
Nucleophosmin Phosphorylation by v-Cyclin-CDK6 Controls KSHV Latency
Nucleophosmin (NPM) is a multifunctional nuclear phosphoprotein and a histone chaperone implicated in chromatin organization and transcription control. Oncogenic Kaposi's sarcoma herpesvirus (KSHV) is the etiological agent of Kaposi's sarcoma, primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD). In the infected host cell KSHV displays two modes of infection, the latency and productive viral replication phases, involving extensive viral DNA replication and gene expression. A sustained balance between latency and reactivation to the productive infection state is essential for viral persistence and KSHV pathogenesis. Our study demonstrates that the KSHV v-cyclin and cellular CDK6 kinase phosphorylate NPM on threonine 199 (Thr199) in de novo and naturally KSHV-infected cells and that NPM is phosphorylated to the same site in primary KS tumors. Furthermore, v-cyclin-mediated phosphorylation of NPM engages the interaction between NPM and the latency-associated nuclear antigen LANA, a KSHV-encoded repressor of viral lytic replication. Strikingly, depletion of NPM in PEL cells leads to viral reactivation, and production of new infectious virus particles. Moreover, the phosphorylation of NPM negatively correlates with the level of spontaneous viral reactivation in PEL cells. This work demonstrates that NPM is a critical regulator of KSHV latency via functional interactions with v-cyclin and LANA.
Published in the journal:
Nucleophosmin Phosphorylation by v-Cyclin-CDK6 Controls KSHV Latency. PLoS Pathog 6(3): e32767. doi:10.1371/journal.ppat.1000818
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000818
Summary
Nucleophosmin (NPM) is a multifunctional nuclear phosphoprotein and a histone chaperone implicated in chromatin organization and transcription control. Oncogenic Kaposi's sarcoma herpesvirus (KSHV) is the etiological agent of Kaposi's sarcoma, primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD). In the infected host cell KSHV displays two modes of infection, the latency and productive viral replication phases, involving extensive viral DNA replication and gene expression. A sustained balance between latency and reactivation to the productive infection state is essential for viral persistence and KSHV pathogenesis. Our study demonstrates that the KSHV v-cyclin and cellular CDK6 kinase phosphorylate NPM on threonine 199 (Thr199) in de novo and naturally KSHV-infected cells and that NPM is phosphorylated to the same site in primary KS tumors. Furthermore, v-cyclin-mediated phosphorylation of NPM engages the interaction between NPM and the latency-associated nuclear antigen LANA, a KSHV-encoded repressor of viral lytic replication. Strikingly, depletion of NPM in PEL cells leads to viral reactivation, and production of new infectious virus particles. Moreover, the phosphorylation of NPM negatively correlates with the level of spontaneous viral reactivation in PEL cells. This work demonstrates that NPM is a critical regulator of KSHV latency via functional interactions with v-cyclin and LANA.
Introduction
Nucleophosmin (NPM) is a multifunctional nucleolar phosphoprotein that constantly shuttles between nucleus and cytoplasm [1]. It functions as a molecular chaperone and has been linked to a number of cellular processes from ribosome maturation and transcriptional control to apoptosis (reviewed in [2]). NPM is reported to associate with unduplicated centrosomes and to dissociate from centrosomes upon phosphorylation on Thr199 by cyclin E (NM001238) -CDK2 (NM001798), which coincides with the initiation of DNA replication and centrosome duplication [3],[4]. It is often highly expressed in tumors, and the NPM1 (M26697) gene is frequently targeted by genetic alterations in various lymphomas and leukemias (reviewed in [2]). NPM can contribute to oncogenesis through various mechanisms, and has been linked to both tumor promoting and suppressing processes.
Kaposi's sarcoma-associated herpesvirus (KSHV) is an oncogenic human DNA virus in the family of γ-herpesviruses. KSHV infection is associated with Kaposi's sarcoma (KS) and certain B-cell malignancies such as an AIDS-related form of non-Hodgkin lymphoma, called primary effusion lymphoma (PEL), and Multicentric Castleman's disease (reviewed in [5]). Similar to other herpesviruses, KSHV life cycle displays latent and lytic phases. Majority of the tumor cells in KS and PELs are latently infected [6]–[8]. Lytic replication phase can be induced (viral reactivation) by a variety of intra - and extracellular factors, including hypoxia, cytokines and chemical agents, such as histone deacetylase (HDAC) inhibitors or protein kinase C agonists (reviewed in [9]). In addition, interactions of the viral machinery with components of the cellular signaling pathways and cellular transcription factors play an important role in the viral reactivation.
Latency-associated nuclear antigen (LANA), the product of ORF73 gene (AF305694) tethers the circular viral DNA to the host chromosomes [10],[11], and is required for the maintenance of the KSHV episomal genome [12],[13]. During latency LANA actively represses transcription of the KSHV lytic reactivator ORF50 gene (YP001129401) as well as several other lytic genes [14]–[18].
Viral cyclin (v-cyclin; U79416) is a latent KSHV gene that is transcribed from the same promoter element as LANA [19]–[21]. v-cyclin is structurally similar to cellular D-type cyclins and forms an active kinase complex with cellular CDK6 kinase (NM001259). The v-cyclin-CDK6 kinase phosphorylates not only the pRb protein (M15400), a common substrate of the cellular cyclin D-CDK6 complex, but also a large repertoire of unique cellular substrates such as p27KIP1 (AF480891), p21CIP1 (U03106), ORC-1 (U40152), CDC6 (U77949), caldesmon (M64110), and Bcl-2 (M14745) (reviewed in [22]–[24]).
We have previously demonstrated that ectopic expression of v-cyclin causes NPM redistribution from the nucleolus to the nucleoplasm [25]. Here we have addressed the functional relationship of v-cyclin and NPM in endothelial cells and naturally KSHV-infected PEL cells.
Results
Phosphorylation of NPM on Thr199 is dependent on v-cyclin-CDK6
To explore a functional relationship of the v-cyclin-NPM association, previously noted by us [25], in patient derived PEL cell lines we size fractionated BC-3 cell lysates and assayed for elution of NPM and v-cyclin. NPM and v-cyclin were found to co-elute at 180–110 kDa (Figure S1A; fractions marked 1–4). Subsequently, v-cyclin containing complexes were purified from these fractions and analyzed for co-precipitated CDK6 and NPM (Figure S1B), and assayed for in vitro kinase activity (Figure S1C). As shown by us before [26], v-cyclin-associated kinase activity was detected toward p27KIP1 and p21CIP1. Interestingly, v-cyclin associated kinase activity was also observed towards a 37 kDa protein most notably in fraction 2 and to a lesser extent in 3, which co-migrated with NPM (Figure S1C). These results suggested that v-cyclin associated kinase phosphorylates co-purified NPM in BC-3 PEL cells.
The notion that v-cyclin phosphorylates NPM in patient derived PEL cells was supported by recent identification of NPM Thr199 CycE-Cdk2 phosphorylation site as a target of v-cyclin-CDK6 in vitro [27]. To confirm that NPM is phosphorylated by v-cyclin on Thr199 in our experiments, U2OS cells were transfected with expression vectors for Myc-tagged v-cyclin or a vector control together with eGFP-tagged wild-type NPM (eGFP-NPM), or its phosphorylation site mutant T4A, with threonine to alanine mutations of NPM codons of 199, 214, 234, 237, and mutant T3A in which all but threonine 199 is replaced by alanine [28]. Phosphorylation of NPM was analyzed by immunoblotting using phospho-NPM antibody (pNPM Thr199), which recognizes NPM phosphorylated on its CDK2 (NM001798), phosphorylation site at threonine 199 [29]. Phosphorylation of NPM was only detected in cells co-transfected with v-cyclin and the eGFP-NPM retaining an intact Thr199 site (i.e. eGFP-NPM or eGFP-NPM T3A; Figure S1D).
To gain more insight into the biological relevance of v-cyclin induced phosphorylation on NPM, we next analyzed NPM phosphorylation in non-infected and KSHV-infected cells. To this end, SLK and EA.hy926 endothelial cells were infected with a recombinant KSHV, rKSHV.219 [30], and analyzed for phosphorylation of NPM by immunoblotting with the pNPM Thr199 antibody. Infection with rKSHV.219 induced prominent phosphorylation of NPM on threonine 199 in both cell lines, which interestingly, was accompanied by an increase in CDK6 protein levels (Figure 1A and B). NPM phosphorylation was detectable only at very low levels in uninfected cells (Figure 1; control lanes, see Figure S2 for longer exposure).

To demonstrate that NPM phosphorylation in KSHV-infected cells was dependent on v-cyclin, we silenced v-cyclin expression in the rKSHV.219-SLK cells using retrovirus-mediated RNA interference. We chose to use rKSHV.219-SLK cells instead of PELs since it has been reported that silencing of v-cyclin in PELs leads to cell death [31]. To this end, rKSHV.219-SLK cells were transduced with retrovirus expressing sh-RNA against v-cyclin (sh-v-cyclin) or a scrambled control sh-RNA (sh-Scr). Silencing of v-cyclin expression was confirmed by immunoblotting after 48 hours, and resulted in a 78% decrease in v-cyclin (Figure 1A). Phospho-NPM signal was markedly attenuated (4.8-fold) in cells expressing the sh-v-cyc as compared to cells expressing the sh-Scr, confirming that v-cyclin is required for NPM Thr199 phosphorylation in KSHV-infected endothelial cells (Figure 1A, top panel).
V-cyclin is transcribed from a common transcription start site for three latency associated genes (v-cyclin, v-FLIP (U90534), and LANA) [32]. The resulting transcript is spliced to yield two messages, one tricistronic encoding LANA, v-cyclin, and v-FLIP and one bicistronic message encoding v-cyclin and v-FLIP. The bicistronic transcript is more abundant of the two [20]. Therefore, targeting of v-cyclin by RNAi results in concurrent depletion of v-FLIP [31]. To rule out the possibility that the inhibition of NPM phosphorylation in sh-v-cyclin expressing cells is due to the loss of v-FLIP, rKSHV.219-SLK cells expressing sh-v-cyclin or sh-Scramble were reconstituted for v-FLIP by transducing them with a retrovirus expressing v-FLIP (v-FLIP-pBMN) or empty vector (pBMN). Consistent with the bicistronic nature of the v-cyclin transcript, expression of sh-v-cyclin reduced the level of v-cyclin and v-FLIP transcripts to 27% and 48%, respectively, of the levels in control cells (sh-v-cyclin -; Figure S3A). This change was accompanied by significant reduction in NPM Thr199 phosphorylation in the sh-v-cyclin cells expressing either pBMN (v-FLIP -) or v-FLIP-PBMN (v-FLIP +), suggesting that reconstitution of v-FLIP had no effect on NPM phosphorylation (Figure S3B). This data further underscores the importance of v-cyclin in phosphorylation of NPM in KSHV-infected endothelial cells. As inhibition of v-cyclin or v-FLIP has been shown to induce apoptosis in PEL cells [31],[33],[34] we analyzed whether RNAi for v-cyclin resulted in changes in the cell cycle or increased cell death. To this end expression of cyclin A (U66838) as a relevant S-phase marker and apoptosis by TUNEL assay were determined. No significant differences in the cyclin A levels (Figure S3B) or in TUNEL positivity (data not shown) were observed in the sh-v-cyclin cells as compared to the sh-Scr cells.
Previous studies have shown that CDK6 is the major kinase partner of v-cyclin in PEL cells [35],[36]. We therefore sought to confirm that also CDK6 is required for NPM phosphorylation in KSHV-infected cells. To this end, we depleted CDK6 in the rKSHV.219-infected endothelial EA.hy926 cells, and, in addition, CDK4 (M14505) and CDK6 (NM 001259) in BC-3 cells using lentivirus-mediated RNA interference. Cells stably expressing sh-RNAs specific for the aforementioned kinases or control sh-RNA (sh-Scr) were first assayed for the levels of respective kinases. Immunoblotting analysis indicated 80% decrease of CDK6 in EA.hy926, and a 98% and a 60% decrease of CDK6 and CDK4 in BC-3, respectively (Figure 1B and 1C). The specificity of silencing was also controlled by reciprocal immunoblotting of the kinases in BC-3 cells (i.e. anti-CDK6 for sh-CDK4-expressing cells and vice versa; Figure 1B). Silencing of CDK4 resulted in a 3.6-fold compensatory increase in CDK6. This is in accordance with previous studies that have illustrated that CDK4 activity is dispensable for most cell types, mostly due to the compensation by the highly related CDK6 [37]–[39]. Suppression of v-cyclin associated kinase activity consequent to CDK6 silencing was confirmed by an in vitro kinase assay in BC-3 cells (Figure S4). Depletion of CDK4/6 neither induced a cell cycle arrest (Figure S5) nor increased apoptosis (data not shown) in the sh-CDK expressing BC-3 cells. Importantly, we observed that NPM phosphorylation was significantly reduced in rKSHV.219-EA.hy926 (4.8-fold) and BC-3 cells (3.7-fold) silenced for CDK6 as compared to cells stably expressing the scrambled control (Figures 1B–D) or sh-CDK4 (Figures 1C and 1D). In contrast, concomitant with an increase in CDK6 level in the CDK4-silenced cells, the phosphorylation of NPM was increased by 1.8-fold. Taken together, the results indicate that v-cyclin phosphorylates NPM on Thr199 by activating CDK6 in the KSHV-infected endothelial and BC-3 cells, which are both biologically relevant cell types in KSHV-infections and malignancies.
NPM interacts with LANA in a v-cyclin dependent manner
A study by Si et al. [40] reported that NPM associates with LANA and the terminal repeats of the KSHV genome in PEL cells. That LANA and NPM interact is supported by gel filtration chromatography and co-immunoprecipitation analysis of LANA and NPM in BC-3 cells (Figure S6 and [37]). Given that NPM is detected in complexes with two KSHV latent proteins, which are transcribed from a common promoter upstream of the LANA gene [19],[32], we considered it reasonable to test whether v-cyclin affects LANA-NPM association. We transfected U2OS cells stably expressing LANA or GFP (Sarek et al., unpublished data) with a Myc-tagged v-cyclin expression vector (Myc-v-cyclin) or with an empty vector as a control. After 48 hours, whole cell extracts were collected and subjected to immunoprecipitation with anti-LANA or -NPM antibodies followed by reciprocal immunoblotting. This revealed that co-expression of v-cyclin facilitated association of NPM with LANA (Figure 2A; middle and rightmost panels). To further address the NPM-LANA interaction in naturally KSHV-infected PEL cells, and the dependency of this interaction on CDK6, the sh-Scr and sh-CDK6 BC-3 cells shown in Figure 1B were subjected to gel filtration chromatography to assess for LANA and NPM co-elution. NPM and LANA were found to co-elute and co-precipitate in fractions marked 1–3, corresponding to ca. 700-440 kDa (Figure S7). Interestingly, silencing of CDK6 reduced co-precipitation of NPM and LANA in these fractions (Figure 2B), further indicating that the v-cyclin-CDK6 kinase activity supports the NPM-LANA interaction.

We then examined the requirement of NPM phosphorylation on Thr199 for NPM-LANA interaction, by performing anti-LANA immunoprecipitation from cell extracts of U2OS cells expressing LANA and transfected with expression vectors for Myc-v-cyclin, empty vector control, and the eGFP-tagged NPM wt or its phosphosite mutant constructs used in Figure S1D. As shown in Figure 2C (right panel), LANA associated with the eGFP-tagged NPM wt and T3A mutant in the presence of v-cyclin, while interaction with the T4A mutant was severely compromised. Together these results indicate that the phosphorylation of Thr199 on NPM by v-cyclin–CDK6 is needed for LANA-NPM interaction.
Interaction of NPM and LANA is affected by acetylation
To explore the role of NPM in KSHV pathobiology we first silenced NPM in BC-3 and BCBL-1 PEL cell lines using lentivirus-mediated RNA interference. NPM protein level was decreased by 77% and 80% in BC-3 and 83% and 80% in BCBL-1 cells stably expressing two different sh-RNAs for NPM, respectively (sh1-NPM and sh2-NPM) as compared to cells expressing a control sh-Scr (Figure 3A). Fibrillarin (AC005393), another nucleolar protein, and tubulin were used as loading controls and were unaffected. There were no major effects on mitotic activity and proliferation (data not shown), or on nucleolar morphology based on localization of fibrillarin (Figure S8), or phase contrast in the selected population of cells silenced for NPM (data not shown). Furthermore, no obvious change was observed for LANA levels in cells expressing sh-RNAs against NPM (as shown for BCBL-1 cells in Figure 3B).

KSHV is predominantly latent in PEL cells, but can be induced to lytic replication phase with chemical HDAC inhibition. NPM was recently reported to recruit HDAC and to exert its repressive effect on transcription by inducing a change in local chromatin structure [41]. To address the function of NPM in PEL cells in this context, we first analyzed the association of NPM and LANA with HDAC1 (D50405) by immunoprecipitation experiments. In agreement with the recently reported role of NPM as an HDAC recruiter, NPM was consistently present in reciprocal co-immunoprecipitations with HDAC1 in latent (uninduced) BCBL-1 cells (Figure 3C) and BC-3 cells (data not shown). Consequently, we tested whether this interaction is affected by inhibition of HDAC activity with sodium butyrate (NaB) that leads to induction of lytic phase (reactivation). The association between NPM and HDAC1 was greatly diminished upon NaB treatment while their protein levels remained unchanged (Figure 3C). Interestingly, we found that LANA complexed with HDAC1 in the latently infected cells and even after HDAC inhibition by NaB (Figure 3D). This interaction, however, was completely abolished upon NPM silencing, suggesting that NPM facilitates interaction of LANA with chromatin modifiers (Figure 3D).
NaB treatment has been shown to increase LANA acetylation and to diminish LANA interactions with chromatin, Sp1 transcription factor, and the early lytic ORF50/RTA promoter [42]. Given that NPM silencing reduced the association of LANA with HDAC1, we decided to assess for acetylation of LANA in both NPM silenced and NaB-treated BCBL-1 cells. To this end we immunoprecipitated cellular extracts with antibody against acetylated lysine (Ac-K) or control IgG antiserum, followed by immunoblotting with anti-LANA antibody. As shown in Figure 3E treatment with NaB as well as silencing of NPM expression resulted in appearance of new bands in the subsequent LANA immunoblot suggesting of an increase in the acetylation of LANA [42]. However, we cannot exclude the possibility that we were detecting other acetylated proteins associated with LANA. Acetylation of NPM by the histone acetyltransferase p300 (NP001420) has been reported to disrupt the nucleosomal structure [43]. We therefore analyzed the level of acetylation on NPM in the NaB-treated BC-3 cells, and found that it was increased upon HDAC inhibition (Figure 3F). In line with this, p300 was co-immunoprecipitated with NPM from extracts of BCBL-1 cells treated with NaB (Figure 3G).
Protein acetylation is one possible mechanism modulating the interaction between LANA and NPM. Thus, we wanted to test whether this interaction is altered by NaB. Whole cell extracts of BCBL-1 cells treated with NaB (1 mM) or vehicle for 24 hours were subjected to immunoprecipitation with anti-LANA followed by immunoblotting for NPM. Intriguingly, we found that NPM association with LANA was abolished in cell extracts pretreated with NaB (Figure 3H). Given that NaB treatment leads to induction of viral reactivation, we considered it reasonable to rule out the possibility that expression of lytic genes would affect the LANA-NPM association. We have recently demonstrated that Pim-1 kinase (NM002648) is a critical regulator of KSHV reactivation, and that depletion of Pim-1 expression by RNAi leads to inhibition of early steps of viral reactivation [44]. To this end we transfected BCBL-1 cells with siRNA oligonucleotides specific for Pim-1 (Pim-1 siRNA) or with a non-targeting control siRNA (Scr siRNA). Cells were subjected to 1 mM NaB or vehicle treatment 48 hours post-transfection and lysates were collected after another 24 hours to confirm silencing of Pim-1 (Figure S9A, left panel). The inhibition of viral reactivation by Pim-1 depletion was confirmed by quantitative real-time PCR (Figure S9B) and immunobloting with vIL-6 (AAB61701) (Figure S9C). Importantly, NPM association with LANA was still abolished by inhibition of HDAC activity in the Pim-1 depleted cell extracts (Figure S9A, right panel), supporting that the NPM-LANA interaction was disrupted due to inhibition of de-acetylation, and not due to induction of lytic gene expression.
Depletion of NPM leads to induction of a complete lytic replication cascade in PEL cells
As silencing of NPM seemed to induce an increase in LANA acetylation in PEL cells, an event previously linked to viral reactivation, we next addressed the expression of lytic replication markers in the NPM silenced BC-3 and BCBL-1 cells. Cells stably expressing a non-target control sh-RNA (sh-Scr), sh1-NPM, and sh2-NPM were analyzed by immunofluorescence using antibodies against an early lytic marker ORF59 (YP001129416). In both cell lines, depletion of NPM expression led to a significant increase in ORF59 positive cells as compared to the sh-Scr expressing control cells (shown for BCBL-1 in Figures 4A and 4B). Viral reactivation in sh1-NPM and sh2-NPM expressing BCBL-1 cells was further confirmed by quantitative real-time PCR (qRT-PCR) for the early lytic genes ORF50 and ORF57 (YP001129410) as well as for a late lytic gene K8.1 (Figure 4C).

Reactivation of the complete lytic cascade of KSHV results in production of infectious progeny virions. We therefore wanted to investigate whether NPM silencing would lead to production of infectious virions in PEL cells. To this end, BCBL-1 cells stably expressing sh1-NPM, sh2-NPM, or sh-Scr were analyzed by immunoblotting for expression of the late lytic capsid glycoprotein K8.1 (AF068829), nine days post-transduction. Expression of K8.1 was induced in cells depleted for expression of NPM with sh1 - or sh2-NPM, but not in cells expressing the control sh-Scr (Figure 4D). To measure production of infectious KSHV particles upon NPM silencing, supernatants from sh1-NPM or sh-Scr expressing BCBL-1 cells were collected six, nine and 12 days post lentiviral transduction, pooled, and used directly or after concentration by ultracentrifugation to infect naive SLK cells. Infection of target cells was monitored by immunofluorescence with anti-LANA antibodies. At 48 hours after infection, 50–90% of the target SLK cells displayed the typical speckled signal from LANA with numerous dots per nucleus thus confirming that silencing of NPM expression led to induction of the full lytic replication cascade and production of infectious virions (Figure 4E and F).
Phosphorylation of NPM correlates with KSHV latency
Considering the essential role of LANA in KSHV latency and suppression of lytic viral transcription, as well as the observation that v-cyclin promotes LANA-NPM interaction, we sought to address whether there is a correlation between NPM Thr199 phosphorylation, v-cyclin expression, and the extent of spontaneous lytic replication in four different patient-derived KSHV-infected PEL lines (BC-3, BCBL-1, BC-1, JSC-1), IHH (a KSHV-positive lymphoblastoid cell line), and IHE (a KSHV-negative cell line). To this end, cell extracts were analyzed by immunoblotting for expression of pNPM Thr199, total NPM (NPM), v-cyclin, and another latent KSHV protein vIRF3 as a control. Interestingly, we found an apparent correlation with the extent of NPM phosphorylation and v-cyclin expression in the KSHV-infected lymphoid cells (Figure 5A). In accordance with our finding that phosphorylation of NPM is needed for the interaction between LANA and NPM (Figure 2), the LANA-NPM interaction was diminished in JSC-1 PEL cells, which had a very low level of NPM phosphorylation, as compared to BC-3 cells with highly phosphorylated NPM and prominent LANA-NPM co-precipitation (Figure 5B). To examine whether NPM phosphorylation correlates with the level of spontaneous, un-induced viral reactivation, we performed qRT-PCR for expression of the early lytic transcripts ORF50 and ORF57 in the KSHV-infected PEL cells (Figure 5C). Reactivation was also analyzed by immunofluorescence using antibodies against the early lytic marker ORF59 (Figure 5D). Intriguingly, we found that the cells with lower phospho-NPM levels had higher level of spontaneous expression of the lytic markers compared to the cells with elevated phospho-NPM, suggesting that NPM is a critical regulator of latency in KSHV-infected lymphoid cells.

To obtain further evidence about the correlation between, v-cyclin expression levels, extent of NPM phosphorylation on Thr199, and spontaneous viral reactivation we over-expressed v-cyclin in BCBL-1 and JSC-1 cells using retroviruses expressing v-cyclin and GFP (KpBMN) or GFP (pBMN) as a control. Exogenous expression of v-cyclin (determined by the number of GFP positive cells) was achieved in 17% and 20% of BCBL-1 and JSC-1 cells, respectively (data not shown), and confirmed by Western blotting at 48 hours post-transduction (Figure 5E). As shown in Figure 5E, NPM phosphorylation on Thr199 increased about 1.8-fold in cells over-expressing v-cyclin as compared to cells infected with the control (v-cyclin + and -, respectively) as evaluated from the luminescence signal, and analyzed by Image J software. In accordance with the increase of pNPM Thr199 levels, qRT-PCR analysis demonstrated a 1.5 to 1.7-fold reduction (in average) in the spontaneous expression of the lytic transcripts ORF50, ORF57 and K8.1 in cells over-expressing v-cyclin (Figure 5F; v-cyclin +). Taken together, these results further support the role of v-cyclin mediated phoshorylation of NPM in controlling spontaneous viral reactivation in PEL cells.
NPM is phosphorylated in KS tumors
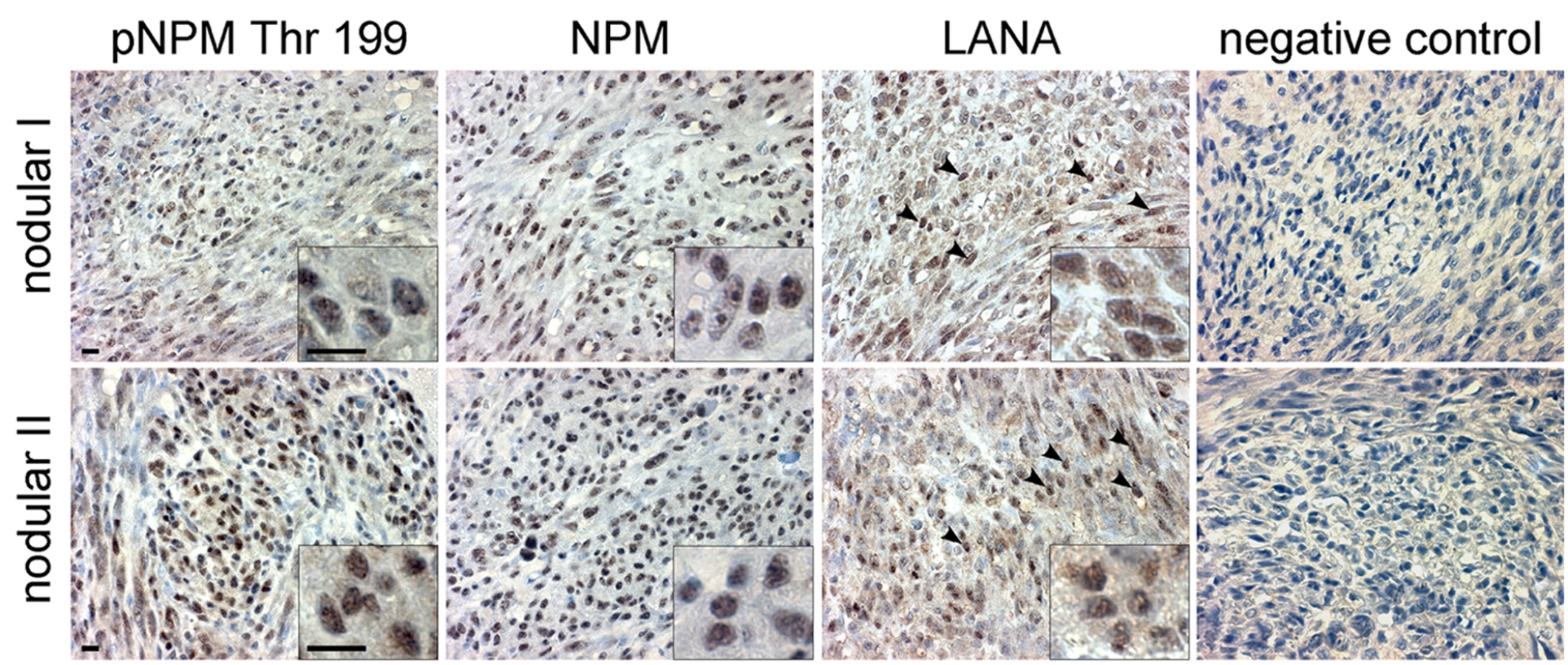
Our results suggest that KSHV infection and more specifically the activity of v-cyclin-CDK6 is responsible for the phosphorylation of NPM on Thr199 in latently infected cells. To determine whether NPM is phosphorylated also in KS tumors we stained primary cutaneous lesions of KS (n = 6) with antibodies against pNPM Thr199 and total NPM. Immunohistochemistry for LANA confirmed KSHV infection and defined the tumor area (Figure 6, LANA). Sections of paraffin-embedded BC-3 cells stably expressing sh-CDK6 or control (sh-Scr) were used to verify the specificity of anti-pNPM Thr199 staining (data not shown). NPM phosphorylation was observed in all of the KS lesions analyzed (Figure 6), suggesting a role for NPM phosphorylation in KSHV pathogenesis.
Discussion
Latency is the predominant mode of viral persistence in KS and PEL tumors, and is considered to have a fundamental impact on KSHV tumorigenesis. In this study, we show that NPM is phosphorylated in KSHV-infected cells by v-cyclin-CDK6. In addition, we show that NPM is phosphorylated in primary human KS tumors. Our study demonstrates the functional interaction between v-cyclin and NPM in a biologically relevant system, and establishes NPM as a substrate for the v-cyclin activated CDK6 in KSHV-infected endothelial cells and PEL cells. Our findings indicate that phosphorylation of NPM is essential for its association with LANA, and thus establishes the first functional link between the latent proteins v-cyclin and LANA. Furthermore, we demonstrate that NPM is a critical key regulator in KSHV latency.
Our data indicates that in latently KSHV-infected cells NPM facilitates interaction of LANA with HDAC1 (Figure 3D) and the core histones (data not shown). This is in accordance with the recent finding identifying NPM as a HDAC recruiter involved in transcriptional repression during differentiation [41], and with earlier data showing that HDACs interact directly or indirectly with viral sequences to inhibit lytic gene expression [45]. A study by Lu et al. (2006) has shown that LANA-mediated repression of viral lytic transcription is mediated by LANA binding to the lytic reactivator ORF50/RTA promoter, which is de-repressed by acetylation of LANA [42], probably due to acetylation disturbing the association of LANA with the chromatin modifiers at the ORF50 promoter. Here our results demonstrate that in cells depleted for NPM LANA is not anymore associated with HDAC1 (Figure 3D). Moreover, depletion of NPM led to an increase in the acetylation of LANA (Figure 3E), and induction of the complete lytic replication cascade (Figure 4). In line with these results, inhibition of HDAC activity by NaB is a widely used mechanism to induce KSHV reactivation. Further supporting the role of acetylation in the LANA-NPM interaction, NaB treatment led to an increase also in lysine acetylation of NPM (Figure 3F), and to dissociation of the NPM-LANA complex (Figure 3H). Interestingly, NaB treatment did not disrupt the interaction of LANA with HDAC1 (Figure 3D), probably because the increase in LANA acetylation was achieved by chemical inhibition of the HDAC enzymatic activity rather than by interfering with its recruitment by NPM. Furthermore, it is possible that acetylation primarily modulates NPM-HDAC1 and NPM-LANA interactions in the ternary complex of NPM-HDAC1-LANA, but does not interfere with the HDAC1-LANA interaction after the recruitment of HDAC1 to the complex has occurred by phosphorylated NPM. However, if de-acetylation by HDAC-1 is chemically suppressed (by NaB) this results in an increase of NPM acetylation (Figure 3F), dissociation of HDAC1 from NPM (Figure 3C), and may allow a more stable interaction between p300 and NPM as suggested by the data in Figure 3G. Although also acetylation on LANA is elevated, this only affects its association to NPM, but not to HDAC-1 that would remain associated with LANA (Figure 3D).
NPM has also been implicated in the replication cycle of other viruses such as adeno-[46],[47], and adeno-associated viruses [48], but the detailed mechanism for the function of NPM in these viral systems is not known. By analyzing several different PEL cell lines and one in vitro KSHV-infected lymphoblastoid cell line we detected significant differences in the level of phosphorylation of NPM, but not in total NPM, which correlated with v-cyclin expression (Figure 5A). Furthermore, we found that the extent of NPM phosphorylation correlated with the low level of spontaneous reactivation in these cells.
Taken together, our study identifies a key role for NPM and its phosphorylation in regulating the KSHV latency in PEL lymphocytes and de novo infected endothelial cells. The data demonstrating a significant reduction in the phosphorylation of NPM upon silencing of CDK6 expression in KSHV-infected endothelial and lymphoid cells (Figure 1) may open up novel opportunities for developing targeted therapies for intervention and treatment of KSHV associated malignancies. Putative approaches could include development of CDK6-specific kinase inhibitors or addressing the effect of small molecule NPM inhibitors or RNA aptamers [49],[50] on disruption of KSHV latency.
Materials and Methods
Cell lines
BC-1, and JSC-1 PEL cell lines [51],[52] were obtained from ATCC (Manassas, VA). BC-3 and BCBL-1 cell lines [8],[53] were kindly provided by E. Cesarman (Cornell Medical College, NY), and IHH and IHE [54] by J. Haas (University of Edinburgh). SLK cells were a kind gift of T. F. Schulz (Hannover Medical School, Germany). PEL cell lines were cultured as described previously [26]. To induce KSHV lytic replication, cells were treated with 1 mM sodium butyrate (NaB; Sigma, St Louis, MO) for 24 hours. U2OS human osteosarcoma cells (ATCC) were grown as detailed in [26]. rKSHV.219-infected endothelial cells were established and maintained as described in [54]. Infected cells were grown in the presence of 1 µg/ml of puromycin for about two weeks to obtain a 100% infected cell population.
Lenti - and retroviral production and transduction
Production of retro - and lentiviral supernatants and CDK silencing in EA.hy926 and SLK cells were performed as described earlier [55]. To silence CDKs and NPM, the PEL cells were seeded at density 5×105/ml and transduced in a 50 ml culture flask using 2 ml lentiviral supernatants in the presence of 8 µg/ml polybrene (Sigma). 24 hours after transduction, the culture was replenished with fresh media, and cells were kept for 48 hours, after which they were subjected to selection with 3.5 µg/ml puromycin (sh-NPM, sh-Scr) or 300 µg/ml hygromycin (sh-CDK4, sh-CDK6, sh-Scr).
For an acute depletion of v-cyclin expression in the rKSHV.219-SLK cells, the cells were spin-transduced (2500 rpm; Heraeus Multifuge 3 S-R; Thermo Scientific) for 30 min at room temperature with fresh amphotropic retroviruses expressing control sh-RNA (sh-Scr) or sh-RNA against v-cyclin (sh-v-cyclin) in the presence of 8 µg/ml polybrene. Cells were then returned to 37°C, 5% CO2, and after 24 h of incubation viral supernatant was removed and replaced with fresh complete media. Cells were harvested for analysis 48 hours post-transduction.
U2OS osteosarcoma cells were spin-transduced as described above with GFP - or LANA-expressing lentiviruses in the presence of 8 µg/ml polybrene, and the cells were incubated for 48 hours. Thereafter LANA and GFP-expressing cells were cultured in the presence of 4.5 µg/ml of blasticidin (Sigma) for at least two weeks. Stable expression of the transduced proteins was assessed by immunofluorescence and Western blotting using anti-GFP or anti-LANA antibodies.
Analysis of production of infectious virions
To study production of infectious virions upon the silencing of NPM expression, BCBL-1 cells (0.5×106/ml) were transduced with sh-Scr and sh1-NPM lentiviral supernatants as described above. The medium was collected at day six, nine, and 12 post-transduction. The supernatants were cleared by centrifugation at 3,000×g for 15 min to remove cell debris and pooled together. The supernatants were concentrated at 21,000 rpm (60,000×g) for 2 hours in a Beckman SW28.1 rotor, resuspended overnight in 1 ml (1/100 of the original volume) of TNE buffer (10 mM Tris-HCl, 0.15 M NaCl, 1 mM EDTA [pH 7.8]), and used to infect naive SLK target cells.
Protein analysis and kinase assay
Immunoblotting, immunoprecipitations, kinase assay and size exclusion chromatography were carried out as detailed [26]. For detection of the p-NPM signal by western blotting see Text S1.
Real-time quantitative PCR
Total RNA was prepared by using the RNAeasy kit according to instructions from the manufacturer (Qiagen, Valencia, CA). RT-PCR was performed with TaqMan Reverse Transcription Reagents kit (Roche Diagnostics, Indianapolis, IN) according to manufacturer's protocol. Real-time PCR conditions and set of primers for LANA, v-cyclin, v-FLIP, ORF50, ORF57, K8.1 and human beta-actin were essentially as described previously [56],[57].
Immunohistochemistry
Sections of paraffin-embedded KS tumors were treated for antigen retrieval by autoclaving in 10 mM sodium citrate (pH 6.0) for 2 min, and then pretreated for peroxidase blocking by incubation in 2.5% H2O2 for 30 min. Sections were processed by using Vectastain Elite ABC rabbit IgG or mouse IgG kits (Vector Laboratories, Burlingame, CA). Sections were blocked for 30 min at room temperature in normal goat serum (1∶50 in PBS) +0.3% BSA. The slides were incubated at +4°C overnight with primary antibodies to pNPM (Thr199) or total NPM diluted 1∶200 or 1∶100, respectively, in PBS +0.3% BSA followed by biotinylated anti-rabbit or anti mouse secondary antibody 1∶200 (Vectastain), respectively in 0.3% BSA in PBS for 30 min at room temperature. Signal was amplified by ABC (Vectastain) and diaminobenzidine (DAB; DakoCytomation) reaction. The slides were then counterstained with hematoxyline and mounted by using GVA mount media (Invitrogen, Carlsbad CA). Immunohistochemistry of LANA in KS tumors was performed essentially as described previously [55]. Signal amplification was performed by using a peroxidase ABC kit (Vector Laboratories) and followed by diaminobenzidine (DAB; DakoCytomation) reaction.
The images were captured with a Zeiss Axioplan 2 microscope equipped with Zeiss Plan-Neofluar x63/0.75NA objective (Carl Zeiss, Oberkochen, Germany). Images were acquired with a Zeiss Axiocam HRc CCD camera, using Zeiss AxioVision 4.6 SP1 software and processed with Adobe Photoshop 8.0 software (Adobe, San Jose, CA).
For complete list of Materials and Methods see Text S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BorerRA
LehnerCF
EppenbergerHM
NiggEA
1989 Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56 379 390
2. GrisendiS
MecucciC
FaliniB
PandolfiPP
2006 Nucleophosmin and cancer. Nat Rev Cancer 6 493 505
3. OkudaM
HornHF
TaraporeP
TokuyamaY
SmulianAG
2000 Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103 127 140
4. TokuyamaY
HornHF
KawamuraK
TaraporeP
FukasawaK
2001 Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem 276 21529 21537
5. AblashiDV
ChatlynneLG
WhitmanJEJr
CesarmanE
2002 Spectrum of Kaposi's sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin Microbiol Rev 15 439 464
6. ZhongW
WangH
HerndierB
GanemD
1996 Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci U S A 93 6641 6646
7. DittmerDP
2003 Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res 63 2010 2015
8. RenneR
ZhongW
HerndierB
McGrathM
AbbeyN
1996 Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2 342 346
9. MillerG
El-GuindyA
CountrymanJ
YeJ
GradovilleL
2007 Lytic cycle switches of oncogenic human gammaherpesviruses(1). Adv Cancer Res 97 81 109
10. BallestasME
KayeKM
2001 Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75 3250 3258
11. BarberaAJ
ChodaparambilJV
Kelley-ClarkeB
JoukovV
WalterJC
2006 The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311 856 861
12. BallestasME
ChatisPA
KayeKM
1999 Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284 641 644
13. YeFC
ZhouFC
YooSM
XieJP
BrowningPJ
2004 Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J Virol 78 11121 11129
14. LanK
KuppersDA
VermaSC
RobertsonES
2004 Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol 78 6585 6594
15. LanK
KuppersDA
RobertsonES
2005 Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol 79 3468 3478
16. GarberAC
ShuMA
HuJ
RenneR
2001 DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 75 7882 7892
17. VermaSC
LanK
ChoudhuriT
RobertsonES
2006 Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J Virol 80 3445 3458
18. AnFQ
CompitelloN
HorwitzE
SramkoskiM
KnudsenES
2005 The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem 280 3862 3874
19. DittmerD
LagunoffM
RenneR
StaskusK
HaaseA
1998 A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72 8309 8315
20. GrundhoffA
GanemD
2001 Mechanisms governing expression of the v-FLIP gene of Kaposi's sarcoma-associated herpesvirus. J Virol 75 1857 1863
21. PearceM
MatsumuraS
WilsonAC
2005 Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J Virol 79 14457 14464
22. MittnachtS
BoshoffC
2000 Viral cyclins. Rev Med Virol 10 175 184
23. VerschurenEW
JonesN
EvanGI
2004 The cell cycle and how it is steered by Kaposi's sarcoma-associated herpesvirus cyclin. J Gen Virol 85 1347 1361
24. JarviluomaA
OjalaPM
2006 Cell signaling pathways engaged by KSHV. Biochim Biophys Acta 1766 140 158
25. KurkiS
PeltonenK
LatonenL
KiviharjuTM
OjalaPM
2004 Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5 465 475
26. SarekG
JarviluomaA
OjalaPM
2006 KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107 725 732
27. CuomoME
KnebelA
MorriceN
PatersonH
CohenP
2008 p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nat Cell Biol 10 723 730
28. OkuwakiM
TsujimotoM
NagataK
2002 The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol Biol Cell 13 2016 2030
29. TaraporeP
ShinmuraK
SuzukiH
TokuyamaY
KimSH
2006 Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett 580 399 409
30. VieiraJ
O'HearnPM
2004 Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325 225 240
31. GodfreyA
AndersonJ
PapanastasiouA
TakeuchiY
BoshoffC
2005 Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105 2510 2518
32. TalbotSJ
WeissRA
KellamP
BoshoffC
1999 Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257 84 94
33. KellerSA
SchattnerEJ
CesarmanE
2000 Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 96 2537 2542
34. GuasparriI
KellerSA
CesarmanE
2004 KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199 993 1003
35. PlattGM
CannellE
CuomoME
SinghS
MittnachtS
2000 Detection of the human herpesvirus 8-encoded cyclin protein in primary effusion lymphoma-derived cell lines. Virology 272 257 266
36. JarviluomaA
KoopalS
RasanenS
MakelaTP
OjalaPM
2004 KSHV viral cyclin binds to p27KIP1 in primary effusion lymphomas. Blood 104 3349 3354
37. RaneSG
DubusP
MettusRV
GalbreathEJ
BodenG
1999 Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 22 44 52
38. TsutsuiT
HesabiB
MoonsDS
PandolfiPP
HanselKS
1999 Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 19 7011 7019
39. MalumbresM
SotilloR
SantamariaD
GalanJ
CerezoA
2004 Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118 493 504
40. SiH
VermaSC
RobertsonES
2006 Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J Virol 80 9017 9030
41. LiuH
TanBC
TsengKH
ChuangCP
YehCW
2007 Nucleophosmin acts as a novel AP2alpha-binding transcriptional corepressor during cell differentiation. EMBO Rep 8 394 400
42. LuF
DayL
GaoSJ
LiebermanPM
2006 Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J Virol 80 5273 5282
43. SwaminathanV
KishoreAH
FebithaKK
KunduTK
2005 Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol Cell Biol 25 7534 7545
44. ChengF
Weidner-GlundeM
VarjosaloM
RainioEM
LehtonenA
2009 KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5 e1000324 doi:10.1371/journal.ppat.1000324
45. GwackY
ByunH
HwangS
LimC
ChoeJ
2001 CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J Virol 75 1909 1917
46. WaltonTH
MoenPTJr
FoxE
BodnarJW
1989 Interactions of minute virus of mice and adenovirus with host nucleoli. J Virol 63 3651 3660
47. MatthewsDA
2001 Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J Virol 75 1031 1038
48. BevingtonJM
NeedhamPG
VerrillKC
CollacoRF
BasrurV
2007 Adeno-associated virus interactions with B23/Nucleophosmin: identification of sub-nucleolar virion regions. Virology 357 102 113
49. QiW
ShakalyaK
StejskalA
GoldmanA
BeeckS
2008 NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene 27 4210 4220
50. JianY
GaoZ
SunJ
ShenQ
FengF
2009 RNA aptamers interfering with nucleophosmin oligomerization induce apoptosis of cancer cells. Oncogene 28 4201 4211
51. CesarmanE
MoorePS
RaoPH
InghiramiG
KnowlesDM
1995 In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86 2708 2714
52. CannonJS
CiufoD
HawkinsAL
GriffinCA
BorowitzMJ
2000 A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J Virol 74 10187 10193
53. ArvanitakisL
MesriEA
NadorRG
SaidJW
AschAS
1996 Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88 2648 2654
54. SarekG
KurkiS
EnbackJ
IotzovaG
HaasJ
2007 Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest 117 1019 1028
55. KoopalS
FuruhjelmJH
JarviluomaA
JaamaaS
PyakurelP
2007 Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog 3 1348 1360
56. DittmerD
StoddartC
RenneR
Linquist-SteppsV
MorenoME
1999 Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J Exp Med 190 1857 1868
57. FakhariFD
DittmerDP
2002 Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76 6213 6223
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2010 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Kaposi's Sarcoma-Associated Herpesvirus ORF57 Protein Binds and Protects a Nuclear Noncoding RNA from Cellular RNA Decay Pathways
- Endocytosis of the Anthrax Toxin Is Mediated by Clathrin, Actin and Unconventional Adaptors
- Perforin and IL-2 Upregulation Define Qualitative Differences among Highly Functional Virus-Specific Human CD8 T Cells
- Exoerythrocytic Parasites Secrete a Cysteine Protease Inhibitor Involved in Sporozoite Invasion and Capable of Blocking Cell Death of Host Hepatocytes