
FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2
Fanconi anemia is a genetic disease characterized by bone marrow failure, congenital malformations and cancer predisposition. Cells derived from Fanconi anemia patients have a dysfunctional FA-BRCA pathway and are deficient in the repair of a specific form of DNA damage, DNA interstrand-crosslinks, that are induced by certain chemotherapeutic drugs. Therefore, the study of FA-BRCA pathway regulation is essential for developing new treatments for Fanconi anemia patients and cancer patients in general. One of the first steps in the pathway is the detection of DNA lesions by the FA core complex. We have optimized a method to visualize the recruitment of the FA core complex to sites of DNA damage and, for the first time, explored how this process occurs. We have uncovered several factors that are required for this recruitment. Among them is a FA core complex substrate, FANCI. We report that non-phosphorylated FANCI, previously believed to be an inactive form, has an important role in the recruitment of the FA core complex and DNA interstrand-crosslink repair. Our findings change the current view of the FA-BRCA pathway and have implications for potential clinical strategies aimed at activating or inhibiting the FA-BRCA pathway.
Published in the journal:
FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005563
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005563
Summary
Fanconi anemia is a genetic disease characterized by bone marrow failure, congenital malformations and cancer predisposition. Cells derived from Fanconi anemia patients have a dysfunctional FA-BRCA pathway and are deficient in the repair of a specific form of DNA damage, DNA interstrand-crosslinks, that are induced by certain chemotherapeutic drugs. Therefore, the study of FA-BRCA pathway regulation is essential for developing new treatments for Fanconi anemia patients and cancer patients in general. One of the first steps in the pathway is the detection of DNA lesions by the FA core complex. We have optimized a method to visualize the recruitment of the FA core complex to sites of DNA damage and, for the first time, explored how this process occurs. We have uncovered several factors that are required for this recruitment. Among them is a FA core complex substrate, FANCI. We report that non-phosphorylated FANCI, previously believed to be an inactive form, has an important role in the recruitment of the FA core complex and DNA interstrand-crosslink repair. Our findings change the current view of the FA-BRCA pathway and have implications for potential clinical strategies aimed at activating or inhibiting the FA-BRCA pathway.
Introduction
Fanconi anemia (FA) is a rare genetic disorder characterized by bone marrow failure, congenital malformations and cancer susceptibility [1]. Eighteen FA genes have been identified (FANC-A, -B, -C, -D1/BRCA2, -D2, -E, -F, -G, -I, -J/BRIP1, -L, -M, -N/PALB2, -O/RAD51C, -P/SLX4, -Q/XPF, -S/BRCA1 and -T/UBE2T) [2–9]. The FA proteins function in a common DNA repair pathway (the FA pathway or the FA-BRCA pathway), which coordinates the repair of interstrand-crosslinks (ICLs). Disruption of this pathway renders cells sensitive to ICL-inducing agents, such as mitomycin C (MMC) [10]. Several FA genes are also breast and ovarian cancer susceptibility genes (FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2, FANCO/RAD51C, and FANCS/BRCA1). Among these, BRCA2, PALB2, RAD51C and BRCA1 have well-defined roles in homologous recombination (HR), linking the FA pathway to HR-mediated repair [11–13].
Eight of the FA proteins (FANC-A, -B, -C, -E, -F, -G, -L and -M) form a ubiquitin ligase complex (the FA core complex) with other associated proteins (FAAP-10/MHF2, -16/MHF1, -20, -24 and -100) in the nucleus [14–16]. Among the FA core complex subunits, FANCM, in complex with FAAP24, is a platform for recruiting the rest of the FA core complex to chromatin [17, 18]. The FA core complex is involved in sensing the DNA lesions and monoubiquitinates FANCD2 and FANCI [14–16, 19]. Monoubiquitination of FANCD2 and FANCI is required for their localization to sites of DNA damage and efficient ICL-repair [20, 21]. FANCD2 and FANCI work together as a protein complex (the ID complex) [21, 22]. Optimal monoubiquitination of FANCD2 and FANCI also requires the ATR-dependent phosphorylation of FANCI at the S/TQ cluster domain [23, 24].
Many proteins involved in DNA repair, including several FA proteins (FANCD2, FANCI, FANCJ, BRCA2, BRCA1, PALB2, SLX4, XPF and BRCA1), accumulate at sites of DNA damage. This accumulation can be visualized as distinct nuclear foci. Visualization of foci has been a fundamental technique for understanding DNA repair pathways: (i) uncovering new players, (ii) identifying sequential steps in the pathways, (iii) understanding the interplay between different DNA repair pathways, and (iv) identifying mechanisms of regulation [25]. However, the recruitment of the FA core complex is poorly understood, due to the difficulty of detecting FA core complex proteins as nuclear foci, although FA core complex accumulation at sites of DNA damage has been sporadically reported [26–28]. To address this, we have optimized the immunocytochemical detection method, allowing the visualization of the FA core complex foci. This enabled us, for the first time, to comprehensively analyze how this early process in the activation of the FA pathway is regulated. Surprisingly, we have found that FANCI, which has been shown to work downstream of the FA core complex, is also required for efficient accumulation of the FA core complex at sites of DNA damage. This FANCI function was independent of its binding partner FANCD2, FANCI monoubiquitination and FANCI phosphorylation. USP1 regulated FA core complex foci formation by deubiquitinating FANCI. Additionally, BRCA1 was required for efficient FA core complex foci formation. Our work challenges the linear, canonical model of the FA-BRCA pathway, and expands on the mechanism of its activation.
Results
FA core complex proteins accumulate at sites of DNA damage in a manner dependent on the whole FA core complex
We carefully optimized immunostaining methods (described in Methods) and successfully detected nuclear foci of FANCA, FANCC, FANCE, FANCF, FANCG and FANCL in U2OS cells treated with MMC (Fig 1A). All of these foci were reduced in FANCA-depleted cells, suggesting that foci formation of the FA core complex proteins is FANCA-dependent and are likely to represent foci formation of the canonical FA core complex. Specificity of the antibodies was also confirmed by western blotting (S1G, S2E, S3C and S3D Figs). The formation of FANCA, FANCG and FANCE foci was induced by treatment with various DNA damaging agents (MMC, cisplatin, hydroxyurea, a PARP inhibitor (AZD2281) and ionizing radiation (IR)), in U2OS cells (Fig 1B and S1A Fig). FANCA and FANCG foci were also observed in HeLa cells and fibroblasts (S1B, S1C and S1E–S1G Fig). FANCA substantially colocalized with FANCD2 and γH2AX, but not TRF1, suggesting that FA core complex proteins localize at sites of DNA damage and not at telomeres (Fig 1C and S1D Fig). Recruitment of FANCA, FANCG, FANCC and FANCF to laser-induced localized DNA damage was also detected (Fig 1D). We were also successful in detecting foci formation of exogenously expressed myc-tagged FANCG (S4 Fig). In this case, detection of the foci was dependent on the level of expression. While high FANCG expression (pMMP-FANCG construct) resulted in pan-nuclear FANCG staining, foci were clearly detected (both with antibodies against FANCG and MYC-tag) when a low expressing (pLentiX1-mycFANCG) was used (S4A and S4B Fig). Both high-expression and low-expression FANCG constructs rescued MMC sensitivity at the same level (S4C Fig).
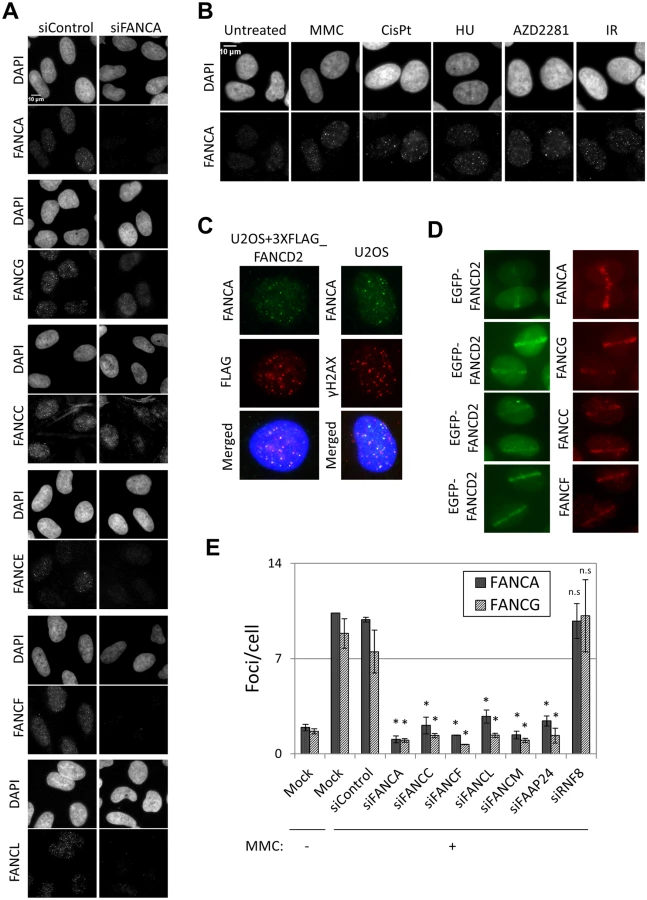
The fact that all FA core complex proteins we tested formed foci at sites of DNA damage and that their recruitment was dependent on FANCA (Fig 1A) suggests that these proteins are present at sites of DNA damage as part of the FA core complex. To test this further, we examined the effects of depleting other components of the FA core complex on FA core complex foci formation. Depletion of FANCA, FANCC, FANCF or FANCL abolished the formation of FANCA, FANCG, FANCC, FANCL and FANCD2 foci, but not γ-H2AX foci (Fig 1E and S2A–S2C Fig). Similarly, formation of FANCA and FANCG foci was impaired in FANCF-deficient cells (TOV21G), but was restored in the corrected cells (S2D and S2E Fig). The FA core complex is loaded onto chromatin through its interaction with FANCM/FAAP24 [17]. RNF8 has also been reported to mediate the recruitment of the FA core complex to psoralen- and laser-induced localized DNA damage [29]. FANCM and FAAP24 depletion abrogated the formation of FANCA, FANCG, FANCC and FANCL foci after MMC without affecting their protein levels (Fig 1E and S2A, S3A and S3B Figs). On the other hand, RNF8 depletion did not affect FANCA, FANCG, FANCC, FANCL or FANCD2 foci, while BRCA1 foci were reduced (Fig 1E and S2A, S2B, S2F and S2G Fig), consistent with the previous reports [30]. These findings suggest that the whole FA core complex including FANCM/FAAP24, but not RNF8, is critical for recruitment of the FA core complex to sites of DNA damage.
FA core complex foci form in S and G2 phases of the cell cycle
The foci formation kinetics of FA core complex was similar to that of FANCD2, both after MMC pulse treatment (Fig 2A) and IR exposure (S5A Fig). Cell synchronization after release from nocodazole arrest revealed that FANCA foci were efficiently induced in S and G2 phases, in untreated cells or after IR, but not in G1 (Fig 2B). Furthermore, more than 95% of cells with FANCA or FANCG foci were cyclin A (a S/G2-phase marker)-positive (Fig 2C). These data indicate that FA core complex foci form during S and G2 phases. Similar results were obtained for FANCD2 foci (Fig 2C).
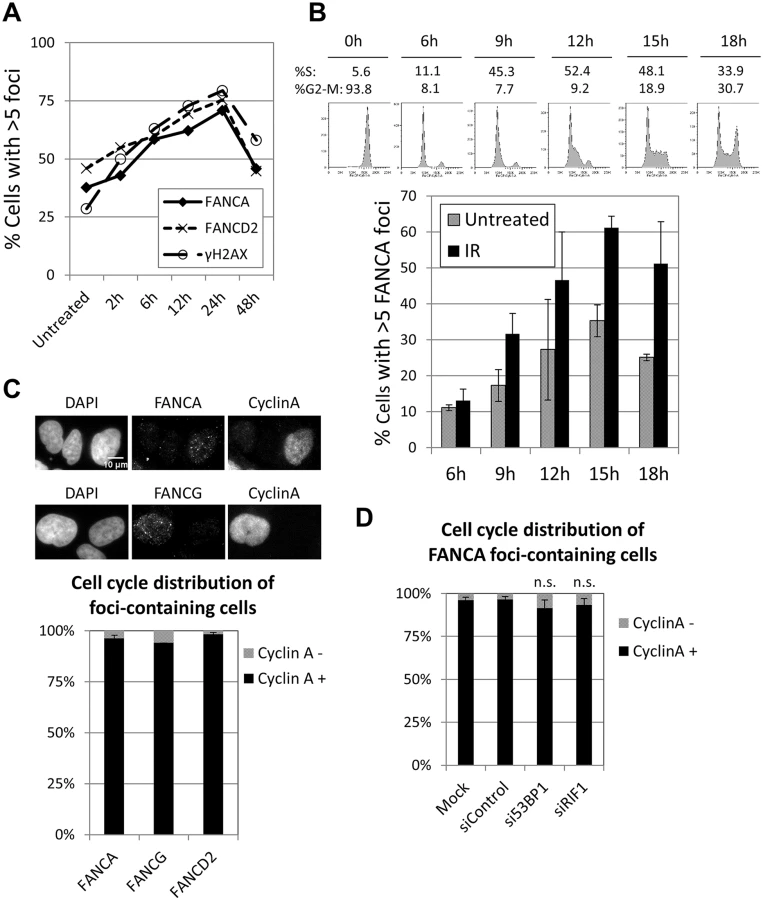
BRCA1, which normally forms foci in S and G2 phases, is able to localize to sites of DNA damage during G1 when 53BP1 or RIF1 are absent [31, 32]. Consistent with these reports, the proportion of BRCA1 foci-containing cells was vastly greater than the proportion of cyclin A-positive cells when 53BP1 or RIF1 were depleted (S5C and S5D Fig), demonstrating that BRCA1 foci formed in G1 in these conditions. In contrast, depletion of 53BP1 or RIF1 did not significantly alter the cell cycle distribution of FANCA (or FANCD2) foci-containing cells, with more than 90% of them corresponding to cyclin A-positive cells (Fig 2D and S5B Fig). These results indicate that the FA core complex and FANCD2 foci form almost exclusively in S and G2, even in the absence of 53BP1 or RIF1, and suggest that the mechanisms of recruitment are distinct from those of BRCA1.
ATR is required for FA core complex foci formation
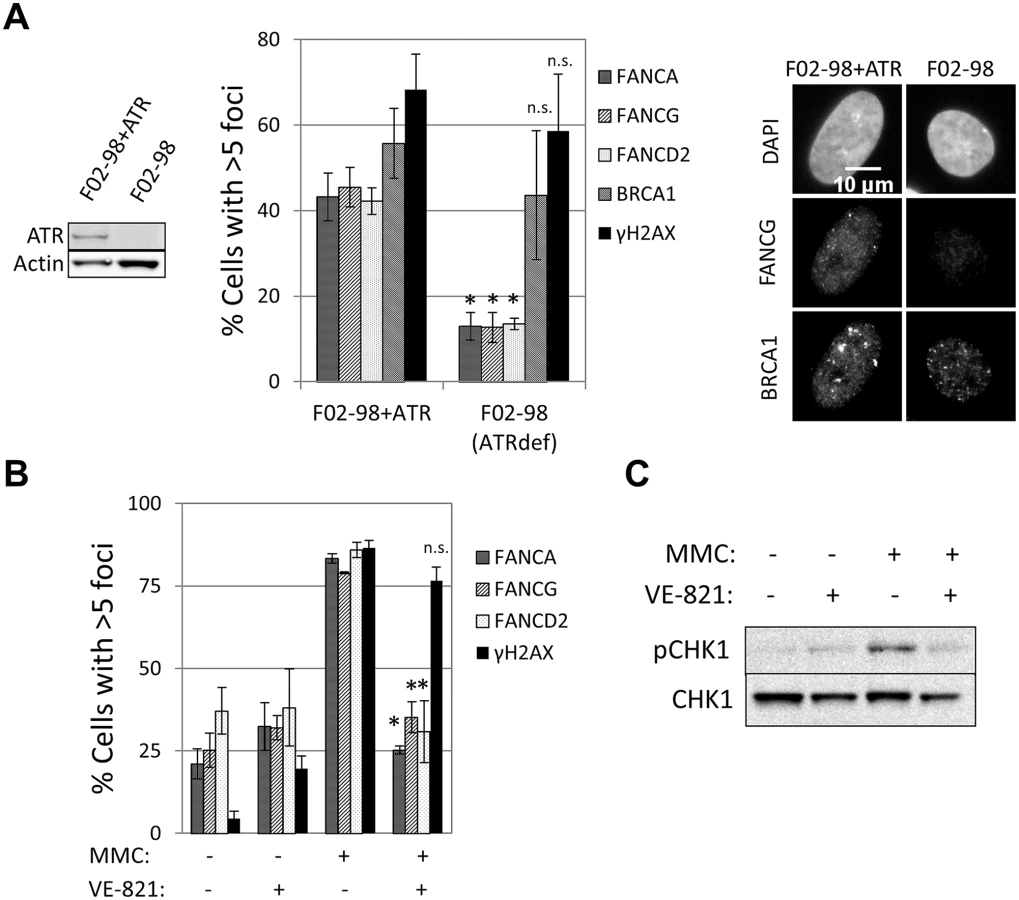
The ability to detect recruitment of FA core complex proteins at sites of DNA damage allowed us to search for factors required for the formation of these foci and therefore gain deeper insight into how the FA pathway is regulated. ATR is the primary kinase that controls FA pathway activation [23, 24, 33]. However, whether ATR is required for FA core complex recruitment is unknown. A strong reduction in the number of cells containing FANCA, FANCG, FANCC, FANCL or FANCD2 foci was observed in the ATR-deficient cells (F02-98 fibroblasts and ATR-depleted U2OS cells), but not in ATR-proficient control cells (Fig 3A and S6A and S6B Fig). ATR specific inhibitor (VE-821) [34], but not ATM inhibitor, impaired the formation of both FA core complex and FANCD2 foci (Fig 3B and S3C, S6C and S6D Fig), indicating that ATR kinase activity is required for FA core complex foci formation.
FANCI, but not FANCD2, is required for FA core complex foci formation
FANCD2 and FANCI function together downstream of the FA core complex [21, 22]. Unexpectedly, we observed a decrease in FANCA, FANCG, FANCC and FANCL foci formation when FANCI, but not FANCD2, was depleted in U2OS cells (Fig 4A and 4B and S7 Fig), suggesting that only FANCI is required for efficient FA core complex foci formation. Consistent with this, FANCD2-deficient fibroblasts (PD20) and PD20 transfected with wild-type FANCD2 or non-ubiquitinatable K561R mutant of FANCD2 showed similar degrees of FANCA foci formation (Fig 4C).

In the absence of FANCD2, FANCI was not ubiquitinated (Fig 4B) and was not bound to chromatin (S10C Fig), suggesting that FANCI ubiquitination and chromatin binding are dispensable for FA core complex foci formation. In a FANCI-deficient fibroblast cell line F010191 [22], wild-type FANCI, but not a non-ubiquitinatable K523R mutant or a DNA-binding mutant K294E/K339E [35], were able to sustain FANCD2 foci (Fig 4D and S7B Fig), as previously described [36]. In contrast, the three FANCI constructs (wild-type, K523R and K294E/K339E) rescued FANCA foci (Fig 4D and S7B–S7D Fig), indicating that FANCI is required for FA core complex foci independently of its ubiquitination and DNA binding.
FANCI promotes FA core complex foci formation independently of ATR
Next we tested if ATR-mediated phosphorylation of FANCI is required for FANCA foci formation. FANCI-deficient F010191 fibroblasts were transduced with wild-type FANCI, a series of FANCI phosphomutants (Ax2-Ax6) or a phosphomimetic mutant (Dx6) (Fig 5A). Consistent with a previous report [24], increasing the number of mutated phosphorylation sites (Ax2-Ax6) resulted in progressively stronger suppression of FANCD2 foci formation and monoubiquitination (Fig 5B and S8 Fig). Expression of the Dx6 mutant resulted in increased basal FANCD2 and FANCI ubiquitination and partial restoration of FANCD2 foci (Fig 5B and S8 Fig). In contrast, all FANCI phosphomutants were able to rescue FANCA foci, while the Dx6 mutant rescued FANCA foci partially (Fig 5B and 5C). Similar results were obtained using FANCI-knockout HCT116 cells (S9 Fig). In this cell line, however, transduction of the Dx6 mutant resulted in a full correction of FANCA foci. These results demonstrate that FANCI promotes FA core complex foci formation independently of the phosphorylation status of the S/TQ cluster domain.
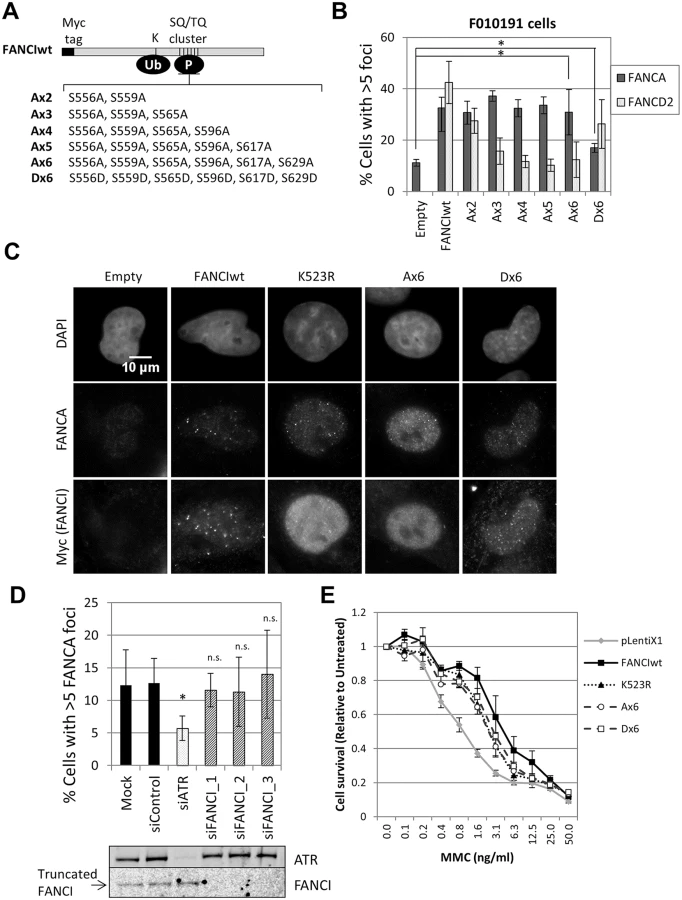
ATR could also promote FA core complex foci formation through FANCI phosphorylation at other sites outside the S/TQ cluster domain. To test if ATR and FANCI promote FA core complex foci formation through shared or distinct mechanisms, we depleted ATR in FANCI-deficient cells (F010191). Compared to FANCI deficiency only, ATR depletion resulted in an additional defect in FANCA foci formation (Fig 5D), suggesting that ATR and FANCI promote FA core complex recruitment through independent mechanisms. Identical results were obtained when ATR and FANCI were co-depleted in U2OS (S10A Fig). F010191 cells express a C-terminal truncated form of FANCI [22, 36]. Depletion of this truncated FANCI did not further suppress FANCA foci, indicating that the truncated FANCI does not support FA core complex foci formation (Fig 5D).
To better understand how ATR and FANCI promote recruitment of the FA core complex to sites of DNA damage, we analyzed the binding of FANCA, FANCC and FANCL to chromatin using cell fractionation. Relative to untreated cells, the amount of chromatin-bound FANCA, FANCC or FANCL was not increased in MMC-treated cells (S10D Fig), indicating that FA core complex chromatin binding and foci formation are two discrete steps. In U2OS cells, FANCI depletion, but not ATR or FANCD2 depletion, significantly reduced the amount of FANCA bound to chromatin (insoluble fraction) (S10B and S10C Fig). This data further supports the previous observations that FANCI regulates FA core complex recruitment at sites of DNA damage in a FANCD2- independent manner, and through a mechanism distinct from ATR-mediated recruitment. However, the same defect in FANCA chromatin binding was not observed when comparing FANCI-knockout and wild-type HCT116 (S10E Fig), although depletion of FANCM did not cause the expected reduction of chromatin-bound FANCA either. The cause of discrepancy between the two cell lines is not obvious. It suggests that FANCI may facilitate two different steps in the regulation of FA core complex recruitment at sites of DNA damage in a context-dependent manner: 1) binding to chromatin, and 2) accumulation at sites of DNA damage (or foci formation).
Non-phosphorylated FANCI contributes to cellular resistance to MMC
Our data indicates that non-phosphorylated FANCI is able to perform part of its functions within the FA pathway by promoting FA core complex recruitment. Therefore, we hypothesized that non-phosphorylated FANCI may contribute to cellular resistance to ICL-inducing agents. To test this, we analyzed MMC sensitivity of FANCI-deficient F010191 cells transduced with wild-type, K523R, Ax6 or Dx6 FANCI constructs. Consistent with our hypothesis, K523R, Ax6 and Dx6 mutants partially rescued MMC resistance, at similar levels (Fig 5E). Similar results were obtained using FANCI-knockout HCT116 cells (S9D Fig). This suggests that FA core complex foci formation may play a role in conferring cellular resistance to ICL-inducing agents.
BRCA1 and USP1 are required for FA core complex foci formation
Our demonstration of a role for FANCI upstream of FA core complex recruitment suggests that the FA pathway may not be as linear as current models suggest. It also raises the possibility that other factors known to work downstream of the FA core complex may also regulate FA core complex recruitment. To test this, we analyzed FA core complex foci formation in cells deficient in several other proteins involved in the FA pathway (BRCA1/FANCS, BRCA2/FANCD1, CtIP, FANCJ/BRIP1, FANCN/PALB2, FANCP/SLX4, FANCQ/XPF and USP1) (S2 Table). Surprisingly, both BRCA1 and USP1 were required for FANCA and FANCG foci formation.
Depletion of BRCA1 resulted in a strong reduction in FANCA, FANCG, FANCC, FANCL and FANCD2 foci formation in U2OS and HeLa cells (Fig 6A and S11A and S11B Fig), without affecting FANCA protein levels or FANCD2 monoubiquitination (S11C Fig). A similar reduction in FA core complex foci-containing cells was observed in BRCA1-deficient HCC1937, when compared to BRCA1-complemented HCC1937 (Fig 6B). To better characterize how BRCA1 promotes FA core complex foci formation, we then tested whether this function was mediated by known BRCA1-interacting proteins (S2 Table and S11D–S11H Fig). No defect in FANCA or FANCG foci was observed in cells deficient in FANCJ/BRIP1, FANCD1/BRCA2, FANCN/PALB2, FAM175A/ABRAXAS, UIMC1/RAP80 or RBBP8/CtIP, suggesting that BRCA1 performs this function independently, or through a binding partner other than the ones tested.
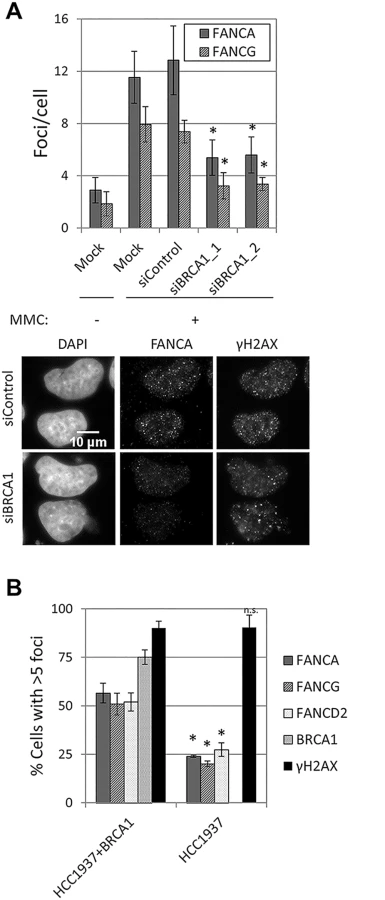
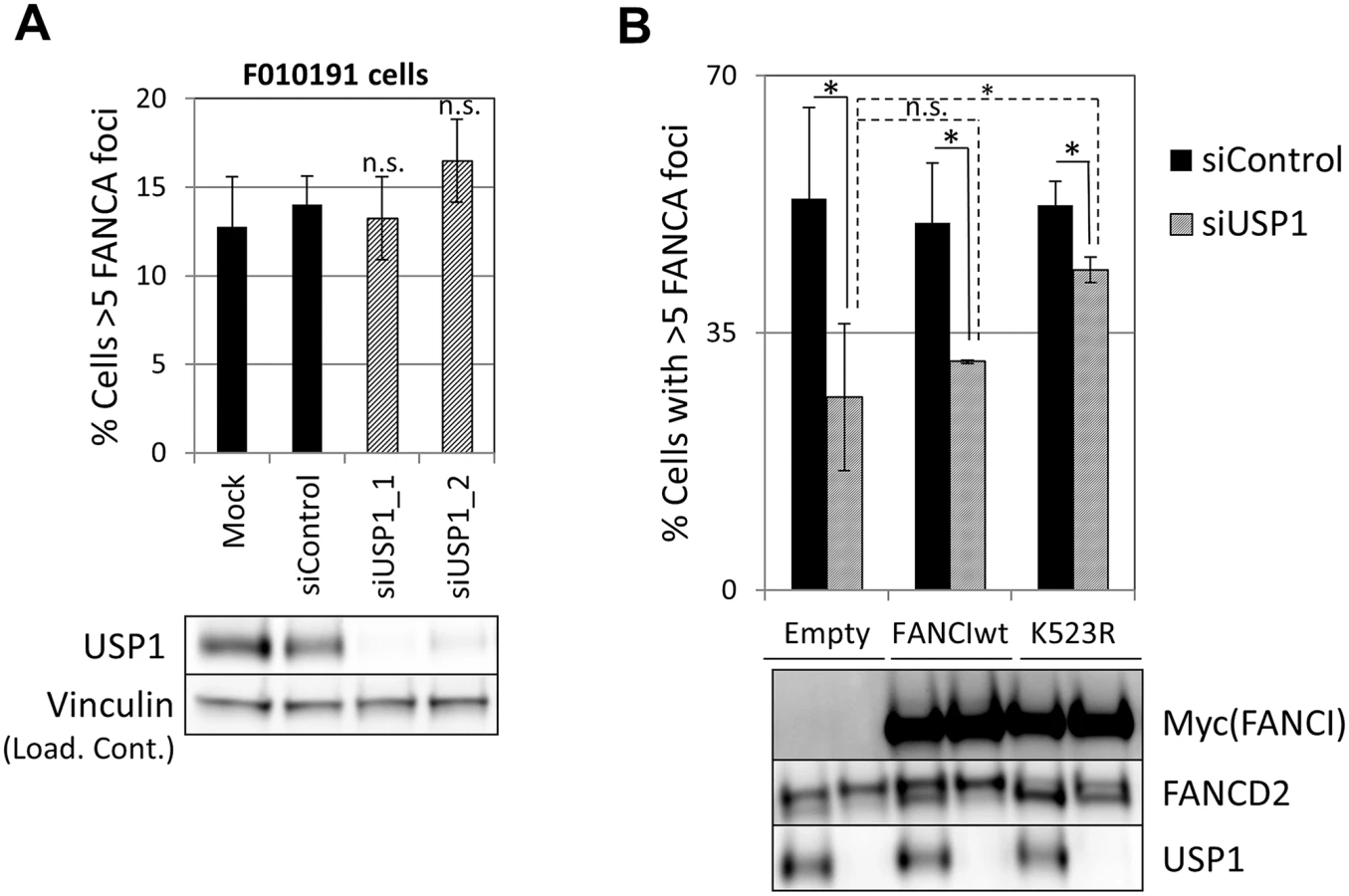
FANCA,FANCG, FANCC and FANCL foci formation were impaired in USP1-depleted U2OS cells and in cells treated with a USP1 specific inhibitor, ML323 [37] (Fig 7A and 7B and S12A Fig). Increasing concentrations of ML323 resulted in decreased FANCA and FANCD2 foci, without affecting BRCA1 or γH2AX foci (Fig 7B). ML323 treatment resulted in increased FANCD2 and FANCI ubiquitination, confirming USP1 inhibition (S12B Fig). To confirm the siRNA and inhibitor data, USP1-depleted cells were complemented using an siRNA-resistant USP1 cDNA. Due to the high instability of overexpressed wild-type USP1 protein, a form of USP1 that is unable to cleave itself, GG670/671AA, (described in [38]) was used. The catalytic function of GG670/671AA mutant of USP1 is comparable to wild-type USP1 [38]. The FA core complex and FANCD2 foci defect was rescued when wild-type USP1, but not the catalytic inactive form (USP1 C90S), was expressed (Fig 7C and 7D and S12C Fig). In both cases, an overall reduction in the number of cells with foci was observed, likely due to cellular toxicity of USP1 overexpression. These data indicate that catalytic activity of USP1 is required to promote efficient recruitment of FA core complex and FANCD2 to sites of DNA damage. They also indicate that impaired FA core complex recruitment at sites of DNA damage does not necessarily translate into deficient FANCD2-FANCI ubiquitination.

Deubiquitination of FANCI by USP1 is required for FA core complex foci formation
Since both non-ubiquitinated FANCI and USP1 catalytic activity promoted FA core complex foci formation, next we tested the possibility that FANCI was the relevant substrate for USP1 in this function. We first tested epistasis between FANCI and USP1. USP1 was depleted using siRNA from FANCI-deficient F010191 cells. As shown in Fig 8A, USP1 depletion did not result in an increased loss of FANCA foci in FANCI-deficient cells, suggesting that FANCI and USP1 promote FA core complex foci formation through the same mechanism. If FANCI is the relevant USP1 substrate to promote FA core complex formation, overexpression of a non-ubiquitinatable FANCI should be able to rescue FA core complex foci formation in USP1-depleted cells. Therefore, we overexpressed wild-type or K523R mutant of FANCI in cells that were either transfected with siRNA control or siUSP1. As shown in Fig 8B, overexpression of the FANCI K523R mutant partially rescued FANCA foci formation in USP1-depleted cells, while the wild-type FANCI was not able to do so. These results suggest that FANCI needs to be deubiquitinated by USP1 to promote FA core complex foci efficiently.
Discussion
Through the analyses of the FA core complex foci formation, we have elucidated several new regulatory mechanisms of the FA core complex recruitment. First, the FA core complex accumulated at sites of DNA damage in a manner dependent on the whole FA core complex, including FANCM/FAAP24, the canonical platform that loads the FA core complex to DNA [17]. An alternative mechanism of FA core complex recruitment at laser-induced localized ICLs, involving FAAP20 binding to RNF8-catalyzed polyubiquitin chains, has been described [29]. However, this mechanism did not make a significant contribution in our system. The discrepancy may be attributable to the different systems used to induce DNA damage and exemplifies the importance of assessing recruitment using foci formation.
Repair of DSBs in mammalian cells occurs mainly by two major different pathways: non-homologous end-joining (NHEJ), which predominates during G1, and homologous recombination (HR), which predominates during S and G2 [39]. As with other proteins involved in the FA pathway and HR, FA core complex foci formed during S and G2. Recently, some light has been shed on the molecular mechanisms that control DNA repair pathway choice, identifying 53BP1-RIF1 and BRCA1-CtIP as central players in this process [31, 32, 40–42]. These studies suggest that enabling BRCA1 to bind to DSBs by depleting 53BP1 or RIF1, allows for CtIP-mediated resection in G1. However, depleting 53BP1 or RIF1 was not enough to promote FA core complex or FANCD2 recruitment in G1. This data is consistent with our observation that, although efficient FA core complex recruitment depended on BRCA1, it did not depend on CtIP. Therefore, alternative activation mechanisms and/or DNA structures, not present in G1, will be needed for FA core complex recruitment at DNA lesions. Consistent with this, in vitro studies showed that FANCM, together with FAAP24 and MHF1-MHF2, have a strong affinity for branched DNA structures that resemble replication forks or Holliday junctions [43–45].
The recruitment of the FA core complex at sites of DNA damage is far from well understood. Our data suggests that the regulation of this process is more complex than initially envisioned. Through a candidate approach directed to proteins that participate in the repair of ICLs, we have identified four proteins that are required for FA core complex foci formation: ATR, FANCI, BRCA1 and USP1. Among these, FANCI, BRCA1 and USP1 are especially interesting, since they have been previously thought to function exclusively downstream of FA core complex. Our findings suggest that they also act upstream, by promoting FA core complex recruitment to sites of DNA damage. We show that FANCI has a function upstream of the FA core complex and independent of FANCD2. FANCD2 and FANCI were previously considered to be obligate partners: they require each other for ubiquitination, foci formation and, partially, protein stability [21, 22, 46]. More recent studies, however, have shown that FANCD2 and FANCI exhibit different responses to DNA damage [47]. Also, a FANCI-independent function of FANCD2 in promoting replication fork recovery through association with the BLMcx complex has been reported [48]. Our study supports the model that FANCD2 and FANCI have both dependent and independent roles in the DNA damage response, and identifies FA core complex foci formation as a novel FANCD2-independent function of FANCI.
Unlike FANCI function in promoting FANCD2 foci formation and ubiquitination, FA core complex recruitment by FANCI was independent of FANCI DNA binding, ubiquitination and phosphorylation of the S/TQ cluster domain, and was also distinct from the ATR-mediated mechanism. All these data together show that FANCI has at least two independent functions within the FA pathway: (i) regulation of FANCD2 foci/ubiquitination and (ii) regulation of FA core complex foci.
Both FANCI phosphomutant (Ax6) and phosphomimetic mutant (Dx6), as well as the non-ubiquitinatable FANCI (K523R), significantly rescued MMC sensitivity in two different human FANCI-deficient cell lines. This data differs from studies in chicken DT40 cells, where the Ax6 mutant did not rescue sensitivity to ICL-inducing agents [24]. The discrepancy may be attributable to differences between human and chicken systems. It is particularly interesting that a phosphomutant FANCI (Ax6), while unable to support FANCD2 foci and ubiquitination, largely rescued MMC sensitivity. This data is consistent with an additional role of non-phosphorylated, non-ubiquitinated FANCI in the repair of ICLs. It is tempting to speculate that the two different pools of FANCI (phosphorylated-ubiquitinated and non-phosphorylated, non-ubiquitinated FANCI) may contribute independently to ICL-resistance by performing different functions. In support of this model, a function of unphosphorylated FANCI in the regulation of dormant origin firing was recently reported [49]. Our findings together with this report emphasize that both phosphorylated FANCI and unphosphorylated FANCI are needed for cellular resistance to ICLs. In the work presented by Chen and coworkers [49], FANCI phosphomimic mutant Dx6 cannot activate dormant origins or reverse MMC sensitivity in FANCI-depleted retinal pigment epithelial (RPE) cells. However, in our studies, the Dx6 mutant partially restored MMC resistance in FANCI-deficient patient-derived fibroblasts and FANCI-knockout HCT116 cells. This discrepancy may be explained by differential dependence on dormant origin firing for ICL tolerance among the cell lines.
Our results indicate that FA core complex binding to chromatin precedes, and is independent of, foci formation (S10D Fig). FANCI may affect the ability of the FA core complex to bind to chromatin in some cell lines such as U2OS. However, this phenomenon was not observed in HCT116 cells where FANCI was still required for the FA core complex to accumulate or persist at sites of DNA damage. Further investigations will be required to understand how this process occurs and to identify the reasons for these differences among cell lines.
ATR kinase activity was required for FA core complex foci formation. This was not unexpected, as ATR is involved in the activation of the FA pathway [23]. However, our findings suggest that ATR mediates FA core complex foci formation independently of FANCI phosphorylation, a well-characterized mechanism of ATR-mediated FA pathway activation [24]. Therefore, ATR will control the FA pathway at, at least, two steps. The relevant substrate of ATR in the FA core complex foci formation remains unknown. Possible candidates include FANCA, FANCG or FANCM, as they are phosphorylated by ATR [33, 50, 51].
The search for other factors with roles upstream of FA core complex foci formation uncovered two additional positive regulators: BRCA1 and USP1. This finding was surprising, since depletion of neither of these two proteins decrease the level of ubiquitination of FANCD2 or FANCI, and their ubiquitination is even increased in the case of USP1 depletion. These findings have, at least, two possible explanations: (i) residual levels of FA core complex foci formation observed in cells deficient for BRCA1 or USP1 are enough to induce normal (or increased) FANCD2-FANCI ubiquitination, or (ii) FA core complex foci formation and FANCD2-FANCI ubiquitination are uncoupled. Regardless of which of the two scenarios is correct, the fact that FA core complex foci and FANCD2-FANCI ubiquitination do not correlate suggests that the accumulation of the FA core complex at sites of DNA damage may serve additional functions. In support of this model, a mutation in FANCA (I939S) that does not impair FANCD2 monoubiquitination was recently described in an FA patient [52]. Also, the fact that depletion of USP1 results in increased ubiquitination of FANCD2-FANCI in the absence of DNA damage (our data and [53]) suggests that ubiquitination may not necessarily happen at sites of DNA damage, and may therefore be disconnected from FA core complex foci formation. It would be interesting to test if FA core complex foci are able to form in FANCA I939S and/or other separation-of-function mutants.
We showed that deubiquitination of FANCI by USP1 is an important step to recruit the FA core complex at sites of DNA damage. This result uncovers the existence of a feedback loop, centered on FANCI: not only ubiquitination, but also deubiquitination, is required for the proper functioning of the FA pathway. We have also, for the first time, uncovered a role of USP1 as a positive regulator of the FA pathway. More experiments are required to assess if deubiquitination of FANCI alone can explain ICL sensitivity of USP1-deficient cells.
It is important to note that BRCA1 or USP1 deficiency also leads to impaired FANCD2 foci formation [20, 54]. How BRCA1 promotes recruitment of both FANCD2 and FA core complex remains enigmatic. A recent in vitro study shows that BRCA1 acts in ICL repair upstream of DSB formation by facilitating eviction of the replicative helicase [55]. Therefore, this step could be required for FANCD2 and FA core complex foci formation
Taken together, we have optimized a protocol to visually detect the recruitment of the FA core complex to sites of DNA damage, which allowed us to identify regulators of FA core complex recruitment, and therefore, elucidate a previously unexplored aspect of the FA pathway. With the remarkable findings that FANCI, USP1 and BRCA1, as well as ATR and FANCM, promote FA core complex foci formation, we provide evidence that the FA pathway is a non-linear, tightly regulated pathway, with several proteins (ATR, FANCI, USP1 and BRCA1) performing roles at multiple stages of its activation (S13 Fig).
Materials and Methods
Cell lines and culture conditions
U2OS, HeLa, HCC1937 and TOV-21G were purchased from the American Type Culture Collections. FANCF-corrected TOV21G [56], SV40 transformed FA fibroblasts (326SV FA-G-/-, GM6914 FA-A-/-, PD20 FA-D2-/-) and their corrected counterparts [20, 57–59], hTERT-immortalized ATR deficient F02-98 fibroblasts and their corrected counterparts [23, 60], VU423 fibroblasts and VU423 corrected with chromosome 13[11], have been described. F010191 transformed fibroblasts [22] were a gift from Dr. Tony Huang (New York University). AG656 fibroblasts and AG656+FANCJ were a gift from Dr. Sharon Cantor (University of Massachusetts). EUFA1341 fibroblasts and EUFA1341+PALB2 were a gift from Dr. Paul Andreassen (Cincinnati Children’s Hospital). HCC1937 cells were cultured in RPMI supplemented with 15% FCS. HCT116 FANCI-/- were grown in McCoy’s 5A media supplemented with 10% FCS and 1% glutamine. All other cell lines were grown in DMEM supplemented with 10% FCS. All cells were maintained in a humidified 5% CO2 atmosphere at 37°C. Cells were treated with MMC (Sigma, M4287), cisplatin (Sigma, P4394), hydroxyurea (HU) (Sigma, H8627), AZD2281 (Selleckchem, S1060) ionizing radiation (IR) (JL Shepherd Mark I Cesium Irradiator (JL Shepherd & Associates)), ATR inhibitor VE-821 (Axon Medchem, 1893), ATM inhibitor KU55933 (Selleckchem, S1092) or USP1 inhibitor, ML323 [37](gift from Dr. Zhihao Zhuang, University of Delaware).
Gene knockdown by RNA interference
siRNA transfections were conducted in 6-well plates using Lipofectamine RNAiMAX (Invitrogen), following the manufacturer’s instructions. 20nM siRNA was used in each transfection. siRNA sequences are provided in S1 Table.
Generation of FANCI-null HCT116 cells
FANCI-null HCT116 cells were generated using recombinant adeno-associated virus (rAAV)-mediated gene targeting [61]. Conditional and knock-out rAAV vectors targeting FANCI exon 10 were constructed as described [62, 63]. The first round of targeting with the conditional vector replaced FANCI exon 10 with a floxed exon 10 along with a neomycin (Neo) drug selection cassette. G418-resistant clones were screened by PCR to confirm correct targeting and the Cre recombinase was subsequently used to remove the Neo selection cassette. Retention of the floxed exon 10 in the conditional allele was confirmed by PCR. The second round of gene targeting was performed with a knock-out vector that replaced exon 10 with a Neo-selection cassette. G418 resistant clones were again screened by PCR for correct targeting. Cre recombinase was subsequently used to remove both the Neo selection cassette and the floxed conditional allele and this resulted in viable FANCI-null clones. The PCR primers flanking FANCI exon 10 that were used to confirm both conditional and null alleles were FancIc_GG_LIF: GCAATGGCACAATCTTGG and FancIcond_GG_loxR: ATAGAACTTTCTGGCTTGCT.
Cell fractionation and western blotting
Cell fractionations were prepared as described [17]. Briefly, cells were resuspended in buffer CSK (10mM PIPES, pH = 6.8, 100mM NaCl, 1mM EGTA, 1mM EDTA, 300mM Sucrose, 1.5mM MgCl2, 0.1% Triton-X-100 and protease inhibitors) and incubated in ice for 5min. Samples were centrifuged at 1500g for 5min. Supernatant was collected and stored (soluble fraction). Pellets (insoluble fraction) were washed once in CSK buffer and then resuspended in sample buffer (0.05 M Tris-HCl (pH 6.8), 2% SDS, 6% β-mercaptoethanol) and boiled for 5min. Western blotting was performed as described [64]. Briefly, cells were lysed in 0.05 M Tris-HCl (pH 6.8), 2% SDS, 6% β-mercaptoethanol and boiled for 5min. SDS-PAGE electrophoresis was done using NuPAGE 3% to 8% Tris-acetate or NuPAGE 4% to 12% Tris-glycine gels (Invitrogen) and proteins were transferred to a nitrocellulose membrane. Primary antibodies were diluted in blocking buffer (5% milk in TBS-Tween 20) and incubated overnight. Horseradish peroxidase—conjugated anti-mouse and anti-rabbit IgG (Amersham) were used as secondary antibodies. Images were acquired using ImageQuant LAS4000 system (GE Healthcare).
Plasmids and cell transduction
Human FANCI coding sequence was amplified by PCR to include a Myc tag sequence at the 5’ end of the gene and cloned into pLentiX1-puro (a gift from Eric Campeau, Addgene plasmid # 20953) using SalI and XbaI sites. Ubiquitin C promoter was extracted from pUB-GFP (a gift from Connie Cepko, Addgene plasmid # 11155) using a SalI digestion. The resulting 1.2 Kb fragment was cloned into pLentiX1-MycFANCI at the SalI site. K523R, Ax2-Ax6, Dx6 and KKEE mutants of FANCI were generated using an overlap extension PCR method. Myc-USP1 and Myc-FANCG were cloned into pLentiX1-puro using the same strategy.
FANCI-containing lentiviruses were produced as described [65]. F010191 fibroblasts or FANCI-deficient HCT116 cells were transduced with fresh lentiviruses-containing supernatants and selected for 2 days with 2μg/ml puromycin. Fresh transductions were used in each experiment, as exogenous FANCI expression was lost when cells underwent prolonged culture after transduction.
Immunofluorescence and microscopy
Cells were grown on coverslips and then fixed and permeabilized for 30 minutes using 4% paraformaldehyde (Santa Cruz, sc-281692) containing 0.5% Triton X-100. After fixation, cells were washed with PBS and then blocked for 15 minutes in PBS containing 3% BSA+ 0.1% Tween20. Primary antibodies were diluted in blocking buffer and incubation was performed at room temperature for one hour on a rocker. After 3 washes with PBS + 0.1% Tween20, cells were incubated with secondary antibodies diluted in blocking buffer for another 45 minutes. AlexaFluor 488 Goat Anti-Rabbit IgG and AlexaFluor 594 Goat Anti-Mouse IgG were used as secondary antibodies (Molecular Probes). At this point, 1μg/ml of 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) was added to the cells and incubated for an additional 15 minutes. Cells were washed 3 times with PBS + 0.1% Tween20 and then coverslips were mounted using Vectashield Mounting Media (VectorLabs, H-1000). Image acquisitions were made with a TE2000 Nikon microscope equipped with a 60X immersion objective and a CCD camera (CoolSNAP ES, Photometrics). Images were acquired and analyzed using MetaVue (Universal Imaging) and ImageJ. Manual counting was used and cells containing more than 5 foci were scored as positive. When possible, cells were seeded in glass-bottom 96 well-plates (Greiner Bio-One, 655892) and images were acquired with Cellomics ArrayScan automated microscope equipped with a 20X objective. In this case, foci counting was automated using Cellomics software and reported as “foci per cell”.
Laser-induced DNA damage
To generate localized DNA lesions, we used a published method [66] with some modifications. Cells were grown on 35mm glass bottom Fluorodish cell culture dishes (World Precision Instruments) for 24 hours before the experiment. One hour before the experiment, cells were placed in warm CO2-independent medium (Life Technologies). Cells were pre-sensitized with 10 μg/ml viable Hoechst dye 33258 (Sigma-Aldrich) for 5 min at 37°C. We performed laser microirradiation using an Nikon Ti fully-motorized inverted spinning disk confocal microscope (UltraView Vox, Perkin Elmer) equipped with a 37°C heating chamber and a 405 nm laser focused through a Nikon Plan Fluor 40x/1.3 NA oil objective. We set the laser output to 100% of maximum power to generate in three iterations in a restricted region detectable localized DNA damage that was restricted to the laser path, dependent on prior pre-sensitization of the cells, and without noticeable cytoxicity. Cells were fixed 30 minutes after generating DNA damage and stained with the indicated antibodies.
Antibodies
The following primary antibodies were used: anti-FANCD2 (Abcam, ab2187), anti-FANCA (Bethyl, A301-980A), anti-Vinculin (Sigma, V9131), anti-ATR (Santa Cruz, sc-1887), anti-FANCI (Santa Cruz, sc-271316), anti-Actin (Santa Cruz, sc-1616), anti-BRCA1 (Santa Cruz, sc-6954), anti-CHK1 (Santa Cruz, sc-8408), anti-pCHK1 S345 (Cell Signalling, 23415), anti-FLAG M2 (Sigma, F1804), anti-γH2AX (Upstate, JBW3001), anti-CyclinA (Abcam, ab16726), anti-MYC tag 9E10 (Upstate, 05–419), anti-TRF1 (Abcam, ab10579), anti-RAD51 (CosmoBio, BAM-70-001-EX), anti-FANCJ (Sigma, B1312), anti-BRCA2 (Calbiochem, OP95), anti-ABRAXAS (Bethyl, A302-180A), anti-RAP80 (Bethyl, A300-763A). Anti-FANCG [67], FANCC [68], FANCE [69], FANCF [70] and FANCD2 pT691 [71] were gifts from Dr. Alan D’Andrea (Dana-Farber Cancer Institute, Boston). Anti-USP1 (C-ter) [38] was a gift from Dr. Tony Huang. Anti-PALB2 [72] was a gift from Drs. David Livingston and Bing Xia. Anti-FANCL and anti-FANCM were gifts from Dr. Weidong Wang.
Cell synchronization and cell cycle analysis
Cell synchronization in M-phase was achieved by incubating cells in nocodazole (Sigma, M1404) at 0.1μg/ml for 16 hours. Floating cells (M-phase cells) were then recovered by shaking the culture flask, and re-seeded in 6 cm dishes and on coverslips in fresh medium without nocodazole. During a period of 21 hours, cells were collected every 3h for cell cycle analysis and immunostaining. For each time point, coverslips were irradiated with 10Gy IR 60min prior the collection. For cell cycle analysis, cells were fixed in ice-cold 70% ethanol for 16 hours. Then, ethanol was removed and cells were incubated in PBS + propidium iodide 40μg/ml + RNase A 0.1mg/ml for 30 minutes at 37°C. After that, cells were kept on ice until analysis by flow cytometry. Flow cytometry analyses were performed on BD FACSCanto II flow cytometers and analyzed with Flow Jo software.
Survival assay
Cell survival was measured by a crystal violet absorbance-based assay. Cells were seeded onto 12-well plates at a density of 9000 cells/well. The next day, cells were treated with increasing concentrations of MMC and incubated for eight more days. After that, plates were processed as described [65].
RT-PCR
mRNA was converted to cDNA using Superscript III Reverse Transcriptase (Invitrogen) following manufacturer’s instructions. To measure specific gene expression, the following sets of primers were used: GAPDH: 5’ GGAGTCAACGGATTTGGTCG 3’ and 5’ CTCCTGGAAGATGGTGATGG 3’; RNF8: 5’ AAGATGGGTGCGAGGTGACT 3’ and 5’ ACGCGCTCTGTTCAGCCAAA 3’.
Statistics
All statistical analyses were done using Student’s t-test (2-tail). P value < 0.05 was considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nature reviews Genetics. 2001;2(6):446–57. Epub 2001/06/05. 11389461
2. Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. American journal of human genetics. 2013;92(5):800–6. Epub 2013/04/30. doi: 10.1016/j.ajhg.2013.04.002 23623386
3. Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nature genetics. 2011;43(2):142–6. Epub 2011/01/18. doi: 10.1038/ng.750 21240275
4. Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian G, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer discovery. 2015;5(2):135–42. Epub 2014/12/05. doi: 10.1158/2159-8290.CD-14-1156 25472942
5. Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nature genetics. 2010;42(5):406–9. Epub 2010/04/20. doi: 10.1038/ng.570 20400963
6. Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature reviews Genetics. 2007;8(10):735–48. Epub 2007/09/05. 17768402
7. Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. American journal of human genetics. 2015;96(6):1001–7. doi: 10.1016/j.ajhg.2015.04.022 26046368
8. Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell reports. 2015;12(1):35–41. doi: 10.1016/j.celrep.2015.06.014 26119737
9. Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL, et al. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet. 2015.
10. Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes & development. 2012;26(13):1393–408. Epub 2012/07/04.
11. Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–9. Epub 2002/06/18. 12065746
12. Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nature genetics. 2010;42(5):410–4. Epub 2010/04/20. doi: 10.1038/ng.569 20400964
13. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature genetics. 2007;39(2):165–7. Epub 2007/01/04. 17200668
14. Medhurst AL, Laghmani el H, Steltenpool J, Ferrer M, Fontaine C, de Groot J, et al. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 2006;108(6):2072–80. Epub 2006/05/25. 16720839
15. Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, et al. Modularized functions of the Fanconi anemia core complex. Cell reports. 2014;7(6):1849–57. Epub 2014/06/10. doi: 10.1016/j.celrep.2014.04.029 24910428
16. Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, et al. The genetic and biochemical basis of FANCD2 monoubiquitination. Molecular cell. 2014;54(5):858–69. Epub 2014/06/07. doi: 10.1016/j.molcel.2014.05.001 24905007
17. Kim JM, Kee Y, Gurtan A, D'Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111(10):5215–22. Epub 2008/01/05. doi: 10.1182/blood-2007-09-113092 18174376
18. Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature genetics. 2005;37(9):958–63. Epub 2005/08/24. 16116422
19. Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nature genetics. 2003;35(2):165–70. Epub 2003/09/16. 12973351
20. Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001;7(2):249–62. Epub 2001/03/10. 11239454
21. Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. Epub 2007/04/07. 17412408
22. Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nature structural & molecular biology. 2007;14(6):564–7. Epub 2007/04/27.
23. Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes & development. 2004;18(16):1958–63. Epub 2004/08/18.
24. Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nature structural & molecular biology. 2008;15(11):1138–46. Epub 2008/10/22.
25. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes & development. 2011;25(5):409–33. Epub 2011/03/03.
26. Castella M, Pujol R, Callen E, Trujillo JP, Casado JA, Gille H, et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood. 2011;117(14):3759–69. Epub 2011/01/29. doi: 10.1182/blood-2010-08-299917 21273304
27. Mi J, Kupfer GM. The Fanconi anemia core complex associates with chromatin during S phase. Blood. 2005;105(2):759–66. Epub 2004/07/17. 15256425
28. Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW. Nonerythroid alphaII spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. Journal of cell science. 2003;116(Pt 5):823–35. Epub 2003/02/07. 12571280
29. Yan Z, Guo R, Paramasivam M, Shen W, Ling C, Fox D 3rd, et al. A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Molecular cell. 2012;47(1):61–75. Epub 2012/06/19. doi: 10.1016/j.molcel.2012.05.026 22705371
30. Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131(5):887–900. Epub 2007/11/16. 18001824
31. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell. 2013;49(5):872–83. Epub 2013/01/22. doi: 10.1016/j.molcel.2013.01.001 23333306
32. Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. The Journal of biological chemistry. 2013;288(16):11135–43. Epub 2013/03/15. doi: 10.1074/jbc.M113.457440 23486525
33. Singh TR, Ali AM, Paramasivam M, Pradhan A, Wahengbam K, Seidman MM, et al. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer research. 2013;73(14):4300–10. Epub 2013/05/24. doi: 10.1158/0008-5472.CAN-12-3976 23698467
34. Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nature chemical biology. 2011;7(7):428–30. Epub 2011/04/15. doi: 10.1038/nchembio.573 21490603
35. Longerich S, Kwon Y, Tsai MS, Hlaing AS, Kupfer GM, Sung P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic acids research. 2014;42(9):5657–70. doi: 10.1093/nar/gku198 24623813
36. Colnaghi L, Jones MJ, Cotto-Rios XM, Schindler D, Hanenberg H, Huang TT. Patient-derived C-terminal mutation of FANCI causes protein mislocalization and reveals putative EDGE motif function in DNA repair. Blood. 2011;117(7):2247–56. Epub 2010/10/26. doi: 10.1182/blood-2010-07-295758 20971953
37. Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nature chemical biology. 2014;10(4):298–304. Epub 2014/02/18. doi: 10.1038/nchembio.1455 24531842
38. Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nature cell biology. 2006;8(4):339–47. Epub 2006/03/15. 16531995
39. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Molecular cell. 2012;47(4):497–510. Epub 2012/08/28. doi: 10.1016/j.molcel.2012.07.029 22920291
40. Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54. Epub 2010/04/07. doi: 10.1016/j.cell.2010.03.012 20362325
41. Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339(6120):711–5. Epub 2013/01/12. doi: 10.1126/science.1230624 23306439
42. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339(6120):700–4. Epub 2013/01/12. doi: 10.1126/science.1231573 23306437
43. Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Molecular cell. 2007;25(3):331–43. Epub 2007/02/10. 17289582
44. Fox D 3rd, Yan Z, Ling C, Zhao Y, Lee DY, Fukagawa T, et al. The histone-fold complex MHF is remodeled by FANCM to recognize branched DNA and protect genome stability. Cell research. 2014;24(5):560–75. Epub 2014/04/05. doi: 10.1038/cr.2014.42 24699063
45. Zhao Q, Saro D, Sachpatzidis A, Singh TR, Schlingman D, Zheng XF, et al. The MHF complex senses branched DNA by binding a pair of crossover DNA duplexes. Nature communications. 2014;5:2987. Epub 2014/01/07. doi: 10.1038/ncomms3987 24390579
46. Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cellular oncology: the official journal of the International Society for Cellular Oncology. 2007;29(3):211–8. Epub 2007/04/25.
47. Sareen A, Chaudhury I, Adams N, Sobeck A. Fanconi anemia proteins FANCD2 and FANCI exhibit different DNA damage responses during S-phase. Nucleic acids research. 2012;40(17):8425–39. Epub 2012/07/04. 22753026
48. Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic acids research. 2013;41(13):6444–59. Epub 2013/05/10. doi: 10.1093/nar/gkt348 23658231
49. Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, et al. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Molecular cell. 2015;58(2):323–38. Epub 2015/04/07. doi: 10.1016/j.molcel.2015.02.031 25843623
50. Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, et al. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113(10):2181–90. Epub 2008/12/26. doi: 10.1182/blood-2008-05-154294 19109555
51. Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, et al. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene. 2008;27(26):3641–52. Epub 2008/01/24. doi: 10.1038/sj.onc.1211034 18212739
52. Xie J, Kim H, Moreau LA, Puhalla S, Garber J, Al Abo M, et al. RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. The Journal of clinical investigation. 2015. Epub 2015/03/10.
53. Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Molecular cell. 2005;17(3):331–9. Epub 2005/02/08. 15694335
54. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Developmental cell. 2009;16(2):314–20. Epub 2009/02/17. doi: 10.1016/j.devcel.2009.01.001 19217432
55. Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Molecular cell. 2014;56(1):174–85. Epub 2014/09/16. doi: 10.1016/j.molcel.2014.08.012 25219499
56. Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nature medicine. 2003;9(5):568–74. Epub 2003/04/15. 12692539
57. Duckworth-Rysiecki G, Toji L, Ng J, Clarke C, Buchwald M. Characterization of a simian virus 40-transformed Fanconi anemia fibroblast cell line. Mutation research. 1986;166(2):207–14. Epub 1986/09/01. 3020399
58. Jakobs PM, Sahaayaruban P, Saito H, Reifsteck C, Olson S, Joenje H, et al. Immortalization of four new Fanconi anemia fibroblast cell lines by an improved procedure. Somatic cell and molecular genetics. 1996;22(2):151–7. Epub 1996/03/01. 8782494
59. Nakanishi K, Moran A, Hays T, Kuang Y, Fox E, Garneau D, et al. Functional analysis of patient-derived mutations in the Fanconi anemia gene, FANCG/XRCC9. Experimental hematology. 2001;29(7):842–9. Epub 2001/07/05. 11438206
60. O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature genetics. 2003;33(4):497–501. Epub 2003/03/18. 12640452
61. Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic acids research. 2004;32(1):e3. Epub 2004/01/06. 14704360
62. Fattah KR, Ruis BL, Hendrickson EA. Mutations to Ku reveal differences in human somatic cell lines. DNA repair. 2008;7(5):762–74. Epub 2008/04/05. doi: 10.1016/j.dnarep.2008.02.008 18387344
63. Oh S, Harvey A, Zimbric J, Wang Y, Nguyen T, Jackson PJ, et al. DNA ligase III and DNA ligase IV carry out genetically distinct forms of end joining in human somatic cells. DNA repair. 2014;21:97–110. Epub 2014/05/20. doi: 10.1016/j.dnarep.2014.04.015 24837021
64. Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer research. 2007;67(15):7395–405. Epub 2007/08/03. 17671210
65. Wang Y, Huang JW, Li M, Cavenee WK, Mitchell PS, Zhou X, et al. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Molecular cancer research: MCR. 2011;9(8):1100–11. Epub 2011/06/23. doi: 10.1158/1541-7786.MCR-11-0007 21693595
66. Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. The Journal of cell biology. 1999;146(5):905–16. Epub 1999/09/09. 10477747
67. Garcia-Higuera I, Kuang Y, Naf D, Wasik J, D'Andrea AD. Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Molecular and cellular biology. 1999;19(7):4866–73. Epub 1999/06/22. 10373536
68. Kupfer GM, Naf D, Suliman A, Pulsipher M, D'Andrea AD. The Fanconi anaemia proteins, FAA and FAC, interact to form a nuclear complex. Nature genetics. 1997;17(4):487–90. Epub 1997/12/17. 9398857
69. Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Molecular and cellular biology. 2007;27(8):3098–108. Epub 2007/02/14. 17296736
70. Siddique MA, Nakanishi K, Taniguchi T, Grompe M, D'Andrea AD. Function of the Fanconi anemia pathway in Fanconi anemia complementation group F and D1 cells. Experimental hematology. 2001;29(12):1448–55. Epub 2001/12/26. 11750104
71. Ho GP, Margossian S, Taniguchi T, D'Andrea AD. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Molecular and cellular biology. 2006;26(18):7005–15. Epub 2006/09/01. 16943440
72. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular cell. 2006;22(6):719–29. Epub 2006/06/24. 16793542
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 10
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice