
Junín Virus Infection of Human Hematopoietic Progenitors Impairs Proplatelet Formation and Platelet Release via a Bystander Effect Involving Type I IFN Signaling
Argentine hemorrhagic fever (AHF) is an endemo-epidemic disease caused by Junín virus (JUNV), a member of the arenaviridae family. Although a recently introduced live attenuated vaccine has proven to be effective, AHF remains a potentially lethal infection. Like in other viral hemorrhagic fevers (VHF), AHF patients present with fever and hemorrhagic complications. Although the causes of the bleeding are poorly understood, impaired hemostasis, endothelial cell dysfunction and low platelet counts have been described. Thrombocytopenia is a common feature in VHF syndromes, and it is a major sign for its diagnosis. However, the underlying pathogenic mechanism has not yet been elucidated. We hypothesized that thrombocytopenia results from a viral-triggered alteration of the megakaryo/thrombopoiesis process. Therefore, we evaluated the impact of JUNV on megakaryopoiesis using an in vitro model of human CD34+ cells stimulated with thrombopoietin. Our results showed that CD34+ cells are infected with JUNV in a restricted fashion. Infection was transferrin receptor 1 (TfR1)-dependent and the surface expression of TfR1 was higher in infected cultures, suggesting a novel arenaviral dissemination strategy in hematopoietic progenitor cells. Although proliferation, survival, and commitment in JUNV-infected cultures were normal, viral infection impaired thrombopoiesis by decreasing in vitro proplatelet formation, platelet release, and P-selectin externalization via a bystander effect. The decrease in platelet release was also TfR1-dependent, mimicked by poly(I:C), and type I interferon (IFN α/β) was implicated as a key paracrine mediator. Among the relevant molecules studied, only the transcription factor NF-E2 showed a moderate decrease in expression in megakaryocytes from either infected cultures or after type I IFN treatment. Moreover, type I IFN-treated megakaryocytes presented ultrastructural abnormalities resembling the reported thrombocytopenic NF-E2−/− mouse phenotype. Our study introduces a potential mechanism for thrombocytopenia in VHF and other diseases associated with increased bone marrow type I IFN levels.
Published in the journal:
Junín Virus Infection of Human Hematopoietic Progenitors Impairs Proplatelet Formation and Platelet Release via a Bystander Effect Involving Type I IFN Signaling. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000847
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000847
Summary
Argentine hemorrhagic fever (AHF) is an endemo-epidemic disease caused by Junín virus (JUNV), a member of the arenaviridae family. Although a recently introduced live attenuated vaccine has proven to be effective, AHF remains a potentially lethal infection. Like in other viral hemorrhagic fevers (VHF), AHF patients present with fever and hemorrhagic complications. Although the causes of the bleeding are poorly understood, impaired hemostasis, endothelial cell dysfunction and low platelet counts have been described. Thrombocytopenia is a common feature in VHF syndromes, and it is a major sign for its diagnosis. However, the underlying pathogenic mechanism has not yet been elucidated. We hypothesized that thrombocytopenia results from a viral-triggered alteration of the megakaryo/thrombopoiesis process. Therefore, we evaluated the impact of JUNV on megakaryopoiesis using an in vitro model of human CD34+ cells stimulated with thrombopoietin. Our results showed that CD34+ cells are infected with JUNV in a restricted fashion. Infection was transferrin receptor 1 (TfR1)-dependent and the surface expression of TfR1 was higher in infected cultures, suggesting a novel arenaviral dissemination strategy in hematopoietic progenitor cells. Although proliferation, survival, and commitment in JUNV-infected cultures were normal, viral infection impaired thrombopoiesis by decreasing in vitro proplatelet formation, platelet release, and P-selectin externalization via a bystander effect. The decrease in platelet release was also TfR1-dependent, mimicked by poly(I:C), and type I interferon (IFN α/β) was implicated as a key paracrine mediator. Among the relevant molecules studied, only the transcription factor NF-E2 showed a moderate decrease in expression in megakaryocytes from either infected cultures or after type I IFN treatment. Moreover, type I IFN-treated megakaryocytes presented ultrastructural abnormalities resembling the reported thrombocytopenic NF-E2−/− mouse phenotype. Our study introduces a potential mechanism for thrombocytopenia in VHF and other diseases associated with increased bone marrow type I IFN levels.
Introduction
Viral hemorrhagic fever (VHF) is an acute systemic febrile syndrome caused by a diverse group of RNA viruses from the Arenaviridae, Bunyaviridae, Filoviridae, and Flaviviridae viral families [1]–[3]. Patients with this syndrome present with a combination of fever, prostration, malaise, and differing degrees of hematological complications [1], [4]. When severe, patients with VHF can present with generalized bleeding that results from alterations of the vascular endothelium, blood coagulation components and platelet levels [5]–[8]. Moreover, a plasma platelet aggregation inhibitor has been described in patients with different types of VHF [9], [10]. These findings suggest that different etiologic agents of VHF may share common processes for disturbing hemostasis during infection, leading to a systemic and frequently fatal disease. In fact, thrombocytopenia is one of the most consistent findings among human patients and experimental animal models of VHF, and it is used as a major diagnostic feature in patients with VHF [11], [12]. However, the causes of the thrombocytopenia associated with VHF remain poorly understood. In this connection, disseminated intravascular coagulation (DIC) could explain platelet consumption; nevertheless, the occurrence of DIC in VHF infections is inconclusive, at least for the arenavirus family [12]. Therefore, a high level of splenic sequestration or impaired megakaryo/thrombopoiesis could be the major physiopathogenic mechanisms responsible for the low platelet count.
We hypothesized that the thrombocytopenia observed in VHF is the result of a viral-triggered alteration of the megakaryo/thrombopoiesis process. Support for this hypothesis comes from studies of experimental murine lymphocytic choriomeningitis virus (LCMV) infection showing an association between thrombocytopenia and reduced megakaryocyte number at the bone marrow level [13] and the observation that the reversible depression of hematopoiesis during early LCMV infection is a direct effect of IFN α/β [14]. In addition, megakaryocyte alterations were found in patients with Argentine hemorrhagic fever (AHF) [15] and Dengue [16], and in experimental studies involving Junín virus (JUNV) infection of guinea pigs [17] and the primates Callithrix jacchus [18] and Rhesus macaques [19].
AHF is an endemo-epidemic disease caused by JUNV, a member of the Arenaviridae family. Although a recently introduced live attenuated vaccine has proven to be effective, AHF remains a potentially lethal infection and JUNV is considered to be a potential biological weapon [20]. Relative to other etiologic agents of VHF, JUNV is a suitable model for studying VHF pathogenesis due to the availability of a relatively large amount of data from both human and experimental infections, viral strains with known nucleotide sequences, several monoclonal antibodies against various components of the virus and an effective protective vaccine [21]–[23].
In this study, we evaluated whether the different megakaryopoiesi/thrombopoiesis stages (including proliferation, commitment, maturation, proplatelet formation and platelet release and function) are affected by JUNV infection of hematopoietic progenitor cells. Our results showed that human CD34+ cells are infected by JUNV. Furthermore, we have identified selective inhibition of platelet release involving transferrin receptor 1 (TfR1) and type I IFN (IFN α/β) as paracrine mediators.
Results
An in vitro model of megakaryopoiesis
To analyze whether the thrombocytopenia observed in AHF patients could be the result of altered megakaryopoiesis, we used an in vitro model of human CD34+ cells stimulated with thrombopoietin (TPO). Under our culture conditions, TPO stimulation promoted proliferation, an increase in cell size and CD41 expression and a decrease in CD34 expression (Figure 1A–C). The formation of long, branching processes called proplatelets, which showed small platelet-sized swellings that represented the developing platelets [24], was also observed on days 11–12 of culture (Figure 1E). In agreement with previous reports, platelets were released into the culture medium [25]. The kinetics of in vitro platelet release started at days 11–13, peaked at day 17 and declined after 21 days, correlating with the apoptosis/senescence of the cell culture (Figure 1D).

JUNV infection selectively impairs in vitro platelet generation
Figure 2 shows that JUNV infection of CD34+ cells influenced neither their apoptosis, their proliferation rate, their maturation (CD42b+ and ploidy levels), megakaryocyte differentiation (CD41+) nor the clonogenic ability of megakaryocyte progenitors. Surprisingly, JUNV infection induced a significant decrease in both proplatelet formation (Figure 3A and B) and platelet release (Figure 3C and D) when compared to mock- or UV-irradiated JUNV-infected cultures. These results indicate that viral infection selectively hindered two key steps in thrombopoiesis that represent the last stage of the megakaryocyte lifespan, and viral replication was necessary to observe this effect.


To gain a deeper insight into the platelet biology of JUNV-infected cultures, we next explored the ability of platelets to express P-selectin. Our flow cytometric analysis showed that 11.1±3.3% of resting platelets generated in UV-irradiated JUNV-infected cultures expressed P-selectin on their surface, and after thrombin stimulation this rose to 49.2±4.1% (n = 3, p<0.05). In contrast, P-selectin externalization was significantly reduced in platelets derived from JUNV-infected cultures (3.3±1.3% and 13.5±4.1% for resting and thrombin-stimulated samples, respectively, n = 3).
Susceptibility of CD34+ cells to JUNV infection
As shown in Figure 4A, CD34+ cells were susceptible to JUNV infection. At ten days post-infection (p.i.), RNA from JUNV-inoculated CD34+ cells displayed a fragment of the viral genome, while RNA from cultures exposed to the UV-irradiated JUNV strain did not. Immunofluorescence and flow cytometric studies revealed that 4.1±0.2 and 4.9±0.5% of the cells were infected, respectively (Figure 4B and C). These results were in sharp contrast to the 68±11% of JUNV-positive cells observed after in-parallel infection of the susceptible Vero-76 cell line [26] (Figure 4D). Although some megakaryocytes were clearly positive for JUNV, most of the susceptible cells were not (Figure 4B and C). These results suggest that viral infection per se is not a direct cause of the observed reduced thrombopoiesis.

Role of TfR1 in JUNV infection and impaired platelet production
In contrast to Lassa and other old world arenaviruses, which use α-dystroglycan to infect cells, it has been reported that TfR1 is the cellular receptor for several new world arenaviruses, including JUNV [27]. Therefore, we evaluated the expression of this protein in JUNV-infected cultures. Interestingly, TfR1 expression was significantly higher in JUNV-infected CD34+ cells compared to UV-irradiated JUNV-infected cells, suggesting that JUNV infection promotes further infection (Figure 5A and B).

Having demonstrated that TfR1 was up-regulated by viral infection, we next examined whether TfR1 has a role in JUNV infection and in the inhibitory effect of JUNV on platelet release. When CD34+ cells were incubated with a specific monoclonal antibody (mAb) against TfR1, neither viral antigens nor the inhibitory effect of JUNV on the number of platelets generated were observed (Figure 5C and D). In contrast, incubation with anti-HLA-ABC (an irrelevant mAb) had no effect. Similar results were obtained when TfR1 was first down-regulated by iron overload. However, up-regulation of TfR1 levels by iron deprivation did not modify the effect of JUNV on platelet generation (Figure 5D). These results not only unveil a potential new mechanism by which JUNV spreads its infection in hematopoietic progenitor cells, but they also provide evidence that the reduced thrombopoiesis is specifically related to JUNV infection.
Involvement of the type I IFN pathway on JUNV-induced impairment of platelet release
Because the number of infected cells was low and only a few were positively identified as megakaryocytes, it was reasonable to consider that the decrease in platelet release was most likely the result of a bystander cell effect rather than a direct effect of JUNV on megakaryocytes. Thus, to further study the biological effects of viral infection on platelet release, we analyzed the effect of poly(I:C), which mimics the double-stranded RNA (dsRNA) product from the replicative cycle of most viruses [28]. Figure 6A shows that the addition of poly(I:C) at early time points to TPO-stimulated CD34+ cell cultures significantly decreased platelet release. Similarly to JUNV infection, this inhibition was not related to apoptosis induction or a decrease in megakaryocyte generation (data not shown). These results suggest that the reduction in platelet release could be a more generalized effect and may not be restricted to JUNV infection.

Because poly(I:C) is a potent inducer of IFN β release in vitro and in vivo [29], we next tested if this was a functional mechanism in our in vitro system. While UV-irradiated JUNV-infected-CD34+ cell cultures stimulated by TPO did not express IFN β mRNA, increasing expression over time was observed in JUNV-infected cultures (Figure 6B). Accordingly, 10 U/ml of IFN β efficiently inhibited platelet release (Figure 6C). However, when used at concentrations of 100 U/ml or higher, it not only impaired platelet generation but also significantly decreased cell number (Figure 6D) without affecting the apoptosis rate (data not shown). These results suggest that IFN β might differentially regulate cellular responses, affecting only platelet production at low levels but affecting the cell cycle at higher levels. That IFN β was responsible for the JUNV-mediated biological effect was further supported by the observation that platelet production was restored to normal values in JUNV-infected cultures when IFN β was blocked by a neutralizing Ab (Figure 6E). To the best of our knowledge, the expression of type I IFN receptor in megakaryocytes has not been previously described. Thus, we examined the presence of mRNAs in purified megakaryocytes. RT-PCR studies revealed that megakaryocyte precursors transcribe both subunits of the IFN α/β receptor mRNAs (Figure 6F).
Because another general feature of the cells' response to viral infection is an increase in IFN α production, as a downstream product of the IFN β pathway, this molecule was also studied. Stimulation of CD34+ cells with IFN α mimicked the results obtained with IFN β (data not shown). These results were not surprising bearing in mind that both IFNs are ligands for the same receptor [30]. Overall, these results strongly indicate that the type I IFN signaling pathway may have a key role in the defective platelet release mediated by JUNV in vitro.
Considering that in vivo studies showed that megakaryocytes from type I IFN receptor knockout mice are more susceptible to LCMV infection [14], we next explored the role of type I IFN in JUNV infection. Figure 6G shows that the percentage of infected cells increased from 5% to 10% when the biological activity of IFN β was neutralized. However, recombinant IFN β did not modify TfR1 expression (Figure 6H) suggesting that viral replication in hematopoietic progenitor cells is controlled, at least in part, by IFN β production in a TfR1-independent manner.
Mechanisms involved in JUNV inhibition of platelet formation
Although the signaling pathways regulating thrombopoiesis are still under study, Src kinases and several transcription factors have been shown to be involved as regulators of this process [31]. To clarify the mechanistic basis of the impaired platelet production in infected cultures, we first determined whether Src kinases were involved by using a specific inhibitor (PP2). Figure 7A shows that although Src inhibition enhanced platelet release in control cells, it did not prevent the JUNV-induced reduction in platelet number.
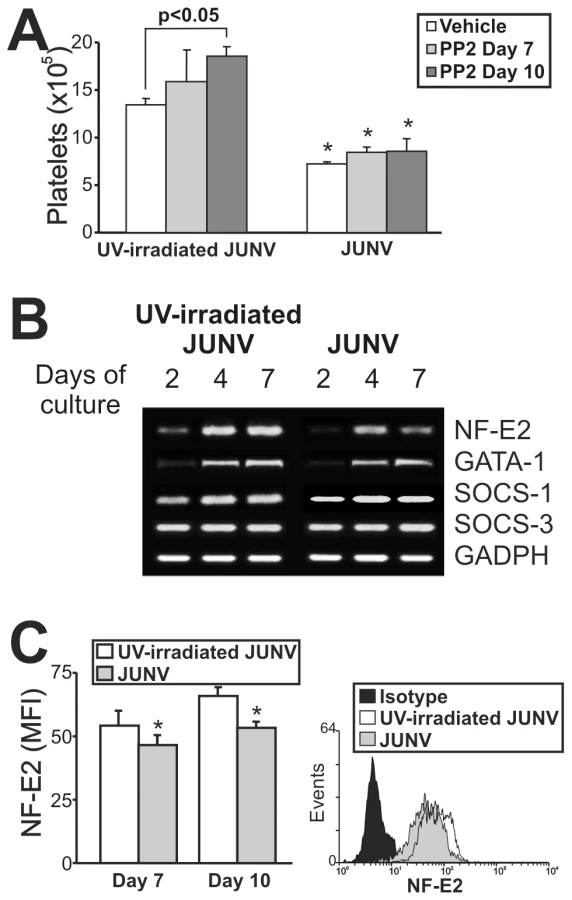
To examine the levels of relevant factors involved in megakaryo/thrombopoiesis, we performed a semi-quantitative RT-PCR assay. We found that the mRNAs of SOCS-1, SOCS-3 and GATA-1, three molecules involved in the initial steps of megakaryocyte development [32], were similar in JUNV- and UV-irradiated JUNV-infected cultures. In contrast, NF-E2, the main transcription factor regulating platelet biogenesis [33], was lower in JUNV-infected samples (Figure 7B). In addition, the total amount of NF-E2 protein was moderately but significantly diminished in the megakaryocytic population by viral infection (Figure 7C).
Type I IFN decreases NF-E2 expression and induces intrinsic megakaryocytic abnormalities
Having demonstrated that JUNV infection triggers the production of type I IFN and impairs platelet formation, in the next experiments we examined whether type I IFN was capable of regulating the expression of NF-E2. The results showed that like viral infection, the exposure of purified megakaryocytes to IFN β (Figure 8A) or α (data not shown) resulted in lower NF-E2 expression compared to control samples.

Because NF-E2−/− mice lack circulating platelets and their megakaryocytes show no cytoplasmic platelet formation [33], we analyzed the effect of type I IFN on megakaryocytes at the ultrastructural level. Electron microscopic studies revealed that in contrast with control megakaryocytes, which had a lobulated nucleus and abundant cytoplasm with a mature demarcation membrane system (DMS), in the IFN β-treated cells the DMS was almost absent and lacked organization and platelet territories (Figure 8B). The nucleus looked normal and there was a large cytoplasm, but the contents of the megakaryocyte were not evenly distributed throughout the cytoplasm, including areas with missing granules and organelles. Finally, we observed patches of released platelets that were produced only by megakaryocytes in the control cultures. Interestingly, all these observations resemble the NF-E2−/− megakaryocyte phenotype [33]. IFN α induced similar ultrastructural abnormalities (data not shown).
Discussion
In this paper, we present evidence that JUNV infects hematopoietic progenitors in a restricted fashion leading to a significant inhibition of thrombopoiesis, including proplatelet formation, platelet release and platelet function.
Previous studies of the effect of viral infection on megakaryocytes derived from CD34+ cells showed that human immunodeficiency virus (HIV), human herpesvirus 6 and human cytomegalovirus negatively affected the survival, differentiation and/or maturation of megakaryocyte progenitors [34]–[36]. In contrast, we here demonstrated that JUNV infection had no significant effect on the proliferation, clonogenic ability or maturation of megakaryocyte progenitors, but it induced a profound decrease in proplatelet formation and platelet release. Although impairment of platelet production has been suggested in HIV patients [37], [38], to the best of our knowledge, this is the first time that a viral infection has been shown to selectively affect the two last key steps of thrombopoiesis.
It has recently been demonstrated that TfR1 is the receptor for some new world arenaviruses, including JUNV [27]. Our results show that blocking or down-regulating TfR1 surface expression in CD34+ cells significantly reduced both JUNV replication as well as the decrease in platelet formation induced by JUNV infection, indicating that viral infection is a necessary event for the inhibition of platelet formation. Moreover, the fact that UV-irradiated virus had no effect on proplatelet production and platelet release indicates that viral replication is also necessary to hinder thrombopoiesis. These data also implicate TfR1 as the main route of viral entry into hematopoietic progenitor cells. This is particularly relevant considering that it has recently been shown that pathogenic strains can use TfR1-dependent or independent pathways [39]. The observation that JUNV infection up-regulates TfR1 in CD34+ cells could represent a viral dissemination strategy at least in hematopoietic bone marrow cells. However, in vivo experiments will be required to determine its relevance.
Remarkably, we found that only 5% of total cells were infected, therefore the observed 50% reduction in platelet production appeared to be due to a selective bystander effect from infected cells rather than a direct effect of viral replication on megakaryocyte biology. Moreover, poly(I:C), a synthetic analogue of the dsRNA associated with the replicative cycle of most viruses [28], mimicked the effect of JUNV infection. Although transfected poly(I:C) can be sensed by the retinoic induced gene (RIG)-I-like RNA helicases that stimulate IFN α and β expression, naked poly(I:C) is recognized by TLR3, which predominantly stimulates the expression of NF-kB and IFN β [40]. Therefore, IFN β appeared to be a suitable mediator responsible for the reduced platelet production mediated by JUNV infection of hematopoietic cells. Our results showing: a) the JUNV-dependent induction of IFN β mRNA synthesis, b) the suppressive effect of IFN β on platelet biogenesis, c) the expression of both subunits of the IFN β receptor in purified megakaryocytes, and d) the reversion of the inhibitory effect of JUNV on platelet formation with a neutralizing Ab against IFN β strongly suggest that IFN β is a central mediator in the JUNV-dependent inhibition of platelet release. Similar results were obtained with IFN α (data not shown). This would not be surprising as both IFNs are ligands for the same receptor and IFN α is a downstream product of IFN β production and signaling [30]. Furthermore, both cytokines may be the mediators involved in the bystander effect, and this supports the hypothesis that JUNV and any other diseases that are associated with increased type I IFN levels in the bone marrow milieu may result in thrombocytopenia.
Surprisingly, despite the low number of infected cells, only few were megakaryocytes. Although we have not yet determined the identity of the vWF negative-infected cells, they could be megakaryocytes with low vWF content because it has been recently shown that treatment of megakaryocytes with IFN α inhibited 70% of vWF RNA expression [41].
In vivo, JUNV may infect other cells in the bone marrow environment, and CD34+ progenitor cells may be exposed to higher levels of type I IFN than produced in our in vitro system. Interestingly, we found that while low IFN β concentrations selectively impaired thrombopoiesis, treatment of CD34+ cells with higher concentrations also reduced megakaryocyte numbers without modifying the apoptosis rate, suggesting an IFN β-mediated cell cycle arrest. Remarkably, both phenomena were independently described by different groups using recombinant IFN α: while Wang et al. demonstrated that IFN α hinders mouse megakaryopoiesis altering proliferation and ploidy [42], Yamane et al. found that IFN α decreases platelet production without altering megakaryocyte growth [41]. Although these findings appears to be controversial, our results suggest that the levels of type I IFN in the bone marrow could define the cell fate of megakaryocytes, affecting either their proliferation or their ability to produce platelets.
Our analysis of the signaling pathways involved in the JUNV-mediated inhibition of platelet production indicated that SOCS-1, SOCS-3, GATA-1, and Src, which are involved in the processes of proliferation, differentiation, and proplatelet formation, respectively [31], [32], [42], were not involved. In contrast, JUNV-infected samples did show lower levels of both NF-E2 mRNA and protein. Interestingly, we also found that type I IFN downregulates NF-E2 in megakaryocytes. As this transcription factor plays a major role in terminal differentiation of megakaryocytes and platelet release [33], it is conceivable that the reduced synthesis of NF-E2 mediated by type I IFN may be one of the molecular pathogenic mechanisms reducing platelet release in JUNV infection (Figure 9). In fact, thrombocytopenia driven by recombinant IFN α has been related to the selective inhibition of cytoplasmic maturation accompanied by downregulation of the expression of the transcription factors NF-E2, GATA-1 and MafG/HPRT [41]. Our ultrastructural studies using IFN β also showed profound alterations in the cytoplasm that could account for the impaired platelet formation. Additionally, we also found a lower number of platelet granules in type I IFN-treated megakaryocytes. Interestingly, NF-E2−/− mice have no circulating platelets and their megakaryocytes present ultrastructural abnormalities that are quite similar to those of our type I IFN-exposed megakaryocytes [33].
As regarding type I IFN's effect on platelet function, elegant studies from Iannacone et al. revealed that mice infected with LCMV exhibit a type I IFN-dependent platelet dysfunction that, if associated with thrombocytopenia below a critical threshold, results in severe bleeding and acute anemia [43]. Because they did not observe a direct effect of IFN β on platelet activation, they suggested that the platelet dysfunction could be the result of a direct effect of IFN β on parent megakaryocytes or due to the release from endothelium of platelet inhibitors. Our results showing that platelets derived from JUNV-infected cultures had less P-selectin expression upon thrombin stimulation together with the observation that type I IFN-treated megakaryocytes have almost no dense or alpha granules strongly support the hypothesis that megakaryocyte exposure to type I IFN driven by viral infection of bone marrow cells results not only in a reduced number of platelets but also in the generation of dysfunctional platelets. Nevertheless, more studies are required to further elucidate the hemostatic status and to determine whether similar structural alterations are observed in platelets from VHF patients.
Do the in vivo data support our hypothesis regarding the critical role of IFN I in the thrombocytopenia present in VHF patients? High levels of circulating IFN a that correlate with virulence and prognosis has been described in different VHF [44]–[46]. Moreover, during systemic viral infection, bone marrow hematopoietic cells appear to be the most important source of type I IFN [47]. In addition, a direct relationship of thrombocyto/neutropenia driven by type I IFN was demonstrated in LCMV infected mice. In that case, however, the condition was mediated by transient bone marrow aplasia [14]. Overall, these data support a role for type I IFN as a pathogenic factor for the thrombocytopenia observed in VHF patients.
In conclusion, we provide the first evidence linking viral infection of human hematopoietic progenitors with selective inhibition of thrombopoiesis through the type I IFN pathway. Our studies highlight a potential mechanism that leads to thrombocytopenia and bleeding in AHF and other diseases as a result of an increase in the levels of type I IFN in the bone marrow milieu. These data should be of use in the effort to find new therapeutic strategies for the thrombocytopenia that is associated with VHF.
Materials and Methods
Ethics statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Institutional Review Board of the National Academy of Medicine, Buenos Aires, Argentina. All patients provided written informed consent for the collection of samples and subsequent analysis.
Reagents
Fluorescein isothiocyanate (FITC)-conjugated monoclonal antibodies (mAbs) against CD34, glycoprotein (GP) IIb (CD41), GP Ib (CD42b), P-selectin (CD62p), phycoerytrin (PE)-conjugated anti-GP IIIa (CD41), unconjugated anti-human HLA-ABC, anti-CD71 Abs, and a fixation/permeabilization kit (BD Cytofix/Cytoperm™) were purchased from BD Biosciences (Franklin Lakes, NJ). FITC-conjugated anti-rabbit immunoglobulins (Igs) and anti-von Willebrand factor (vWF) Abs were obtained from Dako A/S (Glostrup, Denmark). Cy3-conjugated anti-mouse Ig was obtained from Zymed (San Francisco, CA). Recombinant human IFN α2b and rabbit polyclonal anti-human IFN β neutralizing Ab were obtained from Pestka Biomedical Laboratories (Piscataway, NJ). Rabbit polyclonal IgG anti-NF-E2 Ab was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Random hexamers, SuperScript III reverse transcriptase and Trizol were from Invitrogen. The synthetic analog of dsRNA polyriboinosinic polyribocytidylic acid, poly(I:C) and recombinant human IFN β1a were obtained from InvivoGen (San Diego, CA). The Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) was obtained from Biomol International LP (Plymouth Meeting, PA). Thrombopoietin (TPO) was obtained from Peprotech (Veracruz, Mexico). All of the other reagents were obtained from Sigma Chemical Co. (St. Louis, MO).
Isolation of CD34+ cells
Isolation of CD34+ cells was performed as previously described [48]. Briefly, human umbilical cord blood was collected during normal full-term deliveries and was used within 24 hr. After collection, samples were diluted one to three with PBS and centrifuged to remove platelets. Low density mononuclear cells were prepared by centrifugation of the remaining blood over a Ficoll Hypaque. Cells collected from the interface were washed, and CD34+ cells were purified using a magnetic cell-sorting system (Miltenyi Biotec, Bergisch Gladbach, Germany) in accordance with the manufacturer's recommendations. After two Mini-MACS column separations, the purity of the cell suspension was determined by flow cytometry and typically ranged between 95 and 99%. Cell viability was greater than 90%. Fresh CD34+ cells were used for each experiment.
Purification of megakaryocytes
Mature megakaryocytes were purified by immunomagnetic positive selection from TPO-stimulated CD34+ cultures on day 12 using anti-CD41 magnetic beads (Miltenyi Biotec) following the manufacturer's instructions. The purity of the final cell suspension was ≥95% and cell viability was greater than 80%.
Vero-76 cells
Monolayers of Vero-76 cells (CRL-1587, the American Type Culture Collection (ATCC), Manassas, VA) were grown in minimum essential medium (MEM) containing 10% fetal calf serum (FCS) and antibiotics.
Virus
A virulent strain of JUNV originally isolated from an AHF patient (P3441) was kindly provided by Dr. A. Ambrosio of the Instituto Nacional de Enfermedades Virales Humanas “Dr. Julio I. Maiztegui”, Pergamino, Argentina. Virus stocks were grown, identified, and quantified as described previously by using the JUNV-susceptible Vero-76 cell line. Virulence was tested in three-week-old guinea pigs and the median lethal dose, (LD50), assayed between 11 and 14 days p.i., was ≤10 PFU. When required, virus was subjected to UV inactivation for 20 min using a 365 nm UV bulb positioned 5 cm over the stock [49].
Cell infection
CD34+ cells (1×104) or a monolayer of Vero-76 cells were inoculated with JUNV at a multiplicity of infection (MOI) of one or the equivalent volume of UV-irradiated virus for 1 hr at 37°C. Mock-infected controls contained supernatants of Vero cells instead of JUNV. After washing, UV-irradiated JUNV-, JUNV- or mock-infected CD34+ cells were cultured in Iscove's Modified Dulbecco's Medium (IMDM, HyClone) containing 2 mM glutamine, 5% human serum (obtained by re-calcification of citrated platelet-free plasma), 25 ng/ml TPO and antibiotics (growth medium) at 37°C in a humidified atmosphere with 5% CO2 [50]. Fresh TPO (25 ng/ml) was added at day seven. Vero-infected cells were cultured for seven days in MEM containing 2% FCS and antibiotics.
Detection of JUNV proteins
Cells were washed with PBS (Ca2+- and Mg2+-free), cytocentrifuged on silanized glasses, fixed with 1% paraformaldehyde (PFA) for 20 min and permeabilized with 0.1% Tween for 10 min. The slides were incubated overnight at 4°C with a pool of specific mAbs against JUNV [22] and a rabbit-anti-human vWF polyclonal Ab to identify megakaryocytes. FITC-conjugated anti-rabbit and Cy3-conjugated anti-mouse Igs were then applied to the PBS-washed slides for 30 min at room temperature (RT). Abs were diluted with PBS containing 5% fetal bovine serum and 5% goat serum as blocking agents. The slides were counterstained with DAPI and examined under an Olympus BX60 microscope equipped with a 100× PlanApo objective (NA 1.4) and a 100-W mercury lamp. Images were acquired with an Applied Imaging Model 4912–5010/0000 charge coupled device (CCD) camera. Electron image acquisition was under the control of Cytovision software.
For flow cytometric studies, samples were washed, fixed with 1% PFA for 20 min, and permeabilized with 0.1% saponin for 10 min. Cells were then incubated for 1 hr at RT with a pool of specific mAbs against JUNV and a rabbit-anti-human vWF polyclonal Ab. After two washes, FITC-conjugated anti-rabbit and Cy3-conjugated anti-mouse Igs were diluted in 0.1% saponin and added to the samples for 30 min and analyzed on a FACSCalibur flow cytometer using CellQuest software (BD Biosciences).
Negative controls were performed using uninfected cells or omitting primary Abs. JUNV-susceptible Vero cells were used as a positive control.
Evaluation of megakaryocyte development
The levels of CD34, CD41 and CD42b in the TPO-stimulated CD34+ cell cultures were measured by immunostaining with saturating concentrations of specific FITC-labeled mAbs or an isotype-matched control after varying numbers of days. After 30 min of incubation, the samples were fixed with 1% PFA and analyzed by flow cytometry.
To evaluate megakaryocyte ploidy, cells were centrifuged for 10 min at 220×g, washed and fixed in 70% ethanol at −20°C overnight. Cells were then washed, resuspended in PBS, and incubated for 30 min at RT with saturating concentrations of FITC-conjugated anti-CD41 (or isotype control), 1 µg/ml propidium iodide, and 10 U/ml RNaseA. Cell ploidy was analyzed by flow cytometry.
Clonogenic progenitor assays
Aliquots of 5×103 CD34+ cells were resuspended in collagen-based, serum-free medium containing 50 ng/ml TPO (MegaCult, Stem Cell Technology, Vancouver, Canada) and seeded in double-chamber culture slides. After 12 days, megakaryocyte colonies were detected using an anti-CD41 antibody and an alkaline phosphatase detection system and were then counterstained with Evan's Blue. Two categories of colonies were identified: pure megakaryocyte colonies and mixed megakaryocyte colonies (distinguished by the presence of non-megakaryocyte cells within the same colony). Pure megakaryocyte colonies were scored according to their size: small CFU-megakaryocytes (3–20 cells), medium CFU- megakaryocytes (20–50 cells), and large CFU-megakaryocytes (>50 cells).
Proplatelet formation (PPF)
PPF was analyzed as previously described with some modifications [51]. Twelve-mm glass coverslips were coated with 100 µg/ml fibrinogen for 2 hr at RT and subsequently blocked with 1% bovine serum albumin for 1 hr. Cells cultured for 12 days were harvested, plated on coated coverslips in 24-well plates (1×105 cells per well) and allowed to adhere for 4 hr at 37°C and 5% CO2. PPF was then evaluated by fluorescence microscopy by staining cells with TRITC-conjugated phalloidin. DAPI was used as a counterstain. Proplatelet-forming megakaryocytes were identified as large cells exhibiting long filamentous structures. The extent of PPF was calculated as the percentage of proplatelet-bearing cells by counting 500 cells per treatment.
Determination of platelet number produced in culture
Platelets were counted using flow cytometry as previously described [25]. Briefly, cells were incubated with a FITC-labeled anti-CD41 mAb for 15 min and fixed with 1% PFA for 20 min. Cells from each culture condition were distributed in the same volume. For each sample, the acquisition rate was 1 µl/sec for 100 sec. Events were collected without gating using a log scale for size (FSC) and intracellular granularity (SSC). An analytical gate was determined based on the scatter properties of normal blood platelets treated similarly to the culture-derived platelets. This gate excluded large contaminating cells and small debris or microparticles. Culture-derived platelets were counted as CD41+ events with the same scatter properties as blood platelets.
P-selectin expression on in vitro-produced platelets
P-selectin expression on platelets was determined as previously described with minor modifications [52]. Briefly, culture-derived platelets were isolated by centrifugation at 1000×g in the presence of PGI2 to avoid platelet preactivation. The pellet was resuspended in modified Tyrode buffer (134 mM NaCl; 2.9 mM KCl; 0.34 mM Na2HPO412H2O; 12 mM NaHCO3; HEPES 20 mM; MgCl2 1 mM; glucose 5 mM, pH 7.3) and left to rest for 30 min at 37°C. The suspension was stimulated with 1 U/ml thrombin for 10 min at 37°C. After fixation, cells were stained with anti–P-selectin (CD62p)–FITC and anti-CD41-PE mAbs for 30 min and P-selectin expression was measured by flow cytometry using the characteristic forward and side scatter pattern of normal blood platelets treated similarly.
Expression of TfR1 and NF-E2
To determine the level of TfR1 expression on TPO-stimulated CD34+ cell membranes, cells were incubated with a FITC-conjugated anti-CD71 or an isotype-matched control mAb at the indicated days p.i. Samples were washed, fixed, and analyzed by flow cytometry.
CD34-derived megakaryocytes were tested for NF-E2 expression using a double-labeling technique (NF-E2/CD41). Cells were immunostained with a PE-conjugated anti-CD41 mAb or an isotype-matched control and fixed with 1% PFA at 4°C. After washing with PBS containing 0.1% saponin, cells were incubated first for 30 min in the same buffer with anti-NF-E2 polyclonal Ab at 4°C and then with FITC-conjugated swine anti-rabbit Igs. Cells were analyzed by flow cytometry. Non-specific fluorescence was assessed using rabbit serum instead of primary Ab. In selected experiments, NF-E2 expression was determined in purified mature megakaryocytes treated with type I IFN using the anti-NF-E2 polyclonal Ab (or rabbit serum) followed by FITC-conjugated swine anti-rabbit Igs.
RNA isolation and semi-quantitative RT-PCR
Total RNA was isolated from cell pellets using Trizol as recommended by the manufacturer. cDNA was synthesized from 20 ng of total RNA using 15 mM of random hexamers and SuperScript III reverse transcriptase according to the manufacturer's instructions. The cDNA samples were diluted 10-fold, and the PCR reaction was conducted at the annealing temperature of 55°C. All reactions were confirmed to be within the linear range of amplification. The primer sequences and sizes of the amplified fragments are included in Table 1.

Thin-section electron microscopy
Megakaryocytes were fixed with 1.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, for eight hr. Cells were dehydrated through a series of alcohols, infiltrated with propylene oxide, and embedded in epoxy resin in an inverted beam capsule. Ultrathin sections were stained with uranyl acetate and lead citrate and examined with a Tecnai G2 Spirit BioTWIN transmission electron microscope (Hillsboro, OR) at an accelerating voltage of 80 kV, and images were recorded with an AMT 2k CCD camera (Danvers, MA).
Statistical analysis
All results are expressed as the means ± SEM. Student's paired t test was used to determine the significance of differences between means, and p values lower than 0.05 were considered to be statistically significant. When multiple groups were compared, one-way analysis of variance (ANOVA) followed by the Newman-Keuls procedure was used to determine significant differences between groups.
Zdroje
1. MartyAM
JahrlingPB
GeisbertTW
2006 Viral hemorrhagic fevers. Clin Lab Med 26 345 386, viii
2. BrayM
2005 Pathogenesis of viral hemorrhagic fever. Curr Opin Immunol 17 399 403
3. PetersCJ
2002 Human infection with arenaviruses in the Americas. Curr Top Microbiol Immunol 262 65 74
4. VainrubB
SalasR
1994 Latin American hemorrhagic fever. Infect Dis Clin North Am 8 47 59
5. PetersCJ
ZakiSR
2002 Role of the endothelium in viral hemorrhagic fevers. Crit Care Med 30 S268 273
6. HellerMV
MartaRF
SturkA
MaizteguiJI
HackCE
1995 Early markers of blood coagulation and fibrinolysis activation in Argentine hemorrhagic fever. Thromb Haemost 73 368 373
7. GómezRM
PoznerRG
LazzariMA
D'AtriLP
NegrottoS
2003 Endothelial cell function alteration after Junìn virus infection. Thromb Haemost 90 326 333
8. ChenJP
CosgriffTM
2000 Hemorrhagic fever virus-induced changes in hemostasis and vascular biology. Blood Coagul Fibrinolysis 11 461 483
9. CumminsD
MolinasFC
LererG
MaizteguiJI
FaintR
1990 A plasma inhibitor of platelet aggregation in patients with Argentine hemorrhagic fever. Am J Trop Med Hyg 42 470 475
10. CumminsD
Fisher-HochSP
WalsheKJ
MackieIJ
McCormickJB
1989 A plasma inhibitor of platelet aggregation in patients with Lassa fever. Br J Haematol 72 543 548
11. HarrisonLH
HalseyNA
McKeeKTJr
PetersCJ
Barrera OroJG
1999 Clinical case definitions for Argentine hemorrhagic fever. Clin Infect Dis 28 1091 1094
12. GeisbertTW
JahrlingPB
2004 Exotic emerging viral diseases: progress and challenges. Nat Med 10 S110 121
13. BroomhallKS
MorinM
PevearDC
PfauCJ
1987 Severe and transient pancytopenia associated with a chronic arenavirus infection. J Exp Pathol 3 259 269
14. BinderD
FehrJ
HengartnerH
ZinkernagelRM
1997 Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med 185 517 530
15. PonzinibbioC
GonzalezPH
MaizteguiJ
LaguensRP
1979 Morphological study of human bone marrow in Argentinian hemorrhagic fever. Medicina (B Aires) 39 441 446
16. BasuA
JainP
GangodkarSV
ShettyS
GhoshK
2008 Dengue 2 virus inhibits in vitro megakaryocytic colony formation and induces apoptosis in thrombopoietin-inducible megakaryocytic differentiation from cord blood CD34+ cells. FEMS Immunol Med Microbiol 53 46 51
17. CarballalG
CossioPM
LaguensRP
PonzinibbioC
OubinaJR
1981 Junín virus infection of guinea pigs: immunohistochemical and ultrastructural studies of hemopoietic tissue. J Infect Dis 143 7 14
18. GonzalezPH
LaguensRP
FrigerioMJ
CalelloMA
WeissenbacherMC
1983 Junín virus infection of Callithrix jacchus: pathologic features. Am J Trop Med Hyg 32 417 423
19. GreenDE
MahlandtBG
McKeeKTJr
1987 Experimental Argentine hemorrhagic fever in rhesus macaques: virus-specific variations in pathology. J Med Virol 22 113 133
20. BossiP
TegnellA
BakaA
Van LoockF
HendriksJ
2004 Bichat guidelines for the clinical management of haemorrhagic fever viruses and bioterrorism-related haemorrhagic fever viruses. Euro Surveill 9 E11 12
21. GoniSE
IserteJA
AmbrosioAM
RomanowskiV
GhiringhelliPD
2006 Genomic features of attenuated Junín virus vaccine strain candidate. Virus Genes 32 37 41
22. SanchezA
PifatDY
KenyonRH
PetersCJ
McCormickJB
1989 Junín virus monoclonal antibodies: characterization and cross-reactivity with other arenaviruses. J Gen Virol 70(Pt 5) 1125 1132
23. GhiringhelliPD
Rivera-PomarRV
LozanoME
GrauO
RomanowskiV
1991 Molecular organization of Junín virus S RNA: complete nucleotide sequence, relationship with other members of the Arenaviridae and unusual secondary structures. J Gen Virol 72 2129 2141
24. PatelSR
HartwigJH
ItalianoJEJr
2005 The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest 115 3348 3354
25. NorolF
VitratN
CramerE
GuichardJ
BursteinSA
1998 Effects of cytokines on platelet production from blood and marrow CD34+ cells. Blood 91 830 843
26. CandurraNA
ScolaroLA
MersichSE
DamonteEB
CotoCE
1990 A comparison of Junín virus strains: growth characteristics, cytopathogenicity and viral polypeptides. Res Virol 141 505 515
27. RadoshitzkySR
AbrahamJ
SpiropoulouCF
KuhnJH
NguyenD
2007 Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446 92 96
28. JacobsBL
LanglandJO
1996 When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 219 339 349
29. FieldAK
TytellAA
LampsonGP
HillemanMR
1967 Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci U S A 58 1004 1010
30. BekiszJ
SchmeisserH
HernandezJ
GoldmanND
ZoonKC
2004 Human interferons alpha, beta and omega. Growth Factors 22 243 251
31. LarsonMK
WatsonSP
2006 Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood 108 1509 1514
32. ChangY
BluteauD
DebiliN
VainchenkerW
2007 From hematopoietic stem cells to platelets. J Thromb Haemost 5 Suppl 1 318 327
33. ShivdasaniRA
RosenblattMF
Zucker-FranklinD
JacksonCW
HuntP
1995 Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 81 695 704
34. GibelliniD
VitoneF
BuzziM
SchiavoneP
De CrignisE
2007 HIV-1 negatively affects the survival/maturation of cord blood CD34(+) hematopoietic progenitor cells differentiated towards megakaryocytic lineage by HIV-1 gp120/CD4 membrane interaction. J Cell Physiol 210 315 324
35. IsomuraH
YoshidaM
NambaH
YamadaM
2003 Interaction of human herpesvirus 6 with human CD34 positive cells. J Med Virol 70 444 450
36. CrapnellK
ZanjaniED
ChaudhuriA
AscensaoJL
St JeorS
2000 In vitro infection of megakaryocytes and their precursors by human cytomegalovirus. Blood 95 487 493
37. ColeJL
MarzecUM
GunthelCJ
KarpatkinS
WorfordL
1998 Ineffective platelet production in thrombocytopenic human immunodeficiency virus-infected patients. Blood 91 3239 3246
38. SundellIB
KokaPS
2006 Thrombocytopenia in HIV infection: impairment of platelet formation and loss correlates with increased c-Mpl and ligand thrombopoietin expression. Curr HIV Res 4 107 116
39. FlanaganML
OldenburgJ
ReignierT
HoltN
HamiltonGA
2008 New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J Virol 82 938 948
40. KawaiT
AkiraS
2007 Antiviral signaling through pattern recognition receptors. J Biochem 141 137 145
41. YamaneA
NakamuraT
SuzukiH
ItoM
OhnishiY
2008 Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood 112 542 550 Epub 2008 Jun 2003
42. WangQ
MiyakawaY
FoxN
KaushanskyK
2000 Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 96 2093 2099
43. IannaconeM
SitiaG
IsogawaM
WhitmireJK
MarcheseP
2008 Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci U S A 105 629 634
44. LevisSC
SaavedraMC
CeccoliC
FeuilladeMR
EnriaDA
1985 Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J Interferon Res 5 383 389
45. VillingerF
RollinPE
BrarSS
ChikkalaNF
WinterJ
1999 Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis 179 Suppl 1 S188 191
46. DejeanCB
OubinaJR
CarballalG
TeyssieAR
1988 Circulating interferon in the guinea pig infected with the XJ, prototype Junín virus strain. J Med Virol 24 97 99
47. LangPA
Cervantes-BarraganL
VerschoorA
NavariniAA
RecherM
2009 Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 113 1045 1052 Epub 2008 Oct 1029
48. NegrottoS
PacienzaN
D'AtriLP
PoznerRG
MalaverE
2006 Activation of cyclic AMP pathway prevents CD34(+) cell apoptosis. Exp Hematol 34 1420 1428
49. DrillienR
SpehnerD
HanauD
2004 Modified vaccinia virus Ankara induces moderate activation of human dendritic cells. J Gen Virol 85 2167 2175
50. PoznerRG
NegrottoS
D'AtriLP
KotlerML
LazzariMA
2005 Prostacyclin prevents nitric oxide-induced megakaryocyte apoptosis. Br J Pharmacol 145 283 292
51. BalduiniA
PallottaI
MalaraA
LovaP
PecciA
2008 Adhesive receptors, extracellular proteins and myosin IIA orchestrate proplatelet formation by human megakaryocytes. J Thromb Haemost 6 1900 1907
52. DhanjalTS
PendariesC
RossEA
LarsonMK
ProttyMB
2007 A novel role for PECAM-1 in megakaryocytokinesis and recovery of platelet counts in thrombocytopenic mice. Blood 109 4237 4244
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2010 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
- A Timescale for Evolution, Population Expansion, and Spatial Spread of an Emerging Clone of Methicillin-Resistant