
Mutations in a Guanylate Cyclase GCY-35/GCY-36 Modify Bardet-Biedl Syndrome–Associated Phenotypes in
Ciliopathies are pleiotropic and genetically heterogeneous disorders caused by defective development and function of the primary cilium. Bardet-Biedl syndrome (BBS) proteins localize to the base of cilia and undergo intraflagellar transport, and the loss of their functions leads to a multisystemic ciliopathy. Here we report the identification of mutations in guanylate cyclases (GCYs) as modifiers of Caenorhabditis elegans bbs endophenotypes. The loss of GCY-35 or GCY-36 results in suppression of the small body size, developmental delay, and exploration defects exhibited by multiple bbs mutants. Moreover, an effector of cGMP signalling, a cGMP-dependent protein kinase, EGL-4, also modifies bbs mutant defects. We propose that a misregulation of cGMP signalling, which underlies developmental and some behavioural defects of C. elegans bbs mutants, may also contribute to some BBS features in other organisms.
Published in the journal:
Mutations in a Guanylate Cyclase GCY-35/GCY-36 Modify Bardet-Biedl Syndrome–Associated Phenotypes in. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002335
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002335
Summary
Ciliopathies are pleiotropic and genetically heterogeneous disorders caused by defective development and function of the primary cilium. Bardet-Biedl syndrome (BBS) proteins localize to the base of cilia and undergo intraflagellar transport, and the loss of their functions leads to a multisystemic ciliopathy. Here we report the identification of mutations in guanylate cyclases (GCYs) as modifiers of Caenorhabditis elegans bbs endophenotypes. The loss of GCY-35 or GCY-36 results in suppression of the small body size, developmental delay, and exploration defects exhibited by multiple bbs mutants. Moreover, an effector of cGMP signalling, a cGMP-dependent protein kinase, EGL-4, also modifies bbs mutant defects. We propose that a misregulation of cGMP signalling, which underlies developmental and some behavioural defects of C. elegans bbs mutants, may also contribute to some BBS features in other organisms.
Introduction
The cilium plays diverse cellular functions in metazoans which include imparting motility, enabling sensory processes and regulating the activity of cell signalling pathways during development [1]. The biogenesis and maintenance of this evolutionarily conserved organelle relies on intraflagellar transport (IFT) – the bidirectional transportation of diverse cargo proteins along the microtubule-based axoneme. Defective IFT or ciliary dysfunction result in ciliopathies, a growing class of pleiotropic human diseases with overlapping clinical features, some being of significant morbidity [2]. Bardet-Biedl syndrome (BBS, OMIM 209900) is an autosomal recessive and genetically heterogeneous ciliopathy with hallmark clinical features that include photoreceptor degeneration, renal abnormalities, obesity, cognitive impairment, and digit and genital anomalies [3]. To date, sixteen genes are associated with BBS [3]–[5]; of these, eight function mostly as a conserved protein complex (BBSome) [6] to regulate vesicular sorting and packaging [7], IFT [8]–[9], as well as cilium maintenance and function (reviewed in [10]).
Animal models have been instrumental in deciphering the physiological functions of BBS proteins [2], [10]. Initial characterization of Caenorhabditis elegans BBS orthologues led to the discovery of BBS proteins as ciliary components, associating ciliary defects with the loss of BBS protein function [11]–[13]. The loss of BBS-7 and BBS-8 led to shortened cilia, reduced uptake of a lipophilic dye (DiI) by the cilium, and defective chemo- and thermotaxis [13]–[14].
Murine Bbs mutants recapitulate several human BBS features including photoreceptor degeneration, renal anomalies and obesity [15]–[16]. Additionally, these models led to the identification of new features such as neural tube closure defects [17], anosmia [18], and behavioural, mechano- and thermosensory deficits [14] that expanded the diagnostic features of human ciliopathies. Morpholino-mediated knockdown of bbs in zebrafish led to developmental phenotypes such as dorsal thinning, poor somitic definition and Kupffer's Vesicle malformation [19]–[21], while defects characteristic of ciliopathy such as delayed retrograde melanosome transport [20] and vision impairment [22] also manifested.
In addition to a role in sensory transduction, the primary cilium functions as a signalling ‘apparatus’ to regulate development [1]. For example, IFT-dependent localization of Sonic Hedgehog (Shh) receptors to primary cilia is required for Shh signalling [23]–[24]. Disrupting IFT components IFT172, TG737/Polaris and the motor KIF3A in the mouse resulted in phenotypes typical of Shh mutants [25]. Similarly, defective planar cell polarity (PCP) signalling [17], [26] and/or aberrant Wnt signalling [27]–[28] were associated with the inactivation of BBS, Polaris or KIF3A components. These, and others studies [29]–[30] suggest that the cilium may modulate multiple signalling pathways in a tissue-specific manner. Aberrant PCP, Shh and Wnt signalling have been implicated in underlying a number of ciliopathy features, such as neural tube closure, polydactyly and obesity [17], [22], [31]–[32]. The pathology of other features such as photoreceptor degeneration, remains largely unexplained, indicating the presence of unidentified cellular processes that are regulated by the cilium.
C. elegans BBS orthologues are exclusively expressed by 60 ciliated neurons. Localizing at the base of cilia, they undergo active IFT, and their absence results in the destabilization of IFT and sensory defects [8]. C. elegans sensory neurons play key roles in multiple developmental processes [33]. Some chemosensory mutants exhibit a reduced body size [33], indicating that sensory function may influence this developmental process. Another key regulator for body size is the cGMP-dependent protein kinase (PKG) EGL-4 [33]–[36]; a loss of EGL-4 function leads to increased body size that is genetically epistatic to that of chemosensory mutants [33]. The mechanisms for sensory neuron-mediated body size regulation, however, remain to be fully elucidated. In the present study, in order to identify additional cilium-regulated signalling events in C. elegans, we carried out the phenotypic characterization of bbs mutant animals, and identified genetic modifiers that associate aberrant cGMP signalling with a subset of bbs features.
Results
C. elegans bbs mutants share ciliary defects, reduced body size, delayed developmental timing, and reduced roaming
We performed a thorough phenotypic and behavioural assessment of severe or complete loss-of-function (lf) mutants for the C. elegans bbs-1, -2, -7, -8 and -9 genes (Table S1). Consistent with previous reports on bbs-7 and -8 [13], all examined bbs mutants exhibited a failure in the uptake of a lipophilic dye DiI by sensory neurons (Figure 1A), confirming a common structural and functional deficit in the sensory cilia. In addition to sensory defects, we identified three novel bbs-associated phenotypes: decreased body size, altered dwelling/exploration behaviour and delayed developmental timing.

Despite grossly normal body morphology, bbs mutants shared a reduced body length by ∼11–28% when compared to wild-type animals. Defects were visible during early larval stages and persisted throughout adulthood (Figure 1B and Figure S1). In these analyses, we defined a 3.5% or greater difference as biologically relevant in body size change, as this was the upper range for the coefficient of variance in young adult wild-type populations. The reduced body length is caused by the loss of BBS function, as it was fully rescued by introducing a wild-type genomic or cDNA copy of bbs into the respective mutants (Figure 1C). A decrease in body width was also characteristic of bbs mutants (Figure 1D). By DAPI staining of nuclei we did not observe differences in tissue and cell numbers between wild-type and bbs-7 animals (data not shown). The overall decrease in body size is thus best attributed to a smaller, averaged cell size in bbs mutants.
bbs double and triple mutants exhibited smaller body sizes that were no more severe than the smallest bbs single mutant. Although small differences between the body length of bbs single, double and triple mutant strains were observed as animals aged, the effects, however, were not additive (Figure 1B), consistent with BBS proteins functioning in the same biological processes to regulate ciliary development and function [6]–[7], [37].
We further examined the body size of a number of IFT mutants, including che-2, -3, and -11, as well as osm-3, -5, -6 and klp-11, all of which display defects to cilia structure and abnormal dye filling (dyf). Only some exhibited decreases in body size; among them, che-11 mutants exhibited the most significant decrease (by 11% in young adulthood), but not as severely as in age-matched bbs mutants (Figure S2A). Notably, a loss of the IFT motors, KLP-11 and OSM-3 kinesins, and CHE-3 dynein had little or no effect on body size. Therefore while sensory neurons affect body size, the dyf phenotype, caused by defective IFT transport, is not indicative of severe body size defects. The BBSome, exclusively expressed in ciliated sensory neurons, has a greater influence in the regulation of body size, indicating a role beyond bridging IFT motors for BBS proteins in these neurons.
C. elegans exhibits a defined developmental time course [38]. Multiple bbs mutant strains exhibited slower larval development (Figure 1E), resulting in a 2.3–6.2 hour delay between the first (L1) and last (L4) larval stage. During foraging, C. elegans exhibits a combination of dwelling and exploration/roaming behaviours that are altered in some chemosensory defective mutants [33]. Multiple bbs mutants showed a 56% to 76% decrease in overall movement, or roaming (Figure 1F) when compared to wild-type animals. This behavioural change does not reflect a general loss of locomotor activity, as bbs mutant animals exhibit normal locomotion during roaming. These additional phenotypes support a notion that the C. elegans cilia regulate cellular processes in addition to taxis behaviours.
The loss of function of a guanylate cyclase subunit GCY-35 rescues a subset of endophenotypes in bbs mutants
Unlike all other bbs strains, MT3645 bbs-7(n1606) (received from the Caenorhabditis Genetics Center) displayed body size, roaming behaviour and developmental timing characteristics of wild-type animals. Upon genetic outcrossing, we re-isolated a homozygous bbs-7(n1606) strain that exhibited phenotypes characteristic of other bbs mutants. These defects were fully rescued by the expression of the wild-type genomic copy of bbs-7 (Figure 1D). We concluded through genetic analyses that a single modifying locus from the original MT3645 strain rescued a subset of the bbs-7 mutant endophenotypes (Figure 2).

We mapped this modifier allele, hp433, to gcy-35 (Materials and Methods), a gene encoding the α subunit of a soluble guanylate cyclase (sGC). sGC proteins are composed of a heme/NO binding (HNOB), a heme/NO binding associated (HNOBA), and a GC catalytic domain (reviewed in [39]). They are heterodimeric complexes consisting of α and β subunits to catalyze the conversion of GTP to cGMP. hp433 results in a frame-shift in the coding sequence and a premature termination codon in the HNOBA domain, causing a truncation of the GC catalytic domain (Figure 2A). We also saw that a deletion allele in the GC domain of gcy-35, ok769, functions as a recessive suppressor of bbs-7 mutants (Figure 2A and 2B).
While both gcy-35(lf) alleles exhibit similar body length to that of wild-type animals, they suppressed the significant body length defects of multiple bbs mutants (Figure 2B). In contrast, gcy-35(lf);dyf mutants showed little or no improvement to body length (Figure S2B). The consistent suppression observed in bbs mutant animals advocated strongly for the further investigation of gcy-35(lf) as an epistatic suppressor of bbs-mediated phenotypes. gcy-35 also modified the bbs endophenotypes in developmental timing and roaming. Both the developmental timing from the first (L1) to the last (L4) stages, and roaming scores of gcy-35 mutants were comparable to that of wild-type (Figure 2C and 2D). The developmental timing of gcy-35;bbs-7 and gcy-35;bbs-2 mutants was identical to that of wild-type animals, whereas gcy-35;bbs-8 animals showed a partial improvement over that of bbs-8 animals (Figure 2C). Roaming defects of bbs-2, bbs-7, and bbs-8 mutants were partially suppressed by gcy-35 (Figure 2D).
Other bbs phenotypes, such as shortened cilia and defective DiI uptake (Figure 2E and data not shown), were not rescued by gcy-35(lf) mutants. These results suggest that the ciliary structures remain impaired in gcy-35;bbs mutants, and that the cellular pathways regulating body size, developmental timing and roaming behaviours either function genetically downstream of, or differ from those involved in sensation.
The GCY-35/GCY-36 sGC complex influences the body size of bbs mutants through a subset of oxygen sensing body cavity neurons
GCY-35 and its partner GCY-36 form a heterodimeric sGC that modulates C. elegans behaviour in response to ambient oxygen concentrations [40]–[41]. gcy-36(db66) (lf) mutants [40] rescued the body size defect of bbs-7 mutants (Figure 2B). Moreover, gcy-35;gcy-36;bbs-7 animals exhibited a body size no different from either gcy-35;bbs-7 or gcy-36;bbs-7 (Figure 2B), consistent with GCY-35 and GCY-36 also functioning as a heterodimeric sGC to regulate body size.
We examined in which neurons this sGC influences body size using bbs mutants. Both GCY-35 and GCY-36 are expressed in the ciliated body cavity sensory neurons AQR and PQR, and a non-ciliated body cavity neuron URX; GCY-35 is additionally expressed in the non-ciliated ALN, PLN, and SDQ neurons [41]. AQR, PQR and URX expression of wild-type GCY-35 in gcy-35;bbs-7 animals reverted body size similarly to that of bbs-7 mutants, whereas GCY-35 expression in ALN, PLN, and SDQ had no effect (Figure 3A). Similarly, the expression of a GFP-tagged GCY-36 in AQR, PQR and URX neurons in bbs-7;gcy-36 animals was also sufficient to revert body size close to that of bbs-7 mutants (Figure 3A). This suggests a specific requirement of GCY-35 and GCY-36 in body cavity neurons to regulate body size.
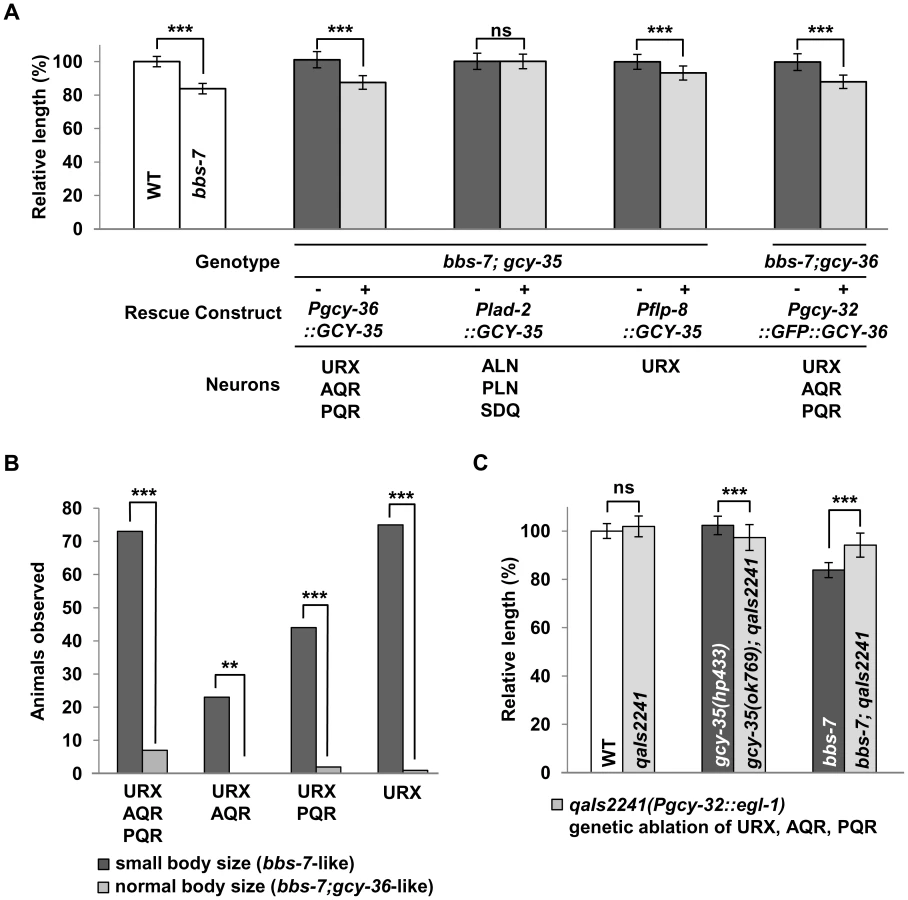
Among them, URX appears to be the most essential neuron, as restoring GCY-35 in URX (and other neurons not normally expressing gcy-35/gcy-36) by an exogenous promoter showed a partial, but significant reversion of the body size in gcy-35;bbs-7 animals (Figure 3A). Similarly, GCY-36 is most essential in URX: we conducted a mosaic analysis of bbs-7;gcy-36 animals carrying a functional GFP::GCY-36 transgene, and we observed expression of GFP::GCY-36 in URX of all rescued animals (Figure 3B). URX expression of the GCY-35/GCY-36 sGC is therefore essential, although not fully sufficient, to regulate the body size of bbs mutants.
Finally, if the body cavity neurons contribute to the small size phenotype of bbs mutants, their ablation by the transgene qaIs2241(Pgcy-36::EGL-1) [42] in a bbs background should also have a rescuing effect. While both qaIs2241 and gcy-35;qaIs2241 animals exhibited normal body sizes, bbs-7;qaIs2241 mutants showed a significantly increased body size when compared to bbs-7 animals (Figure 3C), further supporting that these neurons modulate the body size of bbs mutants.
The expression and localization of GFP::GCY-35 and GFP::GCY-36 are grossly normal in bbs mutants
Our genetic analyses indicate that BBS proteins negatively regulate sGC-mediated signalling activity. We examined if bbs mutants exhibit an elevated expression and/or expanded localization of sGC in these neurons. As reported [40], a functional GFP::GCY-36 localized largely to the soma and along the dendrites of AQR, PQR and URX. We did not observe significant changes in its expression or localization in bbs mutants (Figure S3), nor did we see a loss of ciliary localization as reported for the loss of a putative isoprenylation signal at the C-terminus [40]. bbs mutations did not perturb, at a gross level, the expression and localization of GFP::GCY-35 either (Figure S3). We did observe a high degree of morphological variability in the dendritic endings of URX, AQR, and PQR neurons as previously reported [43] in both wild-type and bbs mutants. However, given the morphological variability, we cannot exclude the possibility of subtle alterations in the cilium length of bbs mutants. Therefore, while AQR, PQR and URX neurons regulate the body size through a process that involves sGC activity, it does not appear to result directly from altered sGC protein level or subcellular localization.
The cGMP-dependent protein kinase (PKG) EGL-4 is a sGC effector in body size regulation
cGMP is a key secondary messenger (reviewed in [44]). In C. elegans, cGMP activates a heteromeric cGMP-gated ion channel TAX-2/TAX-4 for oxygen sensation and other sensory processes [45]. cGMP also activates a PKG, EGL-4, to regulate olfactory adaptation, life span, behavioural states and body size [33]–[34], [46]–[47]. Specifically, egl-4(lf) mutants exhibit a large body size, and are epistatic to the reduced body size and roaming behaviour of some sensory mutants; whereas a constitutively active, gain-of-function (gf) egl-4 mutation, causes a small body size [36]. The shared effects of bbs and egl-4 suggested that EGL-4 could be a downstream effector of sGCs in body size regulation.
We examined the egl-4 mutants (Figure 4) for their genetic interactions with bbs-7 and gcy-35 mutants. egl-4(lf) alleles were epistatic to bbs-7, gcy-35 and bbs-7;gcy-35 for body size and developmental timing (Figure 4B and 4C). egl-4(gf) also exhibited an epistatic effect to bbs-7 and bbs-7;gcy-35 mutants, although egl-4(gf);bbs-7 and egl-4(gf);bbs-7;gcy-35 mutants may exhibit a slightly more severe phenotype than egl-4(gf) that was only distinguished by statistical analyses (Figure 4B). By contrast, lf mutants for another cGMP effector, the cGMP-gated cation channel subunit TAX-2 and TAX-4, did not exert body size suppression for bbs-7 (Figure S4). Moreover, some TGF-β signalling mutants exhibited altered body size [48]–[50]. While EGL-4 was proposed to function genetically downstream of TGF-β signalling [33], body size defects of several TGF-β mutants exhibited additive effects in bbs-7 or bbs-7;gcy-35 backgrounds (Figure 4D), suggesting that TGF-β signalling and BBS-mediated body size regulation likely operates in an additive or parallel manner. Taken together, these results confirm a specific genetic relationship between bbs-7, gcy-35 and egl-4 for cilia-mediated body size regulation and developmental timing.
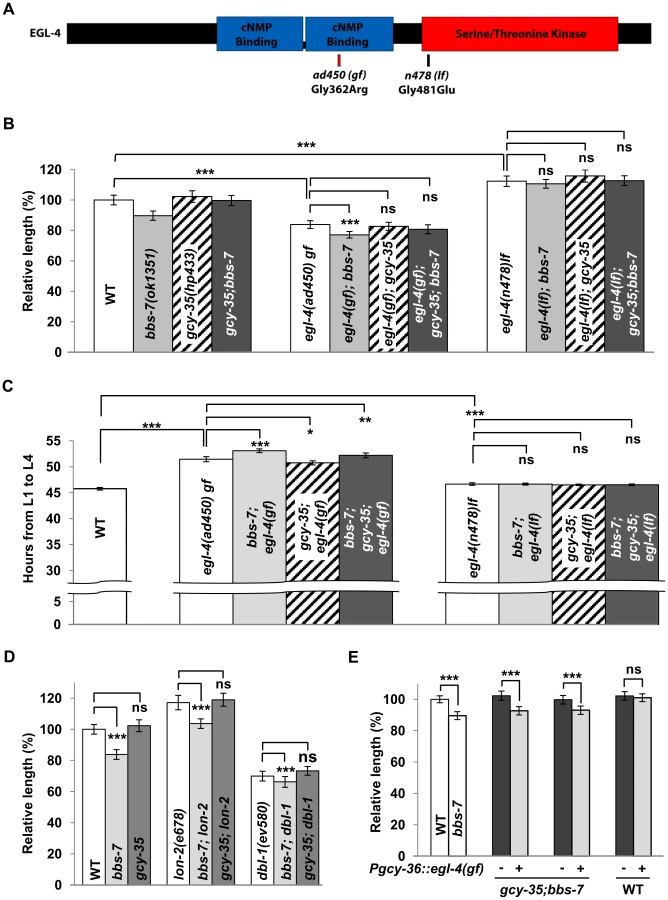
To further test if the body size influence of GCY-35/GCY-36 sGC functions through EGL-4, we expressed egl-4 cDNA harbouring the ad450(gf) mutation in AQR, PQR and URX neurons of gcy-35;bbs-7 animals. If GCY-35 regulates body size through activating EGL-4, a constitutively activated EGL-4(ad450) in these neurons should abrogate the body size suppression by the loss of gcy-35. Indeed, this transgene reverted animals to a bbs-7-like body size (Figure 4E). Wild-type animals expressing the same transgene did not exhibit changes in body size (Figure 4E). Not only is this result consistent with elevated cGMP signalling contributing to the reduced body size in bbs mutants, it further suggests that GCY-35/GCY-36 regulates body size through EGL-4 in the body cavity neurons.
BBS proteins are required in multiple sensory neurons to regulate body size
To investigate whether ciliary functions regulate body size strictly through body cavity neurons like GCY-35/GCY-36, we expressed a functional BBS-7 or GFP::BBS-2 in the AQR, PQR and URX neurons of bbs-7 or bbs-2 mutants, respectively. The small body size of respective bbs mutants was not rescued (Figure 5A). We did, however, observe a complete rescue of the body size with a pan-neuronally expressed GFP::BBS-2 in bbs-2 mutants (Figure 5A). Moreover, we observed a full or partial rescue of the body size defects when a functional BBS-7 was expressed in at least two non-overlapping groups of sensory neurons AWB, AWC, AWA, ADF and ASH, or, ADL, ADF, ASH, PHA and PHB (Figure 5A). All these neurons have sensory cilia exposed to outside of the body cavity. Therefore, while the GCY-35/GCY-36 sGC functions through body cavity neurons to regulate body size, restoring ciliary function in these neurons alone does not sufficiently reduce cGMP signalling to restore body size. Alternatively, other sensory neurons that input onto the body cavity neurons, may serve to regulate body size. Regardless, the observation that BBS proteins can influence body size through different groups of sensory neurons is reminiscent of the previously reported observation that the body size of egl-4(lf) mutants could be rescued by restoring EGL-4 expression in non-overlapping sets of sensory neurons [33].

GCY-35/GCY-36 acts as a predominant effector in cilia-mediated body size regulation
That multiple, non-overlapping and non-body cavity ciliated sensory neurons (CSNs) regulate body size suggests a cumulative effect on cGMP signalling-mediated body size regulation by multiple sensory neurons, or, a predominating effect by body cavity neurons in cilia-mediated body size regulation through GCY-35/GCY-36. The first model predicts that additional GCs in other sensory neurons would epistatically suppress body size. The C. elegans genome encodes 7 sGCs and 27 receptor-like GCs (rGCs) [51]. Single loss-of-function mutations in other sGCs, GCY-31, 32, 33, 34 and 37 all failed to alter bbs-7 body size defects (Figure S5A and data not shown). Of 16 rGC mutants tested, four exhibited modifying effects, two very mildly suppressing and two significantly exacerbating the smaller body size of bbs-7 animals, but none suppressed bbs-7 mutant phenotypes comparably to gcy-35/gcy-36 (Figure 5B and Figure S5A). We examined the effect of several double and triple rGC mutants on bbs-7 body size to explore the possibility that multiple rGCs function redundantly [52] but we did not observe obvious suppression effect in these additional mutants (Figure S5A).
We further examined the combinational effect of gcy-35 and other rGC mutants on bbs-7, including a mildly suppressing allele (gcy-4(tm1653)), two exacerbating alleles (gcy-7(tm901) and gcy-16(ok2538)) and three “neutral” alleles (gcy-23(ok797), gcy-28(tm2411) and gcy-25(tm4300)). We did not observe a significant body size improvement between gcy-35;bbs-7 and these triple mutants (Figure S5B). Therefore with the caveat that we have not exhausted the examination of all single or combinational GC mutants, GCY-35/GCY-36, through the body cavity neurons, act uniquely as a predominating effector for BBS-mediated body size regulation.
Discussion
In the present study, we show that C. elegans bbs mutants exhibit reduced body length, delayed development and altered roaming pattern, in addition to known sensory defects. These endophenotypes depend, fully or in part, on the GCY-35/GCY-36 sGC complex, through its effector EGL-4 PKG, in the AQR, PQR and URX body cavity neurons. On the other hand, body size can also be regulated via multiple, non-overlapping sets of non-body cavity sensory neurons. We propose that the loss of C. elegans BBS function in ciliated sensory neurons leads to non-cell autonomous, aberrant cGMP-PKG signalling in body cavity neurons, which contributes to abnormal body size and delayed development.
C. elegans bbs mutants exhibit non-cell autonomous endophenotypes
Ciliated sensory neurons transduce environmental cues into behavioural responses. In C. elegans bbs mutants, defective IFT and ciliary functions are reflected by chemosensory and thermosensory deficits [13]–[14]. Given the restricted expression of C. elegans BBS proteins in sensory neurons, the additional bbs endophenotypes such as developmental timing, body and inferred cell size, and roaming indicate that in addition to sensory perception, sensory neurons also participate in developmental regulation in a non-cell autonomous manner. These bbs endophenoptypes are not recapitulated by several dyf/IFT motor mutants, further implying that BBS proteins affect sensory neuron function in addition to their role in IFT.
While all bbs mutants share these endophenotypes, they exhibit small differences in the severity of phenotypic expression that could be attributed to specific allelic effects. Alternatively, BBS proteins could possess certain degrees of unknown functional specificity. This may not be so surprising given the difference in phenotypic expression among BBS patient populations [53]–[55], as well as the observation that tissue-specific BBS isoforms are responsible for some syndromic features [22], [56].
The involvement of primary cilia in signalling during development [57] also positions them to affect development in a non-autonomous fashion. For example, mouse BBS proteins are required in the hypothalamus to regulate leptin receptor trafficking and to prevent the onset of obesity [31]. Ciliary dysfunction therefore contributes to increased adiposity partly in a non-cell autonomous manner. The additional phenotypes of C. elegans bbs mutants, highlights the global and non-cell autonomous consequence of sensory ciliary dysfunction, which may also account for some phenotypic features in other ciliopathy models.
The GCY-35/GCY-36 sGC regulates body size through a mechanism divergent from oxygen sensing
Previous studies established that the GCY-35/GCY-36 sGC can regulate oxygen sensation through either the body cavity neurons, or another group of neurons [40]–[42]. Activated by oxygen, this complex catalyzes the conversion of GTP to cGMP, which subsequently activates the cGMP-gated cation channel TAX-2/TAX-4 to initiate hyperoxic avoidance responses [45]. Additional sGCs can act in body cavity neurons or other neurons under specific hypoxic conditions [45], [58].
GCY-35/GCY-36 modifies body size through a mechanism partly divergent from that of hyperoxic avoidance. GCY-35 is only necessary and sufficient in body cavity neurons that either have ciliated dendrites [59] or express some ciliated neuron-specific genes [60]. Furthermore, the loss of EGL-4, but not TAX-2 or TAX-4, suppresses the body size defects of bbs and dyf mutants. The loss of TAX-2 and TAX-4, in fact, slightly exacerbated bbs phenotypes (Figure S4), which may reflect an increased cGMP pool for EGL-4 activation or the loss of a potential EGL-4 phosphorylation target [35]. As well, despite some sGCs having overlapping expression profiles with GCY-35/GCY-36, other oxygen-responsive sGC mutants failed to suppress bbs-7 body size defects under standard culture conditions - possibly due to low activity under normoxia. Therefore, body cavity neurons, through GCY-35/GCY-36 activity, participate in developmental regulation through an alternate cGMP effector.
EGL-4 is present fairly ubiquitously, but the activation of EGL-4 in sensory neurons exerts a dominant influence on body size [33]. The genetic epistasis of both egl-4(lf) and egl-4(gf) alleles over that of bbs and bbs;gcy-35 argues in favour of BBS proteins and EGL-4 functioning through a shared cellular pathway to regulate body size and developmental timing. Expression of EGL-4(gf) in the body cavity neurons of gcy-35;bbs-7 mutants specifically alleviated the rescuing effect on body size, suggesting that increased EGL-4 activity, driven by increased availability of cGMP in body cavity neurons, contributes to the body size defects of some ciliary mutants (Figure 5C).
BBS proteins affect body size indirectly through body cavity neurons
The body size defects of ciliary mutants are rescued by non-overlapping sets of sensory neurons. However, restoring BBS function in body cavity neurons is insufficient to rescue the observed body size defects, giving rise to a possibility that the effect of cGMP signalling by body cavity neurons is indirectly moderated by a non-cell autonomous function of BBS proteins in ciliated sensory neurons. Furthermore, that URX, a pair of non-ciliated neurons, play a necessary role in this suppression indicates that BBS proteins are not directly influencing body size in these neurons. Our genetic analyses of the modifying effect of other GC mutants also support this scenario, as we have not found additional GC mutants that potently restore the body size of bbs mutants.
These results do not exclude the possibility that other GCs function redundantly in non-body cavity sensory neurons to influence body size through EGL-4/PKG (Figure 5D). The overexpression of egl-4(gf) in body cavity neurons was incapable of further reducing the body size of gcy-35;bbs-7 animals beyond that of bbs-7 mutants. This is in concordance with the ablation of body cavity neurons, which did not phenocopy the large body size of EGL-4 loss of function mutants, suggesting additional neuronal groups influence body size through EGL-4/PKG signalling. This study, however, establishes body cavity neurons as a predominating cGMP/PKG effector in body size regulation, and the ciliated sensory neurons as playing a key role in moderating cGMP signalling of these effector neurons.
Mechanisms on how dysfunctional ciliary sensory neurons lead to elevated cGMP/PKG signalling in these neurons are unknown. The body cavity neurons, AQR, PQR and URX do not receive extensive or direct synaptic inputs from sensory neurons where BBS proteins are sufficient to rescue body size. The non-cell autonomous effect of ciliated sensory neurons therefore suggests a potential involvement of indirect synaptic inputs, or other forms of neuronal communications, such as peptidergic and/or hormonal signalling between these neuronal groups. For example, body cavity neurons express the C. elegans homologue of the neuropeptide NPY receptor [61], making their activity susceptible to modulation by neuropeptides, some of which could be secreted by sensory neurons [62]. Sensory neurons also secrete insulin/IGF-like ligands, some of which may systematically affect neuronal states [63]–[64]. Indeed, insulin and leptin have been shown to regulate the activity of specific hypothalamic neurons [65]–[66]. Speculatively, C. elegans BBS proteins could affect the secretion of multiple signals by ciliated sensory neurons to regulate cGMP/EGL-4 signalling in the body cavity neurons.
A potential involvement of aberrant cGMP signalling in ciliopathies
While aberrant PCP, Shh and Wnt signalling underlie a number of ciliopathy features, the biology behind other ciliopathy features such as photoreceptor degeneration, and reduced body size in Bbs mice [15] remains unexplained. cGMP signalling plays key roles in biological processes such as phototransduction, axonal guidance, and synaptic plasticity (reviewed in [67]–[68]). PKGs have also been implicated in photoreceptor degeneration and dwarfism [69]–[70]. It is worth exploring the involvement of cGMP signalling in the underlying pathology of BBS and other ciliopathy features.
Materials and Methods
Strains
All strains were maintained on NGM plates at 20°C. C. elegans bbs, gcy and egl-4 strains were obtained from the CGC. CX7102 was obtained from the Bargmann lab. Genotypes for all strains are listed in Text S1.
Mapping and cloning of hp433
bbs-7(n1606);hp433 mutants were outcrossed twice against N2 by selecting animals that were genotyped for n1606 mutation, but exhibited normal body size. The hp433 mutation was crossed into bbs-7(ok1351) mutants and mapped based on the suppression of small body size and roaming defects using the SNP markers in the CB4856 strain, which placed it at a 93.5 kb interval between the SNPs pkp1133 and uCE1-1426. We conclude that hp433 encodes gcy-35 by: 1) Injection of three overlapping fosmids covering gcy-35, T04D3.5, and T04D3.t2, reverted the body size suppression in hp433; bbs-7 animals. A fragment of WRM641cB09 that encompassed a truncated gcy-35, but complete T04D3.5 and T04D3.t2 failed to revert the hp433 suppression; A genomic fragment containing only gcy-35 fully reverted the suppression. 2) gcy-35(ok769) animals shared the same synthetic phenotypes and genetic interactions with bbs-7 as hp433, while hp433;bbs-7(ok1351) animals also failed to complement gcy-35(ok769);bbs-7(ok1351). 3) Sequencing of gcy-35 identified a 2 bp deletion in exon 8.
Molecular biology, C. elegans phenotype examination
See Text S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BerbariNFO'ConnorAKHaycraftCJYoderBK 2009 The primary cilium as a complex signaling center. Curr Biol 19 R526 535
2. TobinJLBealesPL 2009 The nonmotile ciliopathies. Genet Med 11 386 402
3. BakerKBealesPL 2009 Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 151C 281 295
4. KimSKShindoAParkTJOhECGhoshS 2010 Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 329 1337 1340
5. OttoEAHurdTWAirikRChakiMZhouW 2010 Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet 42 840 850
6. NachuryMVLoktevAVZhangQWestlakeCJPeranenJ 2007 A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129 1201 1213
7. JinHWhiteSRShidaTSchulzSAguiarM 2010 The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141 1208 1219
8. OuGBlacqueOESnowJJLerouxMRScholeyJM 2005 Functional coordination of intraflagellar transport motors. Nature 436 583 587
9. PanXOuGCivelekoglu-ScholeyGBlacqueOEEndresNF 2006 Mechanism of transport of IFT particles in C. elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J Cell Biol 174 1035 1045
10. ZaghloulNAKatsanisN 2009 Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest 119 428 437
11. InglisPNOuGLerouxMRScholeyJM 2007 The sensory cilia of Caenorhabditis elegans. WormBook 1 22
12. AnsleySJBadanoJLBlacqueOEHillJHoskinsBE 2003 Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425 628 633
13. BlacqueOEReardonMJLiCMcCarthyJMahjoubMR 2004 Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 18 1630 1642
14. TanPLBarrTInglisPNMitsumaNHuangSM 2007 Loss of Bardet Biedl syndrome proteins causes defects in peripheral sensory innervation and function. Proc Natl Acad Sci U S A 104 17524 17529
15. NishimuraDYFathMMullinsRFSearbyCAndrewsM 2004 Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A 101 16588 16593
16. EichersERAbd-El-BarrMMPaylorRLewisRABiW 2006 Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum Genet 120 211 226
17. RossAJMay-SimeraHEichersERKaiMHillJ 2005 Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37 1135 1140
18. KulagaHMLeitchCCEichersERBadanoJLLesemannA 2004 Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet 36 994 998
19. BadanoJLLeitchCCAnsleySJMay-SimeraHLawsonS 2006 Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 439 326 330
20. YenHJTayehMKMullinsRFStoneEMSheffieldVC 2006 Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum Mol Genet 15 667 677
21. TayehMKYenHJBeckJSSearbyCCWestfallTA 2008 Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet 17 1956 1967
22. PretoriusPRBayeLMNishimuraDYSearbyCCBuggeK 2010 Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet 6 e1000884 doi:10.1371/journal.pgen.1000884
23. CorbitKCAanstadPSinglaVNormanARStainierDY 2005 Vertebrate Smoothened functions at the primary cilium. Nature 437 1018 1021
24. RohatgiRMilenkovicLScottMP 2007 Patched1 regulates hedgehog signaling at the primary cilium. Science 317 372 376
25. HuangfuDLiuARakemanASMurciaNSNiswanderL 2003 Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426 83 87
26. JonesCRoperVCFoucherIQianDBanizsB 2008 Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40 69 77
27. CorbitKCShyerAEDowdleWEGauldenJSinglaV 2008 Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 10 70 76
28. LinFHiesbergerTCordesKSinclairAMGoldsteinLS 2003 Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A 100 5286 5291
29. SchneiderLClementCATeilmannSCPazourGJHoffmannEK 2005 PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15 1861 1866
30. BerbariNFJohnsonADLewisJSAskwithCCMykytynK 2008 Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 19 1540 1547
31. SeoSGuoDFBuggeKMorganDARahmouniK 2009 Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 18 1323 1331
32. MarionVStoetzelCSchlichtDMessaddeqNKochM 2009 Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci U S A 106 1820 1825
33. FujiwaraMSenguptaPMcIntireSL 2002 Regulation of body size and behavioral state of C. elegans by sensory perception and the EGL-4 cGMP-dependent protein kinase. Neuron 36 1091 1102
34. HiroseTNakanoYNagamatsuYMisumiTOhtaH 2003 Cyclic GMP-dependent protein kinase EGL-4 controls body size and lifespan in C elegans. Development 130 1089 1099
35. L'EtoileNDCoburnCMEasthamJKistlerAGallegosG 2002 The cyclic GMP-dependent protein kinase EGL-4 regulates olfactory adaptation in C. elegans. Neuron 36 1079 1089
36. RaizenDMCullisonKMPackAISundaramMV 2006 A novel gain-of-function mutant of the cyclic GMP-dependent protein kinase egl-4 affects multiple physiological processes in Caenorhabditis elegans. Genetics 173 177 187
37. OuGKogaMBlacqueOEMurayamaTOhshimaY 2007 Sensory ciliogenesis in Caenorhabditis elegans: assignment of IFT components into distinct modules based on transport and phenotypic profiles. Mol Biol Cell 18 1554 1569
38. BrennerS 1973 The genetics of behaviour. Br Med Bull 29 269 271
39. KoeslingDFriebeA 1999 Soluble guanylyl cyclase: structure and regulation. Rev Physiol Biochem Pharmacol 135 41 65
40. CheungBHArellano-CarbajalFRybickiIde BonoM 2004 Soluble guanylate cyclases act in neurons exposed to the body fluid to promote C. elegans aggregation behavior. Curr Biol 14 1105 1111
41. GrayJMKarowDSLuHChangAJChangJS 2004 Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature 430 317 322
42. ChangAJChronisNKarowDSMarlettaMABargmannCI 2006 A distributed chemosensory circuit for oxygen preference in C. elegans. PLoS Biol 4 e274 doi:10.1371/journal.pbio.0040274
43. SwobodaPAdlerHTThomasJH 2000 The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol Cell 5 411 421
44. LucasKAPitariGMKazerounianSRuiz-StewartIParkJ 2000 Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 52 375 414
45. ZimmerMGrayJMPokalaNChangAJKarowDS 2009 Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61 865 879
46. NagamatsuYOhshimaY 2004 Mechanisms for the control of body size by a G-kinase and a downstream TGF-β signal pathway in Caenorhabditis elegans. Genes Cells 9 39 47
47. YouYJKimJRaizenDMAveryL 2008 Insulin, cGMP, and TGF-β signals regulate food intake and quiescence in C. elegans: a model for satiety. Cell Metab 7 249 257
48. GumiennyTLMacNeilLTWangHde BonoMWranaJL 2007 Glypican LON-2 is a conserved negative regulator of BMP-like signaling in Caenorhabditis elegans. Curr Biol 17 159 164
49. MoritaKFlemmingAJSugiharaYMochiiMSuzukiY 2002 A Caenorhabditis elegans TGF-β, DBL-1, controls the expression of LON-1, a PR-related protein, that regulates polyploidization and body length. EMBO J 21 1063 1073
50. PattersonGIPadgettRW 2000 TGF-β-related pathways. Roles in Caenorhabditis elegans development. Trends Genet 16 27 33
51. OrtizCOEtchbergerJFPosySLFrokjaer-JensenCLockeryS 2006 Searching for neuronal left/right asymmetry: genomewide analysis of nematode receptor-type guanylyl cyclases. Genetics 173 131 149
52. InadaHItoHSatterleeJSenguptaPMatsumotoK 2006 Identification of guanylyl cyclases that function in thermosensory neurons of Caenorhabditis elegans. Genetics 172 2239 2252
53. CarmiRElbedourKStoneEMSheffieldVC 1995 Phenotypic differences among patients with Bardet-Biedl syndrome linked to three different chromosome loci. Am J Med Genet 59 199 203
54. BealesPLWarnerAMHitmanGAThakkerRFlinterFA 1997 Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 34 92 98
55. BillingsleyGBinJFieggenKJDuncanJLGerthC 2010 Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J Med Genet 47 453 463
56. PretoriusPRAldahmeshMAAlkurayaFSSheffieldVCSlusarskiDC 2011 Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration. Hum Mol Genet
57. BadanoJLMitsumaNBealesPLKatsanisN 2006 The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7 125 148
58. CheungBHCohenMRogersCAlbayramOde BonoM 2005 Experience-dependent modulation of C. elegans behavior by ambient oxygen. Curr Biol 15 905 917
59. CoatesJCde BonoM 2002 Antagonistic pathways in neurons exposed to body fluid regulate social feeding in Caenorhabditis elegans. Nature 419 925 929
60. KunitomoHUesugiHKoharaYIinoY 2005 Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol 6 R17
61. de BonoMBargmannCI 1998 Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94 679 689
62. RogersCRealeVKimKChatwinHLiC 2003 Inhibition of Caenorhabditis elegans social feeding by FMRFamide-related peptide activation of NPR-1. Nat Neurosci 6 1178 1185
63. PierceSBCostaMWisotzkeyRDevadharSHomburgerSA 2001 Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev 15 672 686
64. CornilsAGloeckMChenZZhangYAlcedoJ 2011 Specific insulin-like peptides encode sensory information to regulate distinct developmental processes. Development 138 1183 1193
65. BelgardtBFHuschARotherEErnstMBWunderlichFT 2008 PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab 7 291 301
66. KlockenerTHessSBelgardtBFPaegerLVerhagenLA 2011 High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci 14 911 918
67. SchlossmannJFeilRHofmannF 2005 Insights into cGMP signalling derived from cGMP kinase knockout mice. Front Biosci 10 1279 1289
68. HofmannFFeilRKleppischTSchlossmannJ 2006 Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 86 1 23
69. PfeiferAAszodiASeidlerURuthPHofmannF 1996 Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science 274 2082 2086
70. Paquet-DurandFHauckSMvan VeenTUeffingMEkstromP 2009 PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J Neurochem 108 796 810
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2011 Číslo 10
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains