
FLCN and AMPK Confer Resistance to Hyperosmotic Stress via Remodeling of Glycogen Stores
The ability of an organism to adapt to sudden changes in environmental osmolarity is critical to ensure growth, propagation, and survival. The synthesis of organic osmolytes is a common adaptive strategy to survive hyperosmotic stress. However, it was not well understood, which biosynthetic pathways and storage strategies were used by organisms to rapidly generate osmolytes upon acute hyperosmotic stress. Here, we demonstrate that glycogen is an essential reservoir that is used upon acute hyperosmotic stress to generate the organic osmolyte glycerol promoting fast and efficient protection. Importantly, we show that this pathway is regulated by FLCN-1, an ortholog of the human tumor suppressor Folliculin responsible for the Birt-Hogg-Dubé cancer syndrome, and by AMPK, the master regulator of energy homeostasis.
Published in the journal:
FLCN and AMPK Confer Resistance to Hyperosmotic Stress via Remodeling of Glycogen Stores. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005520
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005520
Summary
The ability of an organism to adapt to sudden changes in environmental osmolarity is critical to ensure growth, propagation, and survival. The synthesis of organic osmolytes is a common adaptive strategy to survive hyperosmotic stress. However, it was not well understood, which biosynthetic pathways and storage strategies were used by organisms to rapidly generate osmolytes upon acute hyperosmotic stress. Here, we demonstrate that glycogen is an essential reservoir that is used upon acute hyperosmotic stress to generate the organic osmolyte glycerol promoting fast and efficient protection. Importantly, we show that this pathway is regulated by FLCN-1, an ortholog of the human tumor suppressor Folliculin responsible for the Birt-Hogg-Dubé cancer syndrome, and by AMPK, the master regulator of energy homeostasis.
Introduction
Water is a fundamental molecule for life and the ability of an organism to adapt to changes in water content is essential to ensure survival. Hyperosmotic stress promotes water efflux, causing cellular shrinkage, protein and DNA damage, cell cycle arrest and cell death. All living organisms encounter hyperosmotic environments [1,2]. In humans, both renal and non renal tissues are exposed to hyperosmotic stress, a condition that is regarded as a major cause for many chronic and fatal human diseases including diabetes, inflammatory bowel disease, hypernatremia, dry eye syndrome, and cancer [1]. Cells/tissues/organisms have evolved adaptive strategies to cope with threatening hyperosmotic environments [1,2]. Among adaptive strategies, the synthesis of compatible organic osmolytes, which keeps cellular osmotic pressure equal to that of the external environment, is widely used by all organisms [3]. In yeast and C. elegans, hyperosmotic stress triggers glycerol production via transcriptional upregulation of glycerol-3-phosphate dehydrogenase-1 (gpdh-1), a rate-limiting enzyme in glycerol synthesis [4,5]. Moreover, several osmotic stress resistance mutants of divergent signaling pathways exhibit a constitutive transcriptional upregulation of gpdh-1, leading to increased glycerol content [6–10].
Here we define a novel hyperosmotic stress resistance pathway mediated by the 5' AMP-activated protein kinase (AMPK), a key regulator of cellular energy balance [11], which is chronically inactivated by the worm ortholog of the renal tumor suppressor Folliculin (FLCN-1). In humans, FLCN is a tumor suppressor gene responsible for the BHD disease, an autosomal dominantly-inherited syndrome associated with increased susceptibility to the development of several cancerous and non cancerous lesions including kidney cancer, pulmonary, renal, pancreatic and hepatic cysts and skin fibrofolliculomas [12–25]. FLCN has been shown to bind AMPK via the scaffold FLCN-interacting proteins FNIP1 and FNIP2 [26,27]. We have recently demonstrated that FLCN negatively regulates AMPK signaling in the nematode C. elegans and in mammalian cells [28,29]. Moreover, loss of FLCN increased ATP levels via heightened flux of glycolysis, oxidative phosphorylation, and autophagy, which resulted in an AMPK-dependent resistance to several metabolic stresses in C. elegans and mammalian cells [28,29].
Here we identify a pathway involved in the physiological response to hyperosmotic stress resistance in C. elegans mediated by FLCN-1 and AMPK. We demonstrate that glycogen is an essential reservoir that is used upon acute hyperosmotic stress to generate glycerol and promote fast and efficient adaptation to prevent water loss and ensure survival. We show that in flcn-1(ok975) mutant animals, this phenotype is significantly enhanced, due to the robust AMPK-mediated accumulation of glycogen, which is rapidly converted to the osmolyte glycerol upon salt stress. Our results also suggest that the FLCN/AMPK pathway might be an evolutionarily conserved key regulator of glycogen metabolism and stress resistance.
Results
Loss of flcn-1 confers resistance to hyperosmotic stress in C. elegans
Since we have previously observed that loss of flcn-1 in C. elegans increases AMPK-dependent resistance to energy stresses including oxidative stress, heat, and anoxia [28], we asked whether it would also increase resistance to hyperosmotic stress. We measured the survival of wt and flcn-1(ok975) animals (S1A Fig) on plates supplemented with 400mM and 500mM NaCl. Loss of flcn-1 conferred a significant increase in resistance to hyperosmotic stress (Fig 1A and 1B and S1 Table). Although NaCl treatment severely reduced the survival of both wt and flcn-1(ok975) animals as compared to untreated animals (Figs 1A, 1B and S1B), the mean survival of flcn-1(ok975) animals increased by ~2 and ~3 fold upon treatment with 400mM and 500mM NaCl respectively, as compared to wt animals (Fig 1A–1C). Moreover, we did not observe a significant difference in lifespan between untreated wt and flcn-1(ok975) animals, as reported previously [28] (S1B Fig and S1 Table). Importantly, NaCl treatment led to shrinkage and paralysis in both wt and flcn-1(ok975) animals. However, flcn-1(ok975) mutant nematodes recover significantly faster than wt animals after 2 hours of NaCl treatment suggesting that the mechanism of adaptation to salt is more robust upon loss flcn-1 (Fig 1D). We also observed a significantly greater number of wt animals with more than 30% reduction of body size as compared to flcn-1 suggesting that loss of flcn-1 activates pathways that favor body size recovery after hyperosmotic stress (Fig 1E). Importantly, the hyperosmotic stress resistance phenotype can be rescued by transgenic re-expression of C. elegans flcn-1 (S1 Table and Figs 1F and S1A).

In addition, we used Agilent whole genome C. elegans microarrays to determine transcriptional profile differences between wt and flcn-1(ok975) mutant animals [30]. Key genes that were differentially expressed were validated by qRT-PCR (S1C Fig). We compared our data to published transcriptional profiles and found a significant overlap between genes upregulated in untreated flcn-1(ok975) animals versus genes upregulated in wt animals treated with NaCl or osmotic stress resistant strains including osm-7 and osm-11 [8] (S1D, S1E and S1F Fig and S2, S3 and S4 Tables). Altogether, these data suggest that flcn-1 is involved in a mechanism of regulating the resistance to hyperosmotic stress.
Loss of flcn-1 increases glycogen content, which mediates resistance to hyperosmotic stress
To determine how loss of flcn-1 increases resistance to hyperosmotic stress, we assessed the morphological differences between wt and flcn-1(ok975) using electron microscopy with or without NaCl treatment. Interestingly, we observed an increase in the size and number of glycogen stores in adult (Fig 2Ai and 2Aii) and L4 (S2Ai, S2Aii, S2Ci, and S2Cii Fig) flcn-1(ok975) mutant worms as compared to wt. Specifically, our transmission electron data indicate a strong accumulation of glycogen in the hypodermis, muscle, and intestine of flcn-1(ok975) animals as compared to wt (S2C Fig). Glycogen has been previously shown to accumulate in these tissues in C. elegans [31]. Importantly, glycogen stores were barely detectable in wt and flcn-1(ok975) animals after NaCl treatment, suggesting that glycogen degradation is used to protect the animals from hyperosmotic stress (Fig 2Aiii, 2Aiv). Furthermore, we found that the prominent accumulation and salt stress-dependent degradation of glycogen in flcn-1(ok975) adult animals occurs in the hypodermis (Figs 2A, S2A and S2C). We validated and quantified the increase in glycogen levels conferred by loss of flcn-1 using iodine staining which has been previously shown to specifically stain glycogen in C. elegans [32–34] (Fig 2B and 2C). In accordance with the electron microscopy results, glycogen levels were significantly increased in untreated flcn-1(ok975) animals as compared to wt, and NaCl treatment severely reduced glycogen content in both wt and flcn-1(ok975) animals (Fig 2B and 2C).

We then asked whether glycogen is used to protect wt and flcn-1(ok975) animals from damage during hyperosmotic stress. Glycogen synthase (gsy-1) is responsible for the synthesis of glycogen from UDP-glucose molecules and glycogen phosphorylase (pygl-1) catalyzes glycogen breakdown to form glucose-1-phosphate [35]. Importantly, the inhibition of glycogen synthesis or degradation using RNAi against gsy-1 and pygl-1 respectively, strongly reduced the survival in both wt and flcn-1(ok975) animals to an equal level, suggesting that the accumulation of glycogen and its degradation are both required for the resistance of wt and flcn-1(ok975) mutant animals to hyperosmotic stress (Fig 2D and 2E and S1 Table).
Additionally, transcript levels of gsy-1 and pygl-1 with or without 2 hours of 400mM NaCl stress remained unchanged suggesting allosteric regulation of glycogen metabolism (Fig 2F). Altogether, these results demonstrate that the accumulation of glycogen stores and the degradation of glycogen are essential to survive hyperosmotic stress in wt and flcn-1(ok975) mutant animals.
Hyperosmotic stress resistance of flcn-1(ok975) animals is dependent on AMPK
Since we have previously reported that the flcn-1-dependent resistance to energy stresses requires aak-2, the worm ortholog of the AMPKα subunit, we wondered whether the hyperosmotic stress resistance phenotype conferred by loss of flcn-1 is also mediated by AMPK [28]. AMPK is activated by hyperosmotic stress in mammalian systems [36] and its deletion confers sensitivity to NaCl stress in yeast [37]. C. elegans nematodes have two catalytic α subunits aak-1 and aak-2. Loss of aak-2 was shown to mediate lifespan extension and resistance to various stresses including oxidative stress, anoxia, nutrient deprivation, and dietary restriction [38–42]. To determine whether AMPK is involved in the increased resistance of flcn-1(ok975) animals to stress, we crossed aak-2(ok524 and gt33) [39,43] and aak-1(tm1944) [43] loss of function mutants with flcn-1(ok975) animals. Interestingly, loss of aak-2(ok524 and gt33) or aak-1(tm1044) alone conferred stress sensitivity but did not fully suppress the increased survival to hyperosmotic stress conferred by loss of flcn-1 (Fig 3A, 3B and 3C and S1 Table). To control for compensatory effects, we generated the flcn-1(ok975); aak-1(tm1944); aak-2(ok524) triple mutant and compared its survival under high salt conditions to aak-1(tm1944); aak-2(ok524) double mutant animals. Simultaneous loss of aak-1 and aak-2 completely abolished the increased osmotic stress resistance upon loss flcn-1 demonstrating that this phenotype requires both AMPK catalytic subunits (Fig 3D and S1 Table).

The accumulation of glycogen in flcn-1 mutant worms depends on AMPK
AMPK has been shown to regulate glycogen metabolism in different organisms [44–56]. In fact, acute activation of AMPK leads to glycogen degradation [44–47], while chronic AMPK activation results in glycogen accumulation [48–50]. Since we observed an increased constitutive phosphorylation of AMPK upon loss of flcn-1 in nematodes and mammalian cells [28,29], we hypothesized that the chronic AMPK activation in flcn-1(ok975) mutants may lead to increased glycogen levels. We determined glycogen levels in aak-1(tm1944); aak-2(ok524) animals compared to flcn-1(ok975); aak-1(tm1944); aak-2(ok524) triple mutant worms and found that loss of AMPK strongly reduced glycogen levels in both strains (Fig 3E and 3F). This suggests that the chronic AMPK activation in flcn-1 animals is leading to glycogen accumulation. Interestingly, the survival and glycogen accumulation in aak-1(tm1944); aak-2(ok524) mutant animals was also severely reduced as compared to wt (Fig 3E and 3F), suggesting an important role for AMPK in maintaining glycogen stores, which are used for hyperosmotic stress resistance.
Autophagy is not fully required for the hyperosmotic stress resistance conferred by loss of flcn-1
Autophagy is a biological survival process through which cellular components and damaged organelles are degraded to produce energy upon starvation [57]. We reported previously that autophagy was essential for the energy stress resistance of flcn-1(ok975) mutant animals [28]. Therefore, we asked whether autophagy plays a role in osmotic stress resistance. Interestingly, atg-18(gk378) mutant animals were hypersensitive to high salt concentrations suggesting that autophagy is a process involved in the resistance to hyperosmotic stress. However, loss of flcn-1 significantly increased the resistance of atg-18(gk378) animals suggesting that flcn-1-dependent hyperosmotic stress resistance does not require autophagy, which is different from what we observed before during energy stress [28] (S3 Fig and S1 Table).
Glycogen degradation leads to heightened glycerol levels and protects animals from hyperosmotic stress
Degradation of glycogen polymers leads to the formation of glucose-1-phosphate which is converted to glucose-6-phosphate, an important metabolite used in multiple pathways including glycolysis, pentose phosphate pathway, and glycerol production (Fig 4A) [35]. We hypothesized that glycogen degradation may lead to heightened glycerol levels that could protect the animals from hyperosmotic stress. To address this, we measured the mRNA levels of gpdh-1 and gpdh-2. Interestingly, we observed a significant 2-fold increase in gpdh-1 but not gpdh-2 at unstressed conditions in flcn-1(ok975) mutant animals compared to wt, which was consistent with our microarray results (Fig 4B and 4C and S2 Table). Strikingly, after 2 hour treatment with 400mM NaCl, we detected a strong induction of gpdh-1 and gpdh-2 mRNA levels in wt and flcn-1(ok975) mutant animals, which was significantly enhanced in the latter (Fig 4B and 4C). Accordingly, flcn-1(ok975) mutant animals exhibit higher glycerol content at basal level as compared to wt animals which was further increased upon NaCl treatment (Fig 4D).
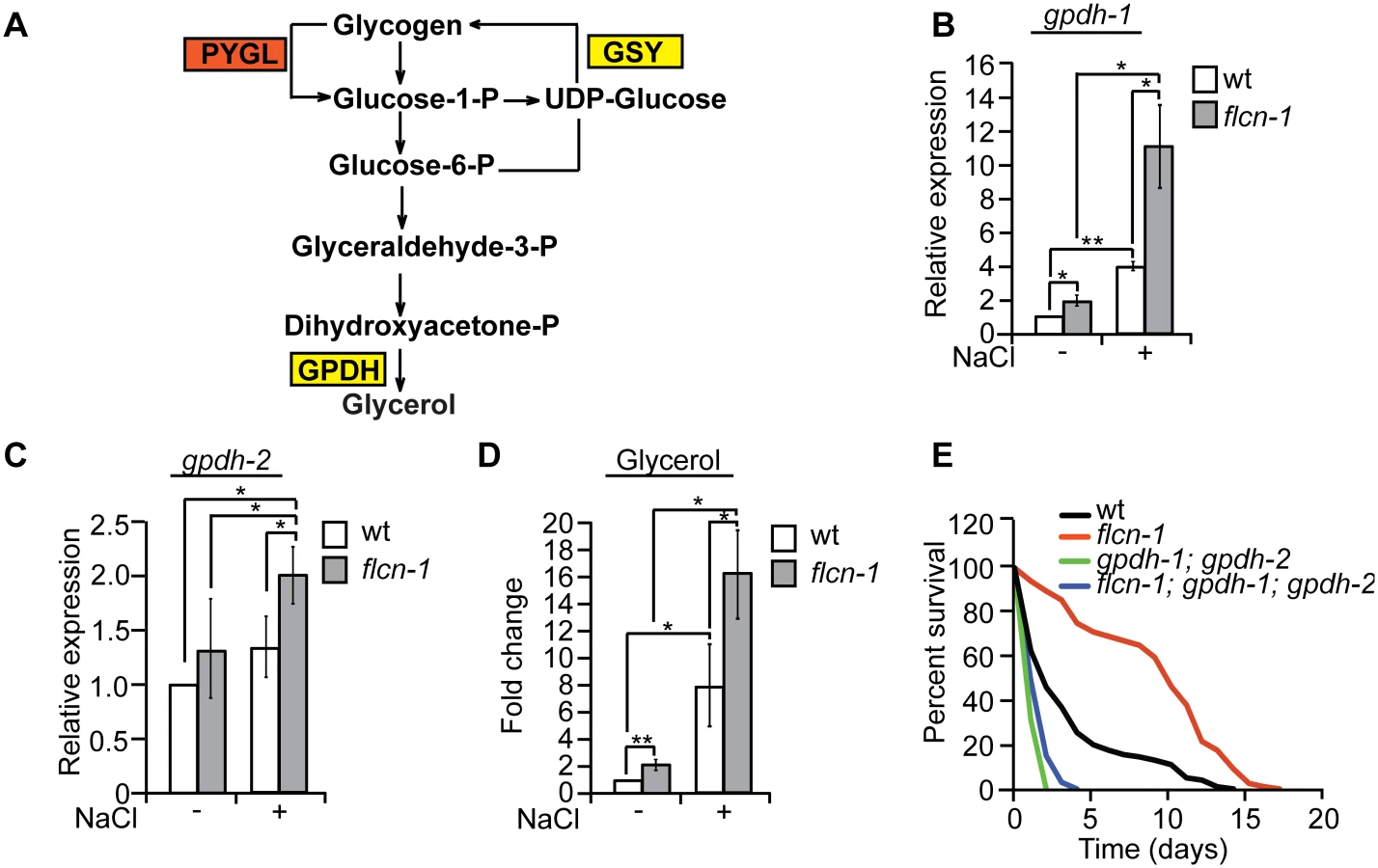
To determine the importance of glycerol in the protection against hyperosmotic stress, we inhibited gpdh-1 and gpdh-2 using RNAi and using mutant strains. Importantly, treatment of flcn-1(ok975) animals with RNAi against either gpdh-1 or gpdh-2 alone did not fully suppress the increased resistance of flcn-1(ok975) animals to hyperosmotic stress (S4A and S4B Fig). We then compared the resistance of flcn-1(ok975); gpdh-1(kb24); gpdh-2(kb33) triple mutant animals to gpdh-1(kb24); gpdh-2(kb33) mutant nematodes. Simultaneous loss of gpdh-1 and gpdh-2 strongly reduced the survival of flcn-1(ok975) mutant animals demonstrating an important role for the osmolyte glycerol in the survival of flcn-1(ok975) and wt animals (Fig 4E and S1 Table). Altogether, these data suggest that upon hyperosmotic stress glycogen stores are metabolized into the osmolyte glycerol via enhanced transcriptional upregulation of gpdh enzymes. This glycerol mediated osmo-protective phenotype is significantly enhanced upon loss of flcn-1 in nematodes.
Loss of pmk-1 does not fully suppress the hyperosmotic stress resistance conferred by loss of flcn-1
HOG/p38/PMK-1 MAP kinase signaling is widely known to control adaptation to hypertonic stresses in multiple organisms [4,9,58]. As expected, pmk-1(km25) mutant worms were highly sensitive to osmotic stress. However, loss of pmk-1 in flcn-1(ok975) mutant animals reduced but did not fully suppress the increased resistance conferred by flcn-1 depletion (S5A Fig and S1 Table). Supporting this result, the expression of gpdh-1 is ~2-fold higher in flcn-1(ok975); pmk-1(km25) mutant animals as compared to pmk-1(km25) alone (S5B Fig). Altogether, this suggests that pmk-1 is not involved in the transcriptional upregulation of gpdh-1 upon loss of flcn-1 and that it acts in parallel to flcn-1 and aak-1/2.
The increased accumulation of glycogen content conferred by loss of FLCN is conserved from C. elegans to humans
Glycogen is linked to the progression and the aggressiveness of multiple cancer types in humans [59,60]. To determine whether loss of FLCN also leads to the accumulation of glycogen in mammalian systems, we used the Flcnflox/flox/Pax8-Cre mouse model where Flcn is specifically deleted in the kidney and determined glycogen content using Periodic-Acid-Schiff (PAS) staining. The Flcnflox/flox/Pax8-Cre mouse was generated by mating Pax8-Cre mice with the Flcn flox/flox C57BL/6 mice. By six months of age, all mice developed visible macroscopic lesions confirmed as cysts that later developed into tumors. Strikingly, kidneys from Flcnflox/flox/Pax8-Cre mice accumulated higher glycogen levels as compared to normal kidneys from Flcnflox/flox mouse littermates (Figs 5A and S6A). Our data show a stronger glycogen accumulation in the kidney cortex, which is due to the fact that Pax8 is expressed in the epithelial cells of the proximal and distal renal tubules, loops of Henle, collecting ducts and the parietal epithelial cells of Bowman’s capsule [61]. Importantly, PAS staining of tumors from BHD patients also indicate a strong accumulation of glycogen as compared to adjacent unaffected kidneys (Figs 5B and S6B). We also compared the expression level of glycogen biosynthesis and degradation genes in 3 different subtypes of kidney cancer, kidney renal papillary cell carcinoma (KIRP), kidney renal clear cell carcinoma (KIRC), and kidney chromophobe (KICH) tumors. Strikingly, we observed a significant upregulation of genes involved in the synthesis and degradation of glycogen (Fig 5C and S5 Table). We also observed that the expression of 46% of these genes are negatively correlated with FLCN expression (Fig 5D). Overall, our data indicate that the accumulation of glycogen upon loss of FLCN is be conserved from nematodes to mammals, and that it might play a role in tumorigenesis.

Discussion
A common mechanism to survive osmotic stress is the synthesis of compatible osmolytes [3]. In yeast and in C. elegans, the rapid accumulation of glycerol after hyperosmotic stress has been demonstrated [4,5]. However, it is not clear what fuels glycerol production upon acute hyperosmotic stress. Here we show that animals have evolved an interesting strategy to maintain glycogen stores, which can serve as fuel for glycerol production to ensure survival to acute hyperosmotic stress (Fig 6). While storage of soluble glucose molecules in cells would lead to osmotic stress, the storage of glucose in the form of insoluble glycogen polymers ensures osmotic homeostasis. Importantly, our data uncover that glycogen stores have a dual role: they can serve as a reservoir for production of energy or osmolytes. Indeed, pretreatment of wt and flcn-1(ok975) animals with oxidative and energy stressor paraquat, depletes glycogen stores rapidly and suppresses survival upon treatment with 400mM NaCl (S2A and S2B Fig).

The regulation of glycogen metabolism by AMPK has long been a paradox [44–50]. Acute activation of AMPK, by in vitro short term treatment of the AMP mimetic drug 5-Aminoimidazole-4-Carboxamide Riboside (AICAR), leads to the phosphorylation and inhibition of glycogen synthase, which favors glycogen degradation for supply of short term energy [44–47]. However, chronic AMPK activation induced by a long term AICAR treatment or by genetic manipulation of AMPK regulatory subunits, results in glycogen accumulation via glucose-6-phosphate-dependent allosteric activation of glycogen synthase, which bypasses the inhibitory effect of the AMPK-mediated phosphorylation [48–50]. In agreement, constitutive AMPK activation through transgenic expression of activating mutations in the γ2 and γ3 subunits in mice and pigs leads to substantial glycogen accumulation in cardiac and skeletal muscles [36,50–53,55,56]. In light of these results, our data indicate that chronic AMPK activation upon loss of flcn-1 leads to glycogen accumulation. Similarly to what has been shown in yeast [54], we demonstrate that AMPK-deficient strains exhibit reduced glycogen content as compared to wt. We further show that the accumulation of glycogen in wt and flcn-1(ok975) mutant animals depends on AMPK. Based on the data presented here together with our recently published reports [28,29], we propose that FLCN is a key regulatory component of AMPK.
Flcn muscle-specific knockout mice and Fnip1 knockout mice exhibited increased glycogen accumulation in muscles and liver, respectively [62,63]. Here we show that loss of FLCN leads to glycogen accumulation in kidneys of mice and in the tumors of BHD patients, suggesting that this pathway is evolutionarily conserved. In agreement with the important role for glycogen in organismal survival to stress, glycogen can be used by tumor cells to survive harsh microenvironments such as hypoxia [59,64]. In fact, glycogen accumulates in many cancer types [64] and inhibition of its degradation led to induction of apoptosis and impaired in vivo growth of tumor xenografts [59].
Importantly, our data might impinge on a novel role for glycogen in tumorigenesis. In addition to its critical role as an energy supplier, we speculate that glycogen degradation might lead to higher osmolyte levels to help survive hyperosmotic tumor microenvironments. In fact, we found that taurine and sorbitol synthesis genes, CSAD and AKR1B1 respectively, are upregulated in many kidney tumors (S5 Table). Supporting this idea, recent evidence shed light on an important role of the nuclear factor of activated T cells 5 (NFAT5), a major transcription factor that regulates osmotic stress resistance genes, in promoting tumorigenesis and metastasis of several cancer types [2,65–67]. In summary, we speculate that the increased glycogen stores in tumors might lead to extended survival of cells under hyperosmotic stress, which could ultimately lead to neoplastic transformation by accumulation of DNA damage [1, 2].
Materials and Methods
C. elegans strains, maintenance, and RNAi treatments
C. elegans strains were obtained from the Caenorhabditis Genetics Center (S6 Table). Nematodes were maintained and synchronized using standard culture methods [68]. The RNAi feeding experiments were performed as described in [69], and bacteria transformed with empty vector were used as control. For all RNAi experiments, phenotypes were scored with the F1 generation.
Osmotic stress resistance assay
To measure osmotic stress resistance, synchronized 1 day adult worms were transferred to high concentration NaCl plates. Survival was measured daily. Worms that responded by movement to touch with the platinum wire were considered as alive.
Percent recovery assay
To measure the percentage of animals that recovered after hyperosmotic shock, 1 day adult animals were transferred to high NaCl plates. Animals shrink and paralyse shortly after exposure to NaCl. After 2 hours, animals that were able to move their entire body forward or backward in response to touch with a platinum wire were considered as “recovered”. Paralyzed animals often look straight and are unable to move.
RNA extraction and real-time PCR
Synchronized young adult nematodes were harvested and total RNA was extracted with Trizol. Reverse transcription and qRT-PCRs were performed as previously described [28]. Transcripts were normalized to cdc-42.
Microarray experiment and gene overlap analysis
Synchronized young adult wt and flcn-1(ok975) animals were harvested and RNA was extracted using Trizol and purified using Qiagen RNeasy columns. Total RNA samples were then hybridized onto Agilent gene chips. Fold change values are calculated using the mean of both data sets. The overlapping genes between flcn-1(ok975) mutant animals and the specified conditions and strains [8] were performed using the “compare two lists” online tool at http://www.nemates.org/MA/progs/Compare.html. The significance of the overlap and enrichment scores were determined via hypergeometric distribution method using http://nemates.org/MA/progs/overlap_stats.html. The number of genes in the C. elegans genome was considered to be19,735.
Transmission electron microscopy
Synchronized 1 day adult nematodes were transferred to 400mM NaCl plates for 16 hours. Recovering animals were picked and transferred for TEM. Immersion fixation and embedding was performed according to [70]. Thin sections were cut on an RMC Powertome XL (Boeckler Instruments) using a diamond knife (DDK) and collected on Pioloform-coated copper slot grids. Grids were post-stained with 4% uranyl acetate and lead citrate and viewed using a Philips CM10 electron microscope (FEI) equipped with a Morada digital camera (Olympus) and iTEM software (Olympus SIS).
Glycogen quantification in C. elegans
Synchronized young adult animals were transferred to agarose pads. For comparisons between strains, different conditions were transferred to the same agarose pad and were exposed to iodine vapor for 30 seconds. Animals were rapidly imaged individually. Quantification of the intensity of the staining was performed using ImageJ software.
Periodic acid Schiff staining
For human normal kidney and BHD tumor samples, slides were rehydrated after deparaffination and treated with 1% periodic acid for 10 minutes. Periodic acid was washed off with H2O and slides were then incubated in Schiff reagent for 20 min. Slides were then rinsed with H2O, counterstained with hematoxylin and embedded in entellan. Images were taken as described in [71].
Glycerol determination in C. elegans
Synchronized L4/young adult animals exposed or not to 400mM NaCl for 2 hours and were harvested and washed with M9 buffer adjusted to match plate salinity. Pellets were flash frozen in liquid nitrogen. Extraction was performed according to [5]. Briefly, frozen pellets were ground using a cold mortar and pestle on dry ice. The worm powder was then resuspended in 1N perchloric acid, and solutions were transferred to 15ml conical tubes and kept on ice for 1 hour. The lysate was then centrifuged and the supernatant was neutralized with 5N KOH containing 61.5mM K2HPO4 and 38.5mM KH2PO4. Glycerol levels were determined using a glycerol determination kit (R-Biopharm, Marshall, MI). Pellets were solubilized in 0.1N NaOH and protein content was determined using BCA. Glycerol levels were normalized to protein content.
Gene expression analysis in kidney cancers from patients
TCGA data including 91 kidney chromophobe gene expression RNASeq (IlluminaHiSeq), 604 kidney renal clear cell carcinoma gene expression RNASeq (IlluminaHiSeq), and 258 kidney renal papillary cell carcinoma gene expression RNASeq (IlluminaHiSeq), were extracted from cancer Genomics Browser (https://genome-cancer.ucsc.edu/proj/site/hgHeatmap). For expression analysis, data were expressed as median fold change and the Mann-Whitney test was used to calculate the p-values between normal and tumor samples. P-values less than 0.05 were considered to be statistically significant. For correlation analysis TCGA expression data (same as expression analysis) were used to calculate the Pearson correlation coefficient, and generate a heat map, using R software 3.1.1 (http://www.r-project.org/). P-values less than 0.05 were considered to be statistically significant.
Statistical analyses
Data are expressed as means ±SEM. Statistical analyses for all data were performed by student's t-test, using Excel (Microsoft, Albuquerque, NM, USA). For hyperosmotic stress survival curve comparisons we used the Log-rank Mantel Cox test using GraphPad software. Statistical significance is indicated in figures (* P<0.05, **P<0.01, ***P<0.001) or included in the supplemental tables.
Supporting Information
Zdroje
1. Brocker C, Thompson DC, Vasiliou V (2012) The role of hyperosmotic stress in inflammation and disease. Biomol Concepts 3: 345–364. 22977648
2. Burg MB, Ferraris JD, Dmitrieva NI (2007) Cellular response to hyperosmotic stresses. Physiol Rev 87: 1441–1474. 17928589
3. Yancey PH (2005) Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol 208: 2819–2830. 16043587
4. O'Rourke SM, Herskowitz I, O'Shea EK (2002) Yeast go the whole HOG for the hyperosmotic response. Trends Genet 18: 405–412. 12142009
5. Lamitina ST, Morrison R, Moeckel GW, Strange K (2004) Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am J Physiol Cell Physiol 286: C785–791. 14644776
6. Lamitina T, Huang CG, Strange K (2006) Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci U S A 103: 12173–12178. 16880390
7. Rohlfing AK, Miteva Y, Moronetti L, He L, Lamitina T (2011) The Caenorhabditis elegans mucin-like protein OSM-8 negatively regulates osmosensitive physiology via the transmembrane protein PTR-23. PLoS Genet 7: e1001267. doi: 10.1371/journal.pgen.1001267 21253570
8. Rohlfing AK, Miteva Y, Hannenhalli S, Lamitina T (2010) Genetic and physiological activation of osmosensitive gene expression mimics transcriptional signatures of pathogen infection in C. elegans. PLoS One 5: e9010. doi: 10.1371/journal.pone.0009010 20126308
9. Solomon A, Bandhakavi S, Jabbar S, Shah R, Beitel GJ, et al. (2004) Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics 167: 161–170. 15166144
10. Wheeler JM, Thomas JH (2006) Identification of a novel gene family involved in osmotic stress response in Caenorhabditis elegans. Genetics 174: 1327–1336.
11. Hardie DG, Ashford ML (2014) AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda) 29: 99–107.
12. Hornstein OP, Knickenberg M (1975) Perifollicular fibromatosis cutis with polyps of the colon—a cutaneo-intestinal syndrome sui generis. Arch Dermatol Res 253: 161–175. 1200700
13. Birt AR, Hogg GR, Dube WJ (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113: 1674–1677. 596896
14. Toro JR, Glenn G, Duray P, Darling T, Weirich G, et al. (1999) Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135: 1195–1202. 10522666
15. Pavlovich CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, et al. (2002) Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol 26: 1542–1552. 12459621
16. Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, et al. (2002) Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev 11: 393–400. 11927500
17. Tobino K, Gunji Y, Kurihara M, Kunogi M, Koike K, et al. (2011) Characteristics of pulmonary cysts in Birt-Hogg-Dube syndrome: thin-section CT findings of the chest in 12 patients. Eur J Radiol 77: 403–409. doi: 10.1016/j.ejrad.2009.09.004 19782489
18. Gupta P, Eshaghi N, Kamba TT, Ghole V, Garcia-Morales F (2007) Radiological findings in Birt-Hogg-Dube syndrome: a rare differential for pulmonary cysts and renal tumors. Clin Imaging 31: 40–43. 17189846
19. Kupres KA, Krivda SJ, Turiansky GW (2003) Numerous asymptomatic facial papules and multiple pulmonary cysts: a case of Birt-Hogg-Dube syndrome. Cutis 72: 127–131. 12953936
20. Furuya M, Tanaka R, Koga S, Yatabe Y, Gotoda H, et al. (2012) Pulmonary cysts of Birt-Hogg-Dube syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am J Surg Pathol 36: 589–600. doi: 10.1097/PAS.0b013e3182475240 22441547
21. Van Denhove A, Guillot-Pouget I, Giraud S, Isaac S, Freymond N, et al. (2011) [Multiple spontaneous pneumothoraces revealing Birt-Hogg-Dube syndrome]. Rev Mal Respir 28: 355–359. doi: 10.1016/j.rmr.2010.08.015 21482341
22. Petersson F, Gatalica Z, Grossmann P, Perez Montiel MD, Alvarado Cabrero I, et al. (2010) Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch 456: 355–365. doi: 10.1007/s00428-010-0898-4 20300772
23. Koga S, Furuya M, Takahashi Y, Tanaka R, Yamaguchi A, et al. (2009) Lung cysts in Birt-Hogg-Dube syndrome: histopathological characteristics and aberrant sequence repeats. Pathol Int 59: 720–728. doi: 10.1111/j.1440-1827.2009.02434.x 19788617
24. Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, et al. (2008) BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet 45: 321–331. doi: 10.1136/jmg.2007.054304 18234728
25. Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, et al. (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2: 157–164. 12204536
26. Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, et al. (2006) Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A 103: 15552–15557. 17028174
27. Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, et al. (2008) Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 415: 60–67. doi: 10.1016/j.gene.2008.02.022 18403135
28. Possik E, Jalali Z, Nouet Y, Yan M, Gingras MC, et al. (2014) Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS Genet 10: e1004273. doi: 10.1371/journal.pgen.1004273 24763318
29. Yan M, Gingras MC, Dunlop EA, Nouet Y, Dupuy F, et al. (2014) The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest.
30. Possik et al. Manuscript in preparation.
31. Wormatlas http://wwwwormatlasorg/dauer/muscle/Musframesethtml.
32. Frazier HN 3rd, Roth MB (2009) Adaptive sugar provisioning controls survival of C. elegans embryos in adverse environments. Curr Biol 19: 859–863. doi: 10.1016/j.cub.2009.03.066 19398339
33. LaMacchia JC, Roth MB (2015) Aquaporins 2 and 4 Regulate Glycogen Metabolism and Survival during Hyposmotic-Anoxic Stress in Caenorhabditis Elegans. Am J Physiol Cell Physiol: ajpcell 00131 02015. doi: 10.1152/ajpcell.00131.2015 26017147
34. LaMacchia JC, Frazier HN 3rd, Roth MB (2015) Glycogen Fuels Survival During Hyposmotic-Anoxic Stress in Caenorhabditis elegans. Genetics. doi: 10.1534/genetics.115.179416 26116152
35. Braeckman BP (2009) Intermediary metabolism. In: Koen Houthoofd JRV, editor. 16 February 2009 ed. wormbook.
36. Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, et al. (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115: 2433–2442. 12006627
37. Sanz P (2003) Snf1 protein kinase: a key player in the response to cellular stress in yeast. Biochem Soc Trans 31: 178–181. 12546680
38. Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, et al. (2011) Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470: 404–408. doi: 10.1038/nature09706 21331044
39. Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18: 3004–3009. 15574588
40. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, et al. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6: 280–293. 17908557
41. Fukuyama M, Sakuma K, Park R, Kasuga H, Nagaya R, et al. (2012) C. elegans AMPKs promote survival and arrest germline development during nutrient stress. Biol Open 1: 929–936. doi: 10.1242/bio.2012836 23213370
42. LaRue BL, Padilla PA (2011) Environmental and genetic preconditioning for long-term anoxia responses requires AMPK in Caenorhabditis elegans. PLoS One 6: e16790. doi: 10.1371/journal.pone.0016790 21304820
43. Lee H, Cho JS, Lambacher N, Lee J, Lee SJ, et al. (2008) The Caenorhabditis elegans AMP-activated protein kinase AAK-2 is phosphorylated by LKB1 and is required for resistance to oxidative stress and for normal motility and foraging behavior. J Biol Chem 283: 14988–14993. doi: 10.1074/jbc.M709115200 18408008
44. Carling D, Hardie DG (1989) The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta 1012: 81–86. 2567185
45. Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, et al. (2004) The alpha2-5'AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53: 3074–3081. 15561936
46. Wojtaszewski JF, Jorgensen SB, Hellsten Y, Hardie DG, Richter EA (2002) Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 51: 284–292. 11812734
47. Miyamoto L, Toyoda T, Hayashi T, Yonemitsu S, Nakano M, et al. (2007) Effect of acute activation of 5'-AMP-activated protein kinase on glycogen regulation in isolated rat skeletal muscle. J Appl Physiol (1985) 102: 1007–1013.
48. Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K (2011) Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60: 766–774. doi: 10.2337/db10-1148 21282366
49. Aschenbach WG, Hirshman MF, Fujii N, Sakamoto K, Howlett KF, et al. (2002) Effect of AICAR treatment on glycogen metabolism in skeletal muscle. Diabetes 51: 567–573. 11872652
50. Luptak I, Shen M, He H, Hirshman MF, Musi N, et al. (2007) Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest 117: 1432–1439. 17431505
51. Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, et al. (2002) Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 109: 357–362. 11827995
52. Ahmad F, Arad M, Musi N, He H, Wolf C, et al. (2005) Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation 112: 3140–3148. 16275868
53. Zou L, Shen M, Arad M, He H, Lofgren B, et al. (2005) N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ Res 97: 323–328. 16051890
54. Wang Z, Wilson WA, Fujino MA, Roach PJ (2001) Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol 21: 5742–5752. 11486014
55. Yu H, Hirshman MF, Fujii N, Pomerleau JM, Peter LE, et al. (2006) Muscle-specific overexpression of wild type and R225Q mutant AMP-activated protein kinase gamma3-subunit differentially regulates glycogen accumulation. Am J Physiol Endocrinol Metab 291: E557–565. 16638825
56. Milan D, Jeon JT, Looft C, Amarger V, Robic A, et al. (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288: 1248–1251. 10818001
57. Lapierre LR, Hansen M (2012) Lessons from C. elegans: signaling pathways for longevity. Trends Endocrinol Metab. doi: 10.1016/j.tem.2012.07.007 22939742
58. Sheikh-Hamad D, Gustin MC (2004) MAP kinases and the adaptive response to hypertonicity: functional preservation from yeast to mammals. Am J Physiol Renal Physiol 287: F1102–1110. 15522988
59. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, et al. (2012) Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 16: 751–764. doi: 10.1016/j.cmet.2012.10.017 23177934
60. Rousset M, Zweibaum A, Fogh J (1981) Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res 41: 1165–1170. 7459858
61. Bouchard M, Souabni A, Busslinger M (2004) Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 38: 105–109. 15048807
62. Hasumi H, Baba M, Hasumi Y, Huang Y, Oh H, et al. (2012) Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J Natl Cancer Inst 104: 1750–1764. doi: 10.1093/jnci/djs418 23150719
63. Baba M, Keller JR, Sun HW, Resch W, Kuchen S, et al. (2012) The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dube syndrome is required for murine B-cell development. Blood 120: 1254–1261. doi: 10.1182/blood-2012-02-410407 22709692
64. Zois CE, Favaro E, Harris AL (2014) Glycogen metabolism in cancer. Biochem Pharmacol 92: 3–11. doi: 10.1016/j.bcp.2014.09.001 25219323
65. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK (2011) Tumor necrosis factor-related genes and colon and rectal cancer. Int J Mol Epidemiol Genet 2: 328–338. 22199996
66. Li JT, Wang LF, Zhao YL, Yang T, Li W, et al. (2014) Nuclear factor of activated T cells 5 maintained by Hotair suppression of miR-568 upregulates S100 calcium binding protein A4 to promote breast cancer metastasis. Breast Cancer Res 16: 454. doi: 10.1186/s13058-014-0454-2 25311085
67. Kuper C, Beck FX, Neuhofer W (2014) NFAT5-mediated expression of S100A4 contributes to proliferation and migration of renal carcinoma cells. Front Physiol 5: 293. doi: 10.3389/fphys.2014.00293 25152734
68. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. 4366476
69. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biology 2.
70. Hall DH (1995) Electron microscopy and three-dimensional image reconstruction. Methods in Cell Biology, Vol 48 : 395–436.
71. Preston RS, Philp A, Claessens T, Gijezen L, Dydensborg AB, et al. (2011) Absence of the Birt-Hogg-Dube gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene 30: 1159–1173. doi: 10.1038/onc.2010.497 21057536
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 10
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice