
p‑ SRM, SWATH a HRM – cílené proteomické přístupy na hmotnostním spektrometru TripleTOF 5600+ a jejich aplikace v onkologickém výzkumu
p ‑ SRM, SWATH and HRM – Targeted Proteomics Approaches on TripleTOF 5600+ Mass Spectrometer and Their Applications in Oncology Research
Development of novel diagnostic and therapeutic approaches in cancer research requires sensitive and quantitative assays for determination of cancer‑associated proteins in clinical samples. Novel quantitative targeted proteomic approaches are overviewed in this communication. A major advantage of selected reaction monitoring (SRM) and pseudo - SRM lies in the selective and sensitive quantification of selected proteins in large sample sets. As such, they represent an alternative to immunochemical approaches. On the other hand, the potential of HRM and SWATH lies in recording of digital fingerprints, which enable post‑acquisition quantitative proteomic data mining on a similar basis to SRM. This article shows applications of targeted proteomics in a number of cancer research studies where they were used for quantification and validation of current or potential protein biomarkers and to study their role in cancer development and progression.
Key words:
proteomics – selected reaction monitoring – oncology – SWATH – biomarkers – molecular diagnostics
This work was supported by the project of Czech Science Foundation No. 14-19250S, by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101) and by MH CZ – DRO (MMCI, 00209805) and BBMRI_CZ (LM2010004).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
3. 2. 2014
Accepted:
26. 3. 2014
Authors:
J. Faktor 1,2; E. Michalová 1; P. Bouchal 1,2
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
1; Ústav biochemie, Přírodovědecká fakulta MU, Brno
2
Published in:
Klin Onkol 2014; 27(Supplementum): 110-115
Overview
Vývoj diagnostických a terapeutických přístupů v onkologii vyžaduje, kromě jiného, citlivé kvantitativní přístupy pro stanovení proteinů souvisejících s nádorovými procesy v klinických vzorcích. V tomto článku jsou představeny nové kvantitativní metody cílené proteomiky. Hlavní potenciál metod monitorování vybraných reakcí (SRM) a pseudo ‑ SRM spočívá v kvantifikaci předem vybraných proteinů ve větších souborech vzorků s vysokou citlivostí a selektivitou, čímž představují alternativu ke stávajícím imunochemickým přístupům. Potenciál HRM a SWATH spočívá naopak v získávání digitálních proteomických fingerprintů, z nichž je následně možné extrahovat kvantitativní proteomická data na podobném principu jako u SRM. Článek představuje aplikace uvedených metod v řadě studií z oblasti onkologického výzkumu, kde byly použity ke stanovení a validaci stávajících i nově navrhovaných proteinových biomarkerů a při studiu jejich úlohy v mechanizmu vzniku a vývoje nádorů.
Klíčová slova:
proteomika – monitorování vybraných reakcí – onkologie – SWATH – biomarkery – molekulární diagnostika
Úvod
Nové poznatky z oblasti molekulární biologie nádorů jdou ruku v ruce s vývojem nových citlivých metod, které umožňují nové analýzy na úrovni genomu, transkriptomu i proteomu a jako celek podávají komplexní obrázek o biologických procesech v buňce. Proteiny jsou nejen stavebními prvky živých organizmů, ale i základními biologickými katalyzátory, regulátory a prostředníky buněčné signalizace. Hladiny proteinů, jejich modifikace a funkce se tak odráží v základním stavu buňky, jejích fyziologických reakcích na okolní podmínky i patologických procesech včetně maligní transformace. Rozvoj proteomických metod a současně porovnávání výsledného stavu proteomu se základní informací obsaženou v genomu a regulací exprese na úrovni transkriptomu umožňují komplexně charakterizovat biologické procesy v buňce. Charakterizace proteinů a jejich kvantifikace je tedy jedním z předpokladů rozvoje nových diagnostických a terapeutických přístupů u nádorových onemocnění založených na nově získaných systémově biologických poznatcích [1,2].
Proteomika je vědní směr, který se zabývá identifikací a kvantifikací proteinů v různých biologických systémech, a představuje tak nástroj k pochopení složitých buněčných procesů. V rámci proteomiky lze rozlišit dva základní směry: 1. tzv. necílenou proteomiku, která se zaměřuje na identifikaci nových proteinových cílů v souvislosti s určitým biologickým stavem, např. onemocněním (srovnání vzorků pacientů s dobře definovanou diagnózou a zdravých jedinců); 2. tzv. cílenou proteomiku, která se zaměřuje na jeden konkrétní protein a jeho biologickou úlohu. Pro rozvoj cílené proteomiky hraje zásadní roli možnost citlivé, specifické a mezilaboratorně dobře reprodukovatelné kvantifikace studovaného proteinu. V této oblasti dosud dominují převážně imunochemické přístupy (western blotting spojený s imunodetekcí, ELISA), které jsou závislé na dostupnosti kvalitních a specifických protilátek. Principiálně zcela odlišná technika hmotnostní spektrometrie (mass spectrometry – MS) v poslední době nabízí alternativní přístupy cílené kvantifikace proteinů na bázi vybraných proteotypických peptidů, jejichž představení je předmětem tohoto článku. Vychází přitom ze zkušeností s technicky ekvivalentním přístupem pro MS kvantifikaci nízkomolekulárních látek, který je řadu let využíván v oblasti farmaceutické a forenzní analýzy [3,4]. Jelikož metodický vývoj v této oblasti dosáhl úrovně potřebné k běžnému uplatnění v biologickém výzkumu včetně onkologického, byla cílená proteomika vyhodnocena časopisem Nature Methods jako metoda roku 2012 [5].
Základní metodou cílené proteomiky je tzv. monitorování vybraných reakcí (selected reaction monitoring – SRM), o níž jsme podrobně referovali již dříve [6], dále viz [7,8]. Pomocí metody SRM lze kvantifikovat až desítky předem vybraných proteinů v rámci jedné analýzy a díky širokému lineárnímu dynamickému rozsahu je tak možno stanovit proteiny obsažené v množství 45 až 1,3 × 106 kopií na buňku v jedné analýze [9]. Je však nezbytné mít pro dané proteiny optimalizované metody, což znamená především vybrané kombinace proteotypických peptidů a jejich specifických fragmentů, které poskytují dobrý signál. SRM se běžně provádí na relativně jednoduchých hmotnostních spektrometrech typu trojitý kvadrupól (QQQ) nebo hybridních systémech typu trojitý kvadrupól‑lineární iontová past (QTRAP). Se zvyšující se citlivostí vysokorozlišovacích hmotnostních spektrometrů (Orbitrap, QExactive, qTOF) se v posledních letech rozšiřují možnosti cílené kvantifikace i na této složitější, ale univerzálnější instrumentaci. V tomto článku se proto budeme věnovat cílené kvantifikaci na hmotnostním spektrometru TripleTOF 5600+, který byl nedávno instalován na pracovišti RECAMO Masarykova onkologického ústavu a který umožňuje provádět také sběr digitálních fingerprintů pomocí metody HRM (hyper reaction monitoring) neboli SWATH (sequential windowed data independent acquisition of the total high resolution mass spectra). Z těchto fingerprintů lze následně, třeba několik let po provedené MS analýze, extrahovat kvantitativní data pro později zvolené proteiny našeho zájmu. To staví proteomiku do zcela nového světla jako nástroje pro získání zcela nových proteomických dat z fyzicky již nedostupných vzorků, což žádná z dosud dostupných metod neumožňuje. Oba přístupy mají velký potenciál při verifikaci a validaci potenciálních biomarkerů nádorových onemocnění a při kvantifikaci vybraných proteinů v rámci studií zaměřených na studium biologické úlohy proteinů in vitro a in vivo.
Příprava vzorků pro cílenou proteomiku a chromatografická separace
Analýza vzorků pomocí cílené proteomiky probíhá podobně jako běžná proteomická LC ‑ MS analýza s tím, že při přípravě vzorku je třeba zohlednit jeho charakter a typ použité kvantifikace. Proteiny se extrahují ze zmražených či archivovaných tkání, případně z biologických tekutin. Vzorky analyzované v rámci onkologického výzkumu jsou obecně velmi komplexní. U vzorků typu sérum nebo plazma se proto někdy provádí imunochemické odstranění proteinů o velmi vysoké koncentraci. Pro proteiny o nízké koncentraci lze provést naopak obohacení pomocí protilátky vůči cílovému proteinu [8], která zvyšuje citlivost přístupu, přičemž specifita zůstává zajištěna MS identifikací; tento přístup se pak označuje jako „immuno ‑ SRM“ [10]. Důležitou otázkou je přídavek izotopově značených peptidů zajišťujících přesnou kvantifikaci. Pro SRM se přidávají izotopově značené proteotypické peptidy o známé koncentraci, které sekvencí odpovídají peptidům z kvantifikovaných proteinů [11]. Pro HRM a SWATH jsou naopak obvykle přidávány směsi ověřených peptidů, jež slouží ke kalibraci retenčních časů u následné chromatografické separace a zvyšují tím spolehlivost celého přístupu. Získaný proteinový extrakt je redukován, alkylován a štěpen trypsinem, který specificky štěpí proteiny na C ‑ straně lysinu a argininu, pokud nenásleduje prolin. Peptidové směsi jsou separovány na principu reverzně fázové chromatografie on‑line propojené do hmotnostního spektrometru. Lze však použít i dvojrozměrnou (2D) chromatografickou separaci [12], která snižuje komplexitu vzorku, interference a limity detekce, prodlužuje ale dobu experimentu. Chromatografií separované peptidy vstupují do (nano)elektrospreje, kde vlivem vysokého napětí dochází k jejich ionizaci, nabité ionty poté vstupují do hmotnostního spektrometru.
Princip SRM a p ‑ SRM
Základní princip SRM měřeného v hmotnostním spektrometru typu trojitý kvadrupól spočívá ve výběru peptidového iontu (prekurzorového iontu) v prvním hmotnostním analyzátoru (kvadrupól Q1).Peptid je následně fragmentován v kvadrupólu Q2, specifické peptidové fragmenty (produktové ionty) jsou pak vyfiltrovány díky nastavení kvadrupólu Q3 (obr. 1A, B), kterým tyto prochází na detektor [13]. Při SRM se tedy kvantifikují určité vybrané peptidy, typické pro daný protein. Z kvantitativních dat těchto peptidů se poté vypočítá kvantifikace celého proteinu. Modifikovaný přístup cílené kvantifikace u vysokorozlišovacího hmotnostního spektrometru typu TripleTOF se označuje jako pseudo ‑ SRM (p ‑ SRM). Modifikace spočívá v tom, že namísto měření jednotlivých předem vybraných fragmentů peptidů je měřeno celé peptidové spektrum pomocí analyzátoru „time ‑ of ‑ flight“ (TOF). Výhodou tohoto přístupu je jednodušší vývoj metod pro kvantifikaci konkrétních peptidů a lepší kontrola selektivity, nevýhodou je horší citlivost ve srovnání s přístroji typu QQQ nebo QTRAP obdobné generace.
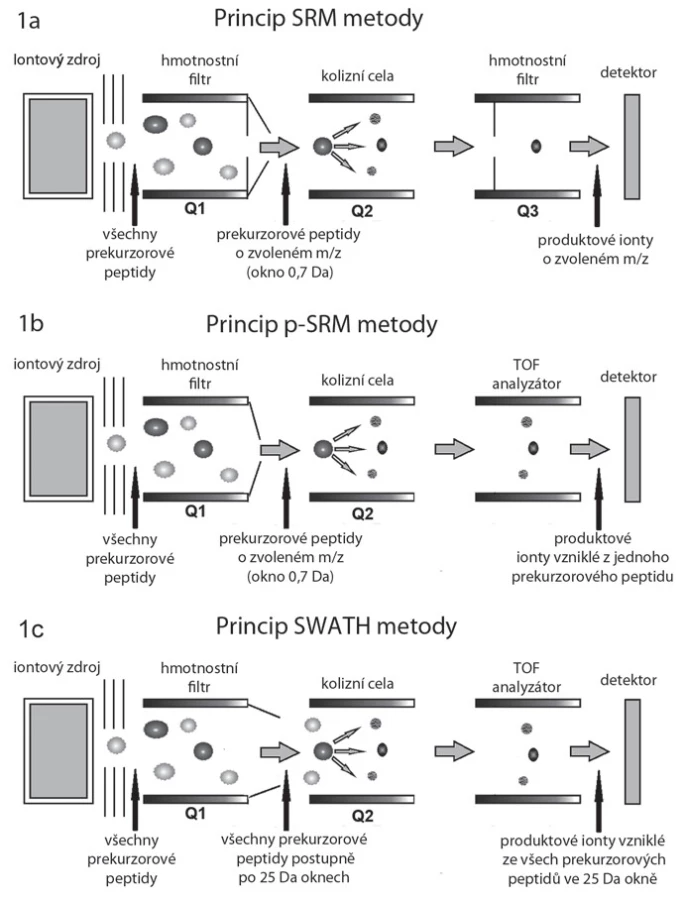
Princip HRM/ SWATH
Metoda HRM neboli SWATH je modifikací metody p ‑ SRM. Zatímco při měření metodou p ‑ SRM jsou pro získávání spekter vybírány pouze určité peptidy typické pro proteiny našeho zájmu, metoda SWATH je navržena k získání digitálního fingerprintu, z něhož lze později vyčíst kvantitativní data až pro několik tisíc peptidů. Pro sběr SWATH digitálních fingerprintů se škála m/ z rozdělí na okna o šířce typicky 25 Da. Kvadrupól Q1 funguje jako hmotnostní filtr, který postupně propouští ionty o určitém rozsahu m/ z do kolizní cely (obr. 1C). Nejedná se zde o jednotlivé ionty, jak je tomu v případě SRM a p ‑ SRM (šířka okna 0,7 Da, obr. 1A, B), ale oblasti m/ z o šířce 25 Da. Takto je v průběhu LC ‑ MS analýzy v krátkých cca 3 s trvajících cyklech opakovaně analyzována vždy celá oblast m/ z spektra, což umožňuje získávat kvantitativní data pro všechny detekovatelné peptidy ve vzorku.
Aplikace cílené proteomiky v onkologickém výzkumu
SRM je jednou z nejcitlivějších kvantitativních metod hmotnostní spektrometrie. V současnosti získává nenahraditelné postavení v oblasti validace proteinů, které by mohly hrát úlohu biomarkerů biologických stavů. Například Fortin et al [14] použili SRM v kombinaci s imunodeplecí vzorku pro stanovení klinicky rutinně užívaného markeru, prostatového specifického antigenu (PSA), v sérech pacientů s benigní hyperplazií prostaty a pacientů s nádory prostaty. Výsledky ukázaly dobrou korelaci výsledků uvedeného přístupu cílené proteomiky a běžně užívaného ELISA testu [14]. Hembrough et al [15] s pomocí SRM přístupu kvantifikovali epidermal growth factor receptor (EGFR) v buňkách linie odvozené z nádoru plic fixované v parafinu a ukázali dobrou shodu mezi daty získanými EGFR ‑ SRM a ELISA daty ze vzorků nefixované kultury. Dále demonstrovali možnost SRM stanovení EGFR ve vzorcích mikrodisekovaných z formalinem fixované tkáně xenograftů a tkání nemalobuněčného karcinomu plic. Bylo prokázáno, že získaná data jsou velmi specifická pro EGFR a jednoznačně odlišují kvantifikaci EGFR od kvantifikace dalších příbuzných proteinů (např. IGF‑1R, cMet, Her2, Her3 a Her4) [15]. Metoda SRM má dále potenciál ve validaci nových potenciálních biomarkerů. Hladiny tří z pěti SRM validovaných proteinů (14 - 3 - 3σ, gelsolin, lumican, transglutaminasa 2 a tissue inhibitor of metalloproteinase 1) vykázaly schopnost odlišení nádorů pankreatu od kontrol v daném souboru pacientů, přičemž nejlepších výsledků bylo dosaženo pro gelsolin [16]. Podobně byla testována včasná detekce hepatocelulárního karcinomu u pacientů po infekci hepatitidou C.V souboru 50 pacientů byly skupiny pacientů s nádorem a bez nádoru jednoznačně rozlišeny pomocí hladin apolipoproteinu A1 stanovených pomocí SRM, přičemž data byla potvrzena dalšími nezávislými metodami [17]. Pro vývoj stanovení nových proteinů pomocí SRM jsou dále vyvíjeny nové rychlejší a efektivnější přístupy. Jeden z nich [18] umožnil vývoj metod pro kvantifikaci jak klinicky rutinně užívaného markeru nádorů vaječníku, CA125, tak i dalších zvažovaných biomarkerů (beta‑2 - mikroglubulin, apolipoprotein A1, transthyretin a transferrin). Vyvinuté metody jsou k dispozici ve formě veřejně přístupné SRM knihovny [18].
V signalizaci u nádorových procesů mají velký význam posttranslační modifikace proteinů, jako je fosforylace, ale také acetylace či glykosylace. Wolf-Yadlin et al [19] demonstrovali velmi efektivní SRM přístup pro analýzu fosforylací na 222 fosforylačních místech proteinů po stimulaci buněk EGF. Bylo přitom identifikováno a sledováno 31 fosforylačních míst, která dosud s EGF stimulací nebyla spojována. Publikovaný přístup umožňuje rutinní analýzu stovek fosforylačních míst za různých volitelných biologických podmínek [19]. Griffiths et al [20] dále zjistili, že metoda SRM je 10násobně citlivější v porovnání s ostatními metodami pro sledování acetylace, a tímto způsobem byli schopni identifikovat pět nových acetylačních míst u cytokeratinu 8 [20]. Hülsmeier et al [21] studovali pomocí SRM vrozenou poruchu glykosylace. Cíleně se zaměřili na glykosylace u α - 1 - antitrypsinu a transferrinu a pomocí SRM zjistili vztah mezi sníženou glykosylací a závažností CDG syndromu [21]. Glykosylace mají rovněž význam z toho důvodu, že řada sekretovaných proteinů použitelných jako neinvazivní biomarkery je glykosylovaných.
Aplikace metody HRM/ SWATH v onkologickém výzkumu
Metoda SWATH umožňuje i zpětnou kvantifikaci všech detekovatelných proteinů ve vzorku [22] z naměřených digitálních fingerprintů. Jelikož se jedná o metodu velmi novou, počet publikovaných prací je zatím omezený a ty se zaměřují převážně na strukturní a funkční studie proteinů na molekulární úrovni. Doposud byla metoda SWATH úspěšně využita ke studiu Her ‑ 2/ neu receptoru (ErbB2) a jeho fosforylované a acetylované formy [23]. Potenciál metody SWATH byl též demonstrován na vzorcích lidské plazmy získaných od zdravých jedinců, v nichž bylo cíleně sledováno 41 N ‑ glykopeptidů pomocí metod SWATH a SRM. V obou posledně jmenovaných studiích bylo zjištěno, že SWATH má sice nižší citlivost v porovnání se SRM, vývoj kvantifikačních metod však trvá kratší dobu a metoda umožňuje kvantifikovat podstatně více proteinů v jednom běhu. Reprodukovatelnost SRM a SWATH byla podobná [24]. SWATH může najít uplatnění také ve funkčním studiu pronádorových procesů, konkrétně při charakterizaci protein‑proteinových interakcí. V této oblasti byl studován vliv stimulace PI ‑ 3K/ AKT signální dráhy na interaktom proteinu 14 - 3 - 3β. Vzorky byly připraveny pomocí „pull ‑ down” afinitní purifikace, která byla prováděna v různých časech po přidání IGF1 (insulin‑like growth factor 1 – stimulátor PI ‑ 3K/ AKT dráhy). Výsledky dynamicky popisují interaktom proteinu 14 - 3 - 3β při aktivaci zmíněné dráhy, přičemž bylo identifikováno několik skupin interagujících proteinů, jež vykazovaly zcela odlišné chování v odpovědi na stimulaci [25]. V jiné studii se autoři zaměřili na charakterizaci změn protein‑proteinových interakcí souvisejících s mutacemi cyklin dependentní kinázy 4 (CDK4), které se vyskytují u melanomů. Metoda AP ‑ SWATH odhalila 17 proteinů, jež ve zvýšené míře interagovaly s mutantními formami CDK4 v porovnání s wild type formou proteinu. Interakce mutantních forem CDK4 s Hsp90 proteiny (Hsp90α, Hsp90β) byly zvýšeny trojnásobně v porovnání s wild type. Mutantní formy CDK4 naopak téměř neinteragovaly s INK proteiny (p15INK, p16INK, p18INK a p19INK – polypeptidové inhibitory CDK4) v porovnání s wild type [26]. Z uvedených výsledků lze říci, že metoda SWATH je robustním přístupem pro charakterizaci změn v posttranslačních modifikacích proteinů a jejich protein‑proteinových interakcích, který nevyžaduje použití značení. V současné době prováděné studie se rovněž zaměřují na koncept digitalizovaných biobank, který by z jedenkrát naměřených dat metodou SWATH umožnil v budoucnu kdykoliv extrahovat jakékoliv dostupné kvantitativní proteinové informace.
![Princip metody SWATH/HRM. Metoda je založená na kvantifikaci produktových iontů vzniklých fragmentací všech prekurzorových iontů (peptidů) v tzv. SWATH oknech. SWATH okna jsou úseky škály <em>m/z</em> nejčastěji o šířce 25 Da (např. při zvoleném hmotnostním rozsahu <em>m/z</em> = 400–1 100 Da se postupně měří 28 SWATH oken v rámci jednoho cca 3 s trvajícího cyklu). Výsledkem měření každého SWATH okna je MS/MS spektrum na pravé straně obrázku. Chromatogram dole znázorňuje koeluci produktových iontů různých peptidů znázorněných různými barvami. Data z chromatogramu se poté využijí ke kvantifikaci peptidů a proteinů ve vzorku. Zdroj: ABSCIEX, se svolením, upraveno dle [27].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0c98f9cf690daa971f75a1bc9192f6ed.jpg)
Závěr
Experimentálně náročné proteomické techniky vykazují v posledních letech metodický rozvoj, který stále více umožňuje splnit představy lékařů, biologů a dalších výzkumných pracovníků. To odráží zejména stále se zvyšující podíl proteomu, jenž může být rutinně sledován. Nové metody cílené kvantifikace přispívají výrazným zlepšením k reprodukovatelnosti, kvantifikovatelnosti a ke zvýšení počtu vzorků, které mohou být zařazeny do proteomických studií. Zde prezentované práce ilustrují úspěšné aplikace cílené proteomiky v oblasti onkologického výzkumu, jež v mnoha případech směřují k reálnému uplatnění v klinické praxi. Laboratoř hmotnostní spektrometrie Masarykova onkologického ústavu s hmotnostními spektrometry TripleTOF 5600+ a Orbitrap ELITE je připravena, ve spolupráci se specialisty napříč obory, k tomuto úsilí významně přispět.
Práce byla podpořena projektem Grantové agentury České republiky č. 14-19250S, Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO, CZ.1.05/2.1.00/03.0101) a MZ ČR – RVO (MOÚ, 00209805) a BBMRI_CZ (LM2010004).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
Mgr. Pavel Bouchal, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: bouchal@mou.cz
Obdrženo: 3. 2. 2014
Přijato: 26. 3. 2014
Sources
1. Faktor J, Dvořáková M, Maryáš J et al. Identification and characterisation of pro‑metastatic targets, pathways and molecular complexes using a toolbox of proteomic technologies. Klin onkol 2012; 25 (Suppl 2): 2S70 – 2S77.
2. Maryáš J, Faktor J, Dvořáková M et al. Proteomics in investigation of cancer metastasis: functional and clinical consequences and methodological challenges. Proteomics 2014; 14(4 – 5): 426 – 440. doi: 10.1002/ pmic.201300264.
3. Humplíková S, Minář J, Kučerová M et al. Stanovení hladiny celkového homocysteinu v plazmě kapalinovou chromatografií s tandemovou hmotnostní spektrometrií. Klin Biochem Metab 2007; 15(36): 31 – 34.
4. Kim K, Kim Y. Preparing multiple reaction monitoring for quantitative clinical proteomics. Expert Rev Proteomics 2009; 6(3): 225 – 229. doi: 10.1586/ epr.09.11.
5. Marx V. Targeted proteomics. Nat Methods 2013; 10(1): 19 – 22.
6. Faktor J, Struhárová I, Fučíková A et al. Kvantifikace proteinových biomarkerů pomocí hmotnostní spektrometrie pracující v režimu monitorování vybraných reakcí. Chemické listy 2011; 105(11): 846 – 850.
7. Kitteringham NR, Jenkins RE, Lane CS et al. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877(13): 1229 – 1239. doi: 10.1016/ j.jchromb.2008.11.013.
8. Gallien S, Duriez E, Domon B. Selected reaction monitoring applied to proteomics. J Mass Spectrom 2011; 46(3): 298 – 312. doi: 10.1002/ jms.1895.
9. Picotti P, Bodenmiller B, Mueller LN et al. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009; 138(4): 795 – 806. doi: 10.1016/ j.cell.2009.05.051.
10. Anderson NL, Anderson NG, Haines LR et al. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti‑peptide antibodies (SISCAPA). J Proteome Res 2004; 3(2): 235 – 244.
11. Lange V, Picotti P, Domon B et al. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol 2008; 4 : 222. doi: 10.1038/ msb.2008.61.
12. Whiteaker JR, Lin C, Kennedy J et al. A targeted proteomics‑based pipeline for verification of biomarkers in plasma. Nat Biotechnol 2011; 29(7): 625 – 634. doi: 10.1038/ nbt.1900.
13. Schiess R, Wollscheid B, Aebersold R. Targeted proteomic strategy for clinical biomarker discovery. Mol Oncol 2009; 3(1): 33 – 44. doi: 10.1016/ j.molonc.2008.12.001.
14. Fortin T, Salvador A, Charrier JP et al. Clinical quantitation of prostate ‑ specific antigen biomarker in the low nanogram/ milliliter range by conventional bore liquid chromatography ‑ tandem mass spectrometry (multiple reaction monitoring) coupling and correlation with ELISA tests. Mol Cell Proteomics 2009; 8(5): 1006 – 1015. doi: 10.1074/ mcp.M800238 - MCP200.
15. Hembrough T, Thyparambil S, Liao WL et al. Selected reaction monitoring (SRM) analysis of epidermal growth factor receptor (EGFR) in formalin fixed tumor tissue. Clin Proteomics 2012; 9(1): 5. doi: 10.1186/ 1559 - 0275 - 9 - 5.
16. Pan S, Chen R, Brand RE et al. Multiplex targeted proteomic assay for biomarker detection in plasma: a pancreatic cancer biomarker case study. J Proteome Res 2012; 11(3): 1937 – 1948. doi: 10.1021/ pr201117w.
17. Mustafa MG, Petersen JR, Ju H et al. Biomarker discovery for early detection of hepatocellular carcinoma in hepatitis C ‑ infected patients. Mol Cell Proteomics 2013; 12(12): 3640 – 3652. doi: 10.1074/ mcp.M113.031252.
18. Hüttenhain R, Soste M, Selevsek N et al. Reproducible quantification of cancer‑associated proteins in body fluids using targeted proteomics. Sci Transl Med 2012; 4(142): 142ra94. doi: 10.1126/ scitranslmed.3003989.
19. Wolf ‑ Yadlin A, Hautaniemi S, Lauffenburger DA et al. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci USA 2007; 104(14): 5860 – 5865.
20. Griffiths JR, Unwin RD, Evans CA et al. The application of a hypothesis‑driven strategy to the sensitive detection and location of acetylated lysine residues. J Am Soc Mass Spectrom 2007; 18(8): 1423 – 1428.
21. Hülsmeier AJ, Paesold ‑ Burda P, Hennet T. N ‑ glycosylation site occupancy in serum glycoproteins using multiple reaction monitoring liquid chromatography‑mass spectrometry. Mol Cell Proteomics 2007; 6(12): 2132 – 2138.
22. Gillet LC, Navarro P, Tate S et al. Targeted data extraction of the MS/ MS spectra generated by data ‑ independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012; 11(6): O111.016717. doi: 10.1074/ mcp.O111.016717.
23. Held JM, Schilling B, D‘Souza AK et al. Label‑free quantitation and mapping of the ErbB2 tumor receptor by multiple protease digestion with data ‑ dependent (MS1) and data ‑ independent (MS2) acquisitions. Int J Proteomics 2013; 2013 : 791985. doi: 10.1155/ 2013/ 791985.
24. Liu Y, Hüttenhain R, Surinova S et al. Quantitative measurements of N‑linked glycoproteins in human plasma by SWATH ‑ MS. Proteomics 2013; 13(8): 1247 – 1256. doi: 10.1002/ pmic.201200417.
25. Collins BC, Gillet LC, Rosenberger G et al. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3 - 3 system. Nat Methods 2013; 10(12): 1246 – 1253. doi: 10.1038/ nmeth.2703.
26. Lambert JP, Ivosev G, Couzens AL et al. Mapping differential interactomes by affinity purification coupled with data ‑ independent mass spectrometry acquisition. Nat Methods 2013; 10(12): 1239 – 1245. doi: 10.1038/ nmeth.2702.
27. Baumann C (ed.). MS/ MSALL with SWATH™ acquisition global quantitative strategies for proteomics & beyond. AB SCIEX Germany; c2012 [cited 2014 February]. Available from: http:/ / proteomics ‑ seminar.imp.ac.at/ fileadmin/ imp/ Images/ Proteomics_seminar/ 2012/ Christian_Baumann_MSMSall_with_SWATH_Acquisition.pdf.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Exprese a purifikace proteinů
- Metody studia buněčné migrace a invazivity nádorových buněk
- Sekvenování nové generace a možnosti jeho využití v onkologické praxi
- Analýza proteinů pomocí hmotnostní spektrometrie