
Programovaná buněčná smrt v nádorových buňkách
Programmed Cell Death in Cancer Cells
Resistance to programmed cell death is one of the hallmarks of cancer cells that affects the process of malignant transformation as well as response to cancer therapy. The goal of this review is to summarize recent information about programmed cell death (PCD) in healthy and cancer cells, as well as new perspectives for anticancer treatments targeting these signaling pathways. Three main types of PCD are described in detail: apoptosis, necrosis/ necroptosis and cell death associated with autophagy. Among them, apoptosis plays the key role in both malignant transformation and response to therapy. In this review, we describe main signaling pathways and molecules participating in apoptosis regulation in healthy cells. In most cancer cells, mutations or aberrant expression of proteins directly or indirectly involved in induction and execution of cell death can be detected – p53, Bcl ‑ 2 family proteins, inhibitors of apoptosis, death receptors/ ligands and other proteins. Mutations or changes in expression of these proteins and their relation to certain types of tumors are described. Finally, we provide a review of recently developed treatments that target and reactivate the machinery of programmed cell death and are currently tested in clinical trials.
Key words:
programmed cell death – apoptosis – necroptosis – autophagy – caspases – Bcl ‑ 2
This work was supported by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101) and by MH CZ – DRO (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
14. 1. 2014
Accepted:
6. 3. 2014
Authors:
E. Ondroušková; B. Vojtěšek
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2014; 27(Supplementum): 7-14
Overview
Rezistence k indukci smrti je jedním z charakteristických znaků nádorové buňky, jež ovlivňuje od počátku jak samotný proces neoplastické transformace, tak i pozdější odpověď na onkologickou léčbu. Cílem tohoto přehledového článku je shrnout recentní informace o programované buněčné smrti zdravých i nádorových buněk a o nových možnostech protinádorové terapie zacílené na tyto signální dráhy. Podrobněji jsou popsány tři hlavní typy: apoptóza, programovaná nekróza a buněčná smrt spojená s autofagií. Především apoptóza hraje významnou roli nejen při neoplastické transformaci buňky, ale i jako jeden z faktorů určujících úspěšnost protinádorové terapie. V textu je podán přehled hlavních signálních drah a molekul podílejících se na regulaci apoptózy ve zdravých buňkách. Většina nádorových buněk nese mutace v proteinech přímo či nepřímo se účastnících indukce a exekuce buněčné smrti, jako jsou proteiny p53, členové rodiny Bcl ‑ 2, proteiny inhibující apoptózu (IAPs), receptory/ ligandy smrti a další. U těchto významných regulátorů jsou popsány jejich nejčastější mutace či změny exprese vyskytující se v buňkách konkrétních typů nádorů. Na závěr je podán přehled některých nových léčiv zaměřených na modulaci programované buněčné smrti, jež právě procházejí klinickými zkouškami. Díky intenzivnímu výzkumu je možné stále detailnější porozumění procesům, které v umírající buňce probíhají, a na jejich základě pak lze navrhovat léčiva nové generace, jež v kombinaci s tradiční terapií umožní významné zlepšení odpovědi na protinádorovou terapii.
Klíčová slova:
programovaná buněčná smrt – apoptóza – nekroptóza – autofagie – kaspázy – Bcl ‑ 2
Úvod
Proces postupné přeměny normální buňky v nádorovou ústí ve fatální změnu jejích vlastností souvisejících především s regulací růstu, schopností přežít a šířit se v organizmu. Tyto základní vlastnosti maligních buněk jsou popsány v respektovaném článku Hanahana a Weinberga [1]. Mezi nejdůležitější charakteristické znaky nádorové buňky tito autoři zařadili i její schopnost uniknout indukci programované buněčné smrti. Nádorové buňky produkují řadu signálů, které za normálních okolností aktivují signální dráhy vedoucí k eliminaci těchto buněk – obsahují poškozenou DNA, aktivované onkogeny, jsou vystaveny oxidativnímu stresu atd. Ty z nich, které získají mutace umožňující zablokovat proces jejich odstranění programovanou buněčnou smrtí, vytváří rezistentní klony a v organizmu dále přežívají a neregulovaně se množí.
Převládající paradigma nádorové terapie předpokládá, že buňky, které jsou citlivé k indukci apoptózy, budou na léčbu odpovídat lépe než ty, jež jsou k indukci apoptózy a tím i k léčbě rezistentní. Základní výzkum, jenž odhaluje signální dráhy a mechanizmy klasické apoptózy i alternativních typů buněčné smrti, poskytuje znalosti, na základě kterých lze vyvíjet nová léčiva a kombinovat je s tradiční protinádorovou terapií. Tato nová léčiva jsou většinou zaměřena na reaktivaci apoptotické dráhy a zvyšují tak úspěšnost eliminace nádorových buněk, jež by jinak byly k indukci smrti více rezistentní.
Typy programované buněčné smrti
Podle toho, jaké morfologické a jiné znaky umírající buňka vykazuje, lze programovanou buněčnou smrt (programmed cell death – PCD) klasifikovat do některého ze základních typů [2]. Morfologie buňky může být apoptotická, nekrotická, autofagická či spojená s mitózou. Historicky nejstarší typ identifikované PCD je apoptóza, popsaná již před více než 40 lety [3]. Nekroptóza a buněčná smrt asociovaná s autofagií jsou další dva typy PCD popsané u nejrůznějších typů buněk. Typická morfologie normální, apoptotické, autofagické a nekrotické buňky, nasnímaná na elektronovém mikroskopu, je na obr. 1. Pro úplnost: čtvrtý oficiálně uznávaný, ač vysoce specifický typ programované buněčné smrti je tzv. kornifikace neboli tvorba zrohovatělé vrstvy kůže z mrtvých keratinocytů v pevném proteinovém obalu [4]. Další typy buněčné smrti, které jsou v literatuře často uváděny (anoikis, excitotoxicita, Wallerova degradace, mitotická katastrofa, paraptóza, pyroptóza, entóza a pyronekróza), se zatím nedoporučuje klasifikovat jako samostatné druhy programované buněčné smrti [2].
![Buňka s normální, apoptotickou, autofagickou a nekrotickou morfologií.
Nasnímáno elektronovým mikroskopem, zvětšení 4 400×, upraveno dle [91]. U zdravé buňky pozorujeme obvyklý tvar jádra i celé buňky (A), u apoptotické chromatin kondenzovaný v jádře a zmenšení objemu buňky (B), u autofagické výrazně vakuolizovanou cytoplazmu a jádro bez kondenzovaného chromatinu (C) a u nekrotické ztrátu integrity membrány a buněčného obsahu (D).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/8d7ca9c4610cd7e4d29966a9b77d8f16.jpg)
Kromě morfologických znaků jsou dalším kritériem funkční aspekty, kterými rozlišujeme buněčnou smrt fyziologickou, patologickou, náhodnou či programovanou. Enzymologicky pak lze určit, do jaké míry jsou aktivovány nukleázy a proteázy různých tříd. Aktivace různých typů buněčné smrti se v jednotlivých buňkách vzájemně nevylučuje, naopak mohou spolupracovat či sdílet některé signální molekuly [5]. Jsou‑li např. makrofágy ve tkáních vystaveny různým stresorům, je většinou současně indukováno více typů buněčné smrti, přičemž apoptóza probíhá nejrychleji, zatímco autofagie nebo nekróza jsou obvykle pozorovatelné až v případě, kdy je apoptóza inhibována [6].
Apoptóza
Termín „apoptosis“ jako první použil J. F. Kerr při objevu a popsání regulované buněčné smrti [3]. Buňka umírající apoptózou vykazuje charakteristické morfologické změny, které ji odlišují od nekrózy – kondenzace a fragmentace chromatinu v jádře, tvorba výběžků cytoplazmatické membrány (membránové puchýřky), celkové zmenšení a zakulacení buňky a v pozdních fázích rozpad celé buňky do tzv. apoptotických tělísek [2]. Tato tělíska obsahují stále funkční organely, fragmenty kondenzovaného jádra a udržují si intaktní membránu, která se od membrány zdravých buněk liší přítomností fosfatidylserinu na její vnější straně [7]. To umožňuje rozpoznání a následné pohlcení apoptotických tělísek sousedními buňkami či buňkami imunitního systému [8]. Z biochemického hlediska jsou v umírající buňce typicky aktivovány proteolytické enzymy – kaspázy, které štěpí řadu cílových proteinů, a nukleázy, degradující DNA.
Aby buňka mohla zahájit proces své vlastní eliminace, musí obdržet odpovídající signály. Na obr. 2 jsou zjednodušeně znázorněny dvě hlavní signální kaskády vedoucí k apoptóze v savčích buňkách. Vnější dráha je aktivována vazbou ligandů smrti (TRAIL, FasL, TNF) na receptory smrti (DR4/ DR5, Fas, TNFR a další), které tvoří trimery, a přes další adaptorové proteiny aktivují prokaspázu ‑ 8, případně prokaspázu ‑ 10 [9,10]. Vnitřní dráha je odpovědná za iniciaci apoptózy v případě neopravitelného poškození DNA. Je to tedy dráha aktivovaná obvykle při onkologické terapii, a proto bude popsána detailněji. Do její regulace se zapojuje celá řada pro ‑ i anti‑apoptotických proteinů. Poškození DNA vede ke stabilizaci a aktivaci proteinu p53, který apoptotický signál dále přenáší především prostřednictvím regulace proteinů rodiny Bcl ‑ 2 [11]. Role proteinu p53 v regulaci apoptotické dráhy je komplexnější a pro podrobnější studium odkazujeme na přehledové články jiných autorů [12,13].
![Vnější a vnitřní signální dráha při apoptotické signalizaci.
Podrobnosti viz text. Upraveno dle [92,93].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ca84a2f2068d5cfd282242e2ae52a9fd.jpg)
Zásadní událostí v klasické apoptotické signalizaci je částečná permeabilizace mitochondriální membrány, jež vede k uvolnění pro‑apoptotických molekul z mezimembránového mitochondriálního prostoru do cytozolu. Do procesu uvolňování těchto proteinů z mitochondrií významně zasahují další proteiny přítomné v membráně mitochondrií nebo v cytoplazmě, především ty z rodiny Bcl ‑ 2 [14,15]. Tato rodina, do které bylo doposud zařazeno 25 proteinů, je charakterizována přítomností BH ‑ domén (Bcl ‑ 2 homology) v jejich struktuře. Podle jejich funkce se rozlišují na anti‑apoptotické (Bcl ‑ 2, Bcl ‑ XL, Mcl ‑ 1, Bcl ‑ W, A1), pro‑apoptotické efektorové (Bak, Bax), pro‑apoptotické přímé aktivátory (Bid, Bim) či nepřímé aktivátory (Bad, Bik, BMF, HRK, Puma, Noxa) [16]. Aktivátory Bid a Bim po přijetí apoptotického signálu indukují oligomerizaci Bak a Bax, což vede k jejich translokaci z cytoplazmy do membrány mitochondrií a k vytvoření pórů ve vnější mitochondriální membráně. Pro‑apoptotické a anti‑apoptotické proteiny rodiny Bcl ‑ 2 mohou tvořit dimery pomocí BH ‑ 3 domény, a tím se navzájem inaktivovat [17,18]. Vzájemný poměr a interakce pro ‑ a anti‑apoptotických členů rodiny Bcl ‑ 2 tedy rozhoduje o tom, zda bude apoptotická dráha inhibována či posilována. Cytochrom c, uvolněný z mezimembránového prostoru mitochondrií, se v cytoplazmě spojuje s proteinem Apaf ‑ 1 a prokaspázou ‑ 9 a v tomto komplexu zvaném apoptozom je prokaspáza ‑ 9 štěpena na aktivní kaspázu ‑ 9 [19,20]. Z mitochondrií jsou dále uvolňovány proteiny SMAC/ DIABLO, ARTS či proteáza Omi/ HtrA2, které vážou a inaktivují anti‑apoptotické proteiny IAPs přítomné v cytoplazmě (inhibitor of apoptosis; NAIP, c ‑ IAP1/ HIAP ‑ 2, c ‑ IAP2/ HIAP ‑ 1, XIAP/ hILP, Survivin and BRUCE ) [21 – 23]. Proteiny AIF (apoptosis ‑ inducing factor) a endonukleáza G, rovněž uvolňované z mitochondrií, mohou realizovat program buněčné smrti i v nepřítomnosti kaspáz [24]. Další organela, která může zasahovat do regulace apoptotické signální dráhy, je endoplazmatické retikulum, jež jako odpověď na stres aktivuje prokaspázu ‑ 12 [25]. Rovněž lyzozomy se mohou účastnit regulace PCD, protože některé stimuly buněčné smrti vedou k částečné permeabilizaci lyzozomální membrány, uvolnění proteolytických enzymů do cytozolu a jejich aktivnímu přispění k aktivaci kaspáz [26,27].
Aktivované signální kaspázy ‑ 8, - 9, - 10 a - 12 dále aktivují tzv. efektorové kaspázy zajišťující inaktivační či aktivační štěpení řady proteinů, jako jsou regulátory apoptózy, proteiny související s buněčnou adhezí, cytoskeletem, strukturou jádra, buněčným cyklem, opravou a syntézou DNA atd. [28], což nakonec vyústí v úspěšnou eliminaci buňky poškozené nebo z jiných důvodů určené k likvidaci.
Buněčná smrt spojená s autofagií
Autofagie je vysoce konzervativní mechanizmus pro degradaci větších objemů buněčného materiálu, včetně celých organel. Poprvé byla popsána Christianem de Duve, který zavedl pojem „autophagy“ jako výstižný popis schopnosti buňky strávit a znovu využít své vlastní části (z řečtiny „autos“ = sebe, „phagein“ = pojídat) [29]. Rozlišujeme makroautofagii, mikroautofagii a autofagii zprostředkovanou chaperony [30]. Při makroautofagii (dále jen „autofagie“) jsou organely, dlouho ‑ žijící proteiny nebo agregáty uzavírány do dvoumembránových útvarů zvaných autofagozomy, které následně fúzují s lyzozomy, a jejich obsah je v nich lyzozomálními proteázami degradován [31]. Katabolické produkty jsou pak buňkou znovu recyklovány v biosyntetických procesech, nebo dále degradovány a využity jako zdroj energie v případě, že buňka hladoví. Celý proces je regulován skupinou genů označovaných ATG (autophagy‑related genes) a je detailně popsán např. v přehledovém článku Rosenfeldta a Ryana [32].
Autofagie je především mechanizmus, který buňce umožňuje přežití stresových podmínek, jakými jsou nedostatek živin nebo indukce nekrotické smrti v rakovinných buňkách či při ischemicko‑reperfúzním poškození tkání [33]. Její role v nádorových buňkách je nejasná a zřejmě závisí na stadiu nádorového onemocnění. Zatímco v raných fázích autofagie spíše suprimuje vývoj nádoru, protože odstraňuje poškozené organely a proteiny s nesprávnou, potenciálně škodlivou konformací [34], v pozdějších stadiích naopak zřejmě pomáhá rakovinným buňkám vyrovnat se se stresovými podmínkami, jako je nedostatek živin či oxidativní stres [35]. Tzv. autofagická buněčná smrt je charakterizována výrazným nárůstem počtu autofagických vakuol v buňce, bez kondenzace jaderného chromatinu, ukončená její smrtí [36]. Ačkoliv je akumulace autofagických vakuol v cytoplazmě často pozorována u buněk umírajících po aplikaci chemoterapie nebo radioterapie, není zcela prokázáno, zda je autofagie v těchto případech příčinou smrti, anebo jen doprovodným jevem [2]. Přímý důkaz, že autofagie může skutečně vést ke smrti konkrétních buněk in vivo, byl zatím podán pouze při vývoji slinných žláz Drosophily, ve kterých zablokování genů nezbytných pro autofagii vedlo k inhibici vývojové degradace těchto buněk [37]. I pokud by ale autofagie nebyla přímý vykonavatel buněčné smrti, vzhledem k časté detekci autofagických vakuol v umírajících buňkách zřejmě hraje v tomto procesu důležitou roli, a proto je i nadále nadějným cílem pro vývoj nových léčiv.
Nekroptóza
Za pasivní formu buněčné smrti byla vždy považována nekróza. Ta je charakterizována rychlou a neregulovanou ztrátou integrity plazmatické membrány a buněčným kolapsem, ačkoliv integrita jádra zůstává poměrně dlouho zachována. V posledních letech se však objevily studie prokazující, že alespoň část buněk s nekrotickou morfologií umírá regulovaným procesem zvaným programovaná nekróza nebo také nekroptóza [38]. Nekrotická smrt je pro buňku nejen záložní postup v případech, kdy je apoptóza z nějakého důvodu zablokována, ale má i svůj fyziologický význam např. jako obranný mechanizmus při virových infekcích. Z hlediska vlivu na nádorové buňky je nekrotická buněčná smrt dvousečnou zbraní. Nekróza vznikající jakožto důsledek chemoterapie přispívá k usmrcování nádorových buněk s defekty v apoptotické dráze. Na druhou stranu lokální zánětlivé procesy způsobené vyplavením obsahu nekrotických buněk např. z hypoxických oblastí nádoru spíše podporují angiogenezi a proliferaci nádorových buněk.
Nekroptózu můžou v buňkách aktivovat různé stimuly: IFNγ, nedostatek ATP, přítomnost patogenů, ischemicko‑reperfúzní poškození tkání, dvouřetězcová RNA (dsRNA) a vazba tzv. ligandů smrti (TNFα, TRAIL, FasL) na příslušný receptor [39]. Za normálních okolností tato vazba receptoru a ligandu vede k sestavení proteinového komplexu obsahujícího kaspázu ‑ 8, adaptorový protein FADD a kinázu RIP1 (receptor ‑ interacting serine ‑ threonine kinase 1) a následně ke spuštění apoptózy prostřednictvím signální kaskády zahrnující aktivovanou kaspázu ‑ 8 [40]. Jestliže jsou však kaspázy z nějakého důvodu inaktivní, je do tohoto proteinového komplexu zahrnuta i kináza RIP3. Kinázy RIP1 a RIP3 vzájemně interagují pomocí RIP ‑ homotypické interakční domény. K této interakci dochází pouze v případě aktivace nekroptotické signální dráhy, jež může být blokována specifickým inhibitorem RIP1 - kinázové aktivity – nekrostatinem [41]. Protein RIP1 je spíše znám svou účastí při aktivaci proteinu NF ‑ κB [42], a je tedy zřejmě důležitý účastník rozhodování, zda buňka přežije díky aktivaci NF ‑ κB nebo zda bude eliminována apoptotickým, resp. nekroptotickým mechanizmem. Síť proteinů zasahujících do regulace programované nekrózy se díky novým poznatkům stále rozrůstá a zahrnuje dále např. PARP1, PAR polymery, NADPH oxidázy a kalpainy [43].
Pro‑nekrotický komplex RIP1 – RIP3 následně interaguje s metabolickými enzymy a zvyšuje uhlovodíkový a glutaminový metabolizmus, což je doprovázeno zvýšením množství reaktivních kyslíkových radikálů (reactive oxygen species – ROS) [44]. Právě ROS se zřejmě u většiny buněčných typů významně podílí na exekuční fázi nekrotického usmrcení buňky [45]. Biochemických změn probíhajících v nekrotické buňce je ale celá řada a většina z nich je fatální. Proto zatím nelze přesně říci, které z nich jsou pro osud buňky nejvíce zásadní a určující [46].
Mutace regulátorů programované buněčné smrti v nádorových buňkách
Úspěch chemoterapeutické léčby, která je často zacílena na DNA, je z podstatné části ovlivněn schopností buněk opravovat poškozenou DNA a také aktivovat eliminační apoptotický proces. V nádorových buňkách je mutována celá řada proteinů, jež přímo či nepřímo ovlivňují rezistenci buněk k indukci buněčné smrti. Pro jejich podrobný popis však není v tomto přehledovém článku dost prostoru. Jsou to např. proteiny p53, Ras, Raf, Src, NF ‑ κB, HSPs a mnoho dalších.
V nádorových buňkách byly detekovány mutace proteinů, které jsou přímou součástí jak vnější apoptotické signální dráhy (vzácněji), tak vnitřní signální kaskády od mitochondriálních regulátorů po kaspázy. Jejich detailnějšímu popisu se budeme dále věnovat.
Receptory smrti
Buněčná smrt indukovaná Fas receptorem může být inhibována tvorbou rozpustného Fas, nedostatečnou expresí Fas na povrchu buňky, nadměrnou expresí inhibičních proteinů (Fas‑associated phosphatase ‑ 1 nebo FLIP) či mutací v primární struktuře Fas [47 – 49]. Mutace ve Fas receptoru byly poprvé popsány u mnohočetného myelomu [50] a jsou detekovány v různých typech lymfomů [51].
TRAILem indukovaná apoptóza může být blokována expresí tzv. decoy receptorů pro TRAIL, nadměrnou expresí inhibičních proteinů (FLIP) nebo mutací v primární struktuře DR4 a DR5 [52]. Mutace TRAIL‑R1 a TRAIL‑R2 receptorů byly detekovány např. u metastáz prsních karcinomů, nemalobuněčných prsních karcinomů či nádorů hlavy a krku [53 – 55].
Rodina Bcl ‑ 2
Bcl ‑ 2 protein byl původně objeven v buňkách B‑lymfomu, v nichž je v důsledku chromozomální translokace t(14;18) jeho gen fúzován se silným promotorem imunoglobulinu. Díky tomu je jeho hladina výrazně zvýšena [56]. Zvýšená hladina Bcl ‑ 2 byla pozorována i v solidních nádorech, jako jsou nádory plic, prsu a mozku. Důvodem zvýšené transkripce jsou pravděpodobně hypometylace promotoru či ztráta příslušných miRNA, které negativně regulují expresi bcl ‑ 2 [57].
Vysoká hladina exprese proteinu Bcl ‑ XL byla zjištěna v buňkách mnohočetných myelomů a lymfomů [58]. Zvýšený počet kopií genů bcl ‑ xL a mcl ‑ 1 byl dále detekován v řadě jiných nádorů, např. plic a kostí [59].
Také pro‑apoptotické proteiny rodiny Bcl ‑ 2 mohou zastávat funkci nádorových supresorů. Jejich snížené hladiny v důsledku mutací (Bim, Puma) či epigenetických modifikací (Puma) byly totiž popsány u B‑lymfomů nebo Burkittova lymfomu [59 – 61]. Mutovaný Bax byl zachycen např. u nádorů gastrointestinálního traktu a v leukemiích [62,63].
Apaf ‑ 1
Snížená hladina Apaf ‑ 1, detekovaná v metastatických melanomech, je spíše důsledek epigenetického umlčení exprese apaf ‑ 1 než mutace v jeho kódující sekvenci [64]. Snížená hladina Apaf ‑ 1 je znak špatné prognózy u pacientů s B typem chronické lymfocytické leukemie, pokud se vyskytuje současně s mutací v proteinu p53 [65].
IAPs
Chromozomální region 11q21 – 22, kódující cIAP1 a cIAP2, je amplifikován v mnoha typech nádorových tkání – hepatocelulárním karcinomu, karcinomu prsu, meduloblastomu, a v karcinomech slinivky, plic, děložního hrdla či jícnu [66]. Asi u 30 % MALT lymfomů je detekována chromozomální translokace t(11;18) (q21;q21), díky které vzniká chimérický protein N‑terminální sekvence cIAP2 spojené s C‑terminální sekvencí MALT1 [67]. V lymfocytech mohou IAP proteiny zřejmě naopak působit protinádorově. Mutace v jejich kódujících sekvencích byly totiž zachyceny v mnohočetných myelomech [68] nebo lymfoproliferativních onemocněních [69].
Kaspázy
Z prozatím testovaných typů nádorových onemocnění byly mutace kaspázy ‑ 8 (v kódující sekvenci, intronech a 3‘ nepřekládané oblasti) nejčastěji zachyceny v pokročilých stadiích nádorů žaludku [70]. Snížená exprese v důsledku hypermetylace promotoru byla popsána u relabujících glioblastomů, což naznačuje, že kaspáza ‑ 8 může mít vliv na vývoj tohoto typu nádorů [71]. Inaktivační mutace kaspáz ‑ 3 a - 7 byly pouze sporadicky zachyceny u solidních nádorů různých typů [72,73]. Polymorfizmus v regionu regulace transkripce kaspázy ‑ 3 byl popsán v buňkách karcinomu skvamózních buněk hlavy a krku [74]. V myších se zablokovanou tvorbou kaspáz nebyla detekována zvýšená incidence nádorových onemocnění. Tyto výsledky tedy nasvědčují spíše tomu, že mutace jednotlivých kaspáz nepatří mezi hlavní příčiny neoplastické transformace buněk [75].
Přehled testovaných léčiv založených na reaktivaci programované buněčné smrti
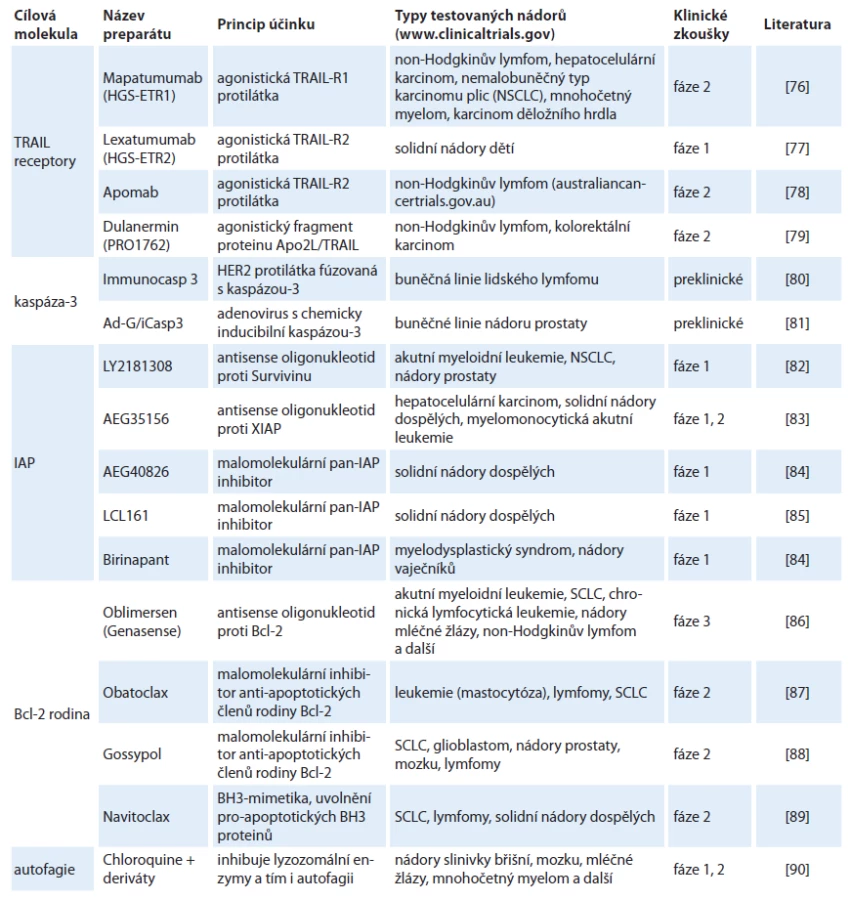
Při onkologické terapii jsou nejčastějším cílem léčiv proliferativní mechanizmy a onkogenní signály podporující přežívání nádorových buněk. Vývoj novějších léků je mimo jiné zaměřen na reaktivaci apoptotických signálních drah, a to jak vnější, tak především vnitřní, jež by mohly vést k úspěšné regresi nádoru. Hlavní cílové molekuly vytipované pro léčbu, princip účinku preparátu a fáze klinických zkoušek, ve kterých jsou nejnadějnější preparáty testovány, jsou shrnuty v tab. 1.
Závěr
Nádorová onemocnění jsou dlouhodobě druhou nejčastější příčinou úmrtí v ČR po nemocech srdce a cév. I přes narůstající incidenci se u nás daří stabilizovat mortalitu díky časnějšímu záchytu onemocnění a také díky novým léčebným metodám. Poznatky základního výzkumu na molekulární a buněčné úrovni přináší nové možnosti přesného a efektivního zacílení léčby, mimo jiné i na proteiny apoptotické signální dráhy, jejíž reaktivace by mohla vést ke zlepšení odpovědi na protinádorovou terapii.
Práce byla podpořena grantem Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO, CZ.1.05/2.1.00/03.0101) a MZ ČR – RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Eva Ondroušková, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: eva.ondrouskova@mou.cz
Obdrženo: 14. 1. 2014
Přijato: 6. 3. 2014
Sources
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5): 646 – 674. doi: 10.1016/ j.cell.2011.02.013.
2. Kroemer G, Galluzzi L, Vandenabeele P et al. Classification of cell death: recommendation of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16(1): 3 – 11. doi: 10.1038/ cdd.2008.150.
3. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ‑ ranging implications in tissue kinetics. Br J Cancer 1972; 26(4): 239 – 257.
4. Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005; 6(4): 328 – 340.
5. Rubinstein AD, Kimchi A. Life in the balance – a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci 2012; 125(Pt 22): 5259 – 5268. doi: 10.1242/ jcs.115865.
6. Martinet W, Schrijvers DM, Herman AG et al. Z ‑ VAD ‑ fmk induced non‑apoptotic cell death of macrophages. Autophagy 2006; 2(4): 312 – 314.
7. Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer 1994; 73(8): 2013 – 2026.
8. Fadok VA, Bratton DL, Frasch SC et al. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 1998; 5(7): 551 – 562.
9. Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science 1998; 281(5381): 1305 – 1308.
10. Wachman K, Pop C, van Raam BJ et al. Activation and specifity of human caspase ‑ 10. Biochemistry 2010; 49(38): 8307 – 8315. doi: 10.1021/ bi100968m.
11. Michalak EM, Villunger A, Adams JM et al. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 2008; 15(6): 1019 – 1029. doi: 10.1038/ cdd.2008.16.
12. Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol 2011; 223(2): 116 – 126. doi: 10.1002/ path.2784.
13. Amaral JD, Xavier JM, Steer CJ et al. The role of p53 in apoptosis. Discov Med 2010; 9(45): 145 – 152.
14. Burlacu A. Regulation of apoptosis by Bcl ‑ 2 family proteins. J Cell Mol Med 2003; 7(3): 249 – 257.
15. Chipuk JE, Kuwana T, Bouchier ‑ Hayes L et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303(5660): 1010 – 1014.
16. Chipuk JE, Moldoveanu T, Llambi F et al. The Bcl ‑ 2 family reunion. Molecular Cell 2010; 37(3): 299 – 310. doi: 10.1016/ j.molcel.2010.01.025.
17. Sedlak TW, Oltvai ZN, Yang E et al. Multiple Bcl ‑ 2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA 1995; 92(17): 7834 – 7838.
18. Vela L, Gonzalo O, Naval J et al. Direct interaction of Bax and Bak proteins with Bcl ‑ 2 homology domain 3(BH3) - only proteins in living cells revealed by fluorescence complementation. J Biol Chem 2013; 288(7): 4935 – 4946. doi: 10.1074/ jbc.M112.422204.
19. Kluck RM, Bossy ‑ Wetzel E, Green DR et at. The release of cytochrome c from mitochondria: A primary site for Bcl ‑ 2 regulation of apoptosis. Science 1997; 275(5303): 1132 – 1136.
20. Zou H, Henzel WJ, Liu A et al. Apaf ‑ 1, a human protein homologous to C. elegans CED ‑ 4, participates in cytochrome c ‑ dependent activation of caspase ‑ 3. Cell 1997; 90(3): 405 – 413.
21. Deveraux QL, Reed JC. IAP family proteins – suppressors of apoptosis. Genes Dev 1999; 13(3): 239 – 252.
22. Du C, Fang M, Li Y et al. Smac, a mitochondrial protein that promotes cytochrome c ‑ dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102(1): 33 – 42.
23. Gottfried Y, Rotem A, Lotan R et al. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J 2004; 23(7): 1627 – 1635.
24. Schafer P, Scholz SR, Gimadutdinow O et al. Structural and functional characterization of mitochondrial EndoG, a sugar non‑specific nuclease which plays an important role during apoptosis. J Mol Biol 2004; 338(2): 217 – 228.
25. Nakagawa T, Zhu H, Morishima N et al. Caspase ‑ 12 mediates endoplasmic ‑ reticulum ‑ specific apoptosis and cytotoxicity by amyloid‑beta. Nature 2000; 403(6765): 98 – 103.
26. Beneš P, Větvička V, Fusek M. Cathepsin D – many functions of one aspartic protease. Crit Rev Oncol Hematol 2008; 68(1): 12 – 28. doi: 10.1016/ j.critrevonc.2008.02.008.
27. Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene 2004; 23(16): 2881 – 2890.
28. Fisher U, Janicke RU, Schulze ‑ Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003; 10(1): 76 – 100.
29. de Duve CH, Wattiaux R. Functions of lysosome. Annu Rev Physiol 1966; 28 : 435 – 492.
30. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147(4): 728 – 741. doi: 10.1016/ j.cell.2011.10.026.
31. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12(9): 814 – 822. doi: 10.1038/ ncb0910 - 814.
32. Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis 2011; 32(7): 955 – 963. doi: 10.1093/ carcin/ bgr031.
33. Esposti DD, Domart MC, Sebagh M et al. Autophagy is induced by ischemic preconditioning in human livers formerly treated by chemotherapy to limit necrosis. Autophagy 2010; 6(1): 172 – 174.
34. Mathew R, Kongara S, Beaudoin B et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21(11): 1367 – 1381.
35. Yang S, Wang X, Contino G et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25(7): 717 – 729. doi: 10.1101/ gad.2016111.
36. Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115(10): 2679 – 2688.
37. Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131(6): 1137 – 1148.
38. Degterev A, Huang Z, Boyce M et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1(2): 112 – 119.
39. Vanlangenakker N, Bertrand MJM, Bogaert P et al. TNF‑induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2011; 2(11): e230. doi: 10.1038/ cddis.2011.111.
40. Scaffidi C, Kirchhoff S, Krammer PH et al. Apoptosis signaling in lymphocytes. Curr Opin Immunol 1999; 11(3): 277 – 285.
41. Degterev A, Hitomi J, Germscheid M et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4(5): 313 – 321. doi: 10.1038/ nchembio.83.
42. Hayden MS, Ghosh S. Shared principles in NF ‑ kappaB signaling. Cell 2008 : 132(3): 344 – 362. doi: 10.1016/ j.cell.2008.01.020.
43. Ouyang L, Shi Z, Zhao S et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012; 45(6): 478 – 498. doi: 10.1111/ j.1365 - 2184.2012.00845.x.
44. Zhang DW, Shao J, Lin J et al. RIP3, an energy metabolism regulator that switches TNF‑induced cell death from apoptosis to necrosis. Science 2009; 325(5938): 332 – 336. doi: 10.1126/ science.1172308.
45. Lin Y, Choksi S, Shen HM et al. Tumor necrosis factor‑induced nonapoptotic cell death requires receptor ‑ interacting protein‑mediated cellular reactive oxygen species accumulation. J Biol Chem 2004; 279(11): 10822 – 10828.
46. Vandenabeele P, Galluzzi L, Vanden Berghe T et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11(10): 700 – 714. doi: 10.1038/ nrm2970.
47. Natoli G, Ianni A, Constanzo A et al. Resistance to Fas ‑ mediated apoptosis in human hepatoma cells. Oncogene 1995; 11(6): 1157 – 1164.
48. Irmler M, Thome M, Hahne M et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388(6638): 190 – 195.
49. Rieux ‑ Laucat F, Le Deist F, Hivroz C et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimunity. Science 1995; 268(5215): 1347 – 1349.
50. Landowsky TH, Qu N, Buyuksal I et al. Mutations in the Fas antigen in patients with multiple myeloma. Blood 1997; 90(11): 4266 – 4270.
51. Gronbaek K, Straten PT, Ralfkiaer E et al. Somatic Fas mutations in non‑Hodgkin‘s lymphoma: association with extranodal disease and autoimmunity. Blood 1998; 92(9): 3018 – 3024.
52. Bin L, Thorburn J, Thomas LR et al. Tumor ‑ derived mutations in the TRAIL receptor DR5 inhibit TRAIL signaling through the DR4 receptor by competing for ligand binding. J Biol Chem 2007; 282(38): 28189 – 28194.
53. Lee SH, Shin MS, Kim HS et al. Alterations of the DR5/ TRAIL receptor 2 gene in non‑small cell lung cancers. Cancer Res 1999; 59(22): 5683 – 5686.
54. Shin MS, Kim HS, Lee SH et al. Mutations of tumor necrosis factor‑related apoptosis ‑ inducing ligand receptor 1(TRAIL‑R1) and receptor 2 (TRAIL‑R2) genes in metastatic breast cancers. Cancer Res 2001; 61(13): 4942 – 4946.
55. Fisher MJ, Virmani LW, Aplenc R et al. Nucleotide substitution in the ectodomain of TRAIL receptor DR4 is associated with lung cancer and head and neck cancer. Clin Cancer Res; 7(6): 1688 – 1697.
56. Tsujimoto Y, Gorham J, Cossman J et al. The t(14;18) chromosome translocations involved in B ‑ cell neoplasms result from mistakes in VDJ joining. Science 1985; 229(4720): 1390 – 1393.
57. Kelly PN, Strasser A. The role of Bcl ‑ 2 and its pro‑survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 2011; 18(9): 1414 – 1424. doi: 10.1038/ cdd.2011.17.
58. Krajewski S, Krajewska M, Shabaik A et al. Immunohistochemical analysis of in vivo patterns of Bcl ‑ X expression. Cancer Res 1994; 54(21): 5501 – 5507.
59. Beroukhim R, Mermel C, Porter D et al. The landscape of somatic copy ‑ number alteration across human cancers. Nature 2010; 463(7283): 899 – 905. doi: 10.1038/ nature08822.
60. Tagawa H, Karnan S, Suzuki R et al. Genome ‑ wide array‑based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005; 24(8): 1348 – 1358.
61. Garrison SP, Jeffers JR, Yang C et al. Selection against PUMA gene expression in Myc‑driven B ‑ cell lymphomagenesis. Mol Cell Biol 2008; 28(17): 5391 – 5402. doi: 10.1128/ MCB.00907 - 07.
62. Rampino N, Yamamoto H, Ionov Y et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997; 275(5302): 967 – 969.
63. Meijerink JP, Smetsers TF, Sloetjes AW et al. Bax mutations in cell lines derived from hematological malignancies. Leukemia 1995; 9(11): 1828 – 1832.
64. Soengas MS, Capoieci P, Polsky D et al. Inactivation of the apoptosis effector Apaf ‑ 1 in malignant melanoma. Nature 2001; 409(6817): 207 – 211.
65. Sturm I, Bosanquet AG, Radetzki S et al. Silencing of Apaf ‑ 1 in B ‑ CLL results in poor prognosis in the case of concomitant p53 mutation. Int J Cancer 2006; 118(9): 2329 – 2336.
66. Dubrez L, Berthelet J, Glorian V. IAP proteins as target for drug development in oncology. Onco Targets Ther 2013; 6 : 1285 – 1304.
67. Garrison JB, Samuel T, Reed JC. TRAF2‑binding BIR1 domain of c ‑ IAP2/ MALT1 fusion protein is essential for activation of NF ‑ kappaB. Oncogene 2009; 28(13): 1584 – 1593. doi: 10.1038/ onc.2009.17.
68. Annunziata CM, Davis RE, Demchenko Y et al. Frequent engagement of the classical and alternative NF ‑ kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12(2): 115 – 130.
69. Filipovich AH, Zhang K, Snow AL et al. X‑linked lymphoproliferative syndromes: brothers or distant cousins? Blood 2010; 116(18): 3398 – 3408. doi: 10.1182/ blood ‑ 2010 - 03 - 275909.
70. Soung YH, Lee JW, Kim SY et al. Caspase ‑ 8 gene is inactivated by somatic mutations in gastric carcinomac. Cancer Res 2005; 65(3): 815 – 821.
71. Martinez R, Setien F, Voelter C et al. CpG island promoter hypermethylation of the pro‑apoptotic gene caspase ‑ 8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis 2007; 28(6): 1264 – 1268.
72. Soung YH, Lee JW, Kim SY et al. Somatic mutations of CASP3 gene in human cancers. Hum Genet 2004; 115(2): 112 – 115.
73. Soung YH, Lee JW, Kim HS et al. Inactivating mutations of CASPASE ‑ 7 gene in human cancers. Oncogene 2003; 22(39): 8048 – 8052.
74. Chen K, Zhao H, Hu Z et al. CASP3 polymorphisms and risk of squamous cell carcinoma of the head and neck. Clin Cancer Res 2008; 14(19): 6343 – 6349. doi: 10.1158/ 1078 - 0432.CCR ‑ 08 - 1198.
75. Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ 2011; 18(9): 1441 – 1449. doi: 10.1038/ cdd.2011.30.
76. Greco FA, Bonomi P, Crawford J et al. Phase 2 study of mapatumumab, a fully human agonistic monoclonal antibody which targets and activates the TRAIL receptor ‑ 1 in patients with advanced non‑small cell lung cancer. Lung Cancer 2008; 61(1): 82 – 90. doi: 10.1016/ j.lungcan.2007.12.011.
77. Zhang I, Zhang X, Barrisford GE et al. Lexatumumab (TRAIL‑receptor 2 mAb) induces expression of DR5 and promotes apoptosis in primary and metastatic renal cell carcinoma in a mouse orthotopic model. Cancer Lett 2007; 251 (1): 146 – 157.
78. Zinonos I, Labrinidis A, Lee M et al. Apomab, a fully human agonistic antibody to DR5, exhibits potent antitumor activity against primary and metastatic breast cancer. Mol Cancer Ther 2009; 8(10): 2969 – 2980. doi: 10.1158/ 1535 - 7163.MCT ‑ 09 - 0745.
79. Soria JC, Mark Z, Zatloukal P et al. Randomized phase IIstudy of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non‑small cell lung cancer. J Clin Oncol 2011; 29(33): 4442 – 4451. doi: 10.1200/ JCO.2011.37.2623.
80. Jia LT, Zhang LH, Yu CJ et al. Specific tumoricidal activity of a secreted proapoptotic protein consisting of HER2 antibody and constitutively active caspase ‑ 3. Cancer Res 2003; 63(12): 3257 – 3262.
81. Shariat SF, Desai S, Song W et al. Adenovirus ‑ mediated transfer of inducible caspases: a novel „death switch“ gene therapeutic approach to prostate cancer. Cancer Res 2001; 61(6): 2562 – 2571.
82. Tanioka M, Nokihara H, Yamamoto N et al. Phase Istudy of LY2181308, an antisense oligonucleotide against survivin, in patients with advanced solid tumors. Cancer Chemother Pharmacol 2011; 68(2): 505 – 511. doi: 10.1007/ s00280 - 010 - 1506 - 7.
83. Schimmer AD, Estey EH, Borthakur G et al. Phase I/ II trial of AEG35156 X‑linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol 2009; 27(28): 4741 – 4746. doi: 10.1200/ JCO.2009.21.8172.
84. Gyrd ‑ Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF ‑ κB, inflammation and cancer. Nat Rev Cancer 2010; 10(8): 561 – 574. doi: 10.1038/ /nrc2889.
85. Qin Q, Zuo Y, Yang X et al. Smac mimetic compound LCL161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumor Biol 2014; 35(3): 2565 – 2574. doi: 10.1007/ s13277 - 013 - 1338-2.
86. O‘Brien S, Moore JO, Boyd TE et al. 5‑year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J Clin Oncol 2009; 27(31): 5208 – 5212. doi: 10.1200/ JCO.2009.22.5748.
87. Goard CA, Schimmer AD. An evidence‑based review of obatoclax mesylate in the treatment of hematological malignancies. Core Evid 2013; 8 : 15 – 26. doi: 10.2147/ CE.S42568.
88. Baggstrom MQ, Qi Y, Koczywas M et al. A phase IIstudy of AT ‑ 101 (Gossypol) in chemotherapy ‑ sensitive recurrent extensive‑stage small cell lung cancer. J Thorac Oncol 2011; 6(10): 1757 – 1760. doi: 10.1097/ JTO.0b013e31822e2941.
89. Rudin CM, Hann CL, Garon EB et al. Phase II study of single‑agent navitoclax (ABT ‑ 263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012; 18(11): 3163 – 3169. doi: 10.1158/ 1078 - 0432.CCR ‑ 11 - 3090.
90. Chaabane W, User SD, El ‑ Gazzah M et al. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp 2013; 61(1): 43 – 58. doi: 10.1007/ s00005 - 012 - 0205 - y.
91. Ondroušková E, Souček K, Horváth V et al. Alternative pathways of programmed cell death are activated in cells with defective caspase ‑ dependent apoptosis. Leuk Res 2008; 32(4): 599 – 609.
92. Walker NI, Harmon BV, Gobé GC et al. Patterns of cell death. Methods Achiev Exp Pathol 1988; 13 : 18 – 54.
93. Brown M, Wilson G. Apoptosis genes and resistance to cancer therapy. Cancer Biol Ther 2003; 2(5): 477 – 490.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Exprese a purifikace proteinů
- Metody studia buněčné migrace a invazivity nádorových buněk
- Sekvenování nové generace a možnosti jeho využití v onkologické praxi
- Analýza proteinů pomocí hmotnostní spektrometrie