
Analýza proteinů pomocí hmotnostní spektrometrie
Analysis of Protein Using Mass Spectrometry
Recently, mass spectrometry has become a powerful tool in cancer research. Mass spectrometry represents the method that allows identification, quantification and characterization of proteins in biological samples. Nowadays, it is mainly used for biomarker discovery that can enable early detection of cancer. This article is focused on protein analysis by mass spectrometry. At first, mass spectrometry and its importance in proteomics are described. Subsequently ionization type and mass analyzers are discussed. This relates to the possibility of on‑line or off‑line analysis connection with separation techniques, such as liquid chromatography and electrophoresis. Different approaches for preparing proteins and methods of analysis of biomolecules using mass spectrometers are described. In addition, the possibility of mass spectrometric analyses of samples and data processing are discussed.
Key words:
mass spectrometry – liquid chromatography – electrophoresis – proteomics
This work was supported by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101) and by MH CZ – DRO (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
31. 1. 2014
Accepted:
25. 3. 2014
:
P. Dvořáková; L. Hernychová; B. Vojtěšek
:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
:
Klin Onkol 2014; 27(Supplementum): 104-109
Rychlý rozvoj hmotnostní spektrometrie spolu s proteomickými přístupy umožňuje detailnější studium biologických systémů. Prvotní problémy hmotnostně spektrometrických analýz biologických makromolekulárních látek byly úspěšně překonány a dnes se běžně tato analytická metoda používá pro identifikaci, kvantifikaci a charakterizaci proteinů. Cílem článku je podat přehled o možnostech analýzy proteinů s využitím hmotnostní spektrometrie. Popisujeme různé typy ionizace a výběr analyzátorů pro hmotnostně spektrometrické měření proteinů, rovněž i on‑line či off‑line spojení analýzy se separačními technikami, jako je kapalinová chromatografie a elektroforéza. Zmiňujeme se i o přípravě proteinů a způsobech analýzy biologických makromolekulárních látek pomocí hmotnostních spektrometrů. Dále jsou uvedeny možnosti hmotnostně spektrometrických analýz vzorků a zpracování naměřených dat.
Klíčová slova:
hmotnostní spektrometrie – chromatografie kapalinová – elektroforéza – proteomika
Hmotnostní spektrometrie
Hmotnostní spektrometrie je analytická metoda, která umožňuje rozlišit ionty na základě jejich poměru hmotnosti k náboji (m/ z, měrná hmotnost). Původně ovšem vznikla na poli fyziky a za její počátek se považuje přelom 19. a 20. století. Jako zakladatel je uváděn fyzik J. J. Thomson a jeho zásadní práce o vychylování katodového záření v elektrickém poli [1]. Analýza malých organických látek se stala rutinní záležitostí až v 80. letech 20. století. V té době však analýza biologických makromolekulárních látek, za které jsou považovány nukleové kyseliny a proteiny, nebyla možná kvůli absenci vhodných ionizačních technik. Od 90. let došlo díky významnému pokroku v oblasti hmotnostní spektrometrie k rozvoji metod identifikace a strukturní analýzy těchto látek. Byly totiž objeveny vhodné techniky, které dokázaly účinně a jemně ionizovat tyto netěkavé makromolekuly, aniž způsobovaly jejich fragmentaci. Za vývoj těchto ionizačních technik byla jejich objevitelům Johnu B. Fennovi [2] a Koichi Tanakovi [3] udělena v roce 2002 Nobelova cena za chemii.
Od té doby se hmotnostní spektrometrie začala v biologických sférách stále více uplatňovat. V posledních 10 letech se pak stala významným nástrojem biologického výzkumu především jako metoda identifikace, charakterizace a kvantifikace proteinů a metabolitů. V analýze proteinů se hmotnostní spektrometrie používá především pro kontrolu kvality rekombinantních proteinů a ostatních makromolekul, k identifikaci proteinů v biologických látkách, hledání biomarkerů pro daná onemocnění a pro detekci a charakterizaci posttranslačních modifikací.
Ionizace a analýza proteinů pomocí hmotnostní spektrometrie
Každý hmotnostní spektrometr (mass spectrometer – MS) se skládá z ionizačního zdroje, hmotnostního analyzátoru dělícího ionizované analyty dle jejich m/ z a z detektoru.
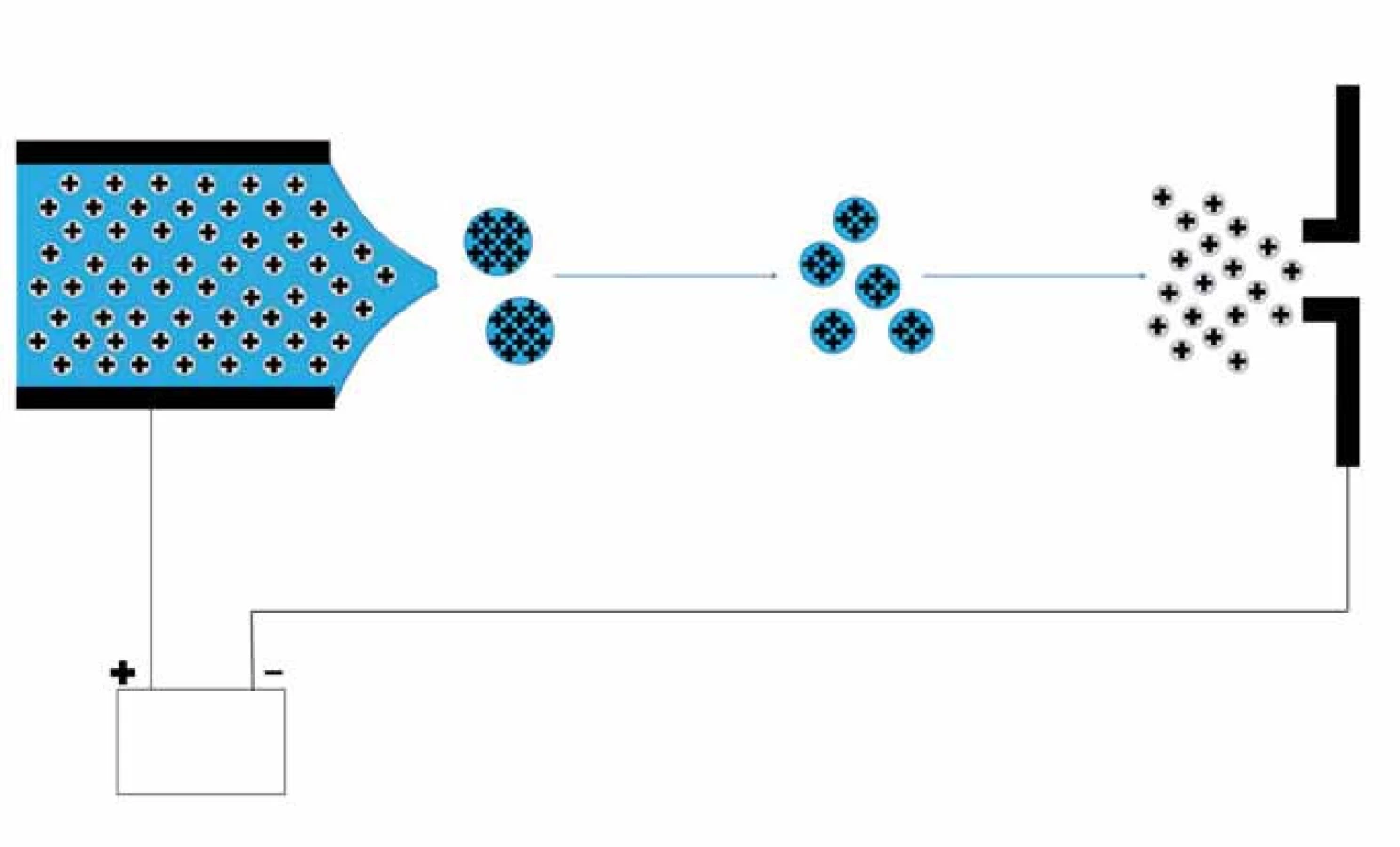
Nejprve je nutné převést analyty pomocí vhodné ionizační techniky na ionty. Pro ionizaci biologických makromolekulárních látek se používají tzv. měkké ionizační techniky, které nezpůsobují jejich fragmentaci. Běžně se setkáváme s ionizací pomocí laseru za účasti matrice (matrix ‑ assisted laser desorption ionization – MALDI) a ionizací elektrosprejem (electrospray ionization – ESI) či nanosprejem (nanoESI) [4]. Při MALDI ionizaci je vzorek nanesen na MALDI destičku a smíchán s vhodnou matricí (pro peptidy se používá např. a ‑ kyano ‑ 4 - hydroxyskořicová kyselina, 2,5 - dihydroxybenzoová kyselina a pro proteiny kyselina sinapová) a ponechán společně krystalizovat. Poté je destička se vzorky umístěna do vakuované části MS. Proces ionizace je uskutečněn krátkými laserovými pulzy, kdy energie záření laseru je pohlcena matricí, což vede k jejímu rychlému zahřátí a následné desorpci včetně iontů analytu. Při tomto procesu dochází ke vzniku převážně jednonásobně nabitých iontů [5]. ESI ionizuje analyty v roztoku, tudíž je obvykle spojován on‑line se separačními technikami, např. s kapalinovou chromatografií (liquid chromatography ‑ ESI – LC ‑ ESI) či kapilární elektroforézou (capillary electrophoresis ‑ ESI – CE ‑ ESI). Při ionizaci ESI je na hrot sprejovací kapiláry přivedeno vysoké napětí (2 – 6 kV) [5], čímž dochází k vytvoření nabitých kapiček analytu obalených rozpouštědlem. V důsledku postupného odpařování rozpouštědla se postupně uvolní vytvořené ionty, které jsou elektricky usměrňovány na vstup do MS (obr. 1). Tato ionizační technika produkuje vícenásobně nabité ionty [M+nH]n+.
Ionty vzniklé v ionizačním zdroji jsou následně usměrňovány iontovou optikou a přechází do hmotnostního analyzátoru. Separace iontů v hmotnostním analyzátoru probíhá za vysokého vakua a je založena na různých fyzikálních principech. Iontová past (ion trap – IT), orbitrap a iontová cyklotrónová rezonance (ion cyclotron resonance – ICR) patří mezi hmotnostní analyzátory, které separují ionty na základě jejich m/ z resonanční frekvence; kvadrupól (quadrupole – Q) používá stability iontů s danou m/ z při aplikování konkrétních hodnot napětí na elektrody v daný časový okamžik; analyzátor doby letu (time of flight – TOF) využívá čas letu iontů [5]. Jednotlivé MS se liší technickými specifikacemi, jako je hmotnostní rozsah, rychlost analýzy, rozlišení, senzitivita či dynamický rozsah, souvisejícími s použitým hmotnostním analyzátorem; více informací lze nalézt v publikaci Holčapek et al (2012) [6].
Po separaci v hmotnostním analyzátoru dopadají ionty na detektor, který zaznamenává počet iontů pro jednotlivé m/ z hodnoty. Nejčastěji se používají elektronové násobiče, kdy ionty dopadají na povrch dynody, z níž vyrazí elektrony, které jsou systémem dynod nebo opakovanými kolizemi zesíleny. Výsledkem měření je hmotnostní spektrum, kde jsou zaznamenány jednotlivé naměřené měrné hmotnosti a jejich intenzity.
Měřeny jsou pseudomolekulární ionty [M+H]+ nebo [M – H]– , kde M představuje hmotnost analytu, k níž byl připojen či z ní byl odloučen atom vodíku. Lze zvolit, zda budou měřeny kladně či záporně nabité ionty. Pro proteiny se zpravidla volí kladný mód, záporný je používán pro speciální experimenty, jako např. pro studium vybraných posttranslačních modifikací. Měří se monoizotopická molekulová hmotnost, která představuje součet přesných hmotností „nejlehčích“ izotopů prvků, a je tedy odlišná od hmotnosti průměrné, jež je dána váženým průměrem všech izotopů daného prvku dle jejich procentuálního zastoupení.
Separační techniky
Biologické vzorky představují komplexní směs obsahující vedle velkého množství proteinů také řadu dalších vysoko ‑ a nízkomolekulárních látek, tuků a cukrů. Po odstranění těchto doprovodných látek získáme vzorek, který je bohatou směsí proteinů lišících se jak koncentrací (abundantní proteiny vs minoritní), tak fyzikálně‑chemickými vlastnostmi, jako polarita (hydrofilicita a hydrofobicita), velikost (molekulová hmotnost) a náboj (izoelektrický bod) apod. Těchto rozdílných vlastností se používá k rozdělení komplexní směsi proteinů na méně komplexní frakce, přičemž platí, že čím kvalitnější frakcionace, tím více proteinů může být identifikováno. Limitující je v tomto případě především koncentrace proteinu ve vzorku a dále dynamický rozsah hmotnostního analyzátoru. V proteomice existují dva hlavní široce používané typy separace. První zahrnuje solubilizaci proteinů pomocí detergentů (nejčastěji sodium dodecyl sulfate – SDS) a jejich následnou separaci gelovou elektroforézou [7]. Druhý přístup nevyužívá gel, nýbrž rozdílné distribuce látek mezi stacionární a mobilní fází, tedy chromatografické separace [8].
Gelová elektroforéza využívá pro separaci proteinů gel. Denaturované proteiny, obalené dodecylsulfátem sodným (SDS) nesoucím silný negativní náboj, jsou naneseny na gel, který je umístěn v mírně alkalickém prostředí. Po aplikaci elektrického pole proteiny začnou migrovat různou rychlostí směrem k anodě. Pohyblivost proteinů v gelu je ovlivněna jednak vlastnostmi proteinu a jednak podmínkami, za nichž separace probíhá. Mezi nejvýznamnější gelové techniky patří jednorozměrná či dvourozměrná polyakrylamidová gelová elektroforéza (1 - DE, 2 - DE) [8]. Tradiční 1 - DE umožňuje za denaturačních podmínek dělit směs proteinů podle molekulových hmotností. Při 2 - DE předchází výše popsané separaci dělení proteinových směsí podle izoelektrických bodů (pI) proteinů. Za výhodu zejména u 2 - DE se považuje, že po rozdělení komplexního vzorku již obvykle není třeba další separační krok. Lze ji tedy spojit s instrumenty umožňujícími přímou analýzu (např. MALDI ‑ TOF). V případě 1 - DE se však vyžaduje zařazení dalšího separačního kroku – kapalinové chromatografie. Gelové techniky jsou omezeny nízkou citlivostí, linearitou, slabou rozpustností membránových proteinů a absencí nízkomolekulárních látek.
Pro chromatografickou separaci se využívá kapalinová chromatografie, kdy jsou nejprve analyty přivedeny na kolonu, která je zadržuje. Následně jsou analyty z kolony postupně vymývány vhodnou mobilní fází. Na rozdíl od gelových technik bývá kapalinová chromatografie přímo spojována s MS a to pomocí ESI [5]. Nejčastěji používanými kolonami jsou kolony s reverzní fází (RP), s kationtově výměnnou fází (strong cation ‑ exchange – SCX), kolony založené na hydrofilní interakci (hydrophilic interaction liquid chromatography – HILIC) nebo na elektrostaticky odpuzované hydrofilní interakci (electrostatic repulsion ‑ hydrophilic interaction chromatography – ERLIC) [9]. Jednotlivé kolony lze použít odděleně před vlastní MS analýzou (off‑line separace) nebo v případě RP kolon je možné přímé spojení s MS (on‑line separace). Pokud jsou vzorky velmi komplexní (např. buněčný lyzát), je vhodné provést multidimenzionální separaci tzv. MudPit strategie. Příkladem multidimenzionální separace může být rozdělení komplexního vzorku off‑line na kapalinovém chromatografu pomocí SCX kolony a jeho následná analýza kapalinovým chromatografem on‑line spojeným s hmotnostním spektrometrem (LC ‑ MS) nebo tandemovým hmotnostním spektrometrem (LC ‑ MS/ MS) s RP kolonou [10].
Proteomické přístupy technik zpracování proteinů
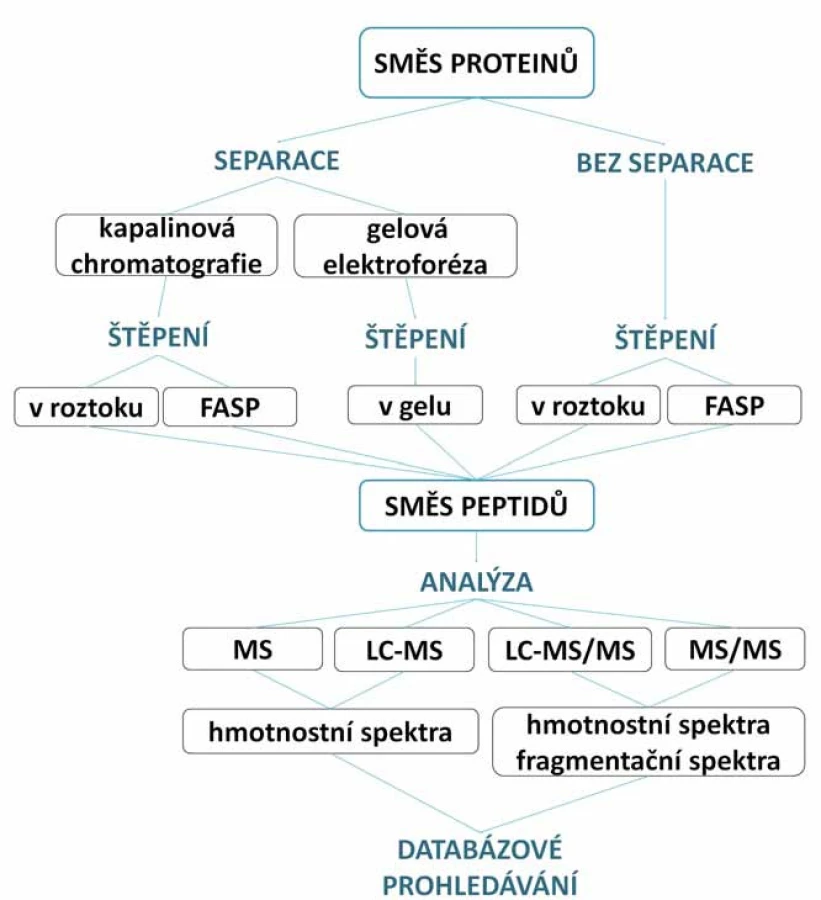
Pro identifikaci a charakterizaci proteinů pomocí hmotnostní spektrometrie se dnes využívají dva hlavní přístupy. První je enzymatické štěpení proteinů na peptidy (bottom ‑ up) (obr. 2), tento přístup je realizován pomocí štěpení proteinu v gelu (tzv. in‑gel štěpení) [11], štěpení proteinů v roztoku (in‑solution štěpení) [12] či na filtrech s regenerovanou celulózou (filter ‑ aided sample preparation – FASP) [7]. Druhý přístup je top ‑ down analýza, kdy se vynechává proteolytické štěpení a přímo se pracuje s intaktními proteiny [13].
Bottom ‑ up přístup
Jedná se o velmi rozšířený proteomický přístup, který je využíván pro identifikaci a kvantifikaci proteinů v komplexních biologických vzorcích. Směs proteinů je před samotným měřením na MS enzymaticky štěpena na peptidy.
Při in‑gel štěpení jsou z gelu vyříznuty pásy obsahující proteiny. Pro efektivnější štěpení proteinů na peptidy je nutné rozrušení třídimenzionální struktury proteinů, která je fixována pomocí disulfidických můstků, aby došlo k zpřístupnění maximálního počtu stérických míst pro následné proteolytické štěpení. Toho se dosahuje přídavkem redukčních činidel (DTT, DTE, TCEP) s následnou řízenou alkylací volných cysteinových zbytků pro zabránění opětovné tvorby disulfidických můstků vlivem vzdušné oxidace. Ke štěpení proteinů na peptidy je možné použít celou řadu enzymů, jejichž volba závisí na požadavcích kladených na výslednou peptidovou směs. Nejčastěji používanou proteázou je trypsin specificky štěpící proteiny na peptidy, které mají na C ‑ konci zbytky bazických aminokyselin argininu nebo lysinu (pokud za nimi nenásleduje prolin) [14]. Jedna z výhod této metody je, že při aplikování vzorku na gel dochází k částečnému odstranění případných nečistot. Naopak nevýhoda je možné zachycení peptidů v matrici gelu a také problematická robotická příprava vzorků.
Metoda štěpení in‑solution umožňuje štěpit komplexní směs proteinů přímo v roztoku. Pro extrakci proteinů jsou využívána silná chaotropní činidla, jako je močovina. Jedná se o metodu, která je mnohem snadněji automatizovatelná oproti in‑gel štěpení. Často je následována dvoudimenzionální chromatografickou separací [7].
FASP kombinuje výhody předchozích dvou metod. Biologický vzorek je kompletně solubilizován za pomocí detergentu. Následně je přenesen na filtr, čímž lze detergent vyměnit za vhodný pufr kompatibilní s proteolytickým štěpením. Protein je posléze štěpen na filtru proteázou a dochází k uvolnění peptidů [7].
Specifickou bottom ‑ up strategií je tzv. shotgun, jehož cílem je získat co největší množství peptidů a tedy i proteinů. Proteinový lyzát bez jakékoliv frakcionace je štěpen v roztoku a následně je separován a analyzován pomocí LC ‑ MS/ MS.
Peptidy získané štěpením proteinů jsou odsoleny a zakoncentrovány, poté je lze měřit pomocí MS. Pro relativně jednoduché peptidové směsi, které vznikly štěpením jednoho či několika proteinů, se obvykle uplatňuje analýza pomocí MALDI ‑ MS/ MS. Pro měření vzorků vzniklých štěpením komplexních proteinových směsí se pak především využívá LC ‑ ESI ‑ MS/ MS [4,11] nebo lze zvolit i LC ‑ MALDI ‑ MS/ MS přístup.
Top ‑ down přístup
Jedná se přístup, který je založen na analýze proteinu či jednoduché proteinové směsi separované z komplexního vzorku bez použití enzymatického štěpení proteinů na peptidy. Obecně je analýza intaktního proteinu méně účinná než měření peptidů. Nabízí však určité možnosti nedosažitelné na úrovni peptidů. Analýzou peptidů je totiž jen zřídka docíleno kompletního pokrytí sekvence proteinů, čímž se omezuje možnost zkoumat místně specifické mutace a posttranslační modifikace jednotlivých proteinů, které mohou být důležité pro biologickou funkci [5,13]. Další výhoda této techniky je, že poskytuje přesnou molekulovou hmotnost; nicméně větší proteiny jsou převážně heterogenní, stanovení přesné molekulové hmotnosti je tedy značně obtížné.
Pro analýzu proteinu je potřeba vysokorozlišovací MS s vysokou přesností měření (např. fourier transform ion cyclotron resonance – FTICR, LTQ ‑ Orbitrap). Správná volba je použití ESI ionizační techniky, přičemž solubilizaci proteinu pro ESI lze podpořit kyselinou mravenčí [4]. MALDI ionizace má pro proteiny větší než 30 kDa obecně nízkou citlivost.
Způsoby hmotnostně spektrometrické analýzy a procesování naměřených dat
Existují dva způsoby měření peptidů a proteinů pomocí hmotnostní spektrometrie. První je přímé měření vzniklých peptidů, získaných štěpením proteinu z gelu. Identifikace následně probíhá metodou peptidového mapování (peptide mass fingerprinting – PMF) [15]. Druhý způsob využívá tandemové hmotnostní spektrometrie, je tedy potřeba náročnější přístrojové vybavení se dvěma analyzátory a kolizní celou. Identifikace je uskutečněna peptidovým sekvencováním [4].
Metoda peptidového mapování
Peptidy vzniklé enzymatickým štěpením proteinu ve vzorku jsou ionizovány, separovány a detekovány pomocí MS,přičemž PMF bývá především spojena s přístroji typu MALDI ‑ TOF. Získané hmotnostní spektrum peptidů představuje specifickou charakteristiku proteinu. Hodnoty m/ z odečtené z hmotnostního spektra peptidů lze porovnat za pomoci speciálních vyhodnocovacích programů s teoreticky předpovězenými hmotnostmi peptidů, jež jsou obsaženy v použité databázi. Na základě shody experimentálních a teoretických databázových hodnot m/ z se dá ke spektru s určitou jistotou přiřadit protein. V některých případech však není možné pouze z hmotnostního spektra peptidů protein identifikovat. V tomto případě se přistupuje k tandemové hmotnostní spektrometrii.
Tandemová hmotnostní spektrometrie
Při tandemové hmotnostní spektrometrii ionty vstupují do prvního hmotnostního analyzátoru, kde nejprve dochází k selekci a izolaci peptidového iontu ze směsi. Následně je vybraný peptidový prekurzor fragmentován v kolizní cele, vzniklé fragmenty jsou separovány a měřeny v druhém hmotnostním analyzátoru (obr. 3) a tam je naměřeno fragmentační spektrum peptidu. Fragmentace může být uskutečněna pomocí kolizní energie (collision induced dissociation – CID, popř. high energy collision induced dissociation – HCD), záchytem elektronů mnohonásobně nabitými ionty peptidů, proteinů (electron capture dissociation – ECD) nebo reakcí peptidových kationtů s fluoranthenovým aniontem (electron transfer dissociation – ETD). Peptidy obvykle bývají fragmentovány pomocí CID fragmentace [16,17]. Pro speciální aplikace např. fosfoproteomika, či kvantifikace se však uplatňuje HCD a ETD fragmentace [18]. Pro proteiny se z fragmentačních technik používá především ECD [19] nebo ETD [20,21] fragmentace.
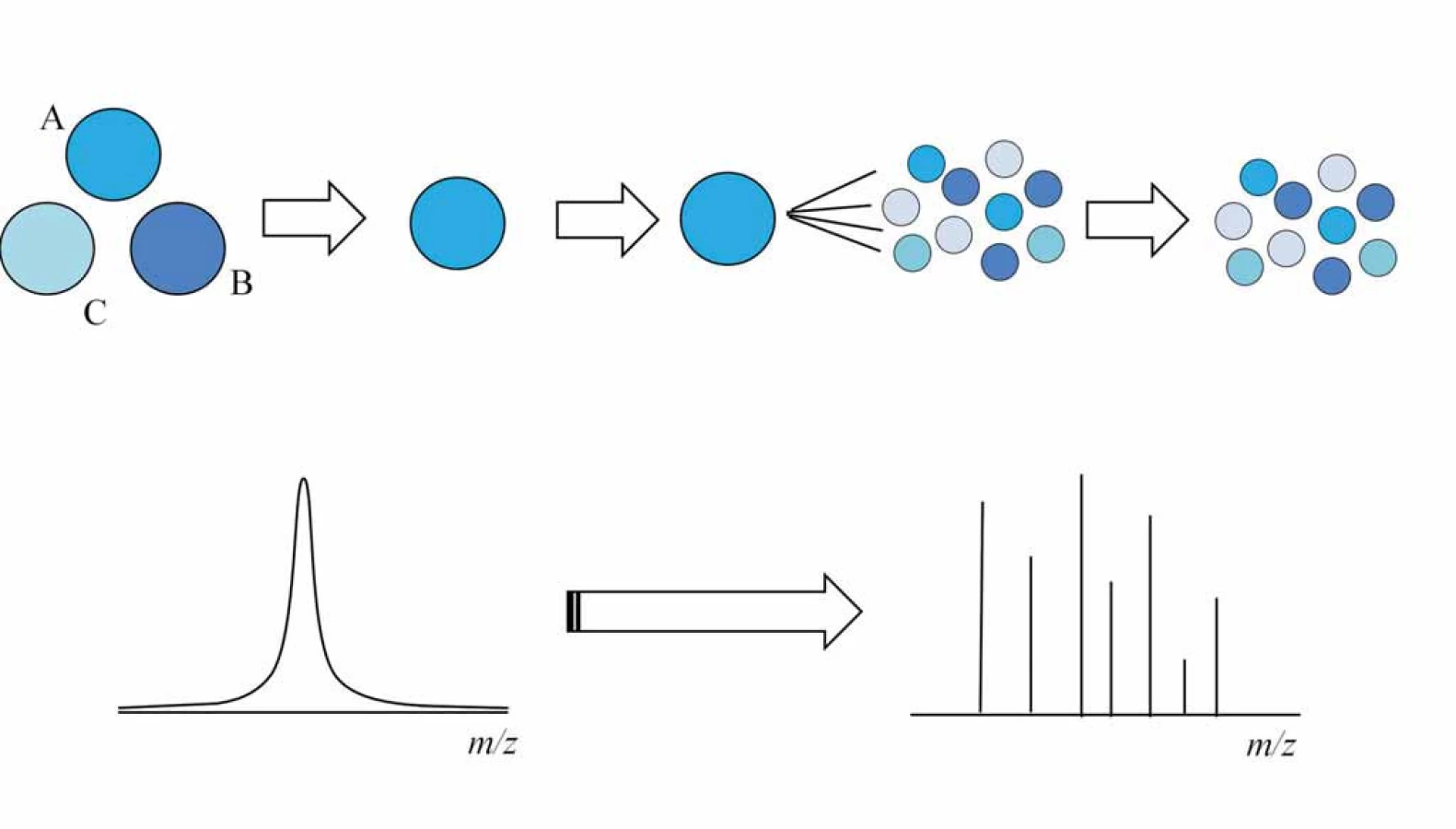
Hmotnostně spektrometrické měření proteomických vzorků je obvykle realizováno ve způsobu data ‑ dependent, kdy izolovány a fragmentovány jsou postupně nejintenzivnější peptidové ionty získané z předešlého MS skenu [5].
Fragmentační spektra peptidů jsou pak procesována pomocí speciálních vyhodnocovacích programů (např. Proteom Discoverer, Protein Pilot). Nejvíce rozšířené je databázové vyhledávání, které experimentální hmotnostně spektrometrická data porovnává s predikovanými [16,17]. K tomuto účelu lze použít vyhledávací algoritmy, jako jsou např. Mascot, Sequest. Velmi důležité je správné zadání všech kritérií týkajících se přípravy vzorku, chemické modifikace, které se mohou vyskytovat během měření a tím modifikovat m/ z hodnotu iontů (např. oxidace, acetylace, karbamidometylace), specifikace MS (např. typ fragmentace, ionizace, analyzátoru, tolerance prekurzorové a fragmentové hmoty) a vybrat databázi (např. NCBI, Swissprot) [22]. Po dokončení prohledávání je vygenerován seznam proteinů, jež byly ve vzorku nalezeny. Dále jsou zde uvedeny jiné informace, mezi něž patří např. skóre, které je vypočteno podle shodných experimentálních a teoretických hmotností peptidu, a dále také procentuální hodnocení pokrytí sekvence proteinu.
Závěr
Hmotnostní spektrometrie od svého počátku do současnosti prošla značným vývojem. V posledních letech se její těžiště přesouvá z pole chemického do biologických sfér, kde se uplatňuje při základní charakterizaci proteomu a nabízí přesnější pohled do biologických procesů buněk či organizmů. Dnes používané proteomické technologie dovolují identifikovat, kvantifikovat a charakterizovat až tisíce proteinů ve vzorku. I přes neustálý pokrok je tato analytická metoda pro biologické účely omezená. Z důvodu vysoké složitosti biologického vzorku totiž není možné identifikovat všechny proteiny podílející se na procesech v buňce. V budoucnu se však dá předpokládat další rozvoj hmotnostní spektrometrie, čímž bude umožněna detailnější identifikace a charakterizace proteomů.
Práce byla podpořena Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO, CZ.1.05/2.1.00/03.0101) a MZ ČR – RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
Ing. Petra Dvořáková, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: petra.dvorakova@mou.cz
Obdrženo: 31. 1. 2014
Přijato: 25. 3. 2014
Sources
1. Griffiths J. A brief history of mass spectrometry. Anal Chem 2008; 80(15): 5678 – 5683. doi: 10.1021/ ac8013065.
2. Fenn JB, Mann M, Meng CK et al. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989; 246(4926): 64 – 71.
3. Tanaka K, Waki H, Ido Y et al. Protein and polymer analyses up to m/ z 100 000 by laser ionization time ‑ of ‑ flight mass spectrometry. Rapid Commun Mass Spectrom 1988; 2(8): 151 – 153.
4. Aebersold R, Mann M. Mass spectrometry‑based proteomics. Nature 2003; 422(6928): 198 – 207.
5. Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advantages, and applications. Annu Rev Biomed Eng 2009; 11 : 49 – 79. doi: 10.1146/ annurev ‑ bioeng ‑ 061008 - 124934.
6. Holčapek M, Jirásko R, Lísa M. Recent developments in liquid chromatography‑mass spectrometry and related techniques. J Chromatogr A 2012; 1259 : 3 – 15. doi: 10.1016/ j.chroma.2012.08.072.
7. Wisniewski JR, Zougman A, Nagaraj N et al. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6(5): 359 – 362. doi: 10.1038/ nmeth.1322.
8. Monteoliva L, Albar JP. Differential proteomics: An overview of gel and non‑gel based approaches. Brief Func Genomic Proteomic 2004; 3(3): 220 – 239.
9. Choi YS. Reaching for the depp proteome: recent nano liquid chromatography coupled with tandem mass spectrometry‑based studies on the deep proteome. Arch Pharm Res 2012; 35(11): 1861 – 1870. doi: 10.1007/ s12272 - 012-1102 - y.
10. van Ulsen P, Kuhn K, Prinz T et al. Identification of proteins Neisseria meningitidis induced under iron‑limiting conditions using the isobaric tandem mass tag (TMT) labeling approach. Proteomics 2009; 9(7): 1771 – 1781. doi: 10.1002/ pmic.200800642.
11. Shevchenko A, Tomas H, Havliš J et al. In ‑ gel digestion for mass spectrometric characterization of protein and proteomes. Nature Publishing Group 2006; 1(6): 2856 – 2860.
12. Olsen JV, Blagoev B, Gnad F et al. Global, in vivo, and site ‑ specific phosphorylation dynamics in signaling networks. Cell 2006; 127(3): 635 – 648.
13. Bogdanov B, Smith RD. Proteomics by FTICR mass spectrometry: top down and bottom up. Mass Spectrom Rev 2005; 24(2): 168 – 200.
14. Lenco J, Stulik J. Identifikace proteinů kombinací peptidového mapování a fragmentace sulfonovaných peptidů. Chem Listy 2004; 98 : 264 – 267.
15. Henzel WJ, Watanabe C. Protein identification: The origins of peptide mass fingerprinting. J Am Soc Mass Spectrom 2003; 14(9): 931 – 942.
16. Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994; 5(11): 976 – 989. doi: 10.1016/ 1044 - 0305(94)80016 - 2.
17. Perkins DN, Pappin DJ, Creasy DM et al. Probability‑based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999; 20(18): 3551 – 3567.
18. Zhang Y, Ficarro SB, Li S et al. Optimized orbitrap HCD for quantitative analysis of phosphopeptides. J Am Soc Mass Spectrom 2009; 20(8): 1425 – 1434. doi: 10.1016/ j.jasms.2009.03.019.
19. Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc 1998; 120(13): 3265 – 3266.
20. Coon JJ, Ueberheide B, Syka JE et al. Protein identification using sequential ion/ ion reactions and tandem mass spectrometry. Proc Natl Acad Sci 2005; 102(27): 9463 – 9468.
21. Syka JE, Coon JJ, Schroeder MJ et al. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci 2004; 101(26): 9528 – 9533.
22. Casado ‑ Vela JC, Cebrián A, Gómez del Pulgar MT et al.Lights and shadows of proteomic technologies for the study of protein species including isoforms, splicing variats and protein post‑translation modifications. Proteomics 2011; 11(4): 590 – 603. doi: 10.1002/ pmic.201000287.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Protein Expression and Purification
- Methods for Studying Tumor Cell Migration and Invasiveness
- Next Generation Sequencing – Application in Clinical Practice
- Analysis of Protein Using Mass Spectrometry