
Chronic Parasitic Infection Maintains High Frequencies of Short-Lived Ly6CCD4 Effector T Cells That Are Required for Protection against Re-infection
Naturally acquired resistance to reinfection by numerous infectious pathogens including Leishmania, Plasmodium, Mycobacterium, and parasitic worms, typically coincides with an ongoing primary infection. This natural resistance to reinfection, termed concomitant immunity, is often referred to as a memory response and provides the rationale for the vaccine effort against these infectious pathogens. However, immune memory is mediated by populations of long-lived cells that do not require an ongoing primary infection to mediate protection. The requirement for chronic infection to maintain concomitant immunity suggests that the critical cells that mediate this immunity are not memory cells. In the present study we define short-lived effector T cells that pre-exist secondary challenge, not memory cells, as the critical cells that mediate concomitant immunity. These observations provide direct evidence on a cellular level that conventional vaccination strategies against chronic infectious diseases, whose development is predicated upon the belief that concomitant immunity can be mediated by long-lived memory cells, are unlikely to succeed.
Published in the journal:
Chronic Parasitic Infection Maintains High Frequencies of Short-Lived Ly6CCD4 Effector T Cells That Are Required for Protection against Re-infection. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004538
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004538
Summary
Naturally acquired resistance to reinfection by numerous infectious pathogens including Leishmania, Plasmodium, Mycobacterium, and parasitic worms, typically coincides with an ongoing primary infection. This natural resistance to reinfection, termed concomitant immunity, is often referred to as a memory response and provides the rationale for the vaccine effort against these infectious pathogens. However, immune memory is mediated by populations of long-lived cells that do not require an ongoing primary infection to mediate protection. The requirement for chronic infection to maintain concomitant immunity suggests that the critical cells that mediate this immunity are not memory cells. In the present study we define short-lived effector T cells that pre-exist secondary challenge, not memory cells, as the critical cells that mediate concomitant immunity. These observations provide direct evidence on a cellular level that conventional vaccination strategies against chronic infectious diseases, whose development is predicated upon the belief that concomitant immunity can be mediated by long-lived memory cells, are unlikely to succeed.
Introduction
Chronic infectious diseases, including those caused by Leishmania, Plasmodium, Mycobacterium, and parasitic worms, continue to be a major cause of morbidity and mortality in the world. Understanding the critical factors mediating protective immunity to these diseases and whether this immunity can be mediated by memory cells is likely to contribute to a long-term solution, including the feasibility of developing a vaccine. Observations employing infection with lymphphocytic choriomeningitis virus (LCMV) and Listeria monocytogenes have established a paradigm of CD8+ T cell memory in which stable populations of central (TCM) and effector (TEM) memory cells are found after clearance of a primary infection. These CD8+ memory cells mediate protective immunity upon secondary infection [1], [2]. In contrast, the nature of CD4+ T cell memory is less clear. This is especially true in cases of CD4-mediated concomitant immunity, where protection against reinfection coincides with the persistence of a primary infection. Infections in which concomitant immunity is thought to play a significant role include Malaria, Leishmaniasis, Tuberculosis, some forms of Salmonellosis, and helminthic infections [3]–[10]. The immune response against re-infection in these settings is often referred to as a memory response, which, as most commonly defined, is constituted by a population of long-lived cells that do not require the continued presence of antigen. However, chronic infection may provide a persistent source of antigen that sustains effector CD4+ T cells. Pre-existing and infection dependent effector cells may be critical to provide protection upon re-infection [6]–[8], [11].
In a murine model of cutaneous leishmaniasis, Zaph et al. [12] demonstrated that CD4+CD62L+ T cells with the functional attributes of TCM cells were maintained in the absence of persistent Leishmania parasites and could mediate delayed protection to re-challenge by needle inoculation. The role of memory cells in Leishmania concomitant immunity has been questioned, however, by studies suggesting that immunity is either completely lost or is suboptimal when the chronic primary infection is eliminated [12]–[15]. Similar observations exist in cancer and malaria [16], [17]. We have recently reported that the clearest correlate of effective concomitant immunity against natural transmission of Leishmania by the bite of an infected sand fly is the rapid recruitment (within 24 hours) of IFN-γ-producing CD4+ T cells to the cutaneous bite site. In contrast, non-living vaccines mediate delayed immunity to needle challenge similar to that reported for TCM cells, and provide no protection to infected sand fly challenge [11], [18]. The rapid CD4+ T cell response is necessary to counteract the disease exacerbating inflammatory response elicited by natural sand fly transmission [11]. The nature of the rapidly responding CD4+ T cells that mediate concomitant immunity, including their frequency and tissue distribution during the course of chronic infection, their migration to and function within the challenge site, and most critically, their life span and memory potential, have not been determined. Here, we define those cells that mediate concomitant immunity as pre-existing, short-lived, T-bethiLy6C+ TEFF cells that, despite their short life span in the absence of infection, are maintained as the dominant population of antigen experienced cells throughout chronic infection.
Results
Analysis of concomitant immunity in cutaneous Leishmaniasis
People with a healed but chronic primary Leishmania infection are highly resistant to reinfection following natural exposure to infected sand flies [19]. The same is true in an experimental setting where mice with a healed but chronic primary infection in the footpad presented with significantly smaller lesions compared to naïve mice starting at 4 weeks post-exposure of the ear dermis to infected sand fly bites (p≤0.04) (Fig. 1A). Chronic mice also had significantly fewer parasites in the sand fly challenged ear (178-fold reduction, p = 0.0011) and ear dLN (27-fold reduction, p≤0.0001) at 6 weeks post-infection (Fig. 1B). This is in contrast to non-living vaccines, which do not protect against sand fly transmitted disease either in mice or humans [11], [18], [20].
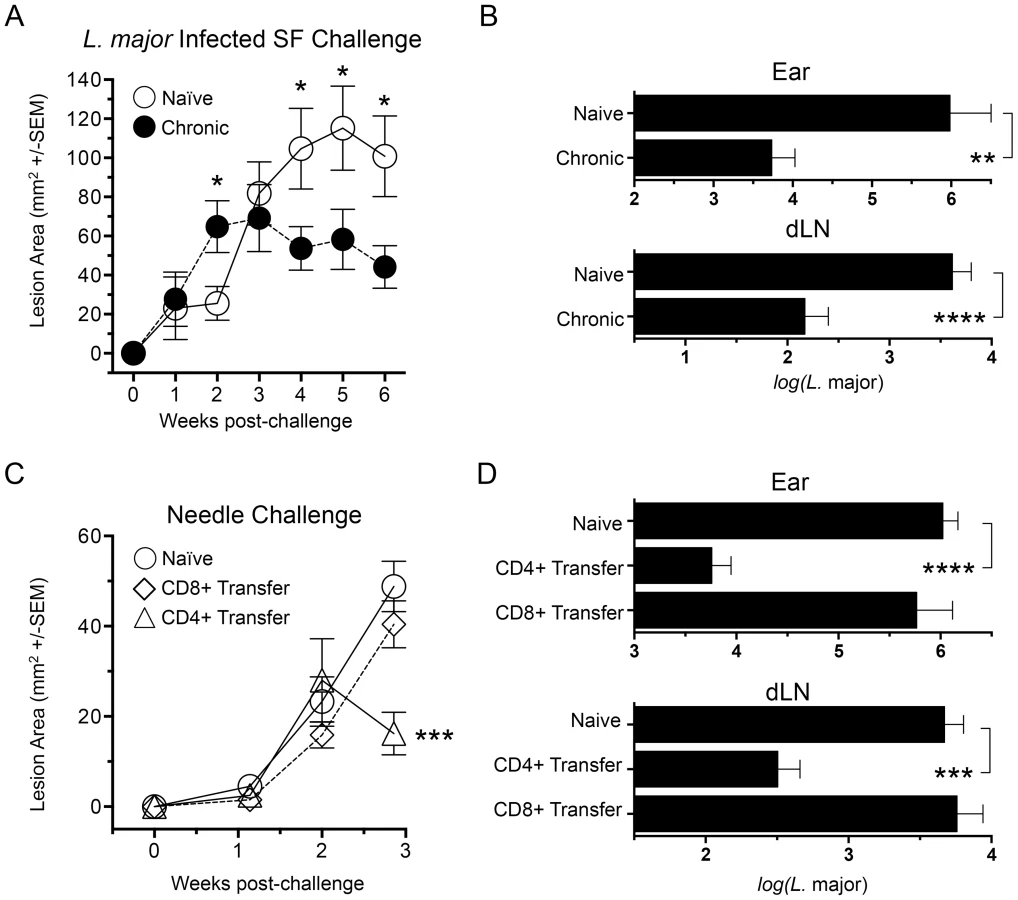
While CD8 cells have been shown to play a role in secondary immune responses to L. major infection [21], protection conferred following adoptive transfer of bulk immune cells versus CD4+ T cells alone has shown that CD4+ T cells alone are sufficient [12]. We expanded upon these observations by determining the protective capacity of CD4+ or CD8+ T cells from chronically infected mice following adoptive transfer into naïve recipients at their pre-existing physiological ratios (Figure 1, C and D). Because of the constraints on the numbers of purified cells available for transfer, in conjunction with the enormous variability of successful transmission by sand fly bite, needle challenge was employed to insure that an infection outcome could be followed in the relatively small numbers of mice per group that we are able to employ in our adoptive transfer system. We measured early control of parasite numbers following needle challenge as this has consistently provided the best correlate of protective immunity against infected sand fly challenge, and protection at later time points against needle challenge does not correlate with protection against sand fly challenge [11], [18]. We found that CD4+ Th cells conferred protection versus control animals at 21 days post-challenge, significantly reducing lesion size (p = 0.0007) (Fig. 1C) and parasite loads in the ear and ear dLN (p≤0.0002) (Fig. 1 D). In contrast, CD8+ T cells conferred no protection versus naïve controls. Subsequent studies were designed to determine the nature of the CD4+ Th cells that confer this protection.
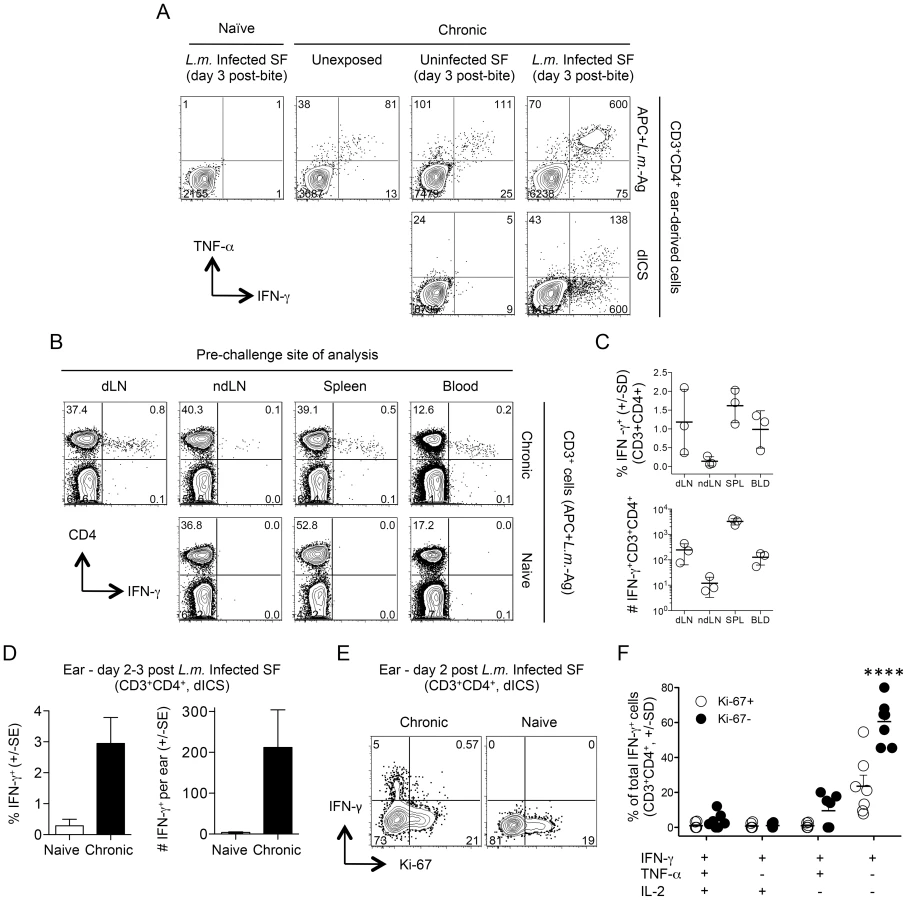
The rapid recruitment (within 3 days) of IFN-γ+CD4+ Th cells to the cutaneous site of infected sand fly bite in mice with a chronic L. major infection is the strongest correlate of protection against natural re-infection [11]. We exposed the ear dermis of mice with a healed chronic infection in the footpad to the bites of L.m.-infected flies and 3 days later dermal derived CD3+CD4+ T cells were analyzed for cytokine production. Following antigen re-stimulation, large numbers of cells produced IFN-γ and TNF-α (Fig. 2A, top, right panel). Exposure of chronic mice to the bites of uninfected sand flies also resulted in greater numbers of cytokine positive cells versus unexposed chronic mice, which contained small numbers of pre-existing CD4+IFN-γ+ cells (Fig. 2A, top, middle panels) [22]. These cells are likely recruited to the skin in an antigen non-specific manner [23] from the blood where significant numbers of T cells with the capacity to make IFN-γ exist before challenge (Fig. 2B and C). Cells recovered from ears exposed to infected sand fly bites and evaluated for cytokine production by direct intracellular staining (dICS, see Figure S1) without antigen or pharmacological re-stimulation contained large numbers of IFN-γ+ cells (Fig. 2A, bottom right panel and 2D). In contrast, exposure to the bites of uninfected flies revealed virtually no IFN-γ+ cells following dICS, likely due to the absence of antigen (Fig. 2A, bottom left panel). Direct ICS is a highly physiological assessment of antigen-specific cells making cytokine in response to in-vivo infection. On day 2 post-infected sand fly exposure, IFN-γ+ cells were highly enriched for Ki-67− cells (Fig. 2E), indicating that these cells had not undergone proliferation in the time since challenge and were therefore unlikely to be derived from memory cells reactivated by secondary antigen exposure [12], [24]. Assessment of IFN-γ, TNF-α and IL-2 production by dICS revealed that IFN-γ+CD4+ T cells were predominantly Ki-67−IFN-γ single producers (Fig. 2F), in striking contrast to the predominant IFN-γ+TNF-α+ phenotype detected following overnight antigen re-stimulation (Fig. 2A).
Rapidly recruited L. major-specific cells are not derived from central memory cells reactivated by secondary challenge
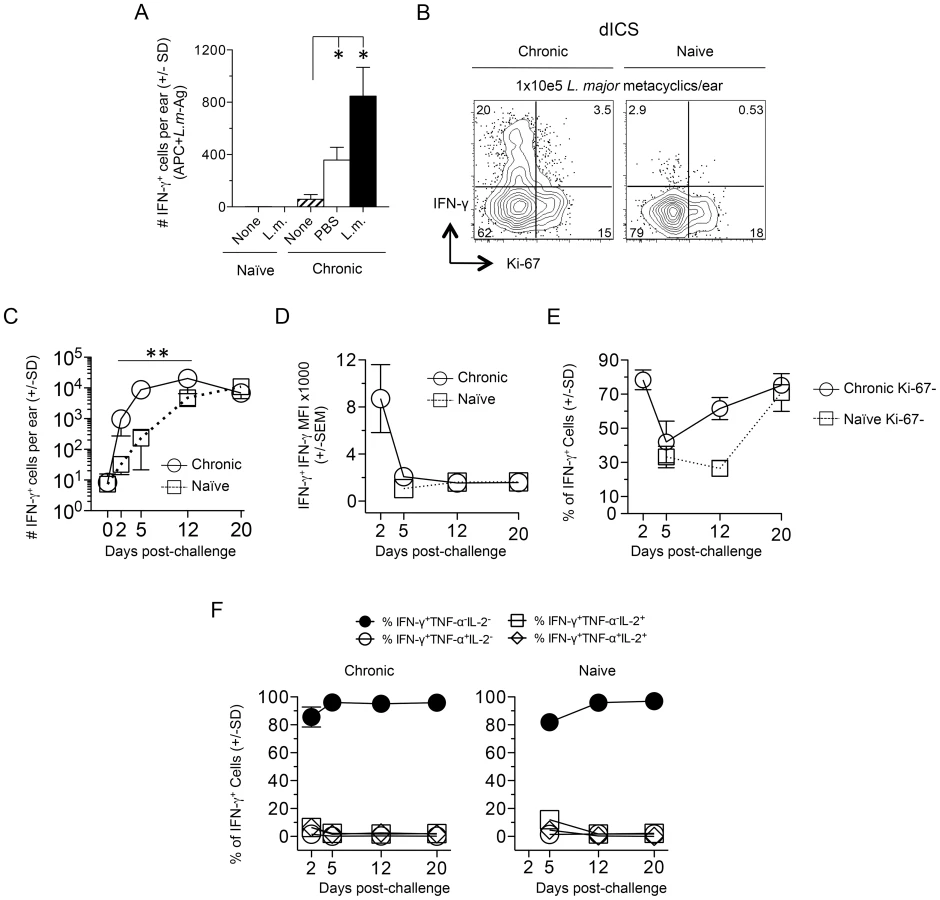
We extended our findings employing needle challenge, which is far more reproducible in terms of inoculum dose and tissue damage. Twenty hours following needle inoculation with PBS, the number of IFN-γ+CD4+ T cells following Ag stimulation was significantly greater in chronic mice versus naïve or non-injected chronic controls (Fig. 3A), again demonstrating the recruitment of these cells to the skin in an antigen non-specific manner, likely from the blood. Employing dICS, rapidly recruited IFN-γ+ cells observed at day 2 post-needle challenge were also highly enriched for Ki-67− cells (Compare Fig. 2E to Fig. 3B). Kinetic analysis of the secondary response by dICS revealed a marked increase in the relative number of CD4+ T cells producing IFN-γ in-vivo by day 2 post challenge in chronic versus naïve mice (p≤0.002) (Fig. 3C). Rapidly recruited IFN-γ+ cells observed at day 2 expressed high levels of IFN-γ relative to later time points (p<0.002) (Fig. 3D) and were highly enriched for Ki-67−, IFN-γ single-producing cells (Fig. 3, E and F).
Infection dependent polyclonal CD4+CD62L− T cells from mice with a chronic primary infection have been shown to mediate protection following adoptive transfer and needle challenge [12]. We employed an adoptive transfer system that for the first time allowed tracking of CD4+CD44+CD62L− cells to the dermal challenge site in recipient mice so that their recruitment, functionality, and lifespan could be assessed. Violet proliferation-dye labeled, CD4+CD44+CD62L+ TCM or CD4+CD44+CD62L− cells, which potentially contain both TEM and TEFF cells, were sorted from the spleens and dLNs of chronic mice and co-transferred into naïve recipients (Fig. 4A and Fig. S2). Prior to transfer both CD44+CD62L+ and CD44+CD62L− Th cells were enriched for the Th1 markers T-bet and CXCR3, and the activation markers CD69, ICOS, and CD54 (ICAM-1) (Fig. S3A). Prior studies have shown IL-7R is rapidly re-expressed on effector CD4+ T cells after a period of non-expression during clonal expansion [17], [25], [26]. Therefore, IL-7R expression does not define a CD4+ T cell as a memory cell. Consistent with these observations we found both CD44+CD62L+ and CD44+CD62L− Th cells were predominantly IL-7R high, but that the CD44+CD62L− population contained a slightly higher frequency of IL-7R negative or low cells (Fig. S3B).
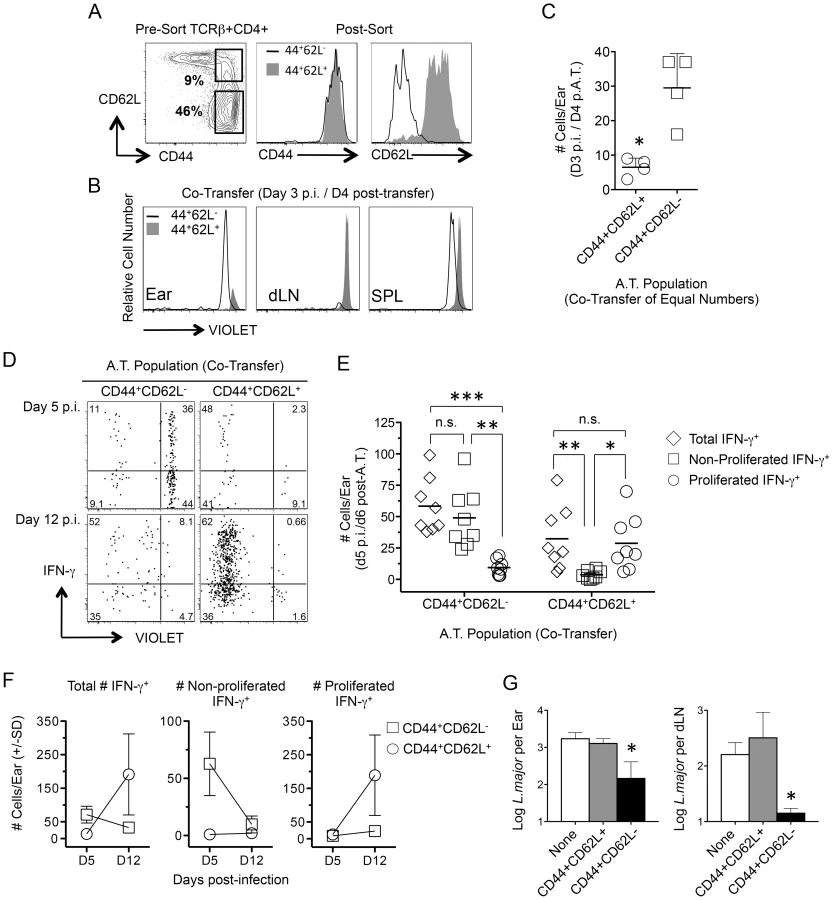
Direct comparison of the tissue homing potential of polyclonal CD44+CD62L+ and CD44+CD62L− Th cells was analyzed following co-transfer of both populations at equivalent numbers and challenge with L. major the next day. On day 3 p.i., CD44+CD62L− cells had homed preferentially to the ear without having undergone division while CD44+CD62L+ cells were found in the dLN but not the ear (Fig. 4B and 4C). While either population could be found in the spleen of naïve or challenged mice, L.m. inoculation was required to observe CD44+CD62L− cells in the ear or the proliferation of CD44+CD62L+ cells in the dLN on day 4 p.i. (Fig. S4). Two-photon imaging of the ear dermis following challenge also revealed the presence of donor CD4+ T cells and the dynamic patrolling behavior of these cells was similar to what has been reported during the early primary response (Movie s1 and Movie s2) [27].
By day 5 p.i., antigen-specific IFN-γ+ T cells derived from either donor population could be found in the ear (Fig. 4D). However, the vast majority of IFN-γ+ cells from the CD44+CD62L− population had not divided while the few IFN-γ+ cells derived from the CD44+CD62L+ TCM population had virtually all divided (Fig. 4E). On day 12 p.i. the number of non-proliferated IFN-γ+CD44+CD62L− donor cells was reduced to virtually zero, while a large and expanded population of proliferated IFN-γ+ CD44+CD62L+ TCM derived cells was detected (TCM, D5 vs. D12 p = 0.028) (Fig. 4F). Similar to previous observations [12], [28], TCM cells acquired effector function only after proliferating in the dLN (Figure S5). Despite the high proliferative capacity of TCM cells and the resulting numeric advantage of TCM-derived cells in the ear at day 12 p.i. (CD44+CD62L− vs. TCM, D12 p = 0.028), analysis of parasite loads in the ear and dLN of recipient animals receiving each population alone revealed that only CD44+CD62L− cells conferred protection at 21 days p.i. (Fig. 4G). These observations directly correlate the early recruitment of IFN-γ-producing, non-dividing CD4+ T cells with protective immunity.
Rapidly-recruited L. major-specific cells are T-bet+Ly6C+
Leishmania-specific cells within the CD44+CD62L− Th cell population are phenotypically heterogenous, and likely include both CD4+ TEFF cells and TEM cells. A high level of T-bet expression has been associated with a TEFF phenotype [26]. On day 1 post-challenge of chronic mice dermal IFN-γ+ cells from the challenge site were highly enriched for T-bet+Ki-67− cells (Fig. 5A and 5B) and were largely T-bet high versus IFN-γ− T cells (Fig. 5B). Recently, LCMV-specific Ly-6C+ cells were identified as T-bet high Th1 TEFF cells [26] that had a shorter lifespan than Ly6C− cells, and expressed a gene expression profile associated with activation rather than memory. Early after challenge dermal-derived CD4+IFN-γ+ T cells were predominantly Ly6C+ and were highly enriched for T-bet and Ly6C co-expressing cells versus CD4+IFN-γ− cells from the same tissue (Fig. 5C and 5D). Both IFN-γ+ and IFN-γ− cells in the ear expressed low levels of CD27, a cell-surface molecule associated with memory cells [29], [30], and comparable levels of IL-7R (Figure S6). Regardless of IFN-γ-production, Ly6C was strongly co-expressed with T-bet (Fig. 5C) and Ly6C+ cells in the ear were highly enriched for IFN-γ+T-bet+Ki-67− cells versus the Ly6C− population (Fig. 5E and 5F).
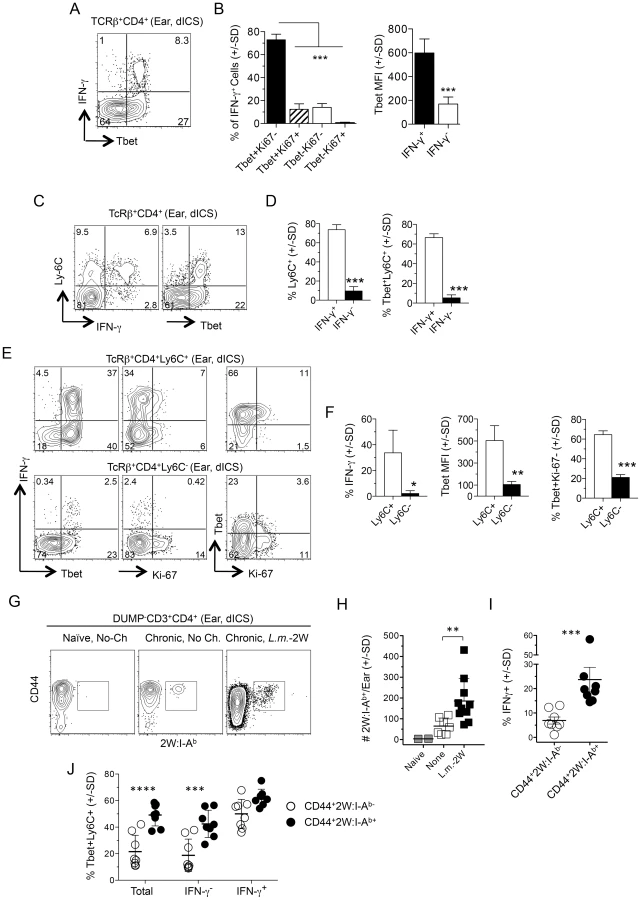
We also employed a 2W peptide:MHC II tetramer that allows quantification of endogenous L. major specific cells following infection with transgenic L. major-2W [31] without relying on antigen driven, intracellular cytokine accumulation. In agreement with our dICS analysis of polyclonal antigen-specific IFN-γ+ cells, i.d. challenge of mice with a chronic L. major-2W infection resulted in a significant increase in 2W:I-Ab tetramer+ cells in the ear dermis by 3 days post-challenge (Fig. 5G and 5H). Note that a few of these CD4+ cells were found patrolling the skin even prior to challenge [22]. Direct ICS also revealed an enrichment of IFN-γ+ cells amongst the rapidly recruited 2W:I-Ab -specific population (Fig. 5I), of which the majority were Ly6C+T-bet+ cells (Fig. 5J). Ly6C+T-bet+ cells were also highly enriched in the total or IFN-γ− 2W:I-Ab-specific population versus the CD44+2W:I-Ab tetramer− cells (Fig. 5J), demonstrating that even amongst non-IFN-γ-producing L. major-specific cells there is a significant enrichment of Ly6C+T-bet+ cells in the re-challenge site.
These data implicate the rapidly recruited L. major-specific cells defined in Figures 2–4 as Ly6C+T-bet+ TEFF cells.
Chronic L. major infection maintains high frequencies of antigen-specific Ly6C+ TEFF cells
Ly6C expression on Th cells at the site of Leishmania challenge could be the result of recruitment or antigen-exposure at the challenge site rather than a marker of the responding cells prior to challenge. Prior to challenge, the CD44+CD62L−Ly6C+ cells in the spleen of chronic mice had characteristics similar to those of dermal CD4+IFN-γ+Ly6C+ T cells following challenge, namely low Ki-67 expression and high expression of T-bet (Fig. 6A). Unlike CD44+CD62L− cells positive for the canonical Th1 marker CXCR3, Ly6C+ cells were exclusively T-bet high (Fig. 6B), and were highly enriched in the circulation compared to the spleen, dLN, or ndLN (Fig. 6C). Prior to challenge, Ly6C+ and Ly6C− polyclonal CD44+CD62L− cells expressed low levels of CD103 and were predominantly IL-7R+, however, only the Ly6C− population contained cells that also expressed CD27 (Fig. S7). In addition, a proportion of the Ly6C+ population in the dLN was proliferating or had recently proliferated based upon Ki-67 expression (Fig. S7B). Sorted CD44+CD62L−Ly6C+ T cells from chronic mice were also enriched for cells with the capacity to produce IFN-γ immediately upon antigen stimulation (Fig. 6D).
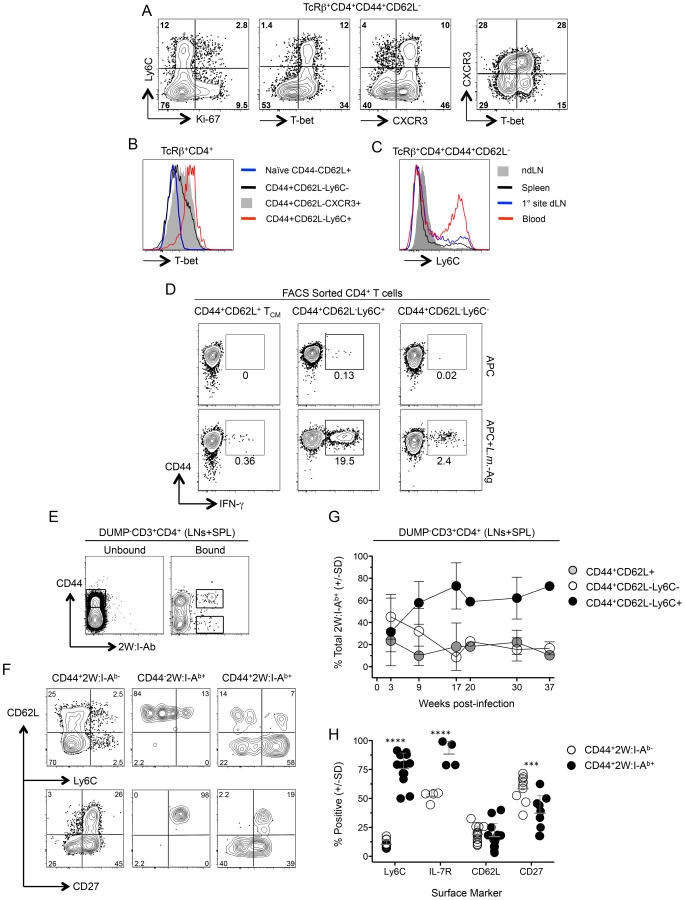
We recently employed the 2W peptide:MHC II tetramer to demonstrate that parasite-specific cells in the spleen and LNs expand during the first weeks of L. major-2W infection in the skin and then contract, maintaining constant numbers and function during the chronic stage of infection [31]. We extend these studies by showing 2W:I-Ab tetramer+ cells from the spleen and LNs were highly enriched for Ly6C+ cells versus the CD44+2W:I-Ab− population (Fig. 6, E–H). Analysis of the frequencies of the CD44+CD62L−Ly6C+, TCM and CD44+CD62L−Ly6C− subsets within the 2W:I-Ab tetramer+ cells over the course of infection revealed that CD44+CD62L−Ly6C+ T cells accounted for 60% of parasite-specific cells throughout the chronic stage of infection (9–37 weeks) (Fig. 6G). The 2W:I-Ab -specific CD4+ T cells were also enriched for IL-7R but not for CD62L, and decreased for expression of the memory marker CD27 during chronic infection (Fig. 6H). These data demonstrate that L. major infection preferentially maintains a high frequency of antigen specific CD44+CD62L−Ly6C+ cells during chronic infection and these cells have properties of circulating TEFF cells.
Ly6C+ TEFF cells emulate the concomitant immune response
We next wished to formally demonstrate that pre-existing CD44+CD62L−Ly6C+ TEFF cells are the source of rapidly recruited IFN-γ+ cells at the site of challenge. Approximately equal numbers of sorted CD44+CD62L−Ly6C+, CD44+CD62L−Ly6C− or CD44+CD62L+ TCM CD4+ T cells derived from chronic mice, see Fig. 7A, were independently transferred with GFP+CD4+ T cells from naïve mice to act as an internal control, into congenic recipients. Mice were challenged with L. major 1 day following transfer. Three days following challenge, ears, and purified CD4+ T cells from the spleen and ear dLNs of recipient mice were analyzed for the presence of adoptively transferred cells (Fig. 7B). CD44+CD62L−Ly6C+ cells preferentially homed to the ear following challenge as compared to both co-transferred CD4+ cells from naïve mice (p = 0.003) and CD44+CD62L−Ly6C− (p = 0.002) or CD44+CD62L+ TCM (p = 0.001) cells. In contrast, CD62L+ TCM cells and naïve CD4+ T cells preferentially homed to the dLN as compared to either CD44+CD62L−Ly6C+ or CD44+CD62L−Ly6C− cells. All populations were found in the spleen in similar numbers. Because the number of naïve GFP+ cells was the same in all recipients, we conclude that differences in the number and location of sorted chronic cells is due to functionality, and not mouse to mouse variation. Analysis of IFN-γ production in the ear employing dICS revealed an average of 44 adoptively transferred Ly6C+ cells were making IFN-γ in-vivo (Fig. 7C). Surprisingly, even when polyclonal CD44+CD4+ T cells from mice infected for 35 weeks were transferred at their pre-sort physiological ratios, see Figure 7D, CD44+CD62L−Ly6C− cells were significantly under represented in the ear versus CD44+CD62L−Ly6C+ cells (p<0.05) and did not produce IFN-γ by dICS (Fig. 7, D and E). These data demonstrate that pre-existing Ly6C+ TEFF cells within the CD44+CD62L− population are the source of the L. major-specific CD4+ T cells that emulate the protective response against secondary challenge.
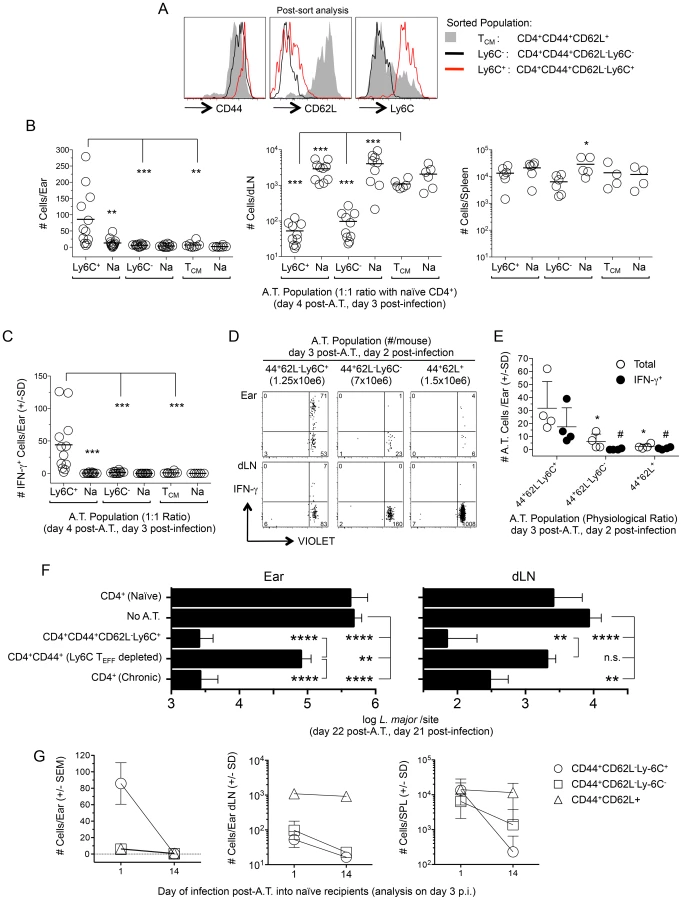
Ly6C+ CD4+ TEFF cells mediate strong protective immunity in the ear and ear dLN upon adoptive transfer
To determine if the Ly6C+ TEFF cells are sufficient and necessary to confer optimal protection, we transferred polyclonal sort-purified CD4+CD44+CD62L−Ly6C+ TEFF cells from chronic mice or the CD4+CD44+ T cells from chronic mice that were sort-depleted of the CD44+CD62L−Ly6C+ TEFF population into naïve recipients at their pre-sort physiological ratios. Mice receiving purified CD4+ T cells from naive or chronic mice were employed as controls. Total CD4+ T cells from chronic mice were transferred in sufficient numbers so as to match the total number of CD44+ cells employed in the CD44+CD62L−Ly6C+ TEFF and CD4+CD44+(Ly6C+ TEFF depleted) adoptive transfers. One day following transfer, mice were challenged in the ear dermis with 5×103 L. major parasites. Three weeks post-challenge, Ly6C+ TEFF cells mediated a highly significant 204-fold reduction in parasite load in the ear (p<0.0001) and a 122-fold reduction in the dLN (p<0.0001) versus non-transferred control animals (Fig. 7F). The protection conferred by the Ly6C+ TEFF population was indistinguishable from that conferred by the total CD4+ chronic population. By contrast, the CD44+CD4+ T cells depleted of the Ly6C+ TEFF cells mediated only a 6.6-fold reduction in parasite load in the ear versus non-transferred controls (p = 0.009) that was 31-fold less than the protection conferred by the Ly6C+ TEFF cells (p<0.0001), and no protection in the dLN. These data demonstrate that Ly6C+ TEFF cells rapidly recruited to the site of L. major challenge mediate and are required for optimal protection.
Ly6C+CD62L−CD44+CD4+ T cells from chronically infected mice are short lived in the absence of infection
Immunological memory, by definition, is mediated by a population of persistent memory cells that do not require the continued presence of the antigen that led to their generation [32]. Therefore, we wished to determine if CD4+CD44+CD62L−Ly6C+ T cells could be maintained in the absence of antigen. Sorted populations of CD44+CD62L−Ly6C+, CD44+CD62L−Ly6C−, or CD44+CD62L+ TCM cells from chronic animals were labeled with a proliferation dye and transferred into naïve recipients at approximately equal numbers. Recipient mice were rested for either 1 or 14 days post-transfer and challenged with L.major in the ear dermis. The small number of cells that non-specifically proliferated during the 14-day rest period were excluded from analysis. On day 3 post-challenge, when antigen-induced proliferation is minimal (Figure 4B), ears, purified CD4+ T cells from the spleen and ear dLNs were analyzed for the presence of adoptively transferred cells (Figure 7G). As expected, recipient mice rested for 1 day contained CD44+CD62L−Ly6C+, but not CD44+CD62L−Ly6C− or TCM cells in the ear. In contrast, if challenge was delayed until 14 days post-transfer, no CD44+CD62L−Ly6C+ cells were detected in the ear (D1 vs. D14 p<0.0001), indicating these cells were no longer available for recruitment to the skin. Remarkably, CD44+CD62L−Ly6C+ cells, which were found in equivalent numbers with TCM cells in the spleen when challenge occurred on day 1 post-transfer, were reduced 58-fold when challenge was delayed to day 14 (D1 vs. D14 p = 0.002), and were detected at significantly lower numbers than TCM cells at this time (D14, TCM vs. Ly6C+ p = 0.009). TCM cells were detected in equal numbers in the dLN and spleen regardless of the time of challenge (dLN p = 0.54; SPL p = 1.0). Interestingly, CD44+CD62L−Ly6C− cells displayed an intermediate, but significant (p = 0.02), five-fold drop in numbers in the spleen when mice were rested for 1 versus 14 days, suggesting this population may contain longer-lived TEM cells, as previously shown [26]. Both Ly6C+ and Ly6C− cells were found in smaller numbers in the dLN when challenge was delayed to day 14 post-transfer (dLN, D1 vs. D14, p≤0.004). Together, these observations confirm that CD44+CD62L+ T cells are true memory cells with the capacity to survive in the naïve recipients, whereas the CD44+CD62L−Ly6C+ cells, which are the cells required for protective immunity, are short-lived.
