
Mechanizmy lékové rezistence a nádorové kmenové buňky
Mechanisms of Drug Resistance and Cancer Stem Cells
Although the success of anticancer treatments has been increasing annually, drug resistance remains the dominant cause of death of cancer patients. Initial therapy often leaves residual disease that leads to repeated tumor development or to loss of its sensitivity to available therapy. One reason of residual disease formation is the presence of cancer stem cells (CSCs). CSCs have been identified as a small population of cells that is capable of self ‑ renewal and differentiation. It is supposed that these cells are responsible for cancer initiation, progression, metastasis, recurrence and drug resistance. Over the past years, much attention has been paid to development of CSCs‑related therapies and to identification of key molecules involved in controlling the specific properties of CSCs populations. This article reviews the basic mechanisms of drug resistance in relation to cancer stem cells.
Key words:
drug resistance − cancer stem cells − membrane transport proteins – epithelial-mesenchymal transition – tumor microenvironment − apoptosis
This work was supported by research program of the Internal Grant Agency, Ministry of Health of the Czech Republic: NT/14602 – 3/2013, by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO CZ.1.05/2.1.00/03.0101) and by Ministry of Health, Czech Republic − conceptual development of research organization (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
3. 2. 2014
Accepted:
7. 5. 2014
Authors:
J. Holčáková; M. Nekulová; P. Orzol; B. Vojtěšek
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2014; 27(Supplementum): 34-41
Overview
Ačkoliv úspěšnost léčby nádorových onemocnění se každoročně zvyšuje, rezistence k léčivům je nadále hlavní příčina úmrtí pacientů s rakovinou. Počáteční léčba často zanechává „zbytkové“ (reziduální) onemocnění, které vede k opětovnému rozvoji nádoru, případně ke ztrátě jeho citlivosti k použité léčbě. Vznik reziduálního onemocnění je ovlivněn přítomností nádorových kmenových buněk (CSCs). Jedná se o malou populaci buněk schopných sebeobnovy a diferenciace. Předpokládá se, že tyto buňky jsou odpovědné za vznik, růst, metastazovaní a udržení nádoru a také za jeho lékovou rezistenci. V poslední době je velká pozornost věnována vývoji terapií zaměřených na eliminaci CSCs a na identifikaci klíčových molekul zapojených do kontroly vlastností charakteristických pro populaci CSCs. Tento článek shrnuje základní poznatky o mechanizmech lékové rezistence ve vztahu k populaci nádorových kmenových buněk.
Klíčová slova:
léková rezistence − nádorové kmenové buňky − membránové transportní proteiny – přeměna epitelové buňky na mezenchymovou – mikroprostředí nádoru − apoptóza
Úvod
Rezistence nádorů k dostupné léčbě (chemorezistence) je jedna z hlavních příčin úmrtí pacientů s rakovinou. Většinou se chemorezistence vyvíjí ve dvou krocích. Nejprve nádor odpovídá na léčbu, ale ne všechny buňky jsou zničeny. Tyto rezistentní buňky mohou poté dát vznik sekundárním nádorům, které již neodpovídají na terapii. Na odolnosti buněk vůči léčbě se podílí mnoho faktorů, jako je snížené vstřebávání léčiv nebo naopak jejich zvýšený výdej, detoxikace, zvýšená intenzita oprav DNA či deregulace drah programované smrti buněk [1,2]. Příčinou chemorezistence u pevných nádorů může být i špatný přísun léčiv z důvodu slabé vaskularizace nádoru či nepropustnosti okolní tkáně. Uvedené mechanizmy lékové rezistence se shodují s původními nebo získanými vlastnostmi charakteristickými pro nádorové kmenové buňky (cancer stem cells – CSCs): stagnace, specifická morfologie, zvýšená produkce antiapoptotických proteinů, transmembránových přenašečů či detoxikačních enzymů. Předpokládá se, že právě tyto buňky odpovídají za lékovou rezistenci, metastazování a recidivu některých nádorů.
Současné strategie léčby proto zahrnují nejen překonávání fyzických bariér a mechanizmů lékové rezistence nádorů, ale také se soustřeďují na cílené potlačení CSCs, aby se zabránilo návratu onemocnění [3].
Nejčastější mechanizmy lékové rezistence
Mikroprostředí nádorů (nika)
Nádor je vysoce komplexní tkáň, kde jsou vlastní nádorové buňky většinou v menšině a koexistují s různými typy buněk v prostředí, které se významně liší od normální tkáně. Nádorové buňky jsou silně provázány s okolním prostředím, jež jim předává a poskytuje onkogenní signály podporující nádorovou progresi [4]. Součástí niky jsou stromální buňky, buňky imunitního systému, endoteliální buňky či pericyty. Převládající stromální buňky jsou fibroblasty, které uvolňují do prostředí komponenty extracelulární matrix, enzymy, růstové faktory, cytosiny a látky stimulující růst nádoru, angiogenezi a infiltraci buněk imunitního systému [5,6]. Infiltrace imunitních buněk je významná pro některé typy nádorů, jako adenokarcinom mléčné žlázy a slinivky. Tyto s nádorem spolupracující makrofágy uvolňují růstové faktory, chemokiny, cytosiny či enzymy degradující matrix, které stimulují angiogenzi [7], růst nádorových buněk, invazivitu, příliv pronádorových imunitních buněk a naopak blokují aktivaci protinádorových T buněk [8]. Dalším příkladem jsou stromální buňky v okolí kostní dřeně nebo sekundárních lymfoidních orgánů, jež podporují růst, proliferaci a chemorezistenci maligních B buněk a napomáhají tak průběhu onemocnění [9]. Nádorové buňky produkují proteolytické enzymy, zejména metaloproteinázy, které mají pro‑migrační a pro‑angiogenní vlastnosti, aktivují povrch buněk a faktory vážící se na extracelulární matrix (ECM). Příkladem je nadměrná produkce aktivovaných forem metaloproteináz u epitelu mléčné žlázy u myší, jež narušují spojitost kostní dřeně, čímž vzniká reaktivní stroma a formují se geneticky nestabilní nádory mléčné žlázy [10]. Složení a uspořádání ECM a stromálních komponent přispívá k výraznému gradientu v koncentraci léků, zvyšuje mezibuněčný tlak kapalin a dochází k metabolickým změnám, což silně přispívá ke zvýšení chemorezistence nádorových buněk [11]. Podobně pomáhá přežívání maligních buněk slabě kyselé pH charakteristické pro okolí pevných nádorů, které snižuje průchod některých bazických chemoterapeutik difúzí přes plazmatickou membránu [12]. Nika obklopující nádor tak vytváří bariéru chránící nádor před účinkem léků. Současné studie ukazují, že enzymatickým narušením stromatu v okolí nádorů a odstraněním jejich ochranné bariéry dochází ke zvýšení vaskularizace a zlepšení účinnosti protinádorové léčby [3].
Chemorezistence navozená buněčnou adhezí (CAM ‑ DR)
Pro růst a přežití normálních buněk je nezbytná adheze (přilnavost) k povrchu, naopak u nádorových buněk je nutná nezávislost na jejich ukotvení. Např. ztráta β1 - integrinu u nemaligních buněk vede k jejich apoptóze, zatímco u nádorových buněk bývá exprese β1 - integrinu zcela potlačena nebo naopak dramaticky zvýšena. Adheze k ECM přes β1 - integriny u buněk mnohočetného myelomu zvyšuje růst nádoru a jeho rezistenci k léčivům jako doxorubicin a melfalan [13]. Zablokování vazby β1 - integrinu k ECM a stromálním buňkám dramaticky snižuje nádorové zatížení a zvyšuje přežití u myších modelů mnohočetného myelomu [14]. Podobně inhibice β1 - integrinu u xenograftů karcinomu mléčné žlázy vede k významnému zmenšení nádorů a zvýšení jejich citlivosti k ozařování [15]. Vazba nádorových buněk k ECM a stromálním buňkám přes β1 - integriny ovlivňuje buněčnou lokalizaci apoptotických regulátorů a kontroluje průběh buněčného cyklu u hematologických a epitelových malignit [16]. Hazlehurst et al popsali, že zástava buněčného cyklu v G1 fázi u buněk myelomu je způsobena vazbou β1 - integrinu na fibronektin, která koreluje se zvýšenou hladinou p27 a zvyšuje rezistenci buněk k etoposidu [17]. Vazba β1 - integrinu k ECM může silně ovlivnit proces rozpoznání a opravy poškozené DNA, kdy adheze nádorových buněk k ECM urychluje a optimalizuje opravy DNA po ozařování a zajišťuje tak stabilnější genom a přežití buněk. Komunikace buněk s EMC také ovlivňují strukturu jejich jádra a uspořádání chromatinu [18,19].
Epiteliální ‑ mezenchymální přeměna
Vliv prostředí nádoru se také podílí na procesu přeměny epiteliální buňky na mezenchymální (EMT). Jedná se o přechodnou změnu buněčného fenotypu, která zahrnuje nárůst fibroidní morfologie, invazivity, odolnosti k apoptóze a navýšení sekrece složek extracelulární matrix [20,21]. Postupně se rozvolňují mezibuněčné spoje, reorganizuje se cytoskelet, buňka ztrácí apikální polaritu a nakonec dochází k její přeměně na buňku s vřetenovitou morfologií [22]. Proces EMT u epitelu různých orgánů je jedním z prvních kroků při vzniku nádoru [23], souvisí s progresí nádoru, s metastazováním a s přítomností nádorových kmenových buněk. Zvýšená hladina markerů EMT je spojována s větší agresivitou nádorů a s jejich rezistencí k různým protinádorovým látkám. Mechanizmus EMT není znám, ale předpokládá se, že dochází k masivnímu „přeprogramování“ genové exprese buněk a k získání nových vlastností souvisejících s rezistencí k léčivům [24]. EMT je pravděpodobně indukována signály z okolního stromatu. Na její regulaci se podílejí růstové faktory jako HGF (hepatocyte growth factor), PDGF (platelet ‑ derived growth factor), TGFβ (transforming growth factor beta), Slug a negativní regulátory ZEB1, 2 (zinc finger E ‑ box binding homeobox 1, 2) či miR ‑ 200 [23]. EMT je také spojena se zvýšenou expresí transmembránových přenašečů a se změnou v opravných mechanizmech DNA [25].
Hypoxie
Hypoxie je jedním ze znaků prostředí obklopujícího pevný nádor a je způsobena nerovnováhou mezi příjmem a spotřebou kyslíku. Přítomnost hypoxie u pevných nádorů je spojována s jejich rezistencí k radioterapii a chemoterapii, s vyšším nádorovým potenciálem buněk a horší prognózou onemocnění [26]. Hlavními regulátory buněčné adaptace k nedostatku kyslíku jsou HIFs (hypoxia inducible factors). Tyto transkripční faktory aktivují geny, které zabraňují buněčné diferenciaci, podporují formování cév, regulují glykolýzu, spotřebu kyslíku, apoptózu, migraci a metastazování [27]. HIFs aktivují enzymy pro opravu dvouřetězcových zlomů DNA a podporují vznik rezistence buněk k látkám poškozujícím DNA. Navíc hypoxie reguluje i angiogenezi, jednu z hnacích sil při vývoji nádoru. Hladina HIFs je regulována koncentrací kyslíku v buňce. Pokud je hladina kyslíku nízká, dochází ke stabilizaci HIFs, jejich translokaci do jádra a k aktivaci cílových genů. Exprese HIFs byla zaznamenána u mnoha typů nádorů a je spojena se špatnou prognózou a odpovědí na léčbu. Preklinické studie potvrdily, že terapie zasahující HIFs (siRNA, inhibitory topoizomeráz) pomáhají překonávat lékovou rezistenci [28,29].
Transmembránové pumpy
Transmembránové pumpy jsou proteiny zajišťující transport širokého spektra látek přes extra ‑ i intracelulární membrány za současné hydrolýzy ATP (adenosine triphosphate) [30]. Velké množství těchto transportérů je zahrnuto do rodiny přenašečů vázajících ATP (ABC přenašeče). Tyto proteiny hrají důležitou úlohu v adsorpci, distribuci a eliminaci léčiv a nadměrná exprese některých z nich vede ke vzniku lékové rezistence. Mezi nejčastěji diskutované proteiny přispívající k chemorezistenci patří P-gp (P-glycoprotein), MRP1, 2 (the multidrug resistance protein 1, 2) a BCRP (breast cancer resistance protein). Zvýšená produkce těchto proteinů vede k rychlejšímu exportu toxických látek z buňky a ke snížení intracelulární akumulace léčiv. Způsobují odolnost buněk k látkám jako taxany, antracykliny, epipodophyllotoxiny, metotrexát, analogy nukleotidů nebo alkylační látky [31,32].
Zablokování programované buněčné smrti
Účinek mnoha protinádorových léčiv je založen na aktivaci programované buněčné smrti (apoptóza). Nádorové buňky, které mají tyto proapoptotické signální dráhy poškozené nebo zablokované, jsou odolnější vůči těmto látkám a získávají čas na opravu poškozené DNA a dalších buněčných struktur. Mechanizmy rezistence k apoptóze zahrnují: 1. porušení rovnováhy mezi pro ‑ a anti‑apoptotickými proteiny, 2. snížení funkce kaspáz a 3. poruchy signálních drah receptorů smrti.
1. Proces apoptózy může být narušen nadměrnou produkcí anti‑apoptotických genů (BCL2, BCL ‑ XL, MCL ‑ 1), snížením produkce pro‑apoptotických genů (BAX, PUMA, NOXA), poškozením p53 či apoptotických signálních drah, deregulací inhibitorů apoptózy (IAPs ‑ NAIP, c ‑ IAP1, c ‑ IAP2, X‑linked IAP, Survivin, Apollon, Livin) nebo proteinů řídících buněčnou odpověď na stresové stimuly (heat shock proteiny – HSPs).
Nadměrná exprese Bcl ‑ 2 je velmi často spojena se špatnou prognózou onemocnění a odolností vůči chemoterapii a radioterapii. Vysoká produkce Bcl ‑ 2 může být způsobena translokací chromozomů t(14, 18), amplifikací genu BCL2 nebo delecí chromozomu, která má za následek ztrátu miRNA specifickou pro BCL2. Vedle nadměrné produkce anti‑apoptotických proteinů nádorové buňky odolávají apoptóze potlačením exprese nebo mutací pro‑apoptotických genů. Příčinou může být ztráta funkce nádorových supresorů jako p53 a tím snížení exprese jejich cílových genů a inaktivace apoptotických signálních drah [33].
IAPs jsou endogenní inhibitory kaspáz, které vazbou na aktivní místo kaspázy iniciují její degradaci nebo brání vazbě se substrátem a blokují spuštění apoptózy. Nadměrná produkce IAPs byla zaznamenána u mnoha typů nádorových onemocnění a bývá spjata se zvýšenou odolností onemocnění k chemoterapii a k podmínkám vyvolávajícím apoptózu [34].
HSPs − jsou produkovány jako odpověď na stres a zvyšují odolnost buňky vůči stresovým podmínkám. Za normálních podmínek zajišťují HSPs v buňce mnoho různých funkcí včetně řízení aktivity enzymů změnou jejich konformace, správné sbalování proteinů při translaci, regulaci tvorby proteinových komplexů, kontrolu degradace proteinů a jejich přenos přes membrány organel. HSPs mohou interagovat s komponentami signálních drah vedoucích k apoptóze, zabraňují tak smrti buněk a umožňují jejich přežití a proliferaci. Inhibice apoptózy probíhá na třech úrovních: 1. modulací signálních drah spouštějících apoptózu, 2. kontrolou uvolňování apoptotických molekul a 3. blokováním pozdních fází apoptózy. Zvýšená hladina HSP byla identifikována u mnoha lidských malignit a je spojována s jejich odolností vůči apoptóze indukované chemoterapeutiky [35].
2. Kaspázy patří k hlavním proteinům, které spouštějí a dokončují apoptózu. Jejich poškození nebo změna funkce vede ke snížení apoptózy a následné karcinogenezi.
3. Receptory smrti (DRs, Fas TRAILs) a jejich ligandy patří mezi vnějších iniciátory apoptózy. Přes doménu smrti aktivují příslušné signální dráhy vedoucí k apoptóze. U různých typů nádorů byla popsána snížená exprese receptorů smrti, poruchy jejich funkce nebo nadměrná exprese analogů receptorů smrti (decoy receptorů), jež váží stejné ligandy, ale nespouští signální apoptotické dráhy [36].
Vliv buněčného cyklu na chemorezistenci
Účinek cytostatik je ovlivněn fází buněčného cyklu, ve kterém se zasažená buňka nachází. Léčiva poškozující DNA postihují primárně buňky v S ‑ fázi buněčného cyklu, zatímco vřeténkové jedy působí na buňky v mitóze. Buňky v G0/ G1 fázi buněčného cyklu jsou relativně rezistentní ke klasické cytotoxické terapii [37]. Současné studie se zabývají možností, že nádory obsahují frakci „klidových“ buněk, které se aktivně (ale reverzibilně) udržují v tomto stavu, kdy snáze odolávají léčivům.
Chemorezistence zprostředkovaná epigenetickými změnami
Je dlouhodobě známo, že nádorová onemocnění jsou spojena s rozsáhlými epigenetickými změnami, např. exprese nádorových supresorů může být potlačena metylacemi jejich promotorů [38]. Tyto změny jsou reverzibilní a mohou být odstraněny použitím látek demetylujících DNA či inhibitorů histonacetyláz (HDAC) [39]. Příkladem jsou buňky nemalobuněčného karcinomu plic (non small cell lung cancer − NSCLC), u kterých se po působení inhibitorů tyrozinkináz vytvořila malá frakce buněk odolná vůči těmto látkám (drug tolerant persisters – DTPs). Tyto buňky, tvořící 0,3−5 % celkové populace buněk, nebyly stabilně rezistentní. Pokud se DTPs kultivovaly bez přítomnosti inhibitorů, opět začaly být k těmto látkám citlivé. Podrobná analýza zjistila velkou variabilitu v genové expresi, včetně zvýšení exprese genů ovlivňujících chromatinové modifikace jako histon H3K demetyláza (KDM5A) nebo HDAC. Potlačení funkce KDM5A nebo působení inhibitorů HDAC redukovalo počet DTPs, což naznačuje, že za odolnost těchto buněk jsou zodpovědné modifikace chromatinu [40].
Nicméně aktivace umlčených genů je dvousečná zbraň, neboť může dojít zároveň k aktivaci nežádoucích genů odpovědných za rezistenci či za nádorovou transformaci [41].
Rezistentní subpopulace buněk
U nádorových onemocnění s vysokým proliferačním indexem, jako jsou leukemie či lymfomy, byly zaznamenány subpopulace buněk odolávající vybraným lékům. Například při léčbě chronické myeloidní leukemie (chronic myelogenous leukemia − CML) imatinibem (inhibitor tyrozinkináz) byla identifikována malá populace leukemických buněk se substitucí aminokyselin v genu pro ABL kinázu, která chránila buňky před inhibičním účinkem imatinibu [42]. Podobné mutace byly zaznamenány i při použití dalších inhibitorů kináz [43]. Některé nádorové buňky jsou navíc schopny blok v jedné signální dráze obejít aktivací alternativní signální dráhy a eliminovat tak efekt léčiva. Na podobném principu fungují rezistence k látkám inhibujícím HER2 nebo B ‑ RAF. Tento jev je možné překonat podáváním inhibitorů kináz druhé či třetí generace [44]. Ačkoliv tento postup zaznamenává částečný úspěch, je tu několik komplikací. Jedna z nich, zvláště u leukemií a lymfomů, je zablokování programované buněčné smrti [45] a také přítomnost malé frakce nádorových „kmenových“ buněk, které se vyskytují v klidovém stadiu a jsou rezistentními k léčivům. Pokud jsou tyto buňky donuceny k proliferaci, např. pomocí interferonu ‑ α, G‑CSF nebo oxidem arzenitým, stávají se citlivými k cytotoxickým látkám [46,47]. Nicméně i terapie založená na tomto přístupu vykazovala nevyrovnané výsledky a nedokázala zabránit návratu onemocnění [48].
Nádorové kmenové buňky − nádor iniciující buňky (CSCs)
Teorie nádorových kmenových buněk byla formulována v roce 1997 pro akutní myeloidní leukemii (AML) [49] a v roce 2003 byla rozšířena na pevné nádory, přesněji na karcinom mléčné žlázy [50]. Nyní již byly CSCs identifikovány v širokém spektru pevných nádorů jako karcinom plic [51], tlustého střeva [52], prostaty [53], vaječníků [54], mozku [55] či u melanomů [56]. Nicméně se stále jedná o teorii kontroverzní, vyvolávající vášnivé diskuze [57,58].
Předpokládá se, že podobně jako u normálních rostoucích tkání (kostní dřeň, kůže či střevní epitel) je i růst a vývoj nádoru „poháněn“ malým množstvím kmenových buněk, charakterizovaných jako „nesmrtelné“ pluripotentní buňky schopné sebeobnovy [59]. Tyto buňky odpovídají za iniciaci, vývoj, metastazování a recidivu nádoru a jako jediné jsou schopny vyvolat tvorbu nového nádoru po přenesení do zvířecího modelu. CSCs tvoří 0,1−30 % nádoru podle jeho typu a stadia onemocnění [60]. Velká část nádoru je naopak tvořena rychle rostoucími, případně post‑mitotickými diferencovanými buňkami, které však nejsou schopny sebeobnovy a jejichž přínos je z hlediska dlouhodobého udržení nádoru nepatrný [58]. Byly formulovány dva odlišné modely vysvětlující růst nádorů a jejich heterogenitu. Model nádorových kmenových buněk předpokládá, že všechny fáze nádoru, jako iniciace, progrese, metastazovaní apod., jsou závislé především na kmenových buňkách. Heterogenita a hierarchie mezi všemi buňkami nádoru je výsledkem asymetrického dělení CSCs. Tento model vidí nádor jako přísně hierarchickou strukturu s unikátní skupinou buněk schopných sebeobnovy na jejím vrcholu. Všechny ostatní buňky tvořící tělo nádoru jsou odvozené od CSCs a vznikají jejich diferenciací [61]. Druhý, evoluční model, předpokládá, že všechny nádorové buňky přispívají k udržení nádoru, i když s různou intenzitou. Mezibuněčné rozdíly jsou dány především subklonálními rozdíly, způsobenými genetickými nebo epigenetickými změnami během vývoje nádoru [62]. Ačkoliv si oba modely vzájemně odporují, nelze podle posledních poznatků ani jeden z nich vyloučit. Naopak se zdá, že se oba modely prolínají a výsledkem je druhá, agresivnější populace CSCs [63].
I když teorie nádorových kmenových buněk má své trhliny, často se používá pro vysvětlení reziduálního onemocnění. Důvodů pro toto tvrzení je několik (obr. 1). Kmenové buňky včetně CSCs produkují velké množství ABC transmembránových přenašečů, jako P ‑ gp a BCPR [64,65]. Populace buněk s vysokým obsahem ABC přenašečů má větší maligní potenciál a je agresivnější [66]. Pro CSCs stejně jako pro ostatní somatické kmenové buňky je charakteristické pomalé tempo dělení. Není zcela zřejmé, zda tato vlastnost je příčina nižší citlivosti CSCs k chemoterapii, ale obecně se předpokládá, že přispívá k odolnosti vůči cytotoxickým látkám [67,68]. CSCs také vykazují větší odolnost k poškození DNA, např. ionizujícím zářením [69,70] a často podléhají EMT. Spojení mezi EMT a CSCs pak často souvisí s metastazováním. Pro CSCs je charakteristická aktivace některých antiapoptotických signálních drah. Nejvýznamnější jsou:
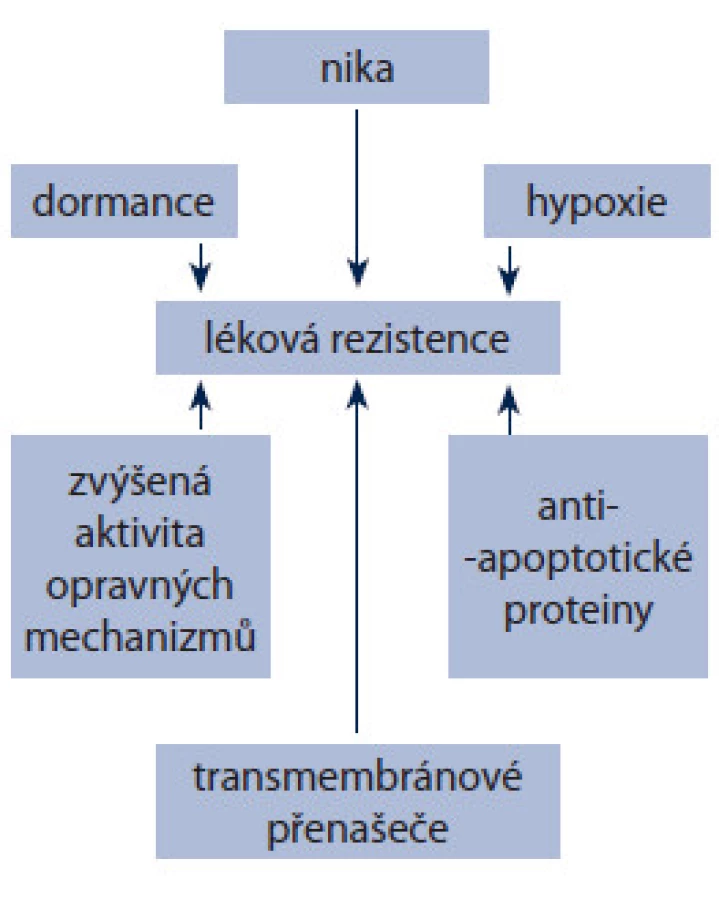
- PI3K/ PTEN/ AKT/ mTOR – aktivace AKT je nezbytná pro buněčnou transformaci a vznik nádoru, byla pozorována např. u buněčné transformace zprostředkované v ‑ Abl u leukemií [71]. Podobně i PTEN a mTOR hrají úlohu při udržení leukemických kmenových buněk. Mutace nebo umlčení exprese PTEN byly pozorovány u různých typů onkologických onemocnění, jako ALL (akutní lymfoblastická leukemie), karcinom prostaty, melanom, glioblastom a karcinom dělohy [72].
- JAK/ STAT signální dráha je zapojena od iniciace nádoru. Chyby v této dráze byly identifikovány zejména u různých typů leukemií. JAK/ STAT signální dráha je negativně regulována rodinou SOCS proteinů. Inhibice těchto proteinů např. fosforylací Bcr ‑ Abl u CML vede k následné buněčné transformaci [73].
- NF ‑ κB – tento transkripční faktor reguluje expresi mnoha genů podílejících se na buněčných odpovědích na různé podněty, jako je působení cytokinů, mikrobů, volných radikálů či ultrafialového záření [74], a také ovlivňuje expresi některých antiapoptotických genů (bcl ‑ 2, bcl ‑ xL, survivin apod.). Poškození signální dráhy NF ‑ κB vede k vývoji nádoru a jeho progresi, chemorezistenci, k chronickým zánětům či autoimunitnímu onemocnění [75,76].
- Wnt/ β ‑ catenin, Hedgehog a Notch signální dráhy hrají zásadní roli v uchování populace CSCs. Dráha Notch ovlivňuje proces sebeobnovy buněk a je nezbytná pro zachování specifické buněčné linie při diferenciaci normálních buněk mléčné žlázy [77]. U pankreatických nádorových buněk rezistentních ke gemcitabinu bylo prokázáno spojení signální dráhy Notch s metastazováním a EMT [78]. Potlačení signalizace Notch pomocí siRNA vedlo částečně ke zvrácení EMT a k zabránění vzniku rezistence ke gemcitabinu [79]. Poruchy regulace signální dráhy receptoru Hedgehog (Hh) jsou kritickým faktorem při léčbě onemocnění a léky zaměřené na tuto dráhu vykazují slibné výsledky v klinických studiích [80]. Aktivní dráha Hh byla identifikována u leukemií, zejména u primárních leukemických buněk s fenotypem CD34+. Monoklonální protilátka anti‑Hh indukuje apoptózu u buněk akutní myeloidní leukemie rezistentních k cytarabinu [81]. Dráha Wnt/ β ‑ catenin hraje významnou úlohu v embryogenezi a podílí se na zachování a proliferaci normálních kmenových buněk. Aktivovaná dráha Wnt/ β ‑ catenin byla identifikována u mnoha typů nádorových onemocnění, jako je leukemie, karcinom tlustého střeva, karcinom mléčné žlázy či kůže, kde navozuje lékovou rezistenci [82,83]. U karcinomu tlustého střeva studie prokázaly mutace genu APC (adenomatous polyposis coli), které vedou ke stabilizaci β ‑ cateninu, následné aktivaci kaskády signální dráhy Wnt a indukci epiteliální transformace buněk [84]. Snížení aktivity dráhy Wnt pomocí siRNA proti β ‑ cateninu vedlo k efektivní inhibici proliferace a lékové rezistence u buněk nádorů plic [85]. Podobně zablokování Wnt dráhy u buněk karcinomu tlustého střeva s expresí CD133+ snížilo jejich odolnost k 5 - fluorouracilu [79].
Antiapoptotické signální dráhy jsou zapojeny do lékové rezistence zprostředkované CSCs. Specifické inhibitory těchto drah, např. siRNA, miRNA apod., v kombinaci s dalšími léky mohou představovat strategii pro překonání lékové rezistence nádorů a pro eliminaci CSCs.
Terapie zaměřené na nádorové kmenové buňky
Dostupné metody radioterapie či chemoterapie ničí především „tělo“ nádoru a často nezasahují CSCs chráněné různými obrannými mechanizmy. U některých typů nádorových onemocnění (glioblastomy, karcinom mléčné žlázy, karcinom tlustého střeva) bylo prokázáno, že reziduální nádory po terapii jsou obohaceny o buňky podobné CSCs [58]. CSCs mohou být snadno detekovány pomocí povrchových receptorů např. kombinací CD34+/ CD38 − u leukemických buněk nebo CD44+/ CD24 − případně CD133+ u pevných nádorů [86].
Pokud jsou základem nádoru opravdu kmenové buňky, je pravděpodobné, že reziduální onemocnění se vyvíjí právě z těchto rezistentních CSCs (obr. 2). Tyto druhotné tumory jsou více maligní, rychle metastazují a jsou odolné vůči lékům použitým v první linii léčby. Cílená terapie zasahující CSCs je klíčová pro úspěšnou léčbu maligních onemocnění a mohla by pomoci předcházet recidivě onemocnění [87].
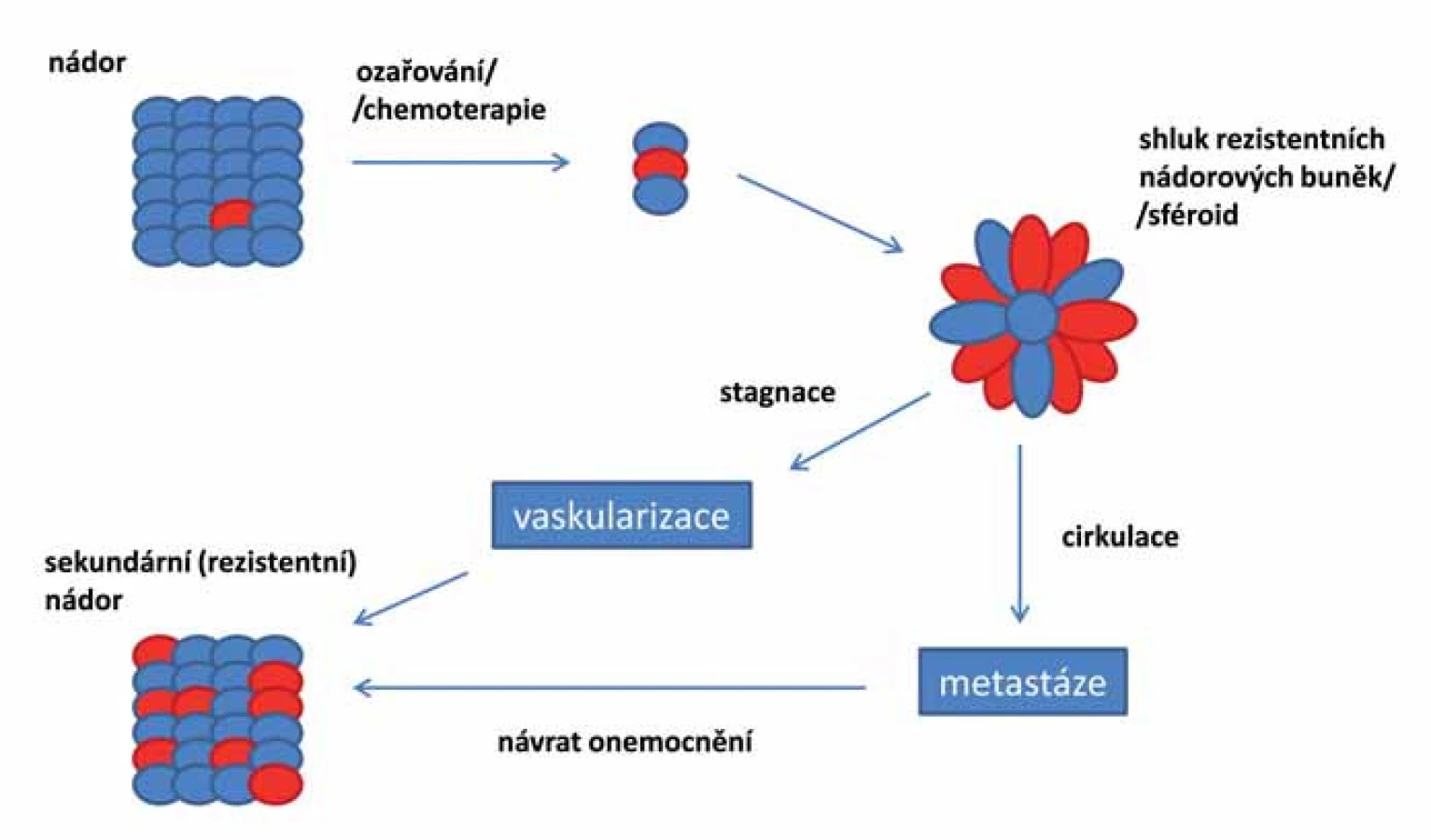
V současné době bylo vyvinuto několik různých terapeutických systémů zaměřených na zničení CSCs, které vykazují slibné výsledky. Můžeme je rozdělit do skupin podle jejich zaměření na:
1. Povrchové markery CSCs
Většinou se jedná o protilátky proti povrchovým markerům nádorových buněk nebo jejich ligandy, (CD44, CD133 apod.), zejména pak o monoklonální protilátky rozeznávající CSCs. Příkladem je gemtuzumab ozogamicin, humanizovaná myší monoklonální protilátka anti‑CD33 konjugovaná s cytotoxickou látkou (kalicheamicinem) používaná při léčbě AML (akutní myeloidní leukemie) [88].
2. Transmembránové pumpy řízené ATP
Sledování hladiny exprese ABC přenašečů pomáhá predikovat úspěšnost chemoterapie a snižovat tak náklady na neúčinnou léčbu. Většina léčebných metod zaměřených na ABC přenašeče se je snaží obejít, neutralizovat nebo jinak využít a předejít tak vzniku rezistence k lékům. Léčba s využitím inhibitorů P ‑ gp či MRP ‑ 1 nebyla příliš úspěšná, proto bylo vyvinuto několik inhibitorů či modulátorů P ‑ gp, které se testují v kombinaci s dalšími protinádorovými léky [89,90]. Alternativní strategie se zaměřuje na regulaci exprese ABC přenašečů např. inhibicí signalizace prostřednictvím receptorů Hedgehog nebo SMO (smoothened) signální dráhy [91].
3. Ovlivnění signálních drah
Aktivní antiapoptotické dráhy a současně inaktivované proapoptotické dráhy jsou dalším oblíbeným cílem nových léčebných strategií. Byly vyvinuty monoklonální protilátky zasahující signální dráhy Notch či Wnt [92,93]. Malé molekuly působící jako antagonisté Hh či inhibitor SMO cyklopamin, inhibující kaskádu Hh, snižují růst, invazivitu a metastazování karcinomů mléčné žlázy, prostaty, slinivky či mozku in vitro i in vivo [94,95]. Inhibice NF ‑ κB zvyšuje citlivost nádorových buněk k lékům. Kombinace doxorubicinu či paclitaxelu s inhibitory NF ‑ κB úspěšně překonává mnohočetnou lékovou rezistenci [96,97]. Slibné výsledky v klinických testech byly získány použitím nízkomolekulárního inhibitoru ABT-137 (inhibitor proteinů Bcl ‑ 2 rodiny), který zvyšoval citlivost nádorových buněk k ozařování a chemoterapii u mnohočetného myelomu, lymfomu a malobuněčného karcinomu plic [98]. Podobně jsou testovány i inhibitory HSP, zejména inhibitory HSP90 jako geldamycin a jeho deriváty.
4. Mikroprostředí nádorů
Změnou mikroprostředí nádorů a poškozením jejich ochranné bariéry může dojít k potlačení či překonání lékové rezistence. Příkladem je redukce krevního řečiště u myšího glioblastomu pomocí bevacizumabu (inhibitor angiogeneze), kdy dochází k narušení mikroprostředí nádoru a ke snížení počtu nádorových kmenových buněk [99]. Podobně působí i kombinace protilátky anti‑VEGF (vascular endothelial growth factor) a cyklofosfamidu [100].
Dalším příkladem je vazba SDF ‑ 1 (stromal cell ‑ derived factor ‑ 1), produkovaným stromálními buňkami, na receptor CXCR4 (chemokine receptor type 4)na povrchu leukemických buněk. Touto vazbou se leukemické buňky dostávají do těsného kontaktu se stromálními buňkami kostní dřeně, současně se aktivuje jejich růst a zvyšuje se jejich odolnost k léčbě. Při aplikaci CXCR4 antagonistů, jako je Plerixafor, dochází k rozpadu vazby CDF ‑ 1/ CXCR4, leukemické buňky se přesouvají mimo kostní dřeň a stávají se citlivější k cytotoxickým lékům [9].
K cílenému transportu léčiv lze také využít mírně kyselé pH prostředí obklopující pevné nádory. Látky citlivé na určité pH konjugované s transportními molekulami a léčivem mohou rychle a přesně dopravit lék na místo s daným pH a podstatně zvýšit jeho účinek [101].
Závěr
Stále se objevují nové důkazy o existenci CSCs u různých typů malignit a o jejich schopnosti sebeobnovy a diferenciace, jež jsou nezbytné pro vznik, růst, udržení a metastazování nádorů. Metody identifikace a izolace CSCs pomocí nově objevených markerů se neustále zlepšují a pozornost je nyní věnována vývoji terapií zaměřených na CSCs. Lze předpokládat, že komplexní strategie léčby bude dosahovat lepších výsledků. Příkladem je vývoj nanočástic konjugovaných se čtyřmi typy látek: 1. ligandy rozpoznávající specificky CSCs, 2. cytotoxické protinádorové látky zasahující CSCs, 3. látky překonávající lékovou rezistenci a 4. látky usnadňující diagnostiku nádoru. Kombinace těchto složek by měla dosáhnout specifického protinádorového efektu s minimem vedlejších účinků. Navíc tento přístup umožní lokalizaci primárního nádoru i jeho metastáz.
Rozšiřující se poznatky o nádorových kmenových buňkách ukazují další směr v boji s malignitami. Nicméně nalezení strategie využívající unikátních vlastností CSCs vyžaduje další výzkum a spolupráci napříč vědními obory.
Tato práce byla realizována za podpory Interní grantové agentury MZ ČR IGA NT/14602 – 3/2013, Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO CZ.1.05/2.1.00/03.0101) a MZ ČR − RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Holčaková Jitka, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: holcakova@mou.cz
Obdrženo: 3. 2. 2014
Přijato: 7. 5. 2014
Sources
1. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 2000; 11(4): 265 – 283.
2. Stavrovskaya AA. Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc) 2000; 65(1): 95 – 106.
3. Provenzano PP, Cuevas C, Chang AE et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21(3): 418 – 429. doi: 10.1016/ j.ccr.2012.01.007.
4. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21(3): 309 – 322. doi: 10.1016/ j.ccr.2012.02.022.
5. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6(5): 392 – 401.
6. Feig C, Gopinathan A, Neesse A et al. The pancreas cancer microenvironment. Clin Cancer Res 2012; 18(16): 4266 – 4276. doi: 10.1158/ 1078 - 0432.CCR ‑ 11-3114.
7. Calabrese C, Poppleton H, Kocak M et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11(1): 69 – 82.
8. Kees T, Egeblad M. Innate immune cells in breast can-cer – from villains to heroes? J Mammary Gland Biol Neoplasia 2011; 16(3): 189 – 203. doi: 10.1007/ s10911 - 011 - 9224 - 2.
9. Konopleva M, Tabe Y, Zeng Z et al. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat 2009; 12(4 – 5): 103 – 113. doi: 10.1016/ j.drup.2009.06.001.
10. Sternlicht MD, Lochter A, Sympson CJ et al. The stromal proteinase MMP3/ stromelysin‑1 promotes mammary carcinogenesis. Cell 1999; 98(2): 137 – 146.
11. Sutherland RM, Eddy HA, Bareham B et al. Resistance to adriamycin in multicellular spheroids. Int J Radiat Oncol Biol Phys 1979; 5(8): 1225 – 1230.
12. Trédan O, Galmarini CM, Patel K et al. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99(19): 1441 – 1454.
13. Sethi T, Rintoul RC, Moore SM et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med 1999; 5(6): 662 – 668.
14. Mori Y, Shimizu N, Dallas M et al. Anti‑alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004; 104(7): 2149 – 2154.
15. Park CC, Zhang H, Pallavicini M et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res 2006; 66(3): 1526 – 1535.
16. Shain KH, Landowski TH, Dalton WS. Adhesion ‑ mediated intracellular redistribution of c ‑ Fas‑associated death domain‑like IL‑1‑converting enzyme‑like inhibitory protein‑long confers resistance to CD95‑induced apoptosis in hematopoietic cancer cell lines. J Immunol 2002; 168(5): 2544 – 2553.
17. Hazlehurst LA, Enkemann SA, Beam CA et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res 2003; 63(22): 7900 – 7906.
18. Sandal T, Valyi ‑ Nagy K, Spencer VA et al. Epigenetic reversion of breast carcinoma phenotype is accompanied by changes in DNA sequestration as measured by AluI restriction enzyme. Am J Pathol 2007; 170(5): 1739 – 1749.
19. Jones CB, McIntosh J, Huang H et al. Regulation of bleomycin‑induced DNA breakage and chromatin structure in lung endothelial cells by integrins and poly(ADP ‑ ribose) polymerase. Mol Pharmacol 2001; 59(1): 69 – 75.
20. Hay ED. An overview of epithelio ‑ mesenchymal transformation. Acta Anat (Basel) 1995; 154(1): 8 – 20.
21. Kalluri R, Neilson EG. Epithelial ‑ mesenchymal transition and its implications for fibrosis. J Clin Invest 2003; 112(12): 1776 – 1784.
22. Lee JM, Dedhar S, Kalluri R et al. The epithelial ‑ mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172(7): 973 – 981.
23. Thiery JP. Epithelial ‑ mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2(6): 442 – 454.
24. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29(34): 4741 – 4751. doi: 10.1038/ onc.2010.215
25. Chiba N, Comaills V, Shiotani B et al. Homeobox B9 induces epithelial ‑ to ‑ mesenchymal transition‑associated radioresistance by accelerating DNA damage responses. Proc Natl Acad Sci USA 2012; 109(8): 2760 – 2765. doi: 10.1073/ pnas.1018867108.
26. Harris AL. Hypoxia – a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2(1): 38 – 47.
27. Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev 2007; 26(2): 223 – 224.
28. Wirthner R, Wrann S, Balamurugan K et al. Impaired DNA double‑strand break repair contributes to chemoresistance in HIF ‑ 1 alpha ‑ deficient mouse embryonic fibroblasts. Carcinogenesis 2008; 29(12): 2306 – 2316. doi: 10.1093/ carcin/ bgn231.
29. Choi YJ, Rho JK, Lee SJ et al. HIF ‑ 1alpha modulation by topoisomerase inhibitors in non‑small cell lung cancer cell lines. J Cancer Res Clin Oncol 2009; 135(8): 1047 – 1053. doi: 10.1007/ s00432 - 009 - 0543-2.
30. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976; 455(1): 152 – 162.
31. Szakacs G, Paterson JK, Ludwig JA et al. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5(3): 219 – 234.
32. Haimeur A, Conseil G, Deeley RG et al. The MRP‑related and BCRP/ ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab 2004; 5(1): 21 – 53.
33. Plati J, Bucur O, Khosravi ‑ Far R. Apoptotic cell signaling in cancer progression and therapy. Integr Biol (Camb) 2011; 3(4): 279 – 296. doi: 10.1039/ c0ib00144a.
34. Wei Y, Fan T, Yu M. Inhibitor of apoptosis proteins and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2008; 40(4): 278 – 288.
35. Lanneau D, Brunet M, Frisan E et al. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med 2008; 12(3): 743 – 761. doi: 10.1111/ j.1582 - 4934.2008.00273.x.
36. Fulda S. Evasion of apoptosis as a cellular stress response in cancer. Int J Cell Biol 2010; 2010 : 370835. doi: 10.1155/ 2010/ 370835.
37. Stewart DJ, Chiritescu G, Dahrouge S et al. Chemotherapy dose – response relationships in non‑small cell lung cancer and implied resistance mechanisms. Cancer Treat Rev 2007; 33(2): 101 – 137.
38. Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358(11): 1148 – 1159. doi: 10.1056/ NEJMra072067.
39. Tsai HC, Li H, Van Neste L et al. Transient low doses of DNA ‑ demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012; 21(3): 430 – 446. doi: 10.1016/ j.ccr.2011.12.029.
40. Sharma SV, Lee DY, Li B et al. A chromatin‑mediated reversible drug‑tolerant state in cancer cell subpopulations. Cell 2010; 141(1): 69 – 80. doi: 10.1016/ j.cell.2010.02.027.
41. Hauswald S, Duque ‑ Afonso J, Wagner MM et al. Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin Cancer Res 2009; 15(11): 3705 – 3715. doi: 10.1158/ 1078-0432.CCR ‑ 08 - 2048.
42. Gorre ME, Mohammed M, Ellwood K et al. Clinical resistance to STI ‑ 571 cancer therapy caused by BCR ‑ ABL gene mutation or amplification. Science 2001; 293(5531): 876 – 880.
43. Carter TA, Wodicka LM, Shah NP et al. Inhibition of drug‑resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 2005; 102(31): 11011 – 11016.
44. Kaiser J. Combining targeted drugs to stop resistant tumors. Science 2011; 331(6024): 1542 – 1545. doi: 10.1126/ science.331.6024.1542.
45. Duy C, Hurtz C, Shojaee S et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR ‑ ABL1 kinase inhibition. Nature 2011; 473(7347): 384 – 388. doi: 10.1038/ nature09883.
46. Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol 2010; 4(5): 443 – 450. doi: 10.1016/ j.molonc.2010.06.001
47. Saito Y, Uchida N, Tanaka S et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 2010; 28(3): 275 – 280. doi: 10.1038/ nbt.1607.
48. Jiang X, Zhao Y, Smith C et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR ‑ ABL targeted therapies. Leukemia 2007; 21(5): 926 – 935.
49. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3(7): 730 – 737.
50. Al ‑ Hajj M, Wicha MS, Benito ‑ Hernandez A et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100(7): 3983 – 3988.
51. Kim CF, Jackson EL, Woolfenden AE et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005; 121(6): 823 – 835.
52. O‘Brien CA, Pollett A, Gallinger S et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445(7123): 106 – 110.
53. Collins AT, Berry PA, Hyde C et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005; 65(23): 10946 – 10951.
54. Szotek PP, Pieretti ‑ Vanmarcke R, Masiakos PT et al. Ovarian cancer side population defines cells with stem cell‑like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci USA 2006; 103(30): 11154 – 11159.
55. Piccirillo SG, Reynolds BA, Zanetti N et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour ‑ initiating cells. Nature 2006; 444(7120): 761 – 765.
56. Fang D, Nguyen TK, Leishear K et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 2005; 65(20): 9328 – 9337.
57. Jordan CT. Cancer stem cells: controversial or just misunderstood? Cell Stem Cell 2009; 4(3): 203 – 205. doi: 10.1016/ j.stem.2009.02.003.
58. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17(3): 313 – 319. doi: 10.1038/ nm.2304.
59. Reya T, Morrison SJ, Clarke MF et al. Stem cells, cancer, and cancer stem cells. Nature 2001; 414(6859): 105 – 111.
60. Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res 2008; 68(11): 4018 – 4021. doi: 10.1158/ 0008 - 5472.CAN ‑ 07 - 6334.
61. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012; 22(3): 457 – 472. doi: 10.1038/ cr.2012.13.
62. Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle 2007; 6(19): 2332 – 2338.
63. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010; 1805(1): 105 – 117. doi: 10.1016/ j.bbcan.2009.11.002.
64. Shackleton M, Vaillant F, Simpson KJ et al. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439(7072): 84 – 88.
65. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8(10): 755 – 768. doi: 10.1038/ nrc2499.
66. Deeley RG, Westlake C, Cole SP. Transmembrane transport of endo ‑ and xenobiotics by mammalian ATP‑binding cassette multidrug resistance proteins. Physiol Rev 2006; 86(3): 849 – 899.
67. Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol 2010; 46(3): 308 – 316. doi: 10.1007/ s12033 - 010-9321 - 2.
68. Aguirre‑Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007; 7(11): 834 – 846.
69. Bao S, Wu Q, McLendon RE et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444(7120): 756 – 760.
70. Diehn M, Cho RW, Lobo NA et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458(7239): 780 – 783. doi: 10.1038/ nature07733.
71. Guo G, Qiu X, Wang S et al. Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v ‑ Abl ‑ mediated pre‑B ‑ cell transformation and survival of Pim ‑ deficient cells. Oncogene 2010; 29(26): 3845 – 3853. doi: 10.1038/ onc.2010.149.
72. Gutierrez A, Sanda T, Grebliunaite R et al. High frequency of PTEN, PI3K, and AKT abnormalities in T ‑ cell acute lymphoblastic leukemia. Blood 2009; 114(3): 647 – 650. doi: 10.1182/ blood ‑ 2009-02 - 206722.
73. Qiu X, Guo G, Chen K et al. A requirement for SOCS ‑ 1 and SOCS ‑ 3 phosphorylation in Bcr ‑ Abl‑induced tumorigenesis. Neoplasia 2012; 14(6): 547 – 558.
74. Baud V, Karin M. Is NF ‑ kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 2009; 8(1): 33 – 40. doi: 10.1038/ nrd2781.
75. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation and cancer. Cell 2010; 140(6): 883 – 899. doi: 10.1016/ j.cell.2010.01.025.
76. Chaturvedi MM, Sung B, Yadav VR et al. NF ‑ kappaB addiction and its role in cancer: ‚one size does not fit all‘. Oncogene 2011; 30(14): 1615 – 1630. doi: 10.1038/ onc.2010.566.
77. Dontu G, Jackson KW, McNicholas E et al. Role of notch signaling in cell ‑ fate determination of human mammary stem/ progenitor cells. Breast Cancer Res 2004; 6(6): R605 – R615.
78. Williams RF, Sims TL, Tracey L et al. Maturation of tumor vasculature by interferon‑beta disrupts the vascular niche of glioma stem cells. Anticancer Res 2010; 30(9): 3301 – 3308.
79. Deng YH, Pu XX, Huang MJ et al. 5 - Fluorouracil upregulates the activity of wnt signaling pathway in CD133 - positive colon cancer stem‑like cells. Chin J Cancer 2010; 29(9): 810 – 815.
80. Merchant AA, Matsui W. Targeting Hedgehog – a cancer stem cell pathway. Clin Cancer Res 2010; 16(12): 3130 – 3140. doi: 10.1158/ 1078 - 0432.CCR ‑ 09- - 2846.
81. Kobune M, Takimoto R, Murase K et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci 2009; 100(5): 948 – 955. doi: 10.1111/ j.1349 – 7006.2009.01111.x.
82. Malanchi I, Peinado H, Kassen D et al. Cutaneous cancer stem cell maintenance is dependent on beta‑catenin signalling. Nature 2008; 452(7187): 650 – 653. doi: 10.1038/ nature06835.
83. Zeng YA, Nusse R. Wnt proteins are self ‑ renewal factors for mammary stem cells and promote their long‑term expansion in culture. Cell Stem Cell 2010; 6(6): 568 – 577. doi: 10.1016/ j.stem.2010.03.020.
84. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434(7035): 843 – 850.
85. Teng Y, Wang X, Wang Y et al. Wnt/ beta‑catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun 2010; 392(3): 373 – 379. doi: 10.1016/ j.bbrc.2010.01.028.
86. Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays 2009; 31(10): 1038 – 1049. doi: 10.1002/ bies.200900058.
87. Zhou BB, Zhang H, Damelin M et al. Tumour ‑ initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov 2009; 8(10): 806 – 823. doi: 10.1038/ nrd2137.
88. Curiel TJ. Immunotherapy: a useful strategy to help combat multidrug resistance. Drug Resist Updat 2012; 15(1 – 2): 106 – 113. doi: 10.1016/ j.drup.2012.03.003.
89. Tsuruo T, Iida H, Tsukagoshi S et al. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981; 41(5): 1967 – 1972.
90. Khdair A, Chen D, Patil Y et al. Nanoparticle ‑ mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J Control Release 2010; 141(2): 137 – 144. doi: 10.1016/ j.jconrel.2009.09.004.
91. Sims ‑ Mourtada J, Izzo JG, Ajani J et al. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007; 26(38): 5674 – 5679.
92. Li K, Li Y, Wu W et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem 2008; 283(12): 8046 – 8054. doi: 10.1074/ jbc.M800170200.
93. He B, Reguart N, You L et al. Blockade of Wnt ‑ 1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005; 24(18): 3054 – 3058.
94. Ramaswamy B, Lu Y, Teng KY et al. Hedgehog signaling is a novel therapeutic target in tamoxifen‑resistant breast cancer aberrantly activated by PI3K/ AKT pathway. Cancer Res 2012; 72(19): 5048 – 5059. doi: 10.1158/ 0008 - 5472.CAN ‑ 12 - 1248.
95. Feldmann G, Habbe N, Dhara S et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008; 57(10): 1420 – 1430. doi: 10.1136/ gut.2007.148189.
96. Fan L, Li F, Zhang H et al. Co ‑ delivery of PDTC and doxorubicin by multifunctional micellar nanoparticles to achieve active targeted drug delivery and overcome multidrug resistance. Biomaterials 2010; 31(21): 5634 – 5642. doi: 10.1016/ j.biomaterials.2010.03.066.
97. Ganta S, Amiji M. Coadministration of Paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol Pharm 2009; 6(3): 928 – 939. doi: 10.1021/ mp800240j.
98. Kang MH, Reynolds CP. Bcl ‑ 2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 2009; 15(4): 1126 – 1132. doi: 10.1158/ 1078 - 0432.CCR ‑ 08-0144.
99. Burkhardt JK, Hofstetter CP, Santillan A et al. Orthotopic glioblastoma stem‑like cell xenograft model in mice to evaluate intra ‑ arterial delivery of bevacizumab: from bedside to bench. J Clin Neurosci 2012; 19(11): 1568 – 1572. doi: 10.1016/ j.jocn.2012.03.012.
100. Folkins C, Man S, Xu P et al. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem‑like cell fraction in glioma xenograft tumors. Cancer Res 2007; 67(8): 3560 – 3564.
101. Lee ES, Gao Z, Kim D et al. Super pH ‑ sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J Control Release 2008; 129(3): 228 – 236. doi: 10.1016/ j.jconrel.2008.04.024.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Exprese a purifikace proteinů
- Metody studia buněčné migrace a invazivity nádorových buněk
- Sekvenování nové generace a možnosti jeho využití v onkologické praxi
- Analýza proteinů pomocí hmotnostní spektrometrie