
Overcomes Stress of Azole Drugs by Formation of Disomy in Specific Multiple Chromosomes
Cryptococcus neoformans is a haploid environmental organism and the major cause of fungal meningoencephalitis in AIDS patients. Fluconazole (FLC), a triazole, is widely used for the maintenance therapy of cryptococcosis. Heteroresistance to FLC, an adaptive mode of azole resistance, was associated with FLC therapy failure cases but the mechanism underlying the resistance was unknown. We used comparative genome hybridization and quantitative real-time PCR in order to show that C. neoformans adapts to high concentrations of FLC by duplication of multiple chromosomes. Formation of disomic chromosomes in response to FLC stress was observed in both serotype A and D strains. Strains that adapted to FLC concentrations higher than their minimal inhibitory concentration (MIC) contained disomies of chromosome 1 and stepwise exposure to even higher drug concentrations induced additional duplications of several other specific chromosomes. The number of disomic chromosomes in each resistant strain directly correlated with the concentration of FLC tolerated by each strain. Upon removal of the drug pressure, strains that had adapted to high concentrations of FLC returned to their original level of susceptibility by initially losing the extra copy of chromosome 1 followed by loss of the extra copies of the remaining disomic chromosomes. The duplication of chromosome 1 was closely associated with two of its resident genes: ERG11, the target of FLC and AFR1, the major transporter of azoles in C. neoformans. This adaptive mechanism in C. neoformans may play an important role in FLC therapy failure of cryptococcosis leading to relapse during azole maintenance therapy.
Published in the journal:
Overcomes Stress of Azole Drugs by Formation of Disomy in Specific Multiple Chromosomes. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000848
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000848
Summary
Cryptococcus neoformans is a haploid environmental organism and the major cause of fungal meningoencephalitis in AIDS patients. Fluconazole (FLC), a triazole, is widely used for the maintenance therapy of cryptococcosis. Heteroresistance to FLC, an adaptive mode of azole resistance, was associated with FLC therapy failure cases but the mechanism underlying the resistance was unknown. We used comparative genome hybridization and quantitative real-time PCR in order to show that C. neoformans adapts to high concentrations of FLC by duplication of multiple chromosomes. Formation of disomic chromosomes in response to FLC stress was observed in both serotype A and D strains. Strains that adapted to FLC concentrations higher than their minimal inhibitory concentration (MIC) contained disomies of chromosome 1 and stepwise exposure to even higher drug concentrations induced additional duplications of several other specific chromosomes. The number of disomic chromosomes in each resistant strain directly correlated with the concentration of FLC tolerated by each strain. Upon removal of the drug pressure, strains that had adapted to high concentrations of FLC returned to their original level of susceptibility by initially losing the extra copy of chromosome 1 followed by loss of the extra copies of the remaining disomic chromosomes. The duplication of chromosome 1 was closely associated with two of its resident genes: ERG11, the target of FLC and AFR1, the major transporter of azoles in C. neoformans. This adaptive mechanism in C. neoformans may play an important role in FLC therapy failure of cryptococcosis leading to relapse during azole maintenance therapy.
Introduction
Cryptococcus neoformans is the most common cause of fungal meningoencephalitis world-wide. A major predisposing factor is the profound cellular immune defect caused by HIV infection or other underlying conditions. Cryptococcal meningoencephalitis is fatal unless treated and even with the most advanced treatment it is known for its high mortality rates [1],[2]. Fluconazole (FLC), a triazole antifungal drug, has been the agent most widely used for prophylactic therapy as well as for the long term management of common mycoses such as candidiasis and cryptococcosis owing to its efficacy and safety [3]. Long-term maintenance therapy with azoles creates favorable conditions for the emergence of resistance to the drug and increased azole resistance in vitro has been shown to be predictive of treatment failures and infection relapses [4].
The molecular basis of resistance to azole antifungals has been studied extensively in Saccharomyces cerevisiae and pathogenic Candida species such as C. albicans and C. glabrata which are phylogenetically distant from C. neoformans [5]–[13]. In these fungi, resistance is known to emerge via (1) increased production of multidrug transporters [14]–[16], (2) mutations in ergosterol biosynthetic pathway genes [17],[18], (3) amplification of genomic regions that contain ergosterol biosynthetic pathway genes and transcription factors that positively regulate a subsets of efflux pump genes [19],[20] and (4) activation of Hsp90 that may facilitate the cells to respond to drug stress [21],[22]. In C. neoformans, FLC resistant strains have rarely been reported and the emergence of resistance has most often been documented with clinical outcomes of AIDS patients receiving azole maintenance therapy [23]–[27]. The mechanism of resistance in C. neoformans during maintenance therapy is poorly understood.
An intriguing pattern of intrinsic azole resistance termed ‘heteroresistance’ was reported in 1999 among C. neoformans strains isolated from AIDS patients undergoing FLC maintenance therapy [28] and has only recently been characterized further [29]. This phenomenon of heteroresistance has been described as the emergence of a resistant minor subpopulation, within the single colony of a susceptible strain, that can tolerate concentrations of FLC higher than the strain's MIC. The resistant subpopulations can adapt to increasing concentrations of the drug in a stepwise manner. However, this acquired resistance to high concentrations of FLC is lost during repeated passage in drug free media and the clones return to their original level of heteroresistance. The level of heteroresistance to FLC (LHF) was defined as the lowest concentration of the azole drug at which resistant subpopulations emerge [29]. All strains of C. neoformans tested in our laboratory thus far have exhibited different LHF regardless of whether they are pre- or post therapy strains and the frequency of resistant subpopulations that emerge at each LHF ranged between 0.3 and 10% depending on strains [29]. Purification of a homogeneously sensitive subpopulation was not achieved at each strain's LHF while a homogeneous population of resistant cells could readily be obtained by exposure to FLC concentrations equal to or higher than its initial LHF. This acquired resistance to high concentrations of FLC, however, was lost during repeated passage in drug free media and the clones returned to the original LHF at which only 0.3 to 10% of the subpopulations grew. The molecular mechanism involved in this unique pattern of azole resistance remains an enigma.
In this paper, we employed a genomic approach to uncover the mechanism by which C. neoformans cells acquire resistance to high concentrations of FLC and then subsequently lose the resistance when the drug stress is removed. We demonstrate that the adaptive resistance to higher concentrations of FLC was achieved by duplications of multiple chromosomes in response to drug pressure. Upon repeated transfer in drug free media, cells with multiple disomic chromosomes lose duplicated copies of the chromosomes sequentially and return to their original levels of drug tolerance. Such genomic fluidity that enables the cells to cope with the drug stress was observed in C. neoformans strains of both serotypes, A and D. Our results provide an explanation as to the mechanism governing the transiently high azole resistance observed in C. neoformans. We propose that this mechanism contributes to the failure of FLC therapy that results in the recurrent infection reported in patients undergoing prolonged azole therapy [28].
Results
Characterization of heteroresistance to FLC in strain H99
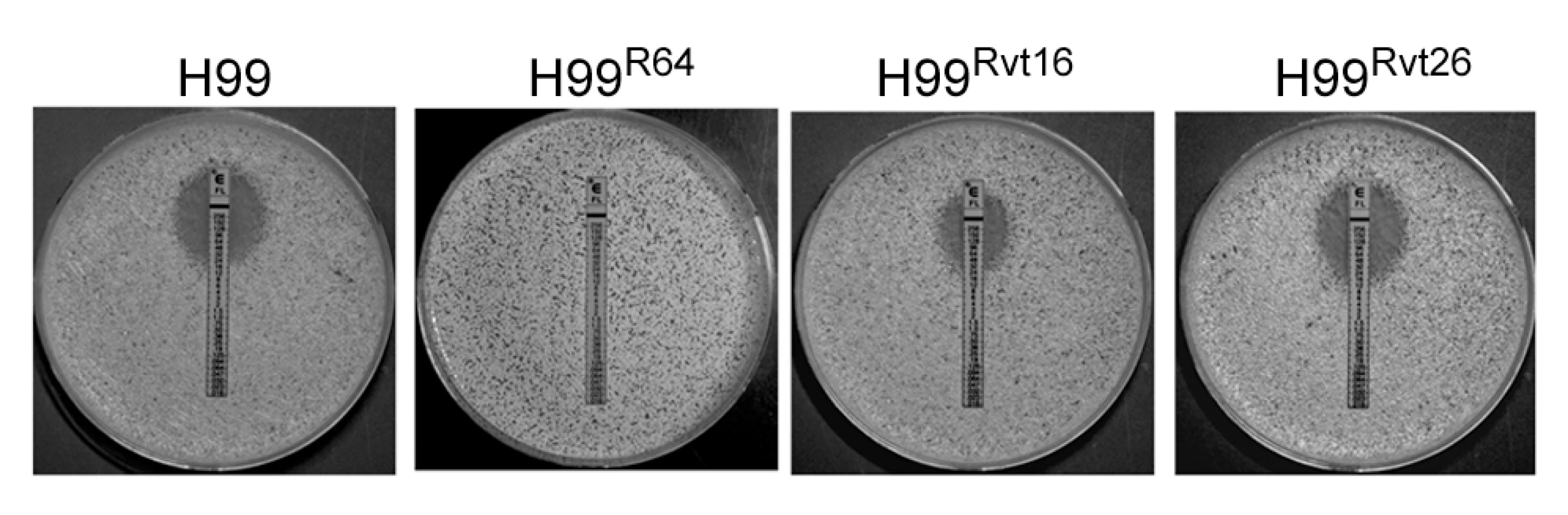
All strains of C. neoformans tested in our laboratory displayed the intrinsic adaptive heteroresistant phenotype to FLC [29]. Since serotype A strains of C.neoformans are the most prevalent of all the four serotypes in clinical settings, we chose the strain H99, a genome sequenced reference strain of serotype A, to study the mechanism of heteroresistance. Equal numbers of colonies were observed on YPD agar media with or without 16 µg/ml FLC. However, growth of the colonies on 16 µg/ml FLC was slightly slower with heterogeneity in size. On YPD media containing 32 µg/ml FLC, only 0.3–0.6% of the input cells consistently formed colonies within 72 h. Therefore, the intrinsic level of H99 FLC heteroresistance was determined to be 32 µg/ml [29]. Exposure of these subclones resistant at 32 µg/ml FLC to stepwise increases in FLC concentration generated clones resistant to 64 µg/ml (strain H99R64) and 128 µg/ml (strain H99R128). Conversely, repeated transfer in drug free media of cells that had adapted to FLC at concentrations >32 µg/ml resulted in their reversal to original levels of heteroresistance. For instance, the H99Rvt16 strain derived from 16 daily transfers of the strain H99R64 in drug-free media displayed a FLC resistance phenotype intermediate between H99R64 and H99. Its colony size on YPD with 32 µg/ml FLC was larger and its FLC E-test value was higher (48 µg/ml) than the parental H99 strain (E-test MIC = 24 µg/ml, Figure 1). In contrast, the H99Rvt26 strain similarly derived from 26 daily transfers of the strain H99R64 in drug free media completely reverted back to the parental type.
Genome analysis of FLC resistant strain H99R64
It is possible that the resistant strain H99R64 may express a different set of genes compared to H99. Thus, we compared the gene expression profiles of H99R64 and H99 using microarray analysis. Of the 6719 detectable genes analyzed, 4149 genes were identified as significant by a mean false discovery rate (FDR) of 5% with significance analysis of microarray (SAM) as described in Material and Methods. We found 763 genes to be up or down regulated at least 1.8-fold in H99R64 compared to H99. As expected, some of the differentially regulated genes are annotated for drug-related functions such as ABC transporter, multidrug resistance protein and enzymes involved in the ergosterol biosynthetic pathway. More significantly, among the 491 genes observed to be upregulated in H99R64, 308 (63%) are located on chromosome 1 (Chr1) and 143 (29%) on chromosome 4 (Chr4), which in collectively comprises 92% of the upregulated genes in H99R64 (Figure 2A). Having a majority of the upregulated genes distinctly clustered in two chromosomes, we suspected some chromosomal anomaly in H99R64. To examine global genomic changes in H99R64, we performed comparative genome hybridization (CGH). Interestingly, CGH analysis of the H99R64 strain revealed that the average log2 ratio of hybridization signals for Chr1 and Chr4 was significantly above zero (0.84 and 0.89, respectively) across the entire chromosome as shown in Figure 2B. The simplest explanation for this observation was that Chr1 and Chr4 had duplicated in the cells of H99R64. We also analyzed H99 and H99R64 by flow cytometry and the data suggested that H99R64 is not a diploid strain harboring trisomic Chr1 and Chr4 (Figure S1). Consequently, the observed overexpression of the genes on Chr1 and Chr4 in cDNA microarray was due to the increase in copy number of the genes on the two chromosomes.
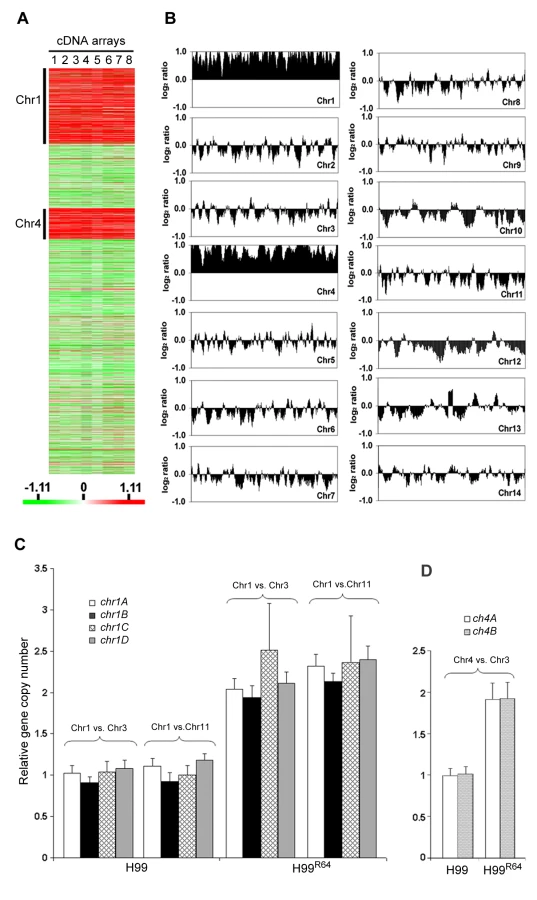
To confirm this phenomenon of multiple chromosome duplications revealed by the CGH data, quantitative real time PCR (qPCR) of genomic DNA was performed. Four probes representing the four genes at different locations of chromosome 1 that span the left and the right arm (chr1A, chr1B, chr1C, and chr1D) were chosen for qPCR using the same genomic DNA used for CGH analysis. qPCR results of each probe on Chr1 were compared to those of the probe on either Chr3 (chr3A) or Chr11 (chr11A), which served as unduplicated internal controls. As shown in Figure 2C, the copy number of all tested genes at different locations on Chr1 was close to two fold higher than the genes on Chr3 or Chr11 in H99R64 (P<0.001), while the relative copy number of those genes was close to 1 in H99. This indicated that H99R64 has two copies for each of the four genes on Chr1. Similarly, the qPCR results from probes representing two genes on Chr4 (chr4A and chr4B) showed the dosage of each gene in H99R64 to be two fold of that in H99 (Figure 2D; P<0.001). These qPCR results corroborated with the CGH data and suggested that chromosomes 1 and 4 in the strain H99R64 were disomic and the disomy of these two chromosomes was associated with resistance at 64 µg/ml FLC.
The number of disomic chromosomes correlates with the level of FLC resistance
Since the resistant clones can adapt to different levels of FLC concentration, it is possible that the FLC resistance level of each clone positively correlates with the number of disomic chromosomes. To test this hypothesis, we analyzed by CGH array six other H99-derived strains that had adapted to different levels of FLC concentration. First, we tested two of the aforementioned reverted strains, H99Rvt16 and H99Rvt26, which had resulted from repeated transfer of H99R64 in drug free media for 16 days and 26 days respectively (Figure 1). CGH data revealed the intermediate revertant H99Rvt16 to be monosomic for Chr1 but still disomic for Chr4 while the complete revertant, H99Rvt26, contained no disomic chromosomes (Figure 3A and Figure S2). These results suggested that removal of drug pressure caused a loss of the duplicated copies of chromosomes in the cells starting with that of Chr1 and then eventually return to the wild type status. Second, we performed CGH analysis of the H99R32 and H99R128 strains which were resistant to 32 µg/ml and 128 µg/ml FLC, respectively. CGH plots revealed that only Chr1 was duplicated in H99R32, while four chromosomes (Chr1, 4, 10, and 14) were duplicated in H99R128 (Figure 3B and Figure S2). Third, we analyzed strain H99R64L, a clone of H99R64 that was maintained for an additional two weeks on the media with 64 ug/ml FLC.. As was the case with H99R64, Chr1 and Chr4 were duplicated in H99R64L. Interestingly, Chr10 was also duplicated in H99R64L and the copy number of many genes on Chr14 increased although not quite two-fold compared to that of H99 (Figure 3B). It appears that prolonged incubation of cells at high FLC concentrations results in the emergence of additional disomic chromosomes.
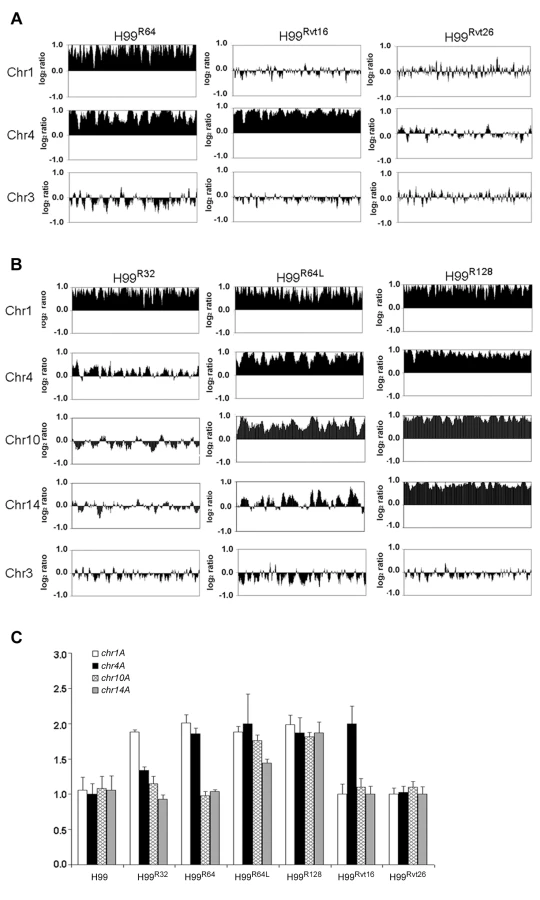
CGH results were confirmed by qPCR analysis of a gene chosen from each of the four chromosomes 1, 4, 10 and 14. As shown in Figure 3C, relative copy numbers of each gene against the internal control gene on Chr3 corroborated the CGH analysis. All four genes located on different chromosomes were duplicated in the strain H99R128 while no gene duplication was evident in the complete revertant strain H99Rvt26 (P<0.001). In the strain H99R64L, the gene copy number on Chr10 and Chr14 was close to 2 and 1.5, respectively. Furthermore, chromosome duplication was also verified by quantitative Southern blot analysis using a probe from each of the four affected chromosomes (Figure S3 and Table S1). Collectively, these data strongly suggested that the number of disomic chromosomes positively correlated with the levels of FLC resistance of the strain and with the duration of exposure to FLC.
Genome fluidity reflected in the gene dosages at colony level
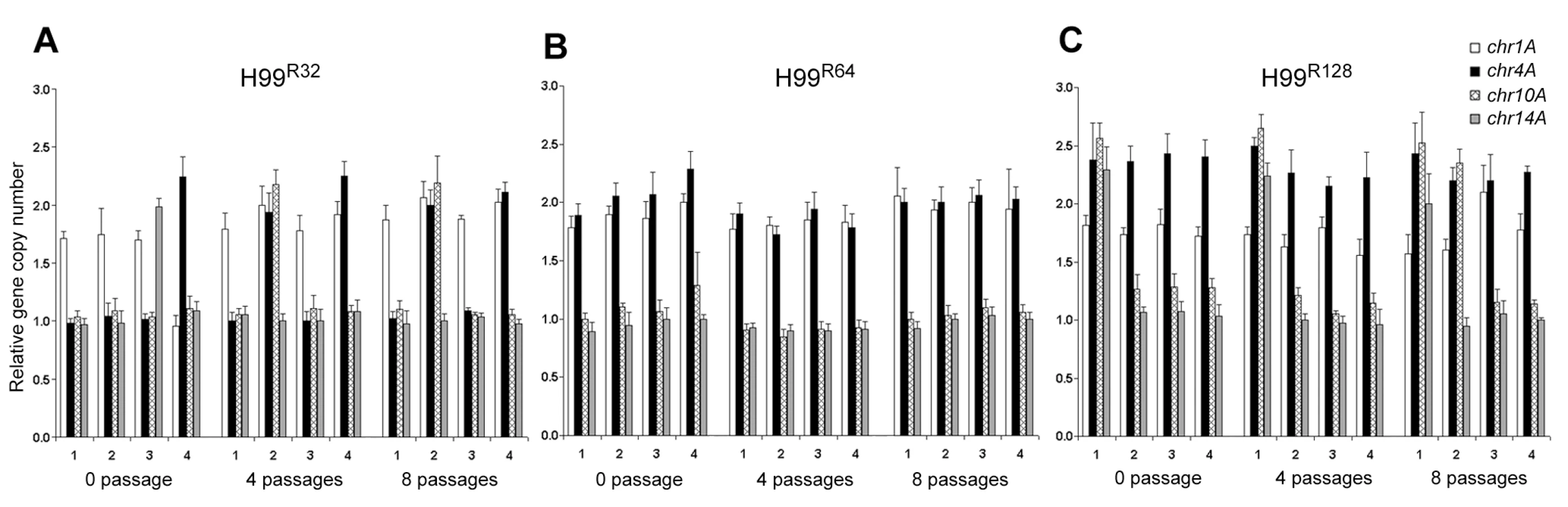
Since CGH experiments require relatively large amounts of genomic DNA, each strain was allowed to proliferate for many generations on the drug media in order to obtain enough cells. The CGH data, therefore, represents the average status of the whole population grown on the media containing a certain concentration of FLC for many generations. qPCR was performed to examine gene dosages in the small number of individual resistant clones immediately after their emergence on plates containing high concentrations of FLC. This would determine whether gene duplication occurred during the early stages of growth in which resistance was initially observed at the single colony level. We chose 4 different colonies that appeared 4 days after plating naive H99 cells on media with 32 µg/ml FLC. Four independent colonies resistant at 64 and/or 128 µg/ml of the drug (derived from four different 32 and/or four different 64 µg/ml FLC resistant clones, respectively) were also isolated and analyzed. The CGH data suggested that the chromosome duplication occurs primarily in Chr1, 4, 10 and 14 and thus we focused our colony qPCR analysis only on these four chromosomes, although the duplication event might not be limited to these chromosomes. Interestingly, variations in the gene duplication events on different chromosomes was observed among the independent colonies grown on 32 µg/ml and 128 µg/ml FLC, respectively, but not on 64 µg/ml FLC (0 passage in Figure 4A, 4B, and 4C). To test whether prolonged drug-exposure would alter the outcome of gene duplication, the same sets of the four clones from each concentration of FLC were streaked on media with the same FLC concentration for 4 and 8 passages. Single colonies were then subjected to qPCR which exposed the tremendous variability in the duplication of genes representing different chromosomes. For example, one clone from 32 µg/ml FLC plate (clone #2) initially had duplication of a gene on Chr1. After 4 passages, genes on Chr4 and Chr10 were also duplicated in addition to Chr1 and the status of gene duplication in these chromosomes was the same when tested after 8 passages (Figure 4A). Clone #3 from the 32 µg/ml plate, however, appeared to have genes on Chr1 and Chr14 duplicated initially, but the genes on Chr14 did not remain duplicated after longer exposure to the drug. In contrast, clone #4 did not show the gene on Chr1 duplicated until after 4 passages on 32 µg/ml FLC media while gene on Chr4 was duplicated from the beginning and remained duplicated throughout the 8 passages. Generally, a more consistent pattern of gene duplication was observed with independent clones isolated from the plates containing FLC 64 µg/ml compared to those isolated from 32 µg/ml FLC plates (Figure 4B). Fluctuations in the pattern of gene duplication, however, were also obvious among the colonies grown on FLC 128 µg/ml (Figure 4C). These data clearly showed the plasticity of gene duplication patterns at the single colony level, which could not be depicted clearly in CGH data. It is likely that CGH results represent the average status of chromosomes in the whole population and the system is not sensitive enough to allow detection of transient chromosomal duplication events in individual colonies. However, the CGH results of H99R64L apparently revealed the intermediate process of Chr14 duplication in which the gene copy number of Chr14 was 1.5 as verified by qPCR using the same batch of DNA (Figure 3B and 3C). Taken together, our data suggested that when C. neoformans was treated with FLC, the process of multiple chromosome duplication may vary among individual cells and the status of chromosome copy number determined by CGH appears to be an average of the whole population from cells grown in the presence of FLC for many generations.
ERG11 is important for chromosome 1 duplication under azole stress
It was plausible that formation of disomic chromosomes in association with FLC resistance was due to the presence of certain genes on the duplicated chromosomes which plays crucial role in the survival of cells under the drug stress. Since Chr1 was universally duplicated in the resistant clones, we focused on Chr1 as the first step to determine whether each duplicated chromosome carries genes that confer selective advantage under azole drug stress. Among the annotated genes on Chr1, AFR1 and ERG11 were the two candidate genes that had already been characterized involving FLC resistance in C. neoformans. AFR1 is an ATP binding cassette (ABC) transporter-encoding gene and have shown to play an important role in azole susceptibility [29],[30]. ERG11 encodes lanosterol-14-α-demethylase, the target of FLC, and increased expression levels of ERG11 is associated with increased FLC resistance in several fungi [15]–[18]. ERG11, the target of FLC, has been proposed to contribute to isochromosome formation in C. albicans [20], so we chose it to address its role in Chr1 duplication. If the presence of ERG11 were the main cause of Chr1 duplication, ERG11-containing chromosome would primarily duplicate regardless of the location of the gene. On the other hand, if other genes besides ERG11 were equally or more important for the survival in the presence of FLC, Chr1 would remain duplicated even if ERG11 is relocated from Chr1 to other chromosomes. Since ERG11 is most likely essential, we first inserted an extra copy of ERG11 on Chr3, which had not duplicated under any level of drug stress and then deleted ERG11 from its native location on Chr1 (Figure 5). Strain C1345, which contained two copies of ERG11 – one on Chr1 and the other on Chr3, exhibited elevated resistance to FLC according to E-test (Figure 5) as well as by growth analysis (100% growth at 32 µg/ml and 0.1% growth on 64 µg/ml in contrast to 0.3–0.6% growth at 32 µg/ml and 0 % growth at 64 µg/ml of H99) compared to H99. These data indicate that the extra copy of ERG11 inserted onto Chr3 conferred increased FLC resistance. Since C1345 had two copies of ERG11 mimicking the effect of Chr1 duplication regarding ERG11 copy number, it was of great interest to determine the status of chromosome duplication in C1345 upon exposure to high concentration of FLC. First, qPCR was performed on two independent colonies of C1345 isolated immediately after emerging on the plate containing 32 µg/ml FLC. We detected close to two copies of ERG11 (chr1A probe) but only one copy of other genes on Chr1, Chr3, and Chr4 suggesting no duplication of chromosomes of C1345 at 32 µg/ml FLC (Figure 6A). This is in contrast to H99 subclones resistant to 32 µg/ml FLC in which Chr1 is duplicated (Figure 3B and 3C). However, qPCR of two C1345 colonies isolated directly from 64 µg/ml of FLC plate showed the existence of three copies of ERG11 (chr1A probe) and two copies of both chr1D and chr4A, but only one copy of chr3A (Figure 6A). These data suggested that both Chr1 and Chr4 were duplicated in the C1345 colonies grown in 64 µg/ml of FLC. CGH analysis of the entire cell population harvested from 64 µg/ml FLC (C1345 R64) clearly showed duplication of Chr1 and Chr4 and not Chr3 (Figure 6B). In addition, C1345R128, the C1345 strain grown on128 µg/ml FLC showed similar chromosome duplication patterns as C1345R64 maintaining duplication of Chr1 and Chr4. It is intriguing, however, to observe intermediate hybridization signal for Chr3 in C1345R128 (average log2 ratio of −0.035 and 0.317 for C1345 R64 and C1345R128, respectively). qPCR using the same DNA showed that the copy number of chr3A probe located on Chr3 was 1.22±0.06, confirming the CGH results. This data suggests that a proportion of the cells in the population of C1345R128 strain may have an extra copy of Chr3. Colony PCR from two independent colonies of C1345R128 supported duplication of the gene on Chr3 (Figure 6A). The second copy of ERG11 resides on Chr3 in C1345R128 which has not been observed to be duplicated in other strains tested so far. These results showed that two copies of ERG11, one on Chr 1 and the other on Chr3 prevented disomy formation of Chr1 at 32 µml FLC but did not prevent disomy formation of Chr1 and Chr4 at FLC 64 µg/ml, a concentration which is 2-fold higher than the level tolerated by C1345. Furthermore, Chr1 was preferentially duplicated over Chr3 at high concentrations of FLC when the ERG11 existed on both chromosomes.
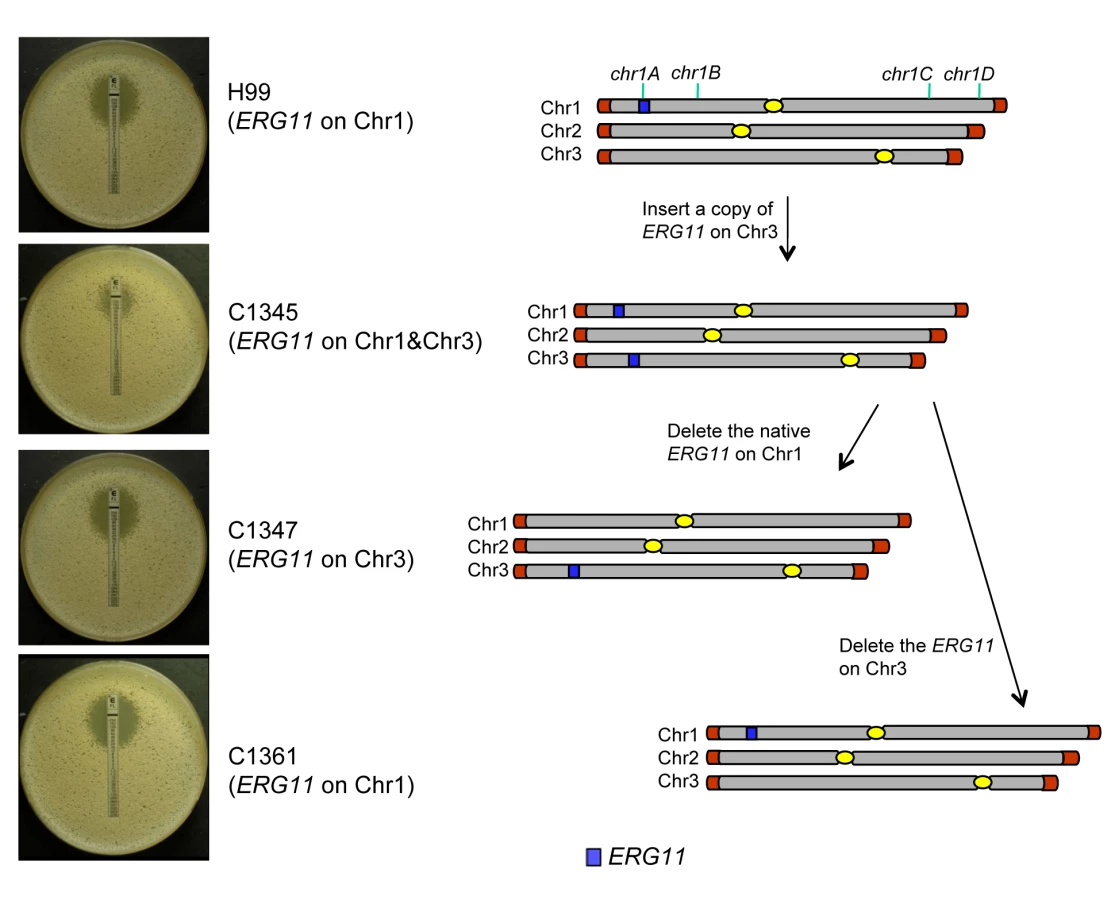
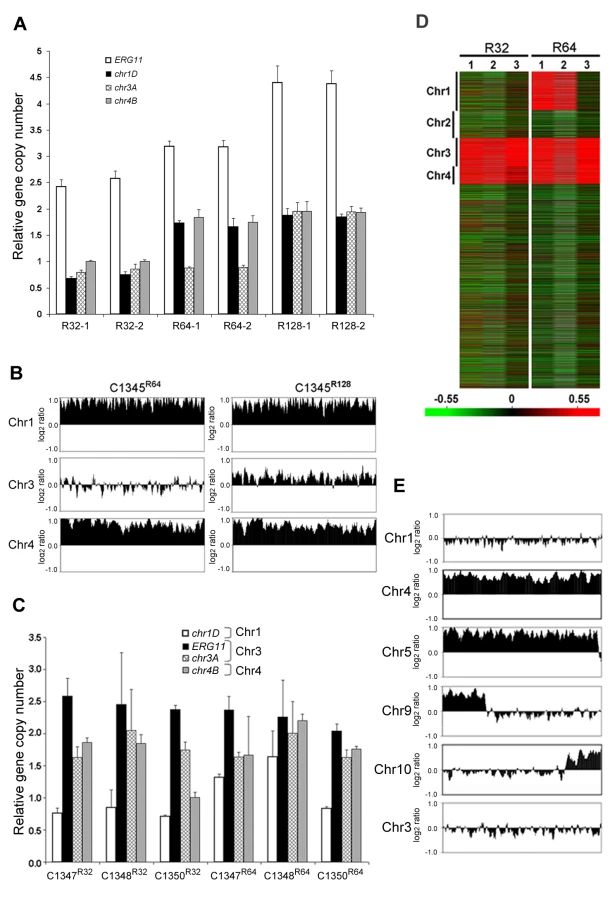
The ERG11 gene on the Chr1 was subsequently deleted from C1345 leaving only one copy of ERG11 inserted on Chr3. The FLC resistance levels of three independent transformants (C1347, C1348, and C1350) were comparable with H99 (100% growth at 16 µg/ml and 0.3–0.6% growth at 32 µg/ml), indicating that translocation of ERG11 from Chr1 to Chr3 did not alter the strain's FLC resistance level. Clones of these three independent transformants grown in 32 µg/ml FLC were subjected to colony qPCR. Noticeably, the copy number of ERG11 (chr1A probe) and chr3A were close to two fold, while the copy number of chr1D remained close to one suggesting duplication of Chr3 but not Chr1 (Figure 6C). CGH analysis of C1347R32, C1348R32, and C1350R32 showed Chr3 was duplicated in all three strains (Figure 6D). Interestingly, Chr4 was also duplicated in all three strains although colony PCR results suggested duplication of a gene on Chr4 only in C1347R32 and C1348R32 (Figure 6C and 6D). This result was different from H9932R in which only Chr1 duplication was observed at 32 µg/ml FLC (Figure 3B). These data suggested that when only one copy of ERG11 is present in the genome, the ERG11 bearing chromosome is the primary one to be duplicated at 32 µg/ml FLC. However, additional chromosome duplication (Chr4) was required to tolerate the stress exerted by FLC when ERG11 was moved from its native location to Chr3. Additional CGH was performed using strains derived from C1347, C1348, and C1350 resistant to 64 µg/ml FLC (Figure 6D). C1347R64 and C1348R64 displayed disomies of Chr1, Chr3 and Chr4 while C1350R64 showed duplication only in Chr3 and Chr4 but not in Chr1. Single colony qPCR of these three strains supported the CGH results although the copy number of chr1D in C1347R64 was only 1.32 (±0.05), suggesting that Chr1 amplification occurred in a certain portion of the clonal population (Figure 6C). These data indicated that when a single copy of ERG11 gene exists in the genome, the chromosome carrying ERG11 is consistently duplicated in all subsequently derived FLC-resistant strains. However, our data also pointed out that ERG11 was not the sole reason for the Chr1 duplication and duplication of other genes on Chr1 and those on Chr4 also appeared to have contributed to the survival of cells at 64 µg/ml FLC.
AFR1 also plays a role in chromosome 1 duplication under FLC stress
In an attempt to investigate other genes on Chr1 that confer resistance to FLC via chromosome duplication, we investigated AFR1. Several lines of evidence have indicated that AFR1 plays an important role in FLC resistance. First, AFR1 expression was upregulated in both H99R64 and the H99 strains treated with FLC. Second, deletion of AFR1 resulted in a drastic decrease in FLC resistance in H99 [29]. Third, high expression level of AFR1 resulted in the increased level of FLC resistance [30]. The afr1Δ strain (C1371) was used to determine the possible involvement of AFR1 in Chr1 duplication under FLC stress. If AFR1 were important for duplication of Chr1, we would not expect Chr1 to be duplicated in afr1Δ strains resistant to FLC. The H99 afr1Δ strain is extremely sensitive to FLC (MIC 0.38 µg/ml) and its level of heteroresistance was reduced from 32 µg/ml to 1 µg/ml [29]. CGH analysis of the subpopulation resistant at 1 µg/ml FLC (afr1ΔR1) clearly showed that Chr1 was not duplicated in the afr1ΔR1 strain (Figure 6E). Instead, Chr4 and Chr5 were duplicated along with short segmental duplications of Chr9 and Chr10. Thus, absence of AFR1 on Chr1 not only abrogated Chr1 duplication but also caused whole duplications or segmental duplication in other chromosomes at 1 µg/ml FLC. Such a chromosomal duplication pattern was presumably due to the presence of genes on these duplicated chromosomes which might compensate for the effect of AFR1 deletion from Chr1 in afr1ΔR1. It is noteworthy that although ERG11 is present on Chr1 in afr1ΔR1, disomy formation of Chr1 does not occur at 1 µg/ml FLC. However, CGH analysis of the subpopulation resistant at 8 µg/ml (afr1ΔR8) showed that Chr1 was duplicated along with an additional four chromosomes (Chr 4, 5, 6, and 10; Figure S4). These findings underscore the importance of both ERG11 and AFR1 in the formation of Chr1 disomy under FLC stress.
Disomic chromosome formation is a common phenomenon in strains of C. neoformans
All strains of C. neoformans tested thus far displayed the FLC heteroresistant phenotype [29]. Although different strains displayed heteroresistance at different concentrations of FLC, the stepwise exposure to higher concentrations of FLC allowed the strains to adapt to levels of FLC that are higher than their original MIC. These resistant strains all reverted to the original level of resistance upon removal of drug pressure. To investigate whether chromosome duplication associated with FLC resistance was an H99-specific event, we analyzed a number of matched pairs of naive vs. FLC-adapted resistant isolates in both serotype A and D backgrounds. Consistent with the observation in H99, FLC-resistant strains derived from both serotype backgrounds contained disomic chromosomes according to CGH analysis (Figure S5), even though the duplicated chromosomes were not always identical in these strains. These results demonstrated that chromosome duplication associated with FLC resistance is a general mechanism employed by C. neoformans to overcome the stress exerted by FLC.
Discussion
We report here that C. neoformans consistently forms disomies in multiple chromosomes in response to high level of azole pressure in both serotype A and D strains. Duplicated copies of the disomic chromosomes are lost as the drug pressure is removed. While there can be minor variations in the number of duplicated chromosomes among individual colonies grown on the same FLC media, the number of disomic chromosomes in the population of the overall cultures positively correlates with the adaptation to stepwise increase in FLC concentration.
Aneuploidy associated with azole resistance was reported in Candida albicans where a substantially higher frequency of aneuploidy was found among azole resistant strains compared to susceptible strains [19]. In addition, chromosome instability, specific segmental aneuploidy, translocation of chromosomal arms and whole chromosome duplication have been previously reported in Candida species [20],[31],[32].
One could argue that the clones with disomy observed in the subpopulation of H99 under FLC stress may comprise a normal population that is selected by the drug rather than the drug induced chromosome amplification. There are three reasons for this argument being unlikely. First, aneuploidy caused by chromosome missegregation occurs once every 5×105 cell divisions in yeast [33] and once every 104 to 105 cell divisions in mammalian cells [34]. The frequency of FLC resistant clones of H99 (0.3 to 0.6%) that emerged on drug containing media is too high to be the result of spontaneous chromosomal missegregation. Furthermore, the frequency of FLC resistant clones in different strains can be as high as 10% [29]. The frequency at which aneuploidy occurs in C. neoformans under FLC stress, therefore, is several logs higher than the frequency of spontaneous aneuploidy formation in other eukaryotes. Second, H99 is the most widely studied strain of C. neoformans and yet a clone derived from H99 that contains disomic chromosomes in a stress-free environment has never been reported. Third, we observed disomy formation in H99 only when exposed to FLC but not other xenobiotics such as trichostatin A, gliotoxin or rhizoxin (data not shown). Aneuploidy is reported to have multiple effects on cellular physiology and cell division in haploid yeast [35]. Consistent with findings in yeast, disomic chromosomes in C. neoformans result in a proliferative disadvantage as evidenced by the retarded growth rate of H99R64 which harbors extra copies of Chr1 and 4, and exhibits lower virulence in mice compared to the wild type strain (Figure S6). Although many fungi undergo chromosome length polymorphisms, chromosomal loss [36] or gain of minichromosomes [37] under different environmental stress, the degree of consistency and reproducibility of genomic fluidity observed in the present work has not been reported in other fungi.
Since genetically identical cells of a single C. neoformans colony exposed to a high concentration of FLC can produce small subpopulations that show a marked difference in FLC susceptibility, we can speculate that this variability is linked to stochasticity in gene expression [38]. The genes that govern the capacity to differentiate into heteroresistant subtypes are not known. Although the CGH data show an increase of specific disomic chromosomes when C. neoformans is challenged by increasing drug pressure, minor variations in duplicated chromosomes appear to occur among individual colonies. Such plastic outcomes of duplication events can be advantageous for C. neoformans since it can provide the flexibility required for the cells to respond to various kinds of sudden stress it encounters either in the environment or in the host. The extra copy of a disomic chromosome may have resulted from non-disjunction, which occurs commonly in eukaryotes under different stresses [39],[40]. In mammalian systems, inhibition of cholesterol biosynthesis by blocking sterol 14 α-demethylase (ERG11 ortholog) induces the formation of polyploid cells and mitotic aberrations [41]. Since ergosterol, the counterpart of cholesterol in fungi, is the essential molecule for maintaining membrane integrity, depletion of ergosterol in nuclear and cell membranes due to FLC treatment may jeopardize normal patterns of cytokinesis and enhance the frequency of chromosomal non-disjunction. For example, the spindle pole body (SPB), a fungal equivalent of the centrosome is closely associated with the outer nuclear membrane in C. neoformans [42]. Once integrity of the nuclear membrane is compromised by depletion of ergosterol in FLC treated cells, segregation of the SPB may become irregular and enhance the chromosomal instability during cell division [43].
Gene duplication is known to be one of the key mechanisms which allows fungi to be selected during evolution [44]. Aneuploidy resulting in gene duplication has been reported to be the initial evolutionary change in S. cerevisiae selected in vitro to overcome loss of the myosin II protein which is crucial for normal cytokinesis [45].
In response to drug pressure, disomic chromosomes that contain genes relevant to ergosterol synthesis and drug transport could be beneficial for the survival of C. neoformans. Our hypothesis on the crucial roles of ERG11 and AFR1 in the occurrence of Chr1 duplication in clones resistant to high drug concentrations was borne out. When grown on 32 ug/ml FLC, the drug level at which Chr1 disomy occurs in H99, the strain with ERG11 translocated from Chr1 to Chr3 showed duplication only in Chr3 but not in Chr1. However, an extra copy of ERG11 on Chr3 in addition to the native copy on Chr1 was not enough to prevent Chr1 duplication at FLC concentrations higher than 32 µg/ml. This indicated that multiple copies of ERG11 alone can not meet the challenge of very high FLC stress. Similarly, Chr1 was not duplicated when AFR1 was deleted and grown on 1 µg/ml FLC (the strain's initial heteroresistance level). However, Chr4 and Chr5 were duplicated along with short segmental duplications of Chr9 and Chr10, which most likely compensate for the loss of AFR1. These findings underscore the important roles of ERG11 and AFR1 in Chr1 duplication under drug stress. Afr1 is related to Snq2 of C. glabrata which is known to function as a transporter for several compounds including FLC [46]. In our test, afr1Δ was also sensitive to cycloheximide and rhizoxin treatment suggesting that AFR1 may function as a transporter for these drugs (data not shown). An ideal experiment to test the hypothesis that duplication of Chr1 causes drug resistance would be to construct a strain in which only Chr1 is duplicated without exposure to azoles and then test the FLC resistance level of the strain. In S. cerevisiae, strains containing duplicated chromosomes could be constructed and the effect of aneuploidy tested [35]. Currently, construction of such strains, however, is technically not feasible in C. neoformans. Duplication of Chr1 has never been observed in H99 prior to the acquisition of FLC resistance. Since the resistance persisted as long as Chr1 disomy remained but was lost simultaneously after prolonged maintenance in drug free media, we are convinced that the two genes contribute to disomy of Chr1.
The C. neoformans genome contains all the genes known to be associated with ergosterol biosynthesis and has twice as many drug-related transporters as S. cerevisiae. These genes are distributed widely among 14 chromosomes and it is possible that some of them play a role in azole tolerance. It remains to be determined whether any other gene and its regulator necessitate duplication of the chromosome on which it resides. C. neoformans strains, regardless of the chronology of isolation either before or after the launch of azole drugs, showed that 0.3 to 10% of the subpopulations consistently resisted FLC concentrations higher than their MICs [29]. This number did not vary significantly during repeated tests. Although FLC resistant strains of C. neoformans have been increasingly reported from azole therapy failure cases [24], [26], [28], [29], [47]–[49], the number of stable FLC resistant mutants among clinical isolates is rare compared to other pathogenic fungi [15],[50]. One reason for the rarity in isolating FLC resistant C. neoformans mutants may be that heteroresistance masks mutation. The regular mutation rate is 10−5 to 10−6 and such a low population would be masked by the adaptive heteroresistant population. Our results provide the foundation for a mechanistic understanding of transient high azole resistance to FLC which might occur during prolonged maintenance therapy with azoles.
Materials and Methods
Strains and media
C. neoformans isolates H99 and NIH376 are serotype A strains; NIH429 is serotype D [29]. Table 1 lists all the H99 derived strains used in this study. Strains were stored in 25% glycerol stocks at −80°C until use and were maintained on YPD (1% yeast extract, 2% peptone, 2% glucose) agar plates at 30°C for routine cultures.
Heteroresistant phenotype
Fluconazole (FLC) was provided as powder by Pfizer Global Research & Development (Groton, CT). Stock solutions were prepared in dimethyl sulfoxide (Sigma) at a concentration of 50 mg/ml. Analysis of FLC heteroresistance was performed by the method described previously [29]. Briefly, cell suspensions (1×103 to 4×103 CFU/ml) in sterile saline were plated on YPD plates containing various concentrations of FLC. Growth was recorded after 72 h incubation at 30°C. Isolates were considered to be heteroresistant when resistant clonal populations were able to grow on a plate containing FLC. Resistant subpopulations were exposed to stepwise increases in FLC concentrations on YPD media.
Gene expression array analysis
Microarray slides were purchased from the Genome Sequencing Center at Washington University, St Louis. For cDNA arrays, overnight cultures were diluted to OD600 ≅ 0.2 and grown in YPD liquid media for 7 hr. RNA was extracted from yeast cells using Trizol (Invitrogen, Carlsbad, CA), and purified with RNeasy MinElute cleanup kit (Qiagen, Valencia, CA). RNA was labeled and hybridized as described previously [51]. Arrays were scanned on a GenePix 4000B scanner and analyzed using GENEPIX PRO 6.0 (Axon Instruments, Foster City, CA). Data were further analyzed in mAdb database at http://madb.niaid.nih.gov. Three biological repeats were performed using three independent RNA sets isolated from cells cultured on different days and the dye-reverse hybridizations were performed for all 3 sets. One set of RNA was also subjected to technical repeats. All statistically significant genes were identified by significance analysis of microarray using a mean false discovery rate of less than 5%. Only statistically significant genes were used for data analysis. Although the microarray slides used in this study were printed with 70-mers that are designed to uniquely represent each gene in C. neoformans serotype D, the oligomers were also optimized for homology to genes predicted in the serotype A strain, H99 (http://genome.wustl.edu/services/microarray/cryptococcus_neoformans).
Comparative genome hybridization
Genomic DNA was prepared from C. neoformans strains grown overnight in 10 ml YPD medium as described previously [52]. 5 µg DNA was digested with DpnII (10 U/µg DNA, New England Biolabs, Ipswich, USA) and labeled with dye according to the BioPrime®Array CGH Genomic Labeling System protocol (Invitrogen Life Technologies, Carlsbad, USA). In all CGH experiments, Alexa647 was used to label DNA from the experimental strains and Alexa555 was used to label DNA from the reference control strain (Invitrogen Life Technologies, Carlsbad, USA). Labeled DNA was purified with the purification kit from the same manufacturer and subjected to competitive hybridization with the 70mers microarray. Sample hybridization and data collection were carried out as described above. Data were further analyzed in mAdb database after applying 50th percentile (Median) normalization.
Two parameters were considered for the CGH experiments. First, we hybridized the slides using H99 genomic DNA as both the experimental and the reference control samples. The scatter plot of the normalized log10 signal intensity of both channels showed tight correlation between two probes attesting to the reliability of the hybridization patterns of H99 genomic DNA to the JEC21-based 70mer slides. Second, we tested the reproducibility between arrays. Data from five independent CGH arrays were obtained from the H99 control set (H99-Alexa 555 vs. H99-Alexa 647) as well as from the H99R64 set (H99-Alexa 555 vs. H99R64-Alexa 647). The data were highly consistent indicating high reproducibility between the arrays. Therefore, in most CGH studies, only one or two arrays per strain were analyzed.
To visualize the CGH array in a chromosomal context, data were imported into Excel format from the mAdb database. CGH data was further normalized by subtracting the average log2 signal ratio of each gene obtained in control experiments (H99 Alexa647 vs. H99 Alexa555) from that of a corresponding gene in the experimental data set to compensate for the dye and background bias. Relative hybridization levels were plotted as a running average over seven ORFs and clipped to the range corresponding to 1–2 copies (log2 ratio of 0–1, respectively). Each ORF was sorted according to their gene number corresponding to its order along each chromosome (plotted on the x-axis). Although the genomes are largely co-linear between the current genomic assemblies of H99 and JEC21, there are several apparent inversions and translocations. Due to these alterations, homologous chromosomes between the H99 and JEC21 assemblies have been assigned different numbers for some chromosomes [53]. The chromosomal number assignment of H99 was adopted in our CGH data. Due to the translocation events in Chr3, Chr4 and Chr11 of H99, the order of genes on these chromosomes was manually arranged according to its JEC21 counterparts.
Quantitative real time PCR
To quantify the gene copy number on specific chromosomes in wild-type and FLC-resistant strains, quantitative real time PCR (qPCR) assays were performed. For confirmation of CGH data, the same genomic DNA from strains used in CGH arrays was used for qPCR assays. For individual colony qPCR, genomic DNA of selected colonies was used. For colony DNA extraction, a single colony was picked with a sterile toothpick, suspended in 40 µl of 10 mM EDTA buffer in a microcentrifuge tube, boiled for 6 min and centrifuged. The supernatant was diluted 1∶10 in TE buffer and 5 µl of diluted DNA template was added to 20 µl of the qPCR mix (Applied Biosystems, Branchburg, NJ). The reaction was performed in an Applied Biosystems 7500 Real-Time PCR System. Each reaction was run in triplicate and the average Ct value was converted to relative amount of DNA using the relative standard curve method. The sequences of the primers and probes used for the qPCR are listed in Table S2. The genes CNAG_02959 on Chr3, CNAG_00869 on Chr5 and /or CNAG_07554 on Chr11 were chosen as endogenous controls. For each specific gene, its copy number was obtained by comparing its qPCR value with the endogenous control and expressed as relative gene copy number.
Gene manipulation
ERG11 was cloned by PCR and sequenced. The NAT selectable marker was cloned into the 5′ flanking region of ERG11 and the resulting construct was inserted in the intergenic region between CNAG_03012 and CNAG_03013 on Chr3 which were generated by PCR and sequenced. The final construct was transformed into H99 and the transformant containing a second copy of ERG11 between the intergenic region of CNAG_03012 and CNAG_03013 on Chr3 was screened by PCR and confirmed by Southern blot analysis. Subsequently, the ERG11 gene on Chr1 was deleted with the NEO gene from the clone containing two copies of ERG11 (C1345) by biolistic transformation.
Statistics
An unpaired t test was used for the statistical analysis of qPCR data. A P value of less than 0.05 was considered to be significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Kwon-ChungKJ
BennettJE
1992 Medical Mycology. Philadelphia Lea & Febiger 866
2. PerfectJR
CasadevallA
2002 Cryptococcosis. Infect Dis Clin North Am 16 837 874
3. ZoniosDI
BennettJE
2008 Update on azole antifungals. Semin Respir Crit Care Med 29 198 210
4. PerfectJR
CoxGM
1999 Drug resistance in Cryptococcus neoformans. Drug Resist Updat 2 259 269
5. KontoyiannisDP
SagarN
HirschiKD
1999 Overexpression of Erg11p by the regulatable GAL1 promoter confers fluconazole resistance in Saccharomyces cerevisiae. Antimicrob Agents Chemother 43 2798 2800
6. LampingE
MonkBC
NiimiK
HolmesAR
TsaoS
2007 Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell 6 1150 1165
7. AkinsRA
2005 An update on antifungal targets and mechanisms of resistance in Candida albicans. Med Mycol 43 285 318
8. BennettJE
IzumikawaK
MarrKA
2004 Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrob Agents Chemother 48 1773 1777
9. BrunS
BergesT
PoupardP
Vauzelle-MoreauC
RenierG
2004 Mechanisms of azole resistance in petite mutants of Candida glabrata. Antimicrob Agents Chemother 48 1788 1796
10. HelmerhorstEJ
VenuleoC
SanglardD
OppenheimFG
2006 Roles of cellular respiration, CgCDR1, and CgCDR2 in Candida glabrata resistance to histatin 5. Antimicrob Agents Chemother 50 1100 1103
11. SanglardD
OddsFC
2002 Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2 73 85
12. TsaiHF
KrolAA
SartiKE
BennettJE
2006 Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50 1384 1392
13. WhiteTC
HollemanS
DyF
MirelsLF
StevensDA
2002 Resistance mechanisms in clinical isolates of Candida albicans. Antimicrob Agents Chemother 46 1704 1713
14. CowenLE
AndersonJB
KohnLM
2002 Evolution of drug resistance in Candida albicans. Annu Rev Microbiol 56 139 165
15. CowenLE
SteinbachWJ
2008 Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell 7 747 764
16. LupettiA
DanesiR
CampaM
Del TaccaM
KellyS
2002 Molecular basis of resistance to azole antifungals. Trends Mol Med 8 76 81
17. MarichalP
KoymansL
WillemsensS
BellensD
VerhasseltP
1999 Contribution of mutations in the cytochrome P450 14alpha-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 145 2701 2713
18. SanglardD
IscherF
CalabreseD
MicheliM
BilleJ
1998 Multiple resistance mechanisms to azole antifungals in yeast clinical isolates. Drug Resist Updat 1 255 265
19. SelmeckiA
ForcheA
BermanJ
2006 Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313 367 370
20. SelmeckiA
Gerami-NejadM
PaulsonC
ForcheA
BermanJ
2008 An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68 624 641
21. CowenLE
LindquistS
2005 Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 309 2185 2189
22. CowenLE
CarpenterAE
MatangkasombutO
FinkGR
LindquistS
2006 Genetic architecture of Hsp90-dependent drug resistance. Eukaryot Cell 5 2184 2188
23. ArmengouA
PorcarC
MascaroJ
Garcia-BragadoF
1996 Possible development of resistance to fluconazole during suppressive therapy for AIDS-associated cryptococcal meningitis. Clin Infect Dis 23 1337 1338
24. BergJ
ClancyCJ
NguyenMH
1998 The hidden danger of primary fluconazole prophylaxis for patients with AIDS. Clin Infect Dis 26 186 187
25. BirleyHD
JohnsonEM
McDonaldP
ParryC
CareyPB
1995 Azole drug resistance as a cause of clinical relapse in AIDS patients with cryptococcal meningitis. Int J STD AIDS 6 353 355
26. PaugamA
Dupouy-CametJ
BlancheP
GangneuxJP
Tourte-SchaeferC
1994 Increased fluconazole resistance of Cryptococcus neoformans isolated from a patient with AIDS and recurrent meningitis. Clin Infect Dis 19 975 976
27. VenkateswarluK
TaylorM
ManningNJ
RinaldiMG
KellySL
1997 Fluconazole tolerance in clinical isolates of Cryptococcus neoformans. Antimicrob Agents Chemother 41 748 751
28. MondonP
PetterR
AmalfitanoG
LuzzatiR
ConciaE
1999 Heteroresistance to fluconazole and voriconazole in Cryptococcus neoformans. Antimicrob Agents Chemother 43 1856 1861
29. SionovE
ChangYC
GarraffoHM
Kwon-ChungKJ
2009 Heteroresistance to fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob Agents Chemother 53 2804 2815
30. PosteraroB
SanguinettiM
SanglardD
La SordaM
BocciaS
2003 Identification and characterization of a Cryptococcus neoformans ATP binding cassette (ABC) transporter-encoding gene, CnAFR1, involved in the resistance to fluconazole. Mol Microbiol 47 357 371
31. PolakovaS
BlumeC
ZarateJA
MentelM
Jorck-RambergD
2009 Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci U S A 106 2688 2693
32. RustchenkoE
ShermanF
2002 Genetic instability of Candida albicans.
HowardDH
Fungi Pathogenic for Humans and Animals New York Marcel Dekker, Inc 723 776
33. HartwellLH
DutcherSK
WoodJS
GarvikB
1982 The fidelity of mitotic chromosome reproduction in S. cerevisiae. Rec Adv Yeast Mol Biol 1 28 38
34. RosenstrausMJ
ChasinLA
1978 Separation of linked markers in Chinese hamster cell hybrids: mitotic recombination is not involved. Genetics 90 735 760
35. TorresEM
SokolskyT
TuckerCM
ChanLY
BoselliM
2007 Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317 916 924
36. ZolanME
1995 Chromosome-length polymorphism in fungi. Microbiol Rev 59 686 698
37. VarmaA
Kwon-ChungKJ
1994 Formation of a minichromosome in Cryptococcus neoformans as a result of electroporative transformation. Curr Genet 26 54 61
38. KaernM
ElstonTC
BlakeWJ
CollinsJJ
2005 Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6 451 464
39. SternC
1958 The nucleus and somatic cell variation. J Cell Physiol Suppl52 1 27; discussion 27-34
40. KaferE
1976 Mitotic crossing over and nondisjunction in translocation heterozygotes of Aspergillus. Genetics 82 605 627
41. FernandezC
Lobo Md MdelV
Gomez-CoronadoD
LasuncionMA
2004 Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp Cell Res 300 109 120
42. YamaguchiM
BiswasSK
OhkusuM
TakeoK
2009 Dynamics of the spindle pole body of the pathogenic yeast Cryptococcus neoformans examined by freeze-substitution electron microscopy. FEMS Microbiol Lett 296 257 265
43. GanemNJ
GodinhoSA
PellmanD
2009 A mechanism linking extra centrosomes to chromosomal instability. Nature 460 278 282
44. WapinskiI
PfefferA
FriedmanN
RegevA
2007 Natural history and evolutionary principles of gene duplication in fungi. Nature 449 54 61
45. RancatiG
PavelkaN
FlehartyB
NollA
TrimbleR
2008 Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135 879 893
46. TorelliR
PosteraroB
FerrariS
La SordaM
FaddaG
2008 The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol Microbiol 68 186 201
47. BicanicT
HarrisonT
NiepiekloA
DyakopuN
MeintjesG
2006 Symptomatic relapse of HIV-associated cryptococcal meningitis after initial fluconazole monotherapy: the role of fluconazole resistance and immune reconstitution. Clin Infect Dis 43 1069 1073
48. FrieseG
DischerT
FussleR
SchmalreckA
LohmeyerJ
2001 Development of azole resistance during fluconazole maintenance therapy for AIDS-associated cryptococcal disease. AIDS 15 2344 2345
49. YamazumiT
PfallerMA
MesserSA
HoustonAK
BoykenL
2003 Characterization of heteroresistance to fluconazole among clinical isolates of Cryptococcus neoformans. J Clin Microbiol 41 267 272
50. CosteA
SelmeckiA
ForcheA
DiogoD
BougnouxME
2007 Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6 1889 1904
51. LeeH
BienCM
HughesAL
EspenshadePJ
Kwon-ChungKJ
2007 Cobalt chloride, a hypoxia-mimicking agent, targets sterol synthesis in the pathogenic fungus Cryptococcus neoformans. Mol Microbiol 65 1018 1033
52. ChangYC
Kwon-ChungKJ
1994 Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol Cell Biol 14 4912 4919
53. KavanaughLA
FraserJA
DietrichFS
2006 Recent evolution of the human pathogen Cryptococcus neoformans by intervarietal transfer of a 14-gene fragment. Mol Biol Evol 23 1879 1890
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2010 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
- A Timescale for Evolution, Population Expansion, and Spatial Spread of an Emerging Clone of Methicillin-Resistant