The source of CD4+CD44+CD62L−Ly6C+Tbet+ T cells during chronic infection
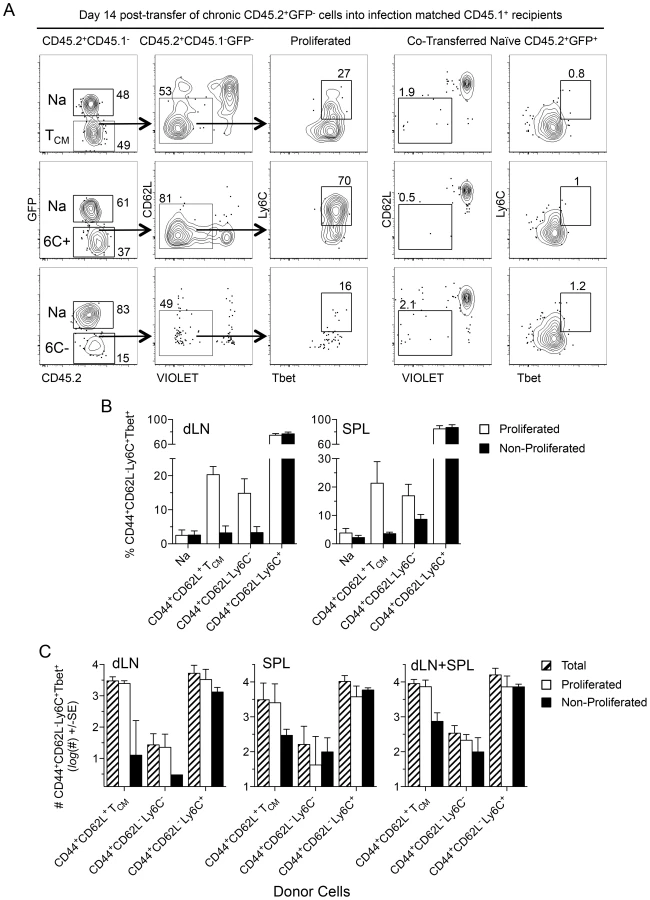
In order to explain how such a high proportion of antigen experienced cells can be maintained as ‘short-lived’ TEFF during chronic infection, we wished to investigate which T cell populations could give rise to these cells. Sorted populations of CD44+CD62L−Ly6C+, CD44+CD62L−Ly6C−, or CD44+CD62L+ TCM T cells from chronic animals were labeled with a proliferation dye and co-transferred with dye-labeled naïve CD62L+GFP+CD4+ cells from naïve mice into infection-matched recipients. Fourteen days later transferred cells in the dLN and spleen were analyzed for Ly6C and Tbet expression (Figure 8). TCM cells underwent robust proliferation whereas co-transferred CD62L+ naïve GFP+CD4+ T cells did not (Figure 8A, top panels). Analysis of proliferated TCM derived cells revealed a significant increase in the frequency of CD44+CD62L−Ly6C+Tbet+ TEFF cells versus non-proliferated TCM cells in both the dLN (p = 0.005) and spleen (p = 0.036) and the same was true for CD44+CD62L−Ly6C− cells in the dLN (p = 0.004) (Figure 8B). Remarkably, a proportion of transferred CD44+CD62L−Ly6C+ cells, which do not need to undergo proliferation to mediate effector function, also underwent proliferation in infection-matched recipients and maintained Ly6C and Tbet co-expression in both the dLN and spleen (Fig. 8A, middle panels, and Fig. 8B). Although the frequency of Ly6C+Tbet+ cells was considerably lower among proliferated TCM versus CD44+CD62L−Ly6C+ derived cells in both the spleen and dLN (p<0.0001), the numerical superiority of the total TCM population (Figure S8) resulted in a non-significant difference in the number of proliferated or total CD44+CD62L−Ly6C+Tbet+ cells derived from either population at 14 days post-transfer in the dLN, spleen, or dLN+spleen (Fig. 8C). In contrast to the low number of CD44+CD62L−Ly6C+ cells found in naïve mice on day 14 post-transfer (Figure 7G), substantial numbers of non-proliferated CD44+CD62L−Ly6C+ cells expressing Tbet were found 14 days following transfer into infection-matched recipients. While CD44+CD62L−Ly6C+Tbet+ cells derived from the CD44+CD62L−Ly6C− population were also found, the total number of these cells was significantly less than that observed in mice receiving TCM (p = 0.003) or CD44+CD62L−Ly6C+ cells (Fig. 8C, right panel, p = 0.001). These observations demonstrate that re-exposure of Leishmania-specific cells to antigen during chronic infection can lead to the generation of CD44+CD62L−Ly6C+Tbet+ cells, thereby contributing to the high frequency of this population prior to challenge.
Discussion
The protective immunity induced by a healed but persistent primary infection remains the ‘gold standard’ of acquired resistance against Leishmania major infection in both mice and humans, and emulating this response remains a key objective of vaccines against all forms of Leishmaniasis. The current studies provide the most detailed analysis to date of the cells mediating this protective response. We found that rapidly recruited (within hours), pre-existing Th1 Ly6C+ TEFF cells mediate immunity at the site of secondary L. major challenge in mice with a chronic primary infection. These findings, along with our previous observation that rapid immunity is a hallmark of protection against infected sand fly challenge in mice [11], [18], strongly suggest that natural immunity in individuals with a chronic primary infection against subsequent infection is dependent upon the presence of pre-existing CD4+ TEFF rather than memory cells.
We employed an intra-dermal ear challenge model that allowed tracking of TEFF cell subsets to the peripheral challenge site. So far as we are aware, this has not been achieved for Leishmania or any other pathogen. We also employed Ly6C [26], [33], a T-bet regulated GPI-anchored surface glycoprotein to further define CD44+CD62L−CD4+ T cells. A recent study also used the differential expression of Ly6C to distinguish between effector and memory Th1 CD4+ T cells responding to acute infection with LCMV, with Ly6CloT-betint and Ly6ChiT-bethi cells defining longer-lived memory and shorter-lived effector populations, respectively [26]. The enrichment of pre-existing Ly6C+ TEFF cells in peripheral blood, their rapid and non-specific homing to sites of tissue damage, their behavior as cells that rapidly secrete cytokine upon antigen encounter in the periphery without undergoing division following challenge, and particularly their short life span are traits that embody differentiated TEFF rather than memory cells. In addition, Ly6C+ TEFF cells were also CD27−, a phenotype associated with short-lived tissue homing cells [29], [30]. Despite their relatively short life span following transfer into naïve recipients, Ly6C+ TEFF cells were the predominant antigen-specific CD4+ T cell population during chronic infection. Transfer of either TCM or Ly6C+ TEFF into infection-matched recipients led to the generation of Ly6C+Tbet+ TEFF cells in the spleen and dLN, suggesting that each of these populations can contribute to the maintenance of the high frequency of Ly6C+ TEFF cells during chronic infection. The continuous renewal of Ly6C+ TEFF cells from thymic precursors [34] or from the activation of cells in a self-renewing pool as we observed here and has been reported by others [9], [35] may explain how the Ly6C+ TEFF population escapes exhaustion that is typically associated at the single-cell level with chronic antigen stimulation [36].
The proliferative capacity of the Ly6C+ TEFF cells in the infection-matched recipients was surprising since they disappeared during the same two weeks time frame in naïve recipients. The findings suggest that while Ly6C expression is indicative of an antigen-dependent effector cell, it does not imply that a cell is terminally differentiated. Rather, Ly6C+ cells may be recently activated effector cells that still retain proliferative capacity and can expand the pool of Ly6C+ TEFF cells when re-exposed to antigen in a chronic infection setting. We also found significant numbers of non-proliferated Ly6C+ TEFF cells in the dLN and spleen of infection-matched recipients, suggesting that their lifespan can be prolonged without undergoing division in the presence of infection. The proliferative capacity and antigen-dependency of the Ly6C+ population suggests these cells are recently activated effector cells, not infection-independent TEM cells.
In mice infected with the dhfr-ts−/− L. major mutant parasite, which is unable to establish chronic infection, only CD62L+CD4+ TCM cells were maintained after parasite clearance and these cells mediated a compromised and delayed immune response upon needle challenge [12]. Akin to the failure of non-living vaccines in human field trials [20], the live mutant vaccine and multiple formulations of non-living vaccines have consistently underperformed the immunity against needle challenge established in chronic mice [11], [12], [18]. Antigen vaccines are especially ineffective against infected sand fly challenge due, we believe, to their inability to maintain cells that can be rapidly recruited and thereby counteract the early immunosuppressive conditions at the site of parasite delivery by sand fly bite [11], [18], [37]–[39]. Indeed, the marginal level of immunity transferred by the CD44+ population depleted of CD44+CD62L−Ly6C+ TEFF cells is reminiscent of the compromised immunity conferred by non-persisting antigen vaccines following needle challenge and may in fact be due to cells that are already transitioning to Ly6C+ TEFF cells just prior to transfer (Figure 8). Thus, while TCM cells can eventually give rise to TEFF cells, the late arrival of these cells to the inoculation site abrogates their efficacy.
The key contribution of pre-existent, short-lived Ly6C+ TEFF cells to the protective response is the likely reason for the critical failure of non-living vaccines against cutaneous leishmaniasis and possibly other chronic infectious diseases for which immunological memory may be an insufficient condition of concomitant immunity [3]–[8], [40]. The primary role of Th1 TCM cells may not be the generation of effector cells following secondary challenge, but rather, the generation of effector cells prior to secondary challenge via re-exposure to antigen provided by the chronic infection. If vaccination is able to generate a stable CD4+ memory population it may be possible to employ boosting or long-term antigen depots to generate the pre-existing TEFF population necessary for protective immunity.
Materials and Methods
Mice
Female C57BL/6, B6.SJL-Ptprca Pepcb/BoyJ and C57BL/6-Tg(UBC-GFP)30Scha/J mice were obtained from Jackson Laboratories. Female C57BL/6 and B6.SJL-Cd45a(Ly5a)/Nai were obtained from Taconic Farms. All mice were maintained in the National Institute of Allergy and Infectious Diseases animal care facility under specific pathogen-free conditions (Animal Study Protocol LPD-68E).
Leishmania parasites and infection of mice
Leishmania major Friedlin (FV1) and L. major RYN were obtained and grown as described previously [41]. L. major Friedlin-RFP [38] and L.m. Friedlin expressing the 2W peptide [31] were generated as described previously. Infective-stage metacyclic promastigotes were isolated from stationary cultures (4–6 day-old) by negative selection of non-infective forms using peanut agglutinin (PNA, Vector Laboratories Inc) [42].
Mice with chronic primary infections (Chronic mice) were generated by infection with 1×104 L. major metacyclic promastigotes subcutaneously (s.c.) in the footpad, and used 10–20 weeks later, unless otherwise indicated, when footpad lesions had completely resolved. L.m.-2W chronic infections were established following inoculation with 1×105 metacyclic promastigotes in the contralateral ear using a 27.5 gauge needle in a volume of 5–10 µl. For secondary needle challenge, mice were inoculated with the indicated number and strain of L. major metacyclic promastigotes in the ear in a volume of 5–10 µl. Transmission of L. major-RYN by exposure to the bites of infected female Phlebotomus duboscqi sand flies was carried out as described previously [41]. Mouse ears were exposed to the bites of 4–8 infected flies.
Parasite load per ear or dLN was determined by limiting dilution analysis as described previously [11].
Sample preparation
Ear, dLN and spleen tissue was prepared as previously described [11]. Briefly for ears, the ventral and dorsal sheets were separated, deposited in 1 ml DMEM containing 160 µg/ml of Liberase TL purified enzyme blend (Roche Diagnostic Corp.), and incubated for 1.5–2 hours at 37°C and 5% CO2. In experiments employing dICS, 20 µg/ml of Brefeldin A was added and media pre-warmed to 37°C. Digested ear sheets were homogenized using the Medicon/Medimachine tissue homogenizer system (Beckton Dickinson). The ear tissue homogenate was then flushed from the medicon with 10 ml RPMI media containing 0.05% DNAse and filtered using a 50 um-pore-size cell strainer and placed on ice. In experiments employing dICS, 2 µg/ml of Brefeldin A was added to DNase media pre-warmed to 37°C and the ear homogenate returned to 37°C and 5% CO2 for an additional 1.5–2 hours. In some experiments, red blood cells were removed from spleen preparations using ACK lysing buffer (Lonza). Blood was obtained by intra-cardiac bleed and lymphocytes were purified using Isopaque-1077 (Sigma).
Purification of CD4+ cells and pMHCII tetramer staining and magnetic enrichment
Purified CD4+ T cells were obtained from spleen or dLN using magnetic bead separation (Miltenyi Biotec). 2W:I-Ab tetramer staining and magnetic enrichment were performed as previously described [31]. Briefly, a single cell suspension of the spleen, dLN, and ndLNs from a single mouse were stained with 10 nM allophycocyanin- or phycoerythrin-labeled 2W:I-Ab-streptavidin tetramers for 1 hour at room temperature. Samples were then chilled to 4°C and incubated with magnetic anti-fluorochrome beads and run through a magnetized LS column (Miltenyi Biotec) in a 4°C cold room. To determine the absolute number of cells, a portion of each sample was removed for counting with AccuCheck Counting Beads (Invitrogen) as described previously [31].
Re-stimulation of T cells
Single-cell suspensions were re-stimulated at 37°C in 5% CO2 for 8 (indicated transfer experiments) or 14 (overnight) total hours in flat-bottom 48-well plates with 0.5–1×106 T cell-depleted (Miltenyi Biotech), irradiated, naïve spleen cells (APCs), with or without 50 µg/ml freeze-thaw Leishmania antigen (L.m.-Ag). During the last 4 hours of culture, 1 µg/ml of Brefeldin A (Golgiplug; BD Biosciences) was added. APC genotype was matched to recipient animals in adoptive transfer experiments. Following culture, washed cells were labeled with VIOLET or AQUA fixable Live/Dead dye (Invitrogen) to exclude dead cells.
Analysis of cells by flow cytometry
All antibodies were from eBioscience or BD-Biosciences unless otherwise noted. Cells were stained with anti-Fc-γ III/II (CD16/32) receptor Ab (2.4G2, BD Biosciences) with or without VIOLET or AQUA fixable LIVE/DEAD dye (Invitrogen) in PBS containing 0.5–1.0% FCS for 10 minutes followed by incubation for 25 minutes with a combination of the following conjugated antibodies: Pacific Blue-, eFluor 450-, or allophycocyanin-eFluor-780-anti-B220 (RA3-6B2), anti-CD11b (MI-70), anti-CD11c (N418), anti-F4/80 (BM8), and/or anti-CD8α (53-6.7); FITC- or PerCP-Cy5.5-anti-Ly6C (HK1.4); V500- or V450-anti-CD3ε (145-2C11); PerCP-Cy5.5-, AlexaFluor 750-, V450-, or PE-Cy7-anti-CD4 (RM4-5); V500-, FITC-, or allophycocyanin-eFluor-780-anti-CD44 (IM7); allophycocyanin- or PE-Cy7-anti-CD127 (A7R34) or anti-CD27 (LG.7F9); allophycocyanin-, PE-Cy7-, or PE-anti-CD62L (MEL-14); Brilliant violet 421-anti-TCRβ (H57-597, Biolegend); PE-anti-CXCR3 (CXCR3-173), PE-anti-CD54(ICAM-1) (3E2), Per-CP Cy5.5-anti-CD69 (H1.2F3); FITC-anti-ICOS (C398.4A). In some experiments, samples were treated with the Foxp3 Fixation/Permeabilization Buffer (eBioscience) per manufacturer's instructions, and then stained with FITC-, or PE-anti-Ki-67 (B56); PE- or eFluor 660-anti-T-bet (eBio4B10); FITC-, PE-, eFluor450-, or allophycocyanin-anti-IFN-γ (XMG1.2), FITC-, PE- or Alexafluor-700 anti-TNF-α (MP6-XT22 or TN3-19); APC-, or Pacific Blue-anti-IL-2 (JES6-5H4). In some experiments, samples were fixed and permeabilized with BD Cytofix/Cytoperm (Becton-Dickinson) according to the manufacturer's instructions, and subsequently stained for 45″-1 hour at 4°C with a combination of the above mentioned anti-cytokine antibodies. Samples were run on LSRII or FACS Canto II flow cytometers (Becton-Dickinson) using FACS DIVA software. Forward-scatter and side-scatter width was employed to exclude cell doublets from analysis. Data was analyzed with FlowJo (Tree Star). Gating was determined by employing isotype controls and/or comparison with the relevant control population.
Cell sorting and adoptive transfer
Pooled single cell suspensions from the spleen, dLN, ndLNs, and in some experiments blood, of chronic animals were stained with a combination of CD44, CD62L, CD4, TCRβ, CD11b, MHC II, NK1.1 and/or Ly6C, as described in the text and purified using FACsAriaIIu (BD Biosciences) cell sorters at the Flow Cytometry Section of the Research Technologies Branch of the NIAID. In some experiments, sorted populations were re-suspended at 1×106 cells/ml in PBS+0.1% FBS at 37°C and labeled with 1 µM VIOLET proliferation dye (Invitrogen) for 14″ at RT. The reaction was stopped with neat FBS, and the cells were washed twice with cold PBS. Sorted populations were transferred independently by intravenous injection into congenic recipients, or, in some experiments, different sorted populations from C57BL/6 or B6.SJL mice were co-transferred into C57BL/6-Tg(UBC-GFP) recipient mice or a derivation thereof. The number of transferred cells is indicated in the text. Recipient mice were challenged one or 14 days following transfer. Congenic mice were source matched.
Statistics
Comparison of cell numbers was done using the Mann-Whitney test. Comparisons of frequency were performed using the unpaired students t-test for comparisons between two groups or a one-way analysis of variance (ANOVA) with Bonferroni's post-test for comparisons between multiple groups. Comparison of cell numbers or parasite loads under conditions of proliferation was performed on log-transformed data using ANOVA. All p-values are two-sided. Statistical calculations were done in Graphpad PRISM 5.0c (www.graphpad.com). Level of significance is reported in the text.
Ethics statement
All animal experiments were performed under the LPD-68E Animal Study Protocol approved by the NIAID Animal Care and Use Committee using guidelines established by the Animal Welfare Act and the PHS Policy on Humane Care and Use of Laboratory Animals.
Supporting Information
Zdroje
1. KaechSM, CuiW (2012) Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12: 749–761.
2. CondottaSA, RicherMJ, BadovinacVP, HartyJT (2012) Probing CD8 T cell responses with Listeria monocytogenes infection. Adv Immunol 113: 51–80.
3. SmithersSR, TerryRJ (1967) Resistance to experimental infection with Schistosoma mansoni in rhesus monkeys induced by the transfer of adult worms. Trans R Soc Trop Med Hyg 61: 517–533.
4. GrenfellBT, MichaelE, DenhamDA (1991) A model for the dynamics of human lymphatic filariasis. Parasitol Today 7: 318–323.
5. MacDonaldAJ, TuragaPS, Harmon-BrownC, TierneyTJ, BennettKE, et al. (2002) Differential cytokine and antibody responses to adult and larval stages of Onchocerca volvulus consistent with the development of concomitant immunity. Infect Immun 70: 2796–2804.
6. ScottP, ArtisD, UzonnaJ, ZaphC (2004) The development of effector and memory T cells in cutaneous leishmaniasis: the implications for vaccine development. Immunol Rev 201: 318–338.
7. SpencePJ, LanghorneJ (2012) T cell control of malaria pathogenesis. Curr Opin Immunol 24: 444–448.
8. UrdahlKB, ShafianiS, ErnstJD (2011) Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol 4: 288–293.
9. NelsonRW, McLachlanJB, KurtzJR, JenkinsMK (2013) CD4+ T Cell Persistence and Function after Infection Are Maintained by Low-Level Peptide:MHC Class II Presentation. J Immunol 190: 2828–2834.
10. Freitas do RosarioAP, MuxelSM, Rodriguez-MalagaSM, SardinhaLR, ZagoCA, et al. (2008) Gradual decline in malaria-specific memory T cell responses leads to failure to maintain long-term protective immunity to Plasmodium chabaudi AS despite persistence of B cell memory and circulating antibody. J Immunol 181: 8344–8355.
11. PetersNC, KimblinN, SecundinoN, KamhawiS, LawyerP, et al. (2009) Vector transmission of leishmania abrogates vaccine-induced protective immunity. PLoS Pathog 5: e1000484.
12. ZaphC, UzonnaJ, BeverleySM, ScottP (2004) Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med 10: 1104–1110.
13. UzonnaJE, WeiG, YurkowskiD, BretscherP (2001) Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J Immunol 167: 6967–6974.
14. BelkaidY, HoffmannKF, MendezS, KamhawiS, UdeyMC, et al. (2001) The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med 194: 1497–1506.
15. BelkaidY, PiccirilloCA, MendezS, ShevachEM, SacksDL (2002) CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420: 502–507.
16. NorthRJ, KirsteinDP (1977) T-cell-mediated concomitant immunity to syngeneic tumors. I. Activated macrophages as the expressors of nonspecific immunity to unrelated tumors and bacterial parasites. J Exp Med 145: 275–292.
17. StephensR, LanghorneJ (2010) Effector memory Th1 CD4 T cells are maintained in a mouse model of chronic malaria. PLoS Pathog 6: e1001208.
18. PetersNC, BertholetS, LawyerPG, CharmoyM, RomanoA, et al. (2012) Evaluation of recombinant Leishmania polyprotein plus glucopyranosyl lipid A stable emulsion vaccines against sand fly-transmitted Leishmania major in C57BL/6 mice. J Immunol 189: 4832–4841.
19. MelbyPC (1991) Experimental leishmaniasis in humans: review. Rev Infect Dis 13: 1009–1017.
20. NoazinS, KhamesipourA, MoultonLH, TannerM, NasseriK, et al. (2009) Efficacy of killed whole-parasite vaccines in the prevention of leishmaniasis: a meta-analysis. Vaccine 27: 4747–4753.
21. MullerI (1992) Role of T cell subsets during the recall of immunologic memory to Leishmania major. Eur J Immunol 22: 3063–3069.
22. GebhardtT, WhitneyPG, ZaidA, MackayLK, BrooksAG, et al. (2011) Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477: 216–219.
23. ReinhardtRL, BullardDC, WeaverCT, JenkinsMK (2003) Preferential accumulation of antigen-specific effector CD4 T cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med 197: 751–762.
24. PitcherCJ, HagenSI, WalkerJM, LumR, MitchellBL, et al. (2002) Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168: 29–43.
25. ColpittsSL, DaltonNM, ScottP (2009) IL-7 receptor expression provides the potential for long-term survival of both CD62Lhigh central memory T cells and Th1 effector cells during Leishmania major infection. J Immunol 182: 5702–5711.
26. MarshallHD, ChandeleA, JungYW, MengH, PoholekAC, et al. (2011) Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35: 633–646.
27. Filipe-SantosO, PescherP, BreartB, LippunerC, AebischerT, et al. (2009) A dynamic map of antigen recognition by CD4 T cells at the site of Leishmania major infection. Cell Host Microbe 6: 23–33.
28. LiuD, UzonnaJE (2010) The p110 delta isoform of phosphatidylinositol 3-kinase controls the quality of secondary anti-Leishmania immunity by regulating expansion and effector function of memory T cell subsets. J Immunol 184: 3098–3105.
29. KapinaMA, ShepelkovaGS, MischenkoVV, SaylesP, BogachevaP, et al. (2007) CD27low CD4 T lymphocytes that accumulate in the mouse lungs during mycobacterial infection differentiate from CD27high precursors in situ, produce IFN-gamma, and protect the host against tuberculosis infection. J Immunol 178: 976–985.
30. PepperM, LinehanJL, PaganAJ, ZellT, DileepanT, et al. (2010) Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol 11: 83–89.
31. PaganAJ, PetersNC, DebrabantA, Ribeiro-GomesF, PepperM, et al. (2012) Tracking antigen-specific CD4(+) T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur J Immunol 43: 427–428.
32. Janeway CAJ, Travers P., Walport M., Murphy K. (2001) Immunobiology:The Immune System in Health and Disease: New York: Garland Science.
33. MatsudaJL, ZhangQ, NdonyeR, RichardsonSK, HowellAR, et al. (2006) T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells. Blood 107: 2797–2805.
34. LinE, KemballCC, HadleyA, WilsonJJ, HofstetterAR, et al. (2010) Heterogeneity among viral antigen-specific CD4+ T cells and their de novo recruitment during persistent polyomavirus infection. J Immunol 185: 1692–1700.
35. ReileyWW, ShafianiS, WittmerST, Tucker-HeardG, MoonJJ, et al. (2010) Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A 107: 19408–19413.
36. WherryEJ (2011) T cell exhaustion. Nat Immunol 12: 492–499.
37. RogersME (2012) The role of leishmania proteophosphoglycans in sand fly transmission and infection of the Mammalian host. Front Microbiol 3: 223.
38. PetersNC, EgenJG, SecundinoN, DebrabantA, KimblinN, et al. (2008) In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321: 970–974.
39. Ribeiro-GomesFL, PetersNC, DebrabantA, SacksDL (2012) Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog 8: e1002536.
40. ZinkernagelRM (2012) Immunological memory not equal protective immunity. Cell Mol Life Sci 69: 1635–1640.
41. StamperLW, PatrickRL, FayMP, LawyerPG, ElnaiemDE, et al. (2011) Infection parameters in the sand fly vector that predict transmission of Leishmania major. PLoS Negl Trop Dis 5: e1288.
42. SacksDL, HienyS, SherA (1985) Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J Immunol 135: 564–569.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 12
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Experimental Cerebral Malaria Pathogenesis—Hemodynamics at the Blood Brain Barrier
- Unique Features of HIV-1 Spread through T Cell Virological Synapses