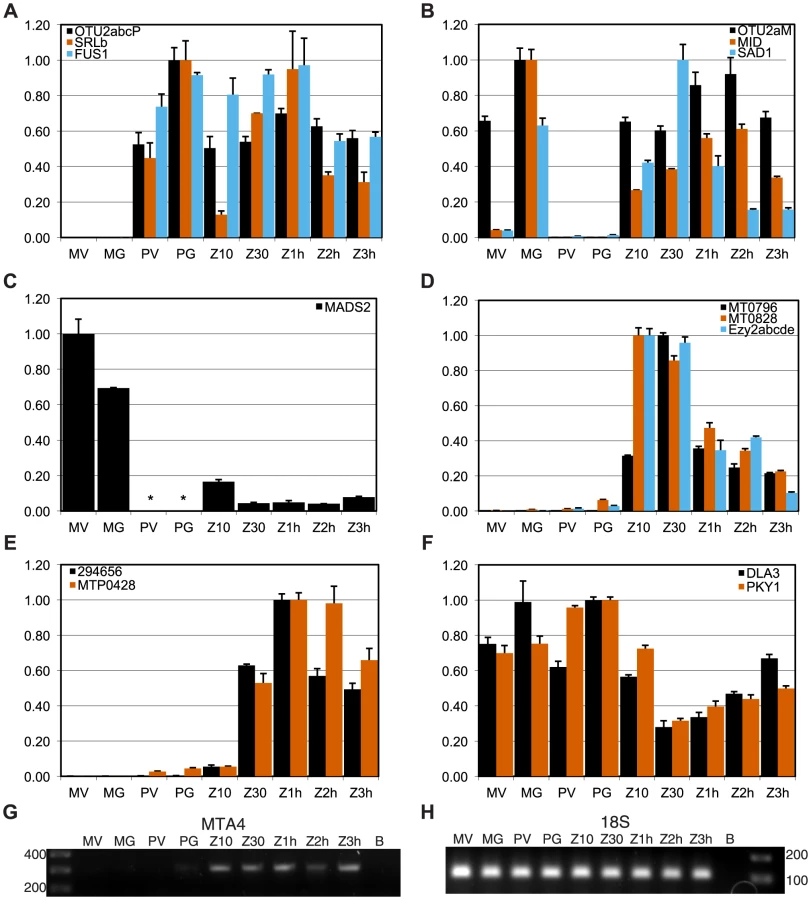
We used quantitative RT-PCR (qRT-PCR) to determine expression patterns during the sexual cycle of uncharacterized MT+ genes along with controls that included SAD1 and MID (minus gametic expression), FUS1 (plus gametic expression) and EZY2 (zygotic expression) [18], [19]. Expression of MTP0428 and its autosomal counterpart, 294656 were both detected in zygotic samples using primers specific for each copy (Figure 4E). MTA2 and MTA3 are probable pseudogenes [18], [19], but expression of MTA4 and MTA5 has not been tested. MTA4 transcript was detected using primers that could not amplify its autosomal paralog, and was expressed in MT+ gametes and zygotes (Figures 4G and S4). Primers specific to the MT+ copies or to the single MT− copy of OTU2 were used to discriminate expression from each mating-type. OTU2 from both haplotypes showed similar patterns of strong gametic and weaker zygotic expression (Figure 4AB, Table S5), but total expression from MT+ was stronger than from MT− probably due to the presence of multiple copies of OTU2 in MT+ versus a single copy in MT− (Figures 1 and S4A). Each of the three SRL genes has the potential to generate an mRNA with an in-frame coding sequence (Figure 2A). We were not able to detect expression of SRLa and SRLc, but we did detect an SRLb mRNA whose transcript showed modest up-regulation in gametic and zygotic stages of the life cycle (Figure 4A, Table S5). SRLb mRNA was also detected in pooled samples from the Chlamydomonas sexual cycle that were subjected to 454 transcriptome sequencing (Figure 2A and [32]).
Shared genes
Most of the shared genes in MT are expressed constitutively and are presumed to have functions that are not sex-related. PKY1, LPS1 and DLA3 fall into this category (Figures 4F, S5F, and Table S5).
However, several shared R-domain genes were found to have sex-regulated expression patterns. The putative MADS-box transcription-factor-encoding gene MADS2 was detected in MT− cells from vegetative and gametic samples as well as in zygotes, but not in MT+ cells (Figures 4C, S5F, and Table S5). PCR primers for detecting MADS2 cDNA match both MT+ and MT− alleles perfectly, so the expression difference between MT+ and MT− strains is due to mating-type-specific differences that could be cis or trans effects. Inspection of the aligned MADS2 sequences revealed a point mutation and two indels of 18 bp and 6 bp in its first intron, as well as several polymorphisms and an indel upstream of the start codon (Figure S2). Primers were designed to discriminate between the MT+ and MT− alleles of MADS2 and were used to determine that the major 18 bp indel in MADS2 was fixed between the two mating-types in 14 independent isolates (Figure S2). It seems likely that polymorphisms in MADS2 contribute to cis-regulatory differences that restrict its expression to MT−, but this idea remains to be directly tested.
Finally, two additional shared R-domain genes, MT0796 and MT0828, were found to have strong zygotic expression with little or no cDNA detected in vegetative and gametic samples (Figures 4D, S5D, and Table S5). Neither predicted protein has a recognizable domain or homology outside of Chlamydomonas.
In summary, we have uncovered several potential new examples of sexual cooption for mating locus genes in Chlamydomonas that acquired sex-regulated expression patterns.
Population genetics of the MT region
Genes in non-recombining sex-determining regions or sex chromosomes of haploid organisms are unsheltered and not expected to undergo loss or degeneration at the same rate that they do on Y and Z chromosomes [33]. Nonetheless, they are still subject to the effects of linkage disequilibrium that reduces the efficiency of natural selection in non-recombining regions (reviewed in [34], [35]) and are also expected undergo genetic differentiation between haplotypes [26], [33]. In a previous study we compared haplotype divergence in the MT locus of Chlamydomonas reinhardtii with its syntenic counterpart in Volvox carteri [11]. That study revealed a large discrepancy in divergence rates between shared genes in Chlamydomonas MT whose rates were low, versus those in Volvox MT whose rates were high, despite the two genomic regions sharing a common origin [14], [21]. The comparatively low rate of haplotype divergence in Chlamydomonas MT might be explained by rare genetic exchanges that cannot be detected in laboratory crosses but which act to reduce inter-haplotype diversity at the mating locus, an effect similar to that which has been observed in “ever-young” tree frog sex chromosomes [36].
Nucleotide diversity in MT and autosomal genes
In order to detect possible evidence of rare genetic exchange in Chlamydomonas MT we investigated patterns of nucleotide diversity in natural isolates. For this analysis we sequenced all or part of seven genes in thirteen wild isolates–seven MT+ and six MT− strains collected from diverse geographic regions (Table S6). We also made use of published data from an additional four genes [37]. Population data were compiled for eleven genes total, including one MT− limited gene (MID), one MT+ limited gene (MTA1), two randomly-selected shared genes in the R-domain (PR46, PDK1), three genes in the C or T domains (SAD1, SPP3, MAT3), and four autosomal genes that are unlinked to MT (GP1, IDA5, CBLP, YPT4). The mating locus genes that were used in this analysis are highlighted in Figure 1. We used these data to ask whether genes in Chlamydomonas MT show patterns of genetic diversity that are indicative of low recombination rates and selective sweeps, and to provide information about genetic exchange that might take place between the two MT haplotypes.
Nucleotide diversity (π) is a function of natural selection, mutation rates, recombination rates and population size/structure [38]. Diversity at synonymous coding sites and non-coding sequences (silent diversity or πsil) is considered neutral or nearly neutral and can be used to assess population structure. Theoretical and empirical data support the expectation of lower nucleotide diversity in non-recombining regions due to selective sweeps, background selection, decreased effective population size and Muller's ratchet effects [34], [39].
πsil (multiplied by 1000 in Table 1) varied about fifty-fold across the genes examined here with values ranging from ∼1 to ∼50, but was lowest for MT+ alleles of R-domain genes PR46 (1.13) and PDK1 (1.27) (Tables 1 and S7). πsil for PR46 and PDK1 in MT− samples (6.08 and 18.8 respectively) was significantly higher than for the MT+ samples, though still lower than πsil for autosomal genes. The low πsil values for PR46 and PDK1 suggest a recent selective sweep of the MT+ haplotype. πsil values for the sex-limited genes MID (11.1) and MTA1 (4.22) were also relatively low and attributable to possible selective sweeps and/or lower effective copy number compared with shared MT genes and autosomal genes. Despite being in a nominally non-recombining region SAD1 and SPP3 had πsil values of ∼30 to 50 that are not distinguishable from those of autosomal genes (GP1, IDA5, CBLP, YPT4). Moreover, the SAD1 and SPP3 πsil values did not show differences between MT+ and MT− when grouped by mating-type as we saw for PR46 and PDK1 (Tables 1 and S7). These data indicate that SPP3 and SAD1 are relatively uncoupled from the effects of presumed selective sweeps in the MT locus. The three MT− isolates of the C domain gene MAT3 had relatively low diversity (5.80) compared with the MT+ MAT3 isolates (24.6) and compared with the other two C/T domain genes SPP3 and SAD1. The low diversity of MAT3 from MT− isolates could be due to a selective sweep, but the nearby gene SAD1 is closer to the R-domain than MAT3 and has a πsil value of ∼50 indicating that the low πsil value for MT− isolates of MAT3 is not associated with the MT region as a whole. Because πsil for MAT3 was based on only three isolates [37], we recalculated πsil for the same three MT− isolates of SAD1 in order to control for sampling bias. However, sub-sampling of SAD1 from the isolates as used for MAT3 increased rather than decreased its πsil value (67.3, standard deviation 9.0) allowing us to rule out sample bias as the cause of low nucleotide diversity in MT− isolates of MAT3. Additional data will be required to resolve whether the low diversity we see for MAT3 from MT− isolates is due to other causes such as a highly localized selective sweep in this gene.

We calculated two indices of gene flow and population structure, dA and FST, to determine the extent to which genetic exchange between MT+ and MT− isolates is constrained [38], [40], [41]. While sequence diversity in autosomal genes (IDA5, CBLP and YPT4, GP1) was independent of mating-type as indicated by dA and FST values near zero (Tables 1 and S7), the R-domain genes PR46 and PDK1 showed strong MT-associated differentiation as evidenced by FST values that are between 0.5 and 1.0 (Table 1) and by dA values that differ significantly from the null value of 0 (Table S7). These findings indicate that genetic exchange between shared R-domain genes is limited compared with autosomal genes that assort freely between MT+ and MT− haplotypes (Tables 1 and S7). Consistent with our findings on nucleotide diversity the C/T domain genes SAD1 and SPP3 showed no evidence of mating-type-linked differentiation, while the C domain gene MAT3 gene showed an intermediate level of mating-type-linked differentiation (FST = 0.45) (Tables 1 and S7).
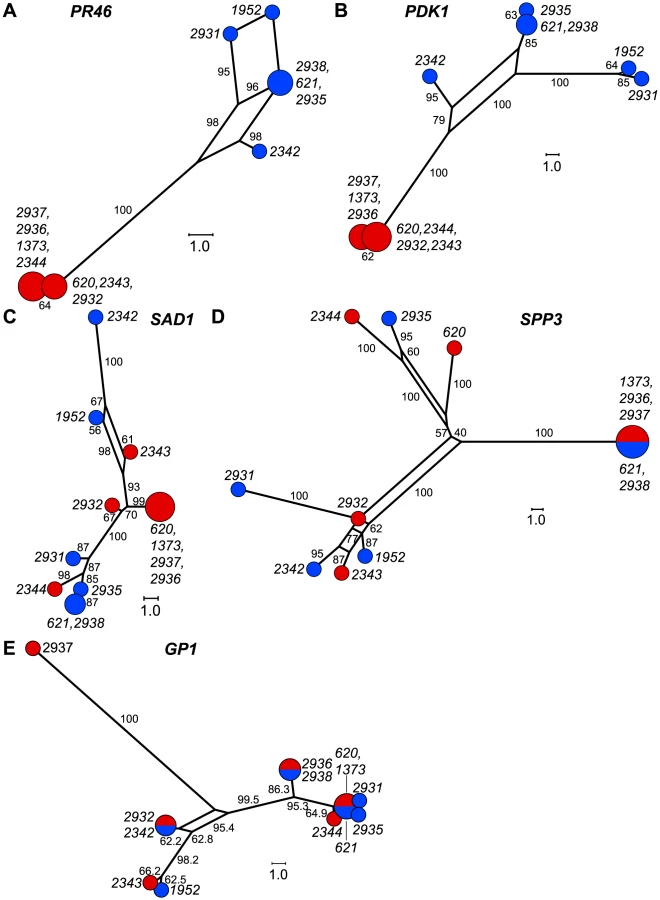
We graphically depicted the genetic relationships between MT+ and MT− allelic diversity by constructing unrooted parsimony-based networks that are similar to phylogenetic trees, but accommodate incongruities by incorporating alternative paths or splits [42]. As suggested above, the networks for the R-domain genes PR46 and PDK1 show clear differentiation between MT+ and MT− isolates (Figure 5A,B) with tight clustering of the MT+ alleles. In contrast, genes in the T- and C-domains (SAD1 and SPP3) show complete intermixing between MT+ and MT− isolates (Figure 5C,D), with no apparent association of specific polymorphisms with mating-type. The MAT3 gene did show some MT-associated differentiation, but this differentiation did not extend to the nearby SAD1 gene (Figure S3) or to the SPP3 gene indicating that these three loci are all separable from each other and from the R-domain by recombination. As expected none of the polymorphisms present in autosomal genes (GP1, YPT4, IDA5, CBLP) showed association with mating haplotype (Figures 5E and S3).
Gene conversion in the R-domain
The preceding data revealed far more genetic exchange in the C and T domains of MT than would be expected based on laboratory tests of recombination. However, these data do not explain why R-domain genes such as PR46 and PDK1 show orders of magnitude lower amounts of sequence differentiation compared with R-domain genes in Volvox MT [11]. Crossovers in the R-domain are likely to be lethal or highly deleterious due to rearrangements and deletions, but a second means of genetic exchange is gene conversion, where tracts of sequence from one allele can be unidirectionally transferred to a homologous partner in the diploid phase of the sexual cycle–most likely during meiosis. Such exchanges are expected to be infrequent, but could still help maintain sequence homogeneity between allelic gene pairs in the R-domain.
Gene conversion can be identified by comparing polymorphisms that are nearly fixed between the two mating-types and then identifying tracts where two or more adjacent polymorphisms have switched their pattern from one haplotype to the other [43]. Here we identified four short regions of gene conversion in the two R-domain genes that were randomly selected for this study–two tracts in PDK1 and two tracts in PR46 (Figures 6 and S6). None of the tracts were in repeat regions or microsatellites (Figure S6), and in all four cases the direction of conversion was from MT+ to MT−. One of the gene conversion tracts in PDK1 is present in both CC1952 and CC2931 (Figure 6) making its occurrence likely to predate the split between these two isolates. These previously undocumented gene conversion events may have important implications for mating locus evolution that are further elaborated below.

Relationship between sequence rearrangements and suppressed recombination in MT
Blocked recombination in sex determining regions is believed to be maintained so that genes in these regions with sex-specific functions can remain tightly linked [4], [44]. Sequence rearrangements in heteromorphic sex chromosomes and in heteromorphic mating loci such as Chlamydomonas MT could accumulate passively as the result of blocked recombination, or they could be the primary cause of blocked recombination [26], [45]. In the latter case normal recombination should be restored in matings with isomorphic MT haplotypes while in the former case restoring collinearity at MT would not relieve suppression of recombination.
Mating between parents with the same MT haplotype in Chlamydomonas provide a means to test whether MT sequences are capable of normal recombination when their meiotic partner is collinear and homologous. Prior work established the basis of mating-type specification in Chlamydomonas and allowed the engineering of strains in which each parent contributes the same MT haplotype in a cross [19], [46]. MT+ strains carrying a Mid transgene (MT+::Mid-T) were used as pseudo-minus parents in MT+::Mid-T×MT+ crosses. MT− mid-1 Fus-T strains were used as pseudo-plus parents in MT− mid-1 Fus-T×MT− crosses (see Materials and Methods). The auxotrophic markers nic7 and thi10 (nicotinamide and thiamine requiring, respectively) flank the mating locus [47] and were used to identify potential crossovers within MT (Figure 1, Table S8). Recombination data for MT+×MT+ and MT−×MT− crosses are summarized in Figure 1 and Tables S8 and S9.
To confirm the absence of recombination across MT in control strains, we crossed the MT+ and MT− strains CC-123 thi10 NIC7 MT+ and CC-2663 THI10 nic7 MT−. Out of 1040 random progeny, none were Nic− Thi−, while ten were Nic+ Thi+ and mated as minus strains. Of those ten, nine were diploid or aneuploid based on the presence of both the nic7 and NIC7 alleles. This leaves at most one true recombinant (0.1% frequency), a value that is consistent with previous data [48].
MT+× MT+ crosses
We performed an MT+×MT+ cross (nic7 THI10 MT+::Mid-T×NIC7 thi10 MT+), and scored 352 random progeny for nicotinamide and thiamine auxotrophy that would be indicative of recombination in or around MT. Thirteen Nic+ Thi+ and three Nic− Thi− putative recombinants were examined further. The NIC7 locus was amplified and scored from the thirteen Nic+ Thi+ strains, two of which were found to contain both parental alleles meaning that they were either diploids or aneuploids. Excluding these two progeny we found 14 recombinants (11 Nic+ Thi+, 3 Nic−Thi−) out of 350 corresponding to a recombination frequency of ∼4% across MT+ and a genetic distance close to the genome-wide average of ∼100 kb per cM [49].
Because the two MT+ strains used above were isogenic, the sites of crossovers could not be determined. Therefore, a second cross was performed using an inter-fertile MT+ wild isolate, CC-2344, as the plus parent and a recombinant nic7 thi10 MT+ Mid-T progeny from the first MT+×MT+ cross as the minus parent. A total of 17 out of 377 random progeny were recombinant: 7 were Nic+ Thi−, and 10 were Nic− Thi+ giving a recombination rate of ∼4.5% that was similar to what we observed in the first cross. The recombinant progeny were further analyzed by scoring several additional polymorphic markers in MT (Figure 1, Table S8). These markers defined a minimum of four different breakpoint intervals, three of which lie entirely within the R-domain of the MT+ haplotype (Figure 1, Table S8). One additional MT-linked marker, MAT3, and three autosomal markers—YPT4, GP1 and MMP1—were scored to confirm normal meiotic segregation in this cross (Table S8). In summary, these data establish that meiotic recombination is possible for the MT+ haplotype and that it is normally suppressed in MT+×MT− crosses.
MT−× MT− crosses
A similar experiment as above was done using MT− strains nic7 MT− and NIC7 MT− mid1 Fus-T as parents. The thi10 marker was not available in this cross, so we instead used the mid1 pseudo-plus mating phenotype as a second MT-linked marker to score recombination (Figure 1). Recombinants in this cross would be Nic+ progeny that mate as minus, or Nic− progeny that mate as plus. 600 progeny from a total of 206 zygotes were scored for mating phenotype and for nicotinamide auxotrophy. 599 of the progeny had the parental markers. A single putative recombinant progeny that was Nic+ and mated as a minus strain (NIC7 MT−) was found to contain both parental NIC7 alleles and is presumed to be a diploid. Therefore, no meiotic recombinants were found between MID and NIC7 in crosses with homologous MT− mating haplotypes (Table S9). The ∼240 kb region of MT− covered by these two markers includes ∼80 kb of collinear sequence flanking MT− (T domain) and ∼160 kb of R-domain sequence. The absence of recombination in this cross is incompatible with an average physical/genetic distance ratio of 100 kb/cM (Chi squared = 14.75, p value = 0.000122). Moreover, this segment of MT− was repressed for recombination at least as much as two previously described autosomal markers that show the largest known physical/genetic distance ratio in Chlamydomonas of 511 kb/cM [49] (Chi squared = 2.81, p value = 0.093).
The absence of recombination between collinear MT− partners could be caused by sequences in MT− that repress recombination in cis, but could also have been caused by the absence of MT+ genes that promote recombination in trans (though no candidates for such genes are known). Both cis and trans effects on recombination have been reported previously in the non-recombining mating type chromosome of Neurospora tetrasperma [50]. To distinguish cis versus trans effects on recombination in MT−×MT− crosses we repeated the above cross with the minus parent CC1952 that has well-characterized molecular markers for mapping [49], [51] and the pseudo-plus strain NIC7 MT− mid1 Fus-T. We first scored a chromosome VI marker, 4121, that was reported to be 27 cM from MT in conventional crosses [49]. 26/96 progeny from the MT−×MT− cross were recombinant for 4121 and MT resulting in a genetic distance of 27 cM. This result is consistent with normal recombination on Chromosome VI outside the mating locus (Table S9). A pair of autosomal markers on Chromosome III, GAR1 and GSAT, also had a normal recombination distance of ∼20 cM (Table S9). However, the MT markers MAT3 and PDK1 had no recombinants (0/146)(Figure 1 and Table S9).
Taken together our data show that the MT− locus is a region of suppressed recombination that inhibits meiotic crossovers even when homologous collinear sequences are available for pairing. In contrast, the MT+ locus shows normal meiotic recombination when it has a collinear pairing partner. This asymmetry between MT+ and MT− may have consequences for other aspects of MT sequence evolution and differentiation that are elaborated in the Discussion.
Discussion
MT and its genetic content redefined
Key findings for our analysis of MT structure were identification of two new autosomal insertions in the MT+ haplotype, MTP0428 and the SRL region, that redefine the borders of MT with ∼30 additional kb of R-domain sequence in the MT+ haplotype. Altogether, the MT+ R-domain is approximately twice the size of the MT− R-domain due to three major autosomal translocations and the 16 kb repeat region (Figure 1). This degree of size asymmetry in a mating locus of a unicellular organism is atypical and has been reported to our knowledge in only one other instance for the smut fungus Microbotryum [9]. On the other hand, X and Y chromosomes of different sizes in haploid bryophytes are well-documented [12], but very little is known about how such size differences evolve in haploid systems. One prediction of Bull's theory of haploid dioecy is that non-recombining haploid X-Y chromosomes would expand by sequence additions rather than deletions and degeneration [33]. Our findings here support the role of sequence insertions causing MT+ expansion, as does previous work on Volvox MT whose increased size relative to Chlamydomonas MT is largely due to accumulation of repeats and transposons with little evidence of gene loss [11]. However, Bull's theory predicts similar overall fates for haploid sex determining chromosomes and does not explain the emergence of size asymmetry that is evident, for example, in around half of the surveyed X-Y chromosome pairs from bryophytes [12]. The size and structural asymmetry of Chlamydomonas MT haplotypes could represent a model for how such size asymmetry evolves. In the last section we speculate on the basis for emergent asymmetry in the Chlamydomonas mating locus.
New mating locus genes with potential functions in the sexual cycle
The SRL region of MT+ is of special interest as it was created from a partial fragment of an autosomal gene, SRR16, which then underwent further fragmentation into three sub-regions. SRLb represents an intriguing example where gene fragmentation, a process typically associated with decay, may lead to the creation of new genes in an environment such as MT where recombination is greatly reduced and where neutral or even slightly deleterious mutations have a greater chance of achieving fixation in the population compared with autosomal regions [35].
In Chlamydomonas MT controls sexual differentiation, fertilization competence and uniparental organellar DNA inheritance [18]. Genes whose presence or expression is limited to only one mating-type are candidates for governing these aspects of the sexual cycle, and in this study we identified several candidates.
Interestingly, within each of the translocations and the 16 kb repeat region of MT+ are candidates. For example MTP0428, MTA4 and EZY2 are zygotically expressed, while SRLb, MTA1 and OTU2 are up-regulated in gametes and zygotes (Figures 1, 4ABDE, S4A, S5A, and Table S5).
We found that two Chlamydomonas-specific genes encoding proteins of unknown function, MT0796 and MT0828 are both expressed zygotically (Figure 4, Table S5) in a pattern similar to the early zygotic genes EZY1 and EZY2 that are speculated to have a role in uniparental chloroplast DNA inheritance [19], [52]. MT0796 and/or MT0828 may also be involved in this process or in other early zygote functions that include zygote wall formation, flagellar resorption, karyogamy and chloroplast fusion [18].
Expression of the putative MADS-box transcription factor-encoding gene MADS2 was restricted to MT− cells and zygotes, and not detectable in MT+ cells (Figures 4C, S5F, and Table S5). The function of MADS2 in MT− cells is unknown, but the potential connection to green algal sexual cycles is intriguing given the major role for MADS box proteins in plant reproductive development [53]. A second shared gene of interest is OTU2 that encodes a putative otubain-related deubiquitylating protease [18], [54]. The OTU2 mRNA in MT+ gametes is expressed at levels several fold higher than that in MT− gametes (Figure S4A), possibly as a result its higher copy number in MT+ cells. This biased expression pattern is consistent with a role for OTU2 in mating-type differentiation or the sexual cycle.
Among the sex-regulated shared genes in MT, only two have Volvox homologs–MADS2 and HRGP1–and these Volvox homologs are either in or adjacent to the mating locus [11]. MADS2 in Volvox shows female-biased expression, which is opposite to the pattern in Chlamydomonas (where MT+ is homologous to Volvox female MT and MT− is homologous to Volvox male MT). It is possible that MADS2 controls a sex-related process such as uniparental mitochondrial DNA inheritance where the inheritance pattern has switched from the MT− parent in Chlamydomonas [18] to the female parent in Volvox [55]. HRGP1 encodes a putative cell wall protein that is up-regulated in gametes of both mating types of Chlamydomonas [11], [19], but which shows male-biased, gametic expression in Volvox [11]. This change from equal expression in both gametes to male-biased expression suggests that HRGP1 participates in Volvox gametogenesis but may be required in higher amounts for spermatogenesis than oogenesis.
Gene conversion and genetic exchange in Chlamydomonas MT
The expectation for genes in non-recombining regions such as MT is allelic differentiation into two haplotypes [26]. Our population data confirm this expectation for shared genes in the R-domain that show overall clustering by mating type (Tables 1 and S7, Figure 5). However, we uncovered evidence of gene conversion between MT+ and MT− alleles of R-domain genes indicating that there is genetic exchange in the rearranged portion of MT that can act as a homogenizing force to counteract the effects of reduced recombination (Figures 5 and 7). We also found evidence for genetic exchange between C and T domain genes that almost never show recombination in laboratory crosses. The observed genetic exchanges in the C/T domains could be from crossovers or from gene conversion. In either case the amount of exchange in the C/T domains is significantly higher than in the R domain and is enough to partially or completely remove linkage between C- or T-domain polymorphisms and mating type (Figure 5, Tables 1 and S7). An important consequence of exchange between MT+ and MT− polymorphisms in the C/T domains is that genes such as SAD1 whose expression and function is limited to one mating type (MT− in the case of SAD1) remain under selection in both mating types, and this explains why the MT+ locus retains a functional copy of SAD1 [56], [57]. Moreover, the data presented here for the first time distinguish the recombination behavior of C and T domain genes that are largely uncoupled from mating type with those in the R-domain that show mating-type associated differentiation (Figure 5, Tables 1 and S7).

The data we obtained on gene conversion in Chlamydomonas MT parallels that found recently for the fungi Cryptococcus neoformans that has a relatively large heteromorphic mating locus [58], and for the non-recombining mat locus of Neurospora tetrasperma [59]. Moreover, infrequent gene conversion between heteromorphic or rearranged regions may be a more general property of sex chromosomes as it has been seen in animal sex chromosomes [60], [61] where has been proposed to act as a means of genetic homogenization [36].
Resolution of the mating locus age paradox in Volvocine algae
Our data documenting genetic exchange in Chlamydomonas MT help resolve a paradox regarding the degree of differentiation between mating haplotypes in the two Volvocine algal species Chlamydomonas reinhardtii and Volvox carteri [14], [21]. We propose that gene conversion in Chlamydomonas MT acts to promote sequence homogeneity between shared genes and thus maintains a “youthful” appearance for such genes despite their time of residence in the MT locus. In contrast, no such mechanism appears to have operated during the recent history of the V. carteri lineage where differentiation of MT genes is orders of magnitude higher and extends back through speciation events [11].
Why do the Chlamydomonas and Volvox MT regions differ in their behavior with respect to genetic exchange? Although their structural organizations are similar, Volvox MT is about five times larger than Chlamydomonas MT, has a much higher repeat content, and retains very little residual synteny or gene order between rearranged genes compared with Chlamydomonas [11]. We speculate that a combination of reduced effective population size and of selection on mating locus genes for oogamous traits in Volvox promoted MT expansion past a critical size/structural threshold where residual exchange between shared genes by gene conversion could no longer occur as it does in Chlamydomonas. Once past such a threshold the differentiation rates between mating haplotypes would be expected to accelerate and further reduce the potential for gene conversion or recombination. Determining the structure of MT in other Volvocine algae with different colony organization and reproductive morphologies may shed light on the parameters that caused MT to evolve so differently between Chlamydomonas and Volvox, and help determine when the recombination dynamics of MT began to diverge in the lineage.
Mechanisms of recombination suppression and emergent asymmetry in MT
While rearrangements in the MT locus may contribute to suppressed recombination, we found evidence here for at least one other mechanism that suppresses recombination in the MT− haplotype even when it has a collinear partner. We propose that one or more sequences within MT− are responsible for suppressing recombination and may have originally evolved to maintain linkage between the MT− sex determining genes MID and MTD [18], [62], similar to what has been proposed to occur during the early evolution of diploid sex chromosomes [4], [45]. Subsequent rearrangements that generated the R-domain could have arisen passively as a result of blocked recombination, or arisen under selection to strengthen linkage between genes in each MT haplotype.
We speculate that MT− mediated recombination suppression (as opposed to rearrangements) is responsible for the extremely low observed recombination rates in the collinear C/T domains of Chlamydomonas MT that flank the R domain. In contrast to the Chlamydomonas C/T domains, recombination in sequences immediately adjacent to Volvox MT is not suppressed [11]. We predict that this difference in recombination behavior for collinear sequences flanking MT in the two species is that Volvox MT lacks sequences that intrinsically repress recombination. If so, recombination would occur normally in Volvox MT for either MTF×MTF or MTM×MTM crosses if such matings could be arranged. Testing this idea will be a goal for future studies.
While Y or W chromosome degeneration is the prevalent mechanism behind heteromorphic sex chromosomes in diploid systems [3], no comparable mechanism explains how heteromorphic sex chromosomes might evolve in a haploid system such as primitive plants [33]. The unique sequence properties of the MT− haplotype that suppress homologous recombination may have generated other asymmetries found in the MT locus. It is striking that of the three independent autosomal insertion events in MT and the 16 kb repeat expansion, all occurred in MT+ that we have demonstrated retains competence for initiation of meiotic recombination. Additionally, all the gene conversion events that we have documented are asymmetric with respect to direction of sequence transfer from MT+ to MT−. While these observations showing asymmetrical behavior of MT+ and MT− haplotypes are limited, they fit a pattern that might be explained in terms of differential access of their sequences to meiotic recombination and DNA repair machinery that could bias the location of non-homologous insertions and gene conversion events.
Interestingly, there are hints of similar types of asymmetry as we have documented for Chlamydomonas MT in mating type chromosomes from other species. In the fungus Microbotryum there is size asymmetry between the two mating type chromosomes that are estimated to be ∼3.3 and ∼4.0 Mb respectively, though detailed sequence information about the two haplotypes is still lacking [63]. Mating locus chromosomes in the heterokaryotic self-fertile fungus Neurospora tetrasperma are blocked for recombination and have rearrangements between the mat a and mat A haplotypes that help ensure linkage between the mat locus and the centromere so that meiotic progeny remain heterokaryotic [7], [50]. Differences in the amount of repeat accumulation in the mat a and mat A chromosomes and in codon usage for genes from the two haplotypes have been reported [7], [31], but the reasons for this intriguing asymmetry are unclear.
Our data indicate that asymmetry in both size and recombination behavior can arise in the evolution of haploid mating systems and perhaps influence the preferential expansion of one mating haplotype over the other. Whether the mechanisms that cause mating locus size asymmetry in Chlamydomonas contribute to the formation of heteromorphic chromosomes in haploid systems such as primitive plants or fungi remains to be determined.
Materials and Methods
Chlamydomonas strains
Strains used for the population studies are listed in Table S6 and were obtained from the Chlamydomonas Stock Center (http://chlamycollection.org/strains/). Strains used to test recombination in the mating locus are as follows: CC-123, thi10 MT+; CC-2663, nic7 MT−. Note that the ac29 mutation present in the original CC-2663 strain reverted [64]; B32, mid-1 MT− with a FUS1 transgene [46]. B32 mates as a plus strain; PF1, nic7 MT+ with a MID transgene. PF1 was created with a MID transgene (3.5 kb ApaI fragment from plasmid pmid7.1 [46]) that was cotransformed into CC-1865 (arg2 fus1-1 MT+) along with pArg7.8 that contains a wild-type argininosuccinate lyase gene [65]. A MID-expressing Arg+ transformant was crossed to CC-85 (nic7 MT+) to create PF1; K33, nic7 MT+ thi10 with a MID transgene. K33 was a progeny from a cross of CC-123 to PF1 that mates as minus, deposited with the Chlamydomonas Stock Center as CC-3947.
Mating and genetic analysis
Chlamydomonas strains were grown on TAP plates supplemented as appropriate with nicotinamide (nic, 4 µg/ml), thiamine (thi, 5 µg/ml), and/or acetylpyridine (AcPy, 15 µl/l) to enhance scoring of the nic- phenotype. Crosses were done by standard procedures [66] and random progeny were scored for auxotrophies by growth on appropriate media, or for polymorphisms using PCR amplification (Table S10). Progeny exhibiting recombinant phenotypes were subcloned and retested to confirm their genotypes.
Mating locus sequences and annotation
Sequences and annotation of the plus and minus mating locus haplotypes are described in [11] and available in Genbank under accession numbers GU814014 and GU814015. Gene models were further refined using predictions available from Phytozome [23] and EST support, and were confirmed where possible using data derived from 454 transcriptome data available at http://genomes.mcdb.ucla.edu/Cre454/project.html and deposited in the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession SRA020135.
Analysis of autosomal duplications
Plus and minus mating locus sequences were aligned to the V4 genome assembly from Phytozome [23] using BLAST in order to identify duplicated regions. Dot plots were generated using the dotmatcher program in the EMBOSS package [67] with default parameters. Putative coding regions were aligned using MUSCLE [68] and then manually verified and adjusted to correct placement of splice junctions. MEGA5 [69] was used to calculate divergence values for the alignments in Table S2 and Figure 3 using the Tamura 3-parameter model to estimate distances. dN and dS values were calculated using yn00 in the PAML package [70], [71]. CAI values were calculated using the CAICal webserver as described in [72].
RNA preparation
C. reinhardtii cultures of CC620 (MT+) and CC621 (MT−) were grown to confluence on TAP plates [66] for one week under continuous light. Cells were washed off of the plates with nitrogen-free (N-free) HSM and placed immediately into either +N (for vegetative samples) or −N (for gametes and zygotes) HSM media [66] at ∼1.0×107 cells/mL at 24°C for 3 hours in large unshaken Erlenmeyer flasks filled to ∼1/4 volume. After resuspension and incubation as described above, vegetative and gametic samples were collected from each culture. To generate zygotes, equal volumes of plus and minus gametes were briefly mixed in an Erlenmeyer flask and samples collected after 10′, 30′ 60′ and 120′. Mating progression was monitored from fixed samples at each time point and had reached ∼90% by 10′ (data not shown). For each sample, 100 mL of cells were collected in 2×50 mL polypropylene conical tubes and Tween-20 was added to a final concentration of 0.005%. The samples were centrifuged at 4,000× g for 3 minutes, the supernatant decanted, and the pellet snap frozen in liquid nitrogen. RNA was extracted with Trizol (Invitrogen, Carlsbad CA) according to the manufacturer's protocol. RNA was further purified using RNAEasy columns (Qiagen) according to the manufacturer's protocol.
cDNA synthesis
DNAseI (Roche) treated total RNA was reverse transcribed using Superscript III (Invitrogen, Carlsbad CA), with the following modifications to the manufacturer's protocol. A 9∶1 mixture of anchored dT20 (TTTTTTTTTTTTTTTTTTTTV) and random hexamer oligos were used to prime first strand cDNA synthesis. cDNA synthesis reactions were incubated as follows: 25C 10′, 42C 10′, 50C 15′, 55C 15′, 60C 15′, 65C 15′, 85C° 5′. RNAse H was subsequently added and reactions incubated 30′ at 37C. cDNAs were diluted 1∶10 in TE (10 mM Tris pH 8.0/1 mM EDTA) and stored at −20C prior to use.
Quantitative RT-PCR (qPCR)
Table S10 lists all primers used. cDNAs were diluted 1∶10 in sterile filtered ddH2O and 10 µL was used for each of the 20 µL qPCR reactions. The reactions were performed in triplicate on each of two biological replicates. Reaction conditions were as described previously [73] and reactions were amplified using a Bio-Rad iCycler iQ Real Time Thermal Cycler w/Optical Module (BioRad, Hercules CA) using the following cycling conditions: 95C 10″, 60C 10″, 72C 30″ for 40 cycles. Melt curves and gel electrophoresis were used to confirm the presence of a single amplification product of the correct size in each reaction. For all primer sets a standard dilution curve was prepared using cDNAs pooled from all samples. Relative cDNA levels were calculated using the best-fit curve from the standard dilution of each primer set and then normalized against the 18S cDNA signal.
Genomic DNA isolation and PCR amplification from C. reinhardtii isolates
Genomic DNA was isolated by CsCl banding [74]. Table S10 lists all primers used for amplification of target genes. PCR products from two independent reactions per sample were sequenced to confirm that no errors were introduced into the sequence during amplification.
Population genetic data and phylogenetic networks
Sequence alignments were done using ClustalX [75] and manually adjusted. DnaSP [76] was used to calculate values in Tables 1 and S7. πsil data were calculated from alignment files with gaps and non-synonymous sites removed. dXY, dA and FST were calculated from full alignments with gaps removed. Three gene conversion tracts were identified by DnaSP using the algorithm of Betran [43]. The fourth tract was present in 2 out of 6 MT− isolates and was identified manually. The manually identified tract meets Betran's criteria for gene conversion since four consecutive occurrences of a polymorphism are present in 1/3 of the MT− isolates with a p-value of .012 (0.334 = 0.012) [43]. Sequences used were derived from this study and from a previous study [37] with strains and accession numbers in Table S6. Phylogenetic networks were constructed using the program SplitsTree [42]. The ParsimonySplits approach was used to calculate the network from ungapped alignments with 1000 bootstrap replicates, and the networks were rendered using the Equal Angle and Convex Hull methods. Network topology was unchanged when calculated using distance-based approaches such as the Neighbor-net method (data not shown).
Supporting Information
Zdroje
1. CharlesworthD (2013) Plant sex chromosome evolution. J Exp Bot doi: 10.1093/jxb/ers322
2. FraserJA, HeitmanJ (2005) Chromosomal sex-determining regions in animals, plants and fungi. Curr Opin Genet Dev 15: 645–651 doi:10.1016/j.gde.2005.09.002
3. BachtrogD, KirkpatrickM, MankJE, McDanielSF, PiresJC, et al. (2011) Are all sex chromosomes created equal? Trends in Genetics 27: 350–357 doi:10.1016/j.tig.2011.05.005
4. CharlesworthD, CharlesworthB, MaraisG (2005) Steps in the evolution of heteromorphic sex chromosomes. Heredity 95: 118–128 doi:10.1038/sj.hdy.6800697
5. BellottDW, SkaletskyH, PyntikovaT, MardisER, GravesT, et al. (2010) Convergent evolution of chicken Z and human X chromosomes by expansion and gene acquisition. Nature 466: 612–616 doi:10.1038/nature09172
6. LeeSC, NiM, LiW, ShertzC, HeitmanJ (2010) The evolution of sex: a perspective from the fungal kingdom. Microbiol Mol Biol Rev 74: 298–340 doi:10.1128/MMBR.00005-10
7. EllisonCE, StajichJE, JacobsonDJ, NatvigDO, LapidusA, et al. (2011) Massive changes in genome architecture accompany the transition to self-fertility in the filamentous fungus Neurospora tetrasperma. Genetics 189: 55–69 doi:10.1534/genetics.111.130690
8. FraserJA, DiezmannS, SubaranRL, AllenA, LengelerKB, et al. (2004) Convergent Evolution of Chromosomal Sex-Determining Regions in the Animal and Fungal Kingdoms. PLoS Biol 2: e384 doi:10.1371/journal.pbio.0020384
9. HoodME (2002) Dimorphic mating-type chromosomes in the fungus Microbotryum violaceum. Genetics 160: 457–461.
10. LeeN, BakkerenG, WongK, SherwoodJE, KronstadJW (1999) The mating-type and pathogenicity locus of the fungus Ustilago hordei spans a 500-kb region. Proc Natl Acad Sci USA 96: 15026–15031.
11. FerrisP, OlsonBJSC, De HoffPL, DouglassS, CaseroD, et al. (2010) Evolution of an expanded sex-determining locus in Volvox. Science 328: 351–354 doi:10.1126/science.1186222
12. AllenCE (1945) The genetics of bryophytes. II. The Botanical Review 11: 260–287.
13. YamatoKT, IshizakiK, FujisawaM, OkadaS, NakayamaS, et al. (2007) Gene organization of the liverwort Y chromosome reveals distinct sex chromosome evolution in a haploid system. Proc Natl Acad Sci USA 104: 6472–6477 doi:10.1073/pnas.0609054104
14. UmenJG (2011) Evolution of sex and mating loci: An expanded view from Volvocine algae. Curr Opin Microbiol 14: 634–641 doi:10.1016/j.mib.2011.10.005
15. NozakiH, MisawaK, KajitaT, KatoM, NoharaS, et al. (2000) Origin and evolution of the colonial volvocales (Chlorophyceae) as inferred from multiple, chloroplast gene sequences. Mol Phylogenet Evol 17: 256–268 doi:10.1006/mpev.2000.0831
16. ColemanA (2012) A Comparative Analysis of the Volvocaceae (Chlorophyta). J Phycol 48: 491–513.
17. NozakiH (1996) Morphology and evolution of sexual reproduction in the Volvocaceae (Chlorophyta). J Plant Res 109: 353–361.
18. GoodenoughU, LinH, LeeJ-H (2007) Sex determination in Chlamydomonas. Seminars in Cell & Developmental Biology 18: 350–361 doi:10.1016/j.semcdb.2007.02.006
19. FerrisPJ, ArmbrustEV, GoodenoughUW (2002) Genetic structure of the mating-type locus of Chlamydomonas reinhardtii. Genetics 160: 181–200.
20. MerchantSS, ProchnikSE, VallonO, HarrisEH, KarpowiczSJ, et al. (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250 doi:10.1126/science.1143609
21. CharlesworthD, CharlesworthB (2010) Evolutionary Biology: The Origins of Two Sexes. Current Biology 20: R519–R521 doi:10.1016/j.cub.2010.05.015
22. FerrisPJ, GoodenoughUW (1994) The mating-type locus of Chlamydomonas reinhardtii contains highly rearranged DNA sequences. Cell 76: 1135–1145.
23. GoodsteinDM, ShuS, HowsonR, NeupaneR, HayesRD, et al. (2011) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40: D1178–D1186 doi:10.1093/nar/gkr944
24. WheelerGL, Miranda-SaavedraD, BartonGJ (2008) Genome Analysis of the Unicellular Green Alga Chlamydomonas reinhardtii Indicates an Ancient Evolutionary Origin for Key Pattern Recognition and Cell-Signaling Protein Families. Genetics 179: 193–197 doi:10.1534/genetics.107.085936
25. LahnBT, PageDC (1999) Four evolutionary strata on the human X chromosome. Science 286: 964–967.
26. BergeroR, CharlesworthD (2009) The evolution of restricted recombination in sex chromosomes. Trends Ecol Evol (Amst) 24: 94–102 doi:10.1016/j.tree.2008.09.010
27. VotintsevaAA, FilatovDA (2009) Evolutionary strata in a small mating-type-specific region of the smut fungus Microbotryum violaceum. Genetics 182: 1391–1396 doi:10.1534/genetics.109.103192
28. MenkisA, JacobsonDJ, GustafssonT, JohannessonH (2008) The mating-type chromosome in the filamentous ascomycete Neurospora tetrasperma represents a model for early evolution of sex chromosomes. PLoS Genet 4: e1000030 doi:10.1371/journal.pgen.1000030
29. PetitE, GiraudT, de VienneDM, CoelhoMA, AguiletaG, et al. (2012) Linkage to the mating-type locus across the genus Microbotryum: insights into nonrecombining chromosomes. Evolution; International Journal of Organic Evolution 66: 3519–3533 doi:10.1111/j.1558-5646.2012.01703.x
30. SharpPM, LiWH (1987) The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295.
31. WhittleCA, SunY, JohannessonH (2011) Degeneration in codon usage within the region of suppressed recombination in the mating-type chromosomes of Neurospora tetrasperma. Eukaryotic Cell 10: 594–603 doi:10.1128/EC.00284-10
32. Merchant S, Pellegrini M (2010) Chlamydomonas 454 Reads. genomes-merchantmcdbuclaedu. Available: http://genomes-merchant.mcdb.ucla.edu/. Accessed 21 January 2013.
33. BullJ (1978) Sex Chromosomes in Haploid Dioecy: A Unique Contrast to Muller's Theory for Diploid Dioecy. The American Naturalist 112: 245–250.
34. CharlesworthB, CharlesworthD (2000) The degeneration of Y chromosomes. Philos Trans R Soc Lond, B, Biol Sci 355: 1563–1572 doi:10.1098/rstb.2000.0717
35. BachtrogD (2006) A dynamic view of sex chromosome evolution. Curr Opin Genet Dev 16: 578–585 doi:10.1016/j.gde.2006.10.007
36. StöckM, HornA, GrossenC, LindtkeD, SermierR, et al. (2011) Ever-young sex chromosomes in European tree frogs. PLoS Biol 9: e1001062 doi:10.1371/journal.pbio.1001062
37. SmithDR, LeeRW (2008) Nucleotide diversity in the mitochondrial and nuclear compartments of Chlamydomonas reinhardtii: investigating the origins of genome architecture. BMC Evol Biol 8: 156 doi:10.1186/1471-2148-8-156
38. Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press. 1 pp.
39. EllegrenH (2009) The different levels of genetic diversity in sex chromosomes and autosomes. Trends Genet 25: 278–284 doi:10.1016/j.tig.2009.04.005
40. NeiM, MillerJC (1990) A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics 125: 873–879.
41. HudsonRR, SlatkinM, MaddisonWP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132: 583–589.
42. HusonDH, BryantD (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267 doi:10.1093/molbev/msj030
43. BetránE, RozasJ, NavarroA, BarbadillaA (1997) The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics 146: 89–99.
44. UyenoyamaMK (2005) Evolution under tight linkage to mating type. New Phytol 165: 63–70 doi:10.1111/j.1469-8137.2004.01246.x
45. IronsideJE (2010) No amicable divorce? Challenging the notion that sexual antagonism drives sex chromosome evolution. BioEssays 32: 718–726 doi:10.1002/bies.200900124
46. FerrisPJ, GoodenoughUW (1997) Mating type in Chlamydomonas is specified by mid, the minus-dominance gene. Genetics 146: 859–869.
47. FerrisPJ (1995) Localization of the Nic-7, Ac-29 and THI-10 Genes within the Mating-Type Locus of Chlamydomonas Reinhardtii. Genetics 141: 543.
48. SmythRD, MartinekGW, EbersoldWT (1975) Linkage of six genes in Chlamydomonas reinhardtii and the construction of linkage test strains. J Bacteriol 124: 1615–1617.
49. RymarquisLA, HandleyJM, ThomasM, SternDB (2005) Beyond complementation. Map-based cloning in Chlamydomonas reinhardtii. PLANT PHYSIOLOGY 137: 557–566 doi:10.1104/pp.104.054221
50. JacobsonDJ (2005) Blocked recombination along the mating-type chromosomes of Neurospora tetrasperma involves both structural heterozygosity and autosomal genes. Genetics 171: 839–843 doi:10.1534/genetics.105.044040
51. KathirP, LaVoieM, BrazeltonW, HaasN (2003) Molecular Map of the Chlamydomonas reinhardtii Nuclear Genome. Eukaryotic Cell 2: 362–379.
52. ArmbrustEV, FerrisPJ, GoodenoughUW (1993) A mating type-linked gene cluster expressed in Chlamydomonas zygotes participates in the uniparental inheritance of the chloroplast genome. Cell 74: 801–811.
53. SmaczniakC, ImminkRGH, AngenentGC, KaufmannK (2012) Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139: 3081–3098 doi:10.1242/dev.074674
54. BalakirevMY, TcherniukSO, JaquinodM, ChroboczekJ (2003) Otubains: a new family of cysteine proteases in the ubiquitin pathway. EMBO Rep 4: 517–522 doi:10.1038/sj.embor.embor824
55. AdamsCR, StamerKA, MillerJK, McNallyJG, KirkMM, et al. (1990) Patterns of organellar and nuclear inheritance among progeny of two geographically isolated strains of Volvox carteri. Curr Genet 18: 141–153.
56. FerrisPJ, WaffenschmidtS, UmenJG, LinH, LeeJ-H, et al. (2005) Plus and minus sexual agglutinins from Chlamydomonas reinhardtii. The Plant Cell 17: 597–615 doi:10.1105/tpc.104.028035
57. HwangCJ, MonkBC, GoodenoughUW (1981) Linkage of Mutations Affecting minus Flagellar Membrane Agglutinability to the mt Mating-Type Locus of Chlamydomonas. Genetics 99: 41–47.
58. SunS, HsuehY-P, HeitmanJ (2012) Gene Conversion Occurs within the Mating-Type Locus of Cryptococcus neoformans during Sexual Reproduction. PLoS Genet 8: e1002810 doi:10.1371/journal.pgen.1002810.t004
59. MenkisA, WhittleCA, JohannessonH (2010) Gene genealogies indicates abundant gene conversions and independent evolutionary histories of the mating-type chromosomes in the evolutionary history of Neurospora tetrasperma. BMC Evol Biol 10: 234 doi:10.1186/1471-2148-10-234
60. Pecon SlatteryJ, Sanner-WachterL, O'BrienSJ (2000) Novel gene conversion between X-Y homologues located in the nonrecombining region of the Y chromosome in Felidae (Mammalia). Proc Natl Acad Sci USA 97: 5307–5312.
61. IwaseM, SattaY, HiraiH, HiraiY, TakahataN (2010) Frequent gene conversion events between the X and Y homologous chromosomal regions in primates. BMC Evol Biol 10: 225 doi:10.1186/1471-2148-10-225
62. LinH, GoodenoughUW (2007) Gametogenesis in the Chlamydomonas reinhardtii minus mating type is controlled by two genes, MID and MTD1. Genetics 176: 913–925 doi:10.1534/genetics.106.066167
63. HoodME, PetitE, GiraudT (2013) Extensive divergence between mating-type chromosomes of the anther-smut fungus. Genetics 193: 309–315 doi:10.1534/genetics.112.146266
64. BellafioreS (2002) Loss of Albino3 Leads to the Specific Depletion of the Light-Harvesting System. The Plant Cell 14: 2303–2314 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=12215522&retmode=ref&cmd=prlinks.
65. DebuchyR, PurtonS, RochaixJD (1989) The argininosuccinate lyase gene of Chlamydomonas reinhardtii: an important tool for nuclear transformation and for correlating the genetic and molecular maps of the ARG7 locus. EMBO J 8: 2803–2809.
66. Harris EH (1989) The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use. Academic Press. 1 pp.
67. RiceP, LongdenI, BleasbyA (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277 Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10827456.
68. EdgarRC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113 doi:10.1186/1471-2105-5-113
69. TamuraK, PetersonD, PetersonN, StecherG, NeiM, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution 28: 2731–2739 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21546353&retmode=ref&cmd=prlinks.
70. YangZ (2007) PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24: 1586–1591 doi:10.1093/molbev/msm088
71. YangZ, NielsenR (2000) Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution 17: 32–43.
72. PuigbòP, BravoI, Garcia-VallveS (2008) CAIcal: A combined set of tools to assess codon usage adaptation. Biology Direct 3: 38.
73. FangS-C, de los ReyesC, UmenJG (2006) Cell size checkpoint control by the retinoblastoma tumor suppressor pathway. PLoS Genet 2: e167 doi:10.1371/journal.pgen.0020167
74. WeeksDP, BeermanN, GriffithOM (1986) A small-scale five-hour procedure for isolating multiple samples of CsCl-purified DNA: application to isolations from mammalian, insect, higher plant, algal, yeast, and bacterial sources. Analytical Biochemistry 152: 376–385 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=3963370&retmode=ref&cmd=prlinks.
75. LarkinMA, BlackshieldsG, BrownNP, ChennaR, McGettiganPA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 doi:10.1093/bioinformatics/btm404
76. LibradoP, RozasJ (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452 doi:10.1093/bioinformatics/btp187
77. TamuraK (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Molecular Biology and Evolution 9: 678–687.
78. KuboT, AbeJ, SaitoT, MatsudaY (2002) Genealogical relationships among laboratory strains of Chlamydomonas reinhardtii as inferred from matrix metalloprotease genes. Curr Genet 41: 115–122 Available: http://www.springerlink.com/openurl.asp?genre=article&id=doi:10.1007/s00294-002-0284-0.
79. LissM, KirkD, BeyserK, FabryS (1997) Intron sequences provide a tool for high-resolution phylogenetic analysis of volvocine algae. Curr Genet 31: 214–27.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2013 Číslo 8
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription