
Kvantitativní hmotnostní spektrometrie a její využití v onkologii
Quantitative Mass Spectrometry and Its Utilization in Oncology
Cancers are genetically and clinically very heterogeneous diseases; therefore, various proteomic studies have been trying to find biomarkers which can facilitate prognosis, diagnosis or treatment of these oncological diseases. The mass spectrometry is an effective tool for identification, quantitation, and characterization of biomolecules in the complex biological samples. The first step suitable for selection of biomarkers called discovery proteomics provides a detailed analysis of the samples contributing to the identification of proteins, comparison of their presence in the samples, and selection of the convenient candidates for the prospective biomarkers. The next step of proteomics analysis is directed towards verification of chosen biomarkers with the approach called targeted proteomics. This technique evaluates presence and quantity of the proteins (biomarkers) in clinically precisely defined samples. This article focuses on the description of various approaches suitable for the quantitative analysis of the proteins connected with mass spectrometry.
Key words:
quantitative proteomics – mass spectrometry – protein – biomarker – oncology – cancer
This work was supported by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101) and by MH CZ – DRO (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
20. 1. 2014
Accepted:
7. 4. 2014
Authors:
L. Hernychová; P. Dvořáková; E. Michalová; B. Vojtěšek
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2014; 27(Supplementum): 98-103
Overview
Nádory jsou geneticky a klinicky velmi různorodá onemocnění, a proto se v poslední době řada proteomických studií zabývá hledáním biomarkerů, které by usnadnily prognostiku, diagnostiku či léčbu onkologických onemocnění. Účinným nástrojem je hmotnostní spektrometrie umožňující identifikaci, kvantifikaci a charakterizaci biomolekul v komplexních biologických vzorcích. Prvním krokem vedoucím k selekci biomarkerů je tzv. discovery proteomika, jejímž cílem je detailní analýza vzorků směřující ke zmapování a porovnání přítomných proteinů a výběru vhodných kandidátů na potenciální biomarkery. V dalším kroku proteomické analýzy probíhá verifikace vybraných biomarkerů tzv. cílenou (targeted) proteomikou, jejímž úkolem je ověření přítomnosti a kvantity daného proteinu v analyzovaných, přesně klinicky definovaných vzorcích. Předložený článek je zaměřen na popis různých typů metod vhodných pro kvantitativní analýzu proteinů využívající hmotnostní spektrometrií.
Klíčová slova:
kvantitativní proteomika – hmotnostní spektrometrie – protein – biomarker – onkologie – nádor
Úvod
Hmotnostní spektrometrie je jedním z proteomických nástrojů umožňujících identifikovat, kvantifikovat a charakterizovat proteiny v komplexních biologických vzorcích. Vývoj proteomických přístupů začínal identifikací vybraných purifikovaných proteinů a verifikací genomických dat a přes komparativní proteomové analýzy se rozvinul až po charakterizaci posttranslačních modifikací proteinů, jejich dynamických změn a protein‑proteinových interakcí.
V současné době jsou výsledky hmotnostně spektrometrických analýz dále rozšířeny o predikované bioinformatické údaje, které jsou teprve následně experimentálně ověřovány. Mezi bio-informatické nástroje patří programy předpovídající lokalizaci proteinu v eukaryotické nebo prokaryotické buňce (program PSORT [1]), jeho funkci (programy COG [2], KEGG [3], DAVID [4]), součinnost s ostatními proteiny či ligandy, zapojení v signálních dráhách anebo jeho úlohu v biologickém celku např. v rámci buňky (programy IPA [5], PathVisio [6]). Je známo, že mutace proteinu způsobená třeba záměnou aminokyseliny či přítomností/ ztrátou posttranslačních modifikací může způsobit chybné sbalování proteinu a tím ovlivnit jeho funkci. Ta se dále může projevit změnami v interakci proteinu a narušení buněčné signalizace vedoucí k změnám v proliferaci buněk, apoptóze či nekróze. Vzhledem k tomu, že tyto faktory přispívají ke vzniku nádorově transformovaných buněk, budeme se v další části této práce detailněji zabývat metodami vhodnými ke studiu kvantitativních změn proteinů, detekcí a charakterizací posttranslačních modifikací proteinů a jejich interakcí s ligandy. Všechny uvedené metody mají společný jmenovatel, kterým je hmotnostní spektrometrie.
Jaké biologické materiály lze analyzovat?
Proteomové analýzy lze aplikovat na všechny typy biologických materiálů, které však musí být přesně klinicky vymezeny a definovány. Pro srovnávací proteomové analýzy se pak vždy definují experimentální skupiny analyzovaných vzorků, mezi nimiž se hledají rozdíly na proteinové úrovni. K nejčastěji analyzovaným klinickým biologickým materiálům patří tkáně, tělní tekutiny (moč, mozkomíšní nebo ascitická tekutina), krev (sérum, plazma), v případě laboratorních experimentů se pak používají buněčné linie či rekombinantní proteiny. Komplexita biologického materiálu představuje pro proteomickou analýzu vždy velký problém. Proto hmotnostně spektrometrické analýze předcházejí separační metody schopné materiál frakcionovat, a snížit tak jeho komplexitu. Je tedy výhodné předem určit skupiny proteinů např. se stejnou lokalizací v buňce nebo funkcí či strukturními motivy (membránové, jaderné, fosforylované, glykosylované proteiny) a účel, který má analýza splnit (identifikace, kvantifikace proteinů, určení místa posttranslační modifikace či změny proteinové konformace). Z těchto požadavků následně vychází vlastní protokol zpracování vzorků, který analýzu maximálně zjednoduší, avšak bez ztráty požadovaných informací.
Kvantifikace proteinů
S technologickým pokrokem lze již nyní identifikovat a kvantifikovat tisíce proteinů v jednom hmotnostně spektrometrickém měření, a to i z velmi omezeného množství výchozího materiálu (koncentrace proteinů se pohybuje v rozmezí 10 – 100 mg/ ml). Samotná kvantifikace změn na proteinové úrovni poskytuje řadu informací o stavu biologického systému v čase nebo za definovaných biologických podmínek, jež přispívají k objasnění biochemických a fyziologických mechanizmů na molekulární úrovni. V proteomických přístupech se v poslední době začaly používat subproteomové analýzy, které jsou schopné podat podrobnější informace o proteinovém složení ve vybrané izolované frakci analyzovaného materiálu, jež se při globální proteomové analýze nerozděleného vzorku ztrácí. Mezi subproteomové analýzy patří např. analýza membránových proteinů (membranom), imunoreaktivních proteinů (imunom), proteinů s proteolytickou aktivitou (proteazom), fosforylovaných či glykosylovaných proteinů (fosfoproteom či glykoproteom).
Kvantifikace proteinů může probíhat ve dvou základních uspořádáních. V prvním případě lze porovnávat kvantitu proteinů dvou či více biologických systémů (relativní kvantifikace), ve druhém uspořádání je možné přímo určit koncentraci daného proteinu v analyzovaném vzorku (absolutní kvantifikace). Přístupy používané pro relativní kvantifikace se dají rozdělit do dvou skupin: 1. metody závislé na izotopovém značení (label‑based) a 2. nezávislé na značení (label‑free). Důležitým požadavkem u metod závislých na značení je jasné oddělení značených peptidů/ proteinů od neznačených. Mezi často využívané metody závislé na izotopovém značení patří stable isotope labeling by amino acids in cell culture (SILAC) postup [7], který lze aplikovat pouze na živé organizmy in vitro, in vivo nebo ex vivo. Ke značení se používají stabilní izotopy neradioaktivních těžkých aminokyselin, jež jsou v průběhu metabolického procesu (např. při kultivaci buněčných kultur) inkorporovány do nově syntetizovaných proteinů. Ve standardním experimentálním uspořádání jsou kontrolní a ovlivněné buňky (např. normální a nádorové buňky) kultivovány v lehkém a těžkém kultivačním médiu lišící se přítomností neznačených aminokyselin, např. arginin, lysin (lehké médium), a stejných značených aminokyselin (těžké médium), které obsahují izotopy 13C a/ nebo 15N. Po kontrole inkorporace těžkých aminokyselin do proteinů buněk kultivovaných v těžkém médiu jsou z obou buněčných linií izolovány proteiny, smíchány v poměru 1 : 1, proteolyticky štěpeny a analyzovány na hmotnostním spektrometru tzv. shotgun postupem (obr. 1A). Na základě hmotnostních spekter jsou pak identifikovány píky stejných peptidů lišících se přesně definovanými hmotnostními posuny jejich efektivních hmot (m/ z) a z poměrů ploch pod těmito píky (resp. jejich intenzit) je odečítána relativní kvantifikace (obr. 1B). Identifikace proteinů je potom odvozena z fragmentačních spekter. Výhody tohoto postupu lze shrnout do následujících bodů: 1. získání dostatečného množství značeného materiálu pro další analýzy bez variací v účinnosti značení mezi jednotlivými vzorky (dochází vždy ke stejné inkorporaci těžkých aminokyselin), 2. možnost sledování proteinových změn v čase a 3. od izolace proteinů, kdy jsou vzorky smíchány 1 : 1, jsou všechny další úkony se značeným a neznačeným vzorkem prováděny stejně (nedochází k variabilitám způsobeným přípravou vzorků pro MS analýzu). Velká nevýhoda postupu je však vysoká finanční i časová náročnost, možnost porovnání nejvýše tří různých stavů a nemožnost aplikace na lidské vzorky tkání. V hmotnostních spektrech často dochází k překryvu peptidových píků a výskytu satelitních píků vlivem metabolické konverze [8,9], což způsobuje problémy v interpretaci spekter, a tím i samotnou kvantifikaci proteinů.
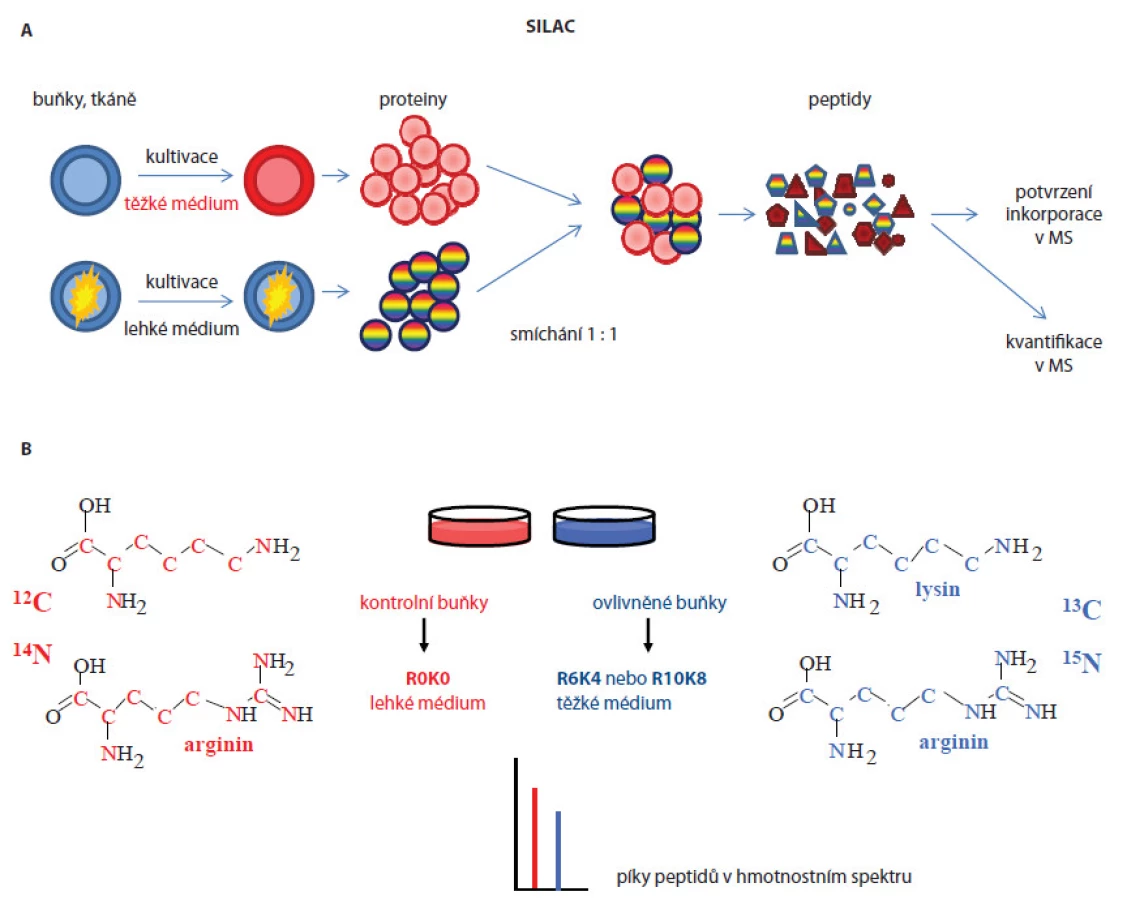
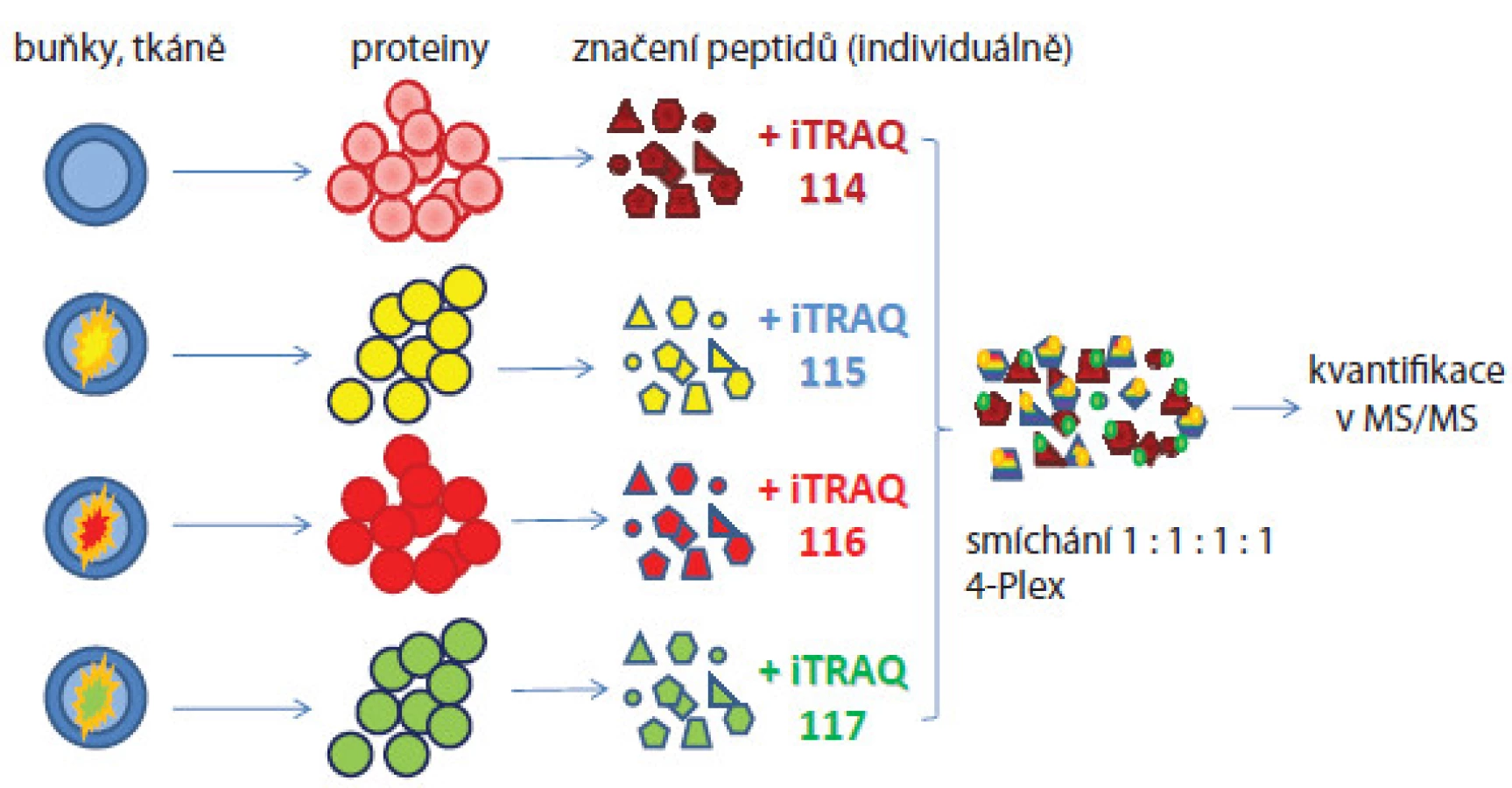
Selekce biomarkerů analýzou sér či tkání pacientů výše popisovaným postupem je náročné z hlediska dostupnosti a variability vzorků. Proto analýzy nádorových buněčných linií byly využívány jako výhodná alternativa pro zjištění biologicky zajímavých molekul, které jsou pak verifikovány na vzorcích pacientů. Jedním z příkladů je práce Geigera et al [10], v níž byly použity definované buněčné linie odvozené z různých stadií karcinomu mléčné žlázy. Metodou SILAC byly identifikovány biomarkery mitochondrial matrix protein isocitrate dehydrogenase 2 (IDH2), cellular retinoic acid binding protein 2 (CRABP2) a alpha ‑ tocopherol‑associated protein (SEC14L2), jež byly validovány cílenou hmotnostně spektrometrickou analýzou a imunohistochemicky na vzorcích tkání pacientek. Tyto proteiny se ukázaly být vhodnými biomarkery pro monitorování progrese a prognózy karcinomu mléčné žlázy. SILAC technika byla také použita in vivo – experimentální myši byly krmeny granulemi obsahujícími lysin se stabilním izotopem 13C nebo řasu spirulinu se stabilním izotopem 15N [11]. Všechny nově syntetizované proteiny byly označeny a mohly být na proteomové úrovni porovnávány s kontrolními neznačenými vzorky. Tohoto modelu lze využívat pro monitorování funkcí genů u knockout myší. Vylepšenou verzí SILAC metody je super ‑ SILAC postup [12], který se snaží řešit problém heterogenity nádorových tkání a značení proteinů (nelze kultivovat v těžkém médiu). Z tohoto důvodu byl problém elegantně vyřešen přidáním SILAC značených proteinů získaných buněčných linií do lyzátu analyzované tkáně. Boersema et al [13] použili tuto metodu pro identifikaci N ‑ glykosylovaných proteinů přítomných v sekretomu buněčných nádorových linií a vzorcích krve pacientů. Do 11 buněčných linií odvozených z karcinomu mléčné žlázy přidali interní standard představující značené proteiny reprezentující tzv. super ‑ SILAC mix, který umožnil kvantifikaci proteinů v analyzované tkáni. Proteom obohacený o N ‑ glykoproteiny byl analyzován hmotnostně spektrometrickými metodami. Následně byla přítomnost proteinů identifikovaných v tkáni validována ve vzorcích lidské krve. Další populární postupy používané v relativní kvantifikaci jsou založeny na značení peptidů proteolyticky štěpených proteinů. Jako příklad lze uvést isobaric tags for relative and absolute quantitation (iTRAQ, obr. 2 [14]) nebo tandem mass tag (TMT [15]). Jedná se o chemické značení primárních aminů peptidů, k nimž je připojena molekula reagentu složená ze tří skupin (reportérová, vyrovnávací a reagující s peptidem). Hmotnostní rozdíly molekuly reagentu jsou ovlivněny začleněním izotopů atomů 13C, 15N a 18O a tím umožňují hmotnostní posuny píků značených peptidů ve spektrech. Výhoda přístupu je, že lze značit až osm různých biologických stavů a jejich kvantifikaci odečítat najednou z jednoho spektra a identifikovat proteiny přítomné ve vzorku v rozdílné koncentraci (široký dynamický rozsah). Další výhoda je současná identifikace a kvantifikace proteinů v jednom měření ve fragmentačních spektrech. Nevýhoda metody je možné zkreslení informace o kvantitě proteinů ve vzorcích z důvodu značení peptidů až po proteolytickém štěpení proteinů, neboť před tímto krokem se manipulovalo se vzorky odděleně. Tuto metodu použili Rehman et al [16] pro identifikaci potenciálních prognostických biomarkerů nádoru prostaty spojených s progresí onemocnění a metastazováním. K analýze použili buněčné linie odvozené od nádoru prostaty a směsné vzorky sér pacientů. Protein eukaryotic translation elongation factor 1 alpha 1(eEF1A1) byl identifikován jako nový kandidátní biomarker se signifikantně zvýšenou expresí u pacientů zařazených do studie. V jiné studii zaměřené na analýzu fosforylace na tyrozinu v imortalizované buněčné linii odvozené z epiteliálních buněk mléčné žlázy MCF ‑ 10A bylo identifikováno 57 unikátních proteinů zahrnujících tyrozinové kinázy, fosfatázy a proteiny účastnící se buněčné signalizace. Poprvé byly identifikovány proteiny SLC4A7 (sodium bicarbonate cotransporter) a TOLLIP (toll interacting protein) jako nové tyrozin kinázové substráty spojené s vývojem karcinomu mléčné žlázy [17].
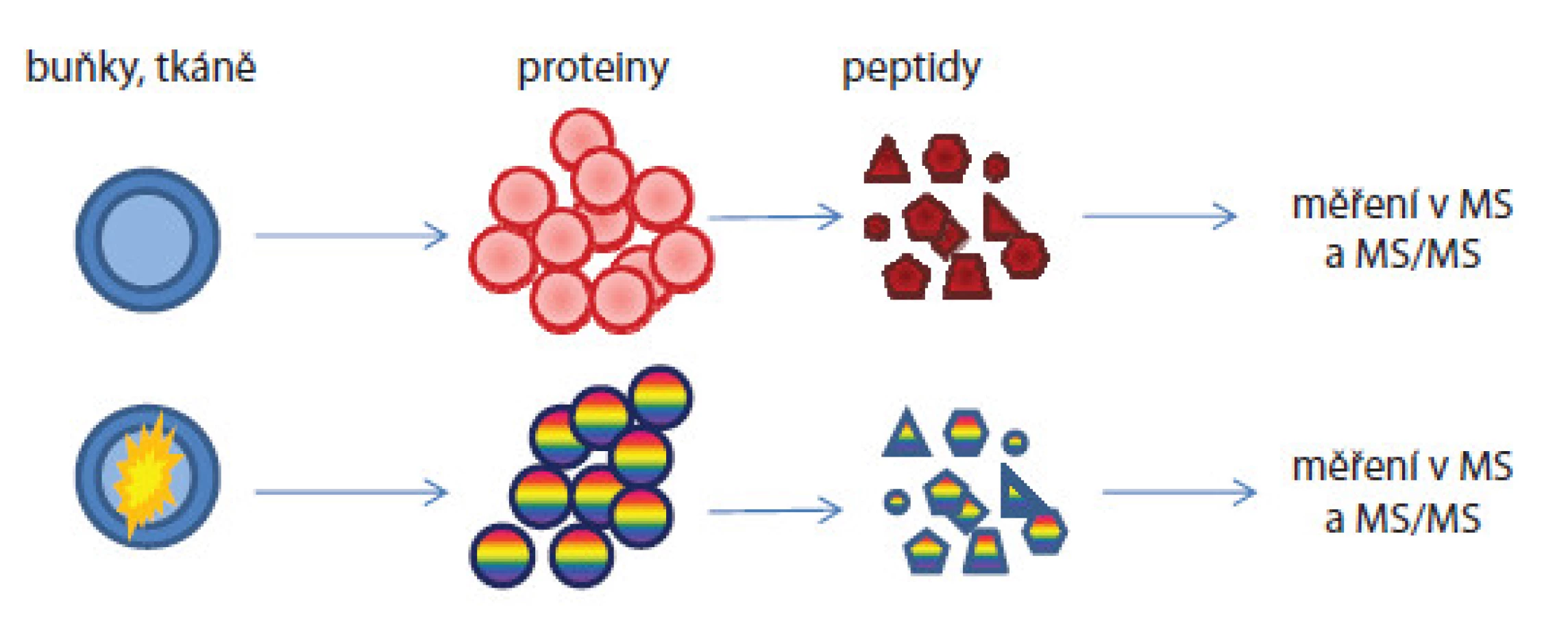
U label‑free postupu nejsou vzorky značeny žádnými značkami [18]. Metodaje rychlá, cenově dostupná, avšak méně přesná a nevhodná pro proteiny přítomné ve vzorcích v nízkých koncentracích. Další problém je náročné hodnocení hmotnostně spektrometrických dat, pro něž musí být použity vysoce sofistikované programy. V průběhu přípravy i měření se pracuje se vzorky odděleně, ale za stejných experimentálních podmínek (obr. 3). Předností je analýza neomezeného počtu vzorků, které mohou být mezi sebou porovnávány. Informace o kvantitě jsou získávány: 1. z intenzit píků peptidových iontů či ploch pod píky v hmotnostních spektrech nebo v chromatogramech nebo 2. z počtu MS/ MS spekter přiřazených danému peptidu/ proteinu nazývané spectral counting. Empiricky bylo zjištěno, že peptidy přítomné ve vzorcích ve vyšších koncentracích vykazují častější výskyt iontů v hmotnostních spektrech.
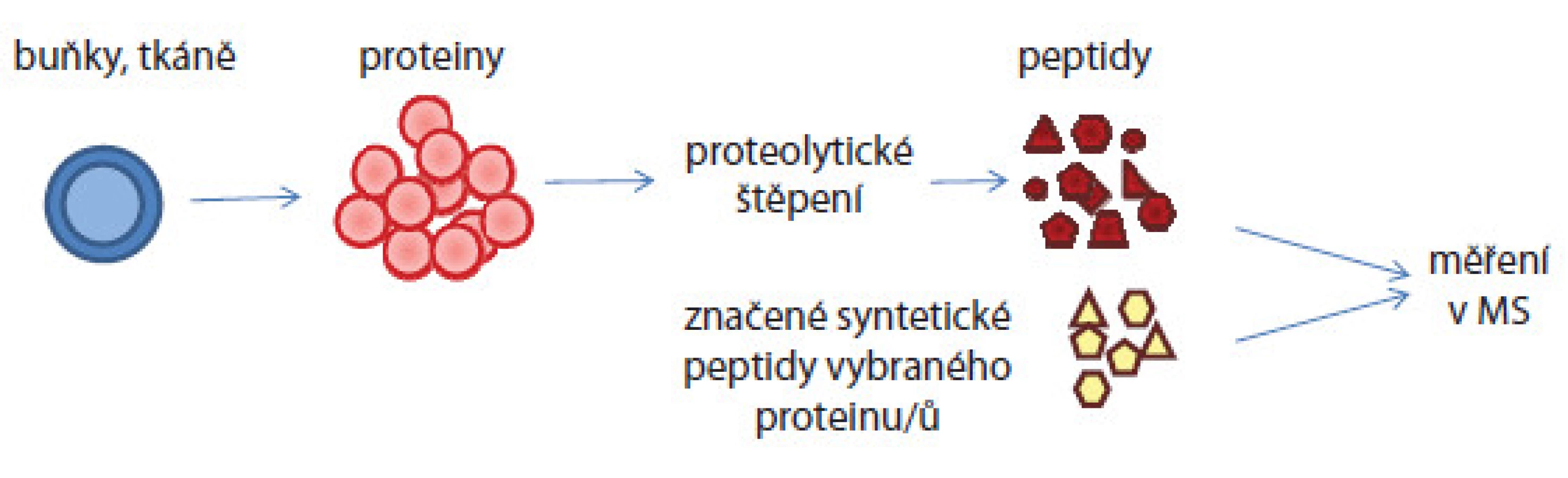
V případě absolutní kvantifikace musí být již známy proteiny/ peptidy, které ve vzorcích chceme cíleně kvantifikovat. Absolutní kvantifikaci tedy musí předcházet tzv. discovery analýza vzorku. Aminokyselinová sekvence proteinu či jeho části je pak syntetizována a je obohacena o značku, která umožní jeho odlišení v hmotnostních spektrech od nativního proteinu. Syntetický konstrukt je přidáván do vzorku v několika známých koncentracích pokrývajících předpokládanou koncentraci nativního proteinu. Koncentrace nativního proteinu je pak odvozena porovnáním intenzit píků syntetického konstruktu a nativního peptidu v hmotnostních spektrech (obr. 4).
Závěr
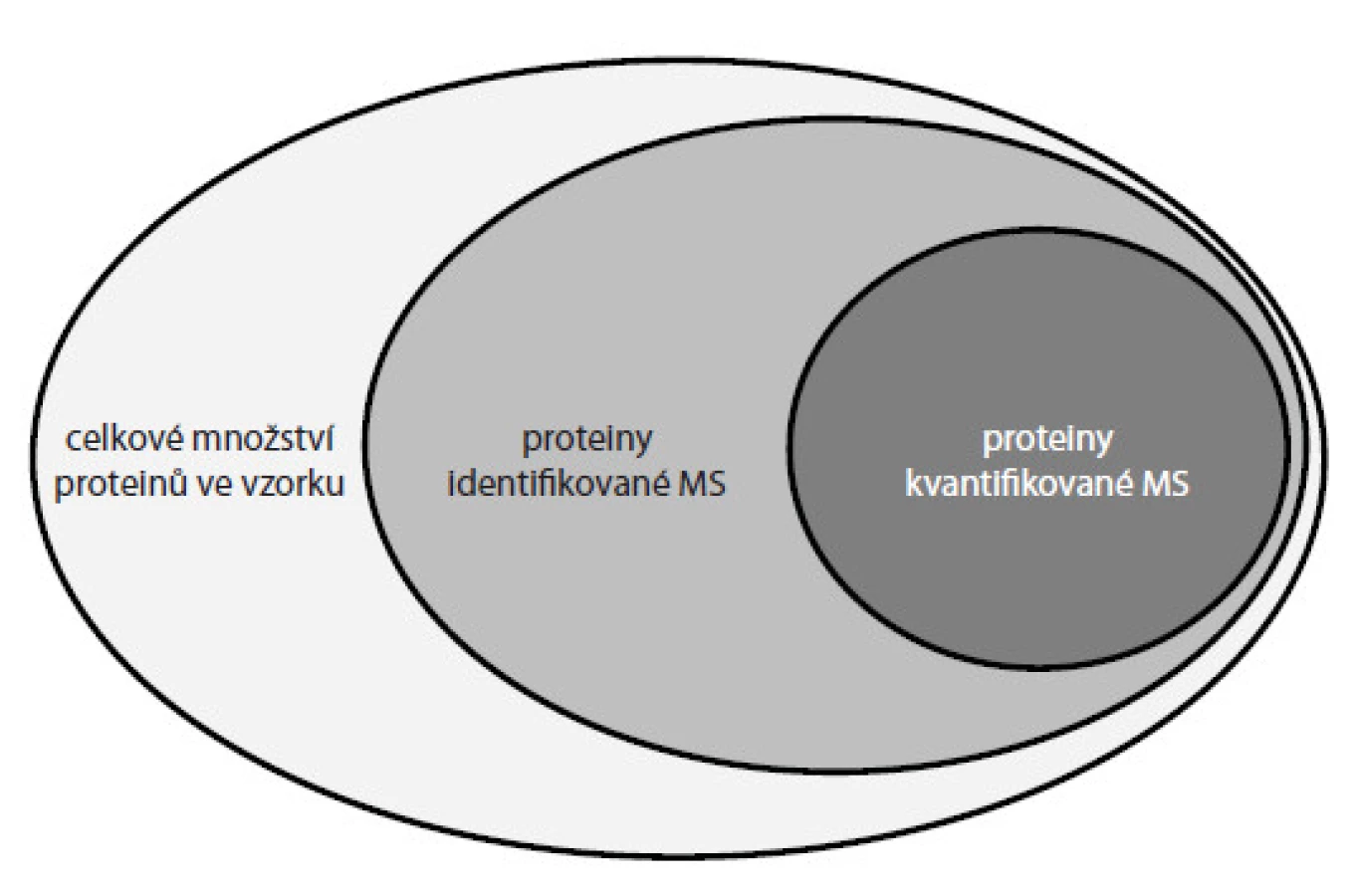
Všechny popisované metody však mají stále svá omezení vedoucí ke kvantifikaci jen určité části proteinů přítomných ve vzorku (obr. 5). Limitujícími jsou metodické i technologické faktory. Z hlediska metodiky často dochází při přípravě vzorků ke ztrátám části analyzovaného materiálu a v případě technologie (např. hmotnostní spektrometr) stále není dostačující přístrojové vybavení pro zachycení iontů všech proteinových komponent přítomných v analyzovaném vzorku. V návaznosti na tento proces je nutný i vývoj zpracování a vyhodnocení obsáhlých dat získaných z hmotnostních spektrometrů. Firmy i akademická pracoviště investují nemalé úsilí do vývoje dokonalejších algoritmů schopných zpracovat obrovské množství informací tak, aby se k uživateli dostaly ve formě snadno interpretovatelných výsledků. Cílem proteomiky je systematická kvantifikace proteinů v buňkách nebo tkáních, a tím sledování změn mezi různými biologickými stavy, např. zdravý vs nemocný. Detekce těchto změn přispívá ke stanovení specifických proteinových biomarkerů, které je pak možné využít k diagnostice nebo prognostice různých typů nádorových onemocnění. V posledních letech se stále více diskutuje o personalizované léčbě, kdy by každému pacientovi mohla být ordinována účinná léčba právě na základě detekovaných biomarkerů přítomných v krvi či tkáni pacienta. Kvantifikace proteinů hmotnostní spektrometrií je metoda náročná na přípravu a zpracování vzorků a závislá na finančně nákladném přístrojovém vybavení. Není tedy vhodná pro rutinní testování vzorků odebraných pacientům v rámci jejich vyšetření. Dává však možnost primárně odhalit nové biomarkery nádorových onemocnění a detekovat změny v biologických systémech, které jsou základem pro vývoj jednodušších a levnějších přístupů a jejich aplikací v běžné klinické praxi.
Práce byla podpořena Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI – RECAMO, CZ.1.05/2.1.00/03.0101) a MZ ČR – RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. Ing. Lenka Hernychová, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: lenka.hernychova@mou.cz
Obdrženo: 20. 1. 2014
Přijato: 7. 4. 2014
Sources
1. Psort.hgc.jp [homepage on the Internet]. PSORT www Server. University of Tokio, Japan; c2007 [cited 2014 February 17]. Available from: http:/ / psort.hgc.jp/ .
2. Tatusov RL, Galperin MY, Natale DA et al. The COG database: a tool for genome ‑ scale analysis of protein functions and evolution. Nucleic Acids Res 2000; 28(1): 33 – 36.
3. Genome.jp/ kegg [homepage on the Internet]. KEGG: Kyoto Encyclopedia of Genes and Genomes. Kanehisa Laboratories, Japan; c1995 – 2014 [cited 2014 February 17]. Available from: http:/ / www.genome.jp/ kegg/ .
4. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008; 4(1): 44 – 57. doi: 10.1038/ nprot.2008.211.
5. Hsls.pitt.edu/ molbio/ ipa [homepage on the Internet]. Search.HSLS.MolBio, Health Sciences Library system; c1996 – 2014 [cited 2014 February 17]. Available from: http:/ / www.hsls.pitt.edu/ molbio/ ipa.
6. van Iersel MP, Kelder T, Pico AR et al. Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 2008; 9(1): 399. doi: 10.1186/ 1471 - 2105 - 9 - 399.
7. Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 2006; 7(12): 952 – 958.
8. Ong SE, Blagoev B, Kratchmarova I et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002; 1(5): 376 – 386.
9. Marcilla M, Alpizar A, Paradela A et al. A systematic approach to assess amino acid conversions in SILAC experiments. Talanta 2011; 84(2): 430 – 436. doi: 10.1016/ j.talanta.2011.01.050.
10. Geiger T, Madden SF, Gallagher MW et al. Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res 2012; 72(9): 2428 – 2439. doi: 10.1158/ 0008 - 5472.CAN ‑ 11 - 3711.
11. Krüger M, Moser M, Ussar S et al. SILAC mouse for quantitative proteomics uncovers kindlin‑3 as an essential factor for red blood cell function. Cell 2008; 134(2): 353 – 364. doi: 10.1016/ j.cell.2008.05.033.
12. Geiger T, Cox J, Ostasiewicz P et al. Super ‑ SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods 2010; 7(5): 383 – 385. doi: 10.1038/ nmeth.1446.
13. Boersema PJ, Geiger T, Wisniewski JR et al. Quantification of the N ‑ glycosylated secretome by super ‑ SILAC during breast cancer progression and in human blood samples. Mol Cell Proteomics MCP 2013; 12(1): 158 – 171. doi: 10.1074/ mcp.M112.023614.
14. Ross PL, Huang YN, Marchese JN et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine ‑ reactive isobaric tagging reagents. Mol Cell Proteomics MCP 2004; 3(12): 1154 – 1169.
15. Rauniyar N, Gao B, McClatchy DB et al. Comparison of protein expression ratios observed by sixplex and duplex TMT labeling method. J Proteome Res 2013; 12(2): 1031 – 1039. doi: 10.1021/ pr3008896.
16. Rehman I, Evans CA, Glen A et al. iTRAQ identification of candidate serum biomarkers associated with metastatic progression of human prostate cancer. PloS One 2012; 7(2): e30885. doi: 10.1371/ journal.pone.0030885.
17. Chen Y, Choong LY, Lin Q et al. Differential expression of novel tyrosine kinase substrates during breast cancer development. Mol Cell Proteomics MCP 2007; 6(12): 2072 – 2087.
18. Liu NQ, Dekker LJ, Stingl C et al. Quantitative proteomic analysis of microdissected breast cancer tissues: comparison of label‑free and SILAC‑based quantification with shotgun, directed, and targeted MS approaches. J Proteome Res 2013; 12(10): 4627 – 4641. doi: 10.1021/ pr4005794.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Exprese a purifikace proteinů
- Metody studia buněčné migrace a invazivity nádorových buněk
- Sekvenování nové generace a možnosti jeho využití v onkologické praxi
- Analýza proteinů pomocí hmotnostní spektrometrie