
Region-Specific Activation of mRNA Translation by Inhibition of Bruno-Mediated Repression
Proteins are often enriched to specific regions within cells via localization of mRNAs. This phenomenon serves a variety of roles, both bringing together factors involved in particular cellular processes to enhance their efficiency, and in restricting proteins that could do harm if deployed at inappropriate positions. In the latter situation, translational repression prevents expression before mRNA localization, and there must be activation mechanisms to inhibit or override repression. How the processes of mRNA localization and translation are coordinated is not well understood, in part because cellular extracts prepared to study mechanisms in vitro do not retain the spatial information present in the intact cell. We developed an in vivo assay to monitor the pattern of translation in the Drosophila oocyte, where several patterning determinants must be localized to specific regions. Using this assay, we showed that repression of translation by the Bruno protein is inhibited, and we could visualize when and where this occurs during oogenesis. Regional activation occurs not only at the site of mRNA localization, but more broadly in a graded fashion, and it does not require an activation element in the mRNA. We also show that Bruno dimerizes, and that dimerization is important for translational activation.
Published in the journal:
Region-Specific Activation of mRNA Translation by Inhibition of Bruno-Mediated Repression. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004992
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004992
Summary
Proteins are often enriched to specific regions within cells via localization of mRNAs. This phenomenon serves a variety of roles, both bringing together factors involved in particular cellular processes to enhance their efficiency, and in restricting proteins that could do harm if deployed at inappropriate positions. In the latter situation, translational repression prevents expression before mRNA localization, and there must be activation mechanisms to inhibit or override repression. How the processes of mRNA localization and translation are coordinated is not well understood, in part because cellular extracts prepared to study mechanisms in vitro do not retain the spatial information present in the intact cell. We developed an in vivo assay to monitor the pattern of translation in the Drosophila oocyte, where several patterning determinants must be localized to specific regions. Using this assay, we showed that repression of translation by the Bruno protein is inhibited, and we could visualize when and where this occurs during oogenesis. Regional activation occurs not only at the site of mRNA localization, but more broadly in a graded fashion, and it does not require an activation element in the mRNA. We also show that Bruno dimerizes, and that dimerization is important for translational activation.
Introduction
Localized mRNAs function in many biological settings to facilitate region-specific protein synthesis [1–3]. Translational repression of these mRNAs helps restrict distribution of the encoded proteins, an essential property if the protein has adverse effects at inappropriate locations. In addition, translational repression could be a prerequisite for mRNA localization, if the act of translation interferes with that process. With repression comes the need for translational activation, either by disrupting repression or by a separate mechanism. The Drosophila oskar (osk) mRNA is subject to an extensive program of regulation and provides a model for elucidation of the mechanisms of repression, localization and activation, and how these events are coordinated [4].
The Osk protein, whose distribution during oogenesis is restricted to the extreme posterior region of later stage oocytes, acts as a posterior determinant responsible for posterior patterning of the embryos and formation of the embryonic germline (reviewed in [5]). In the absence of Osk, the abdominal segments are missing and no germ cells form [6]. Conversely, mis- or overexpression of Osk, such that the protein is not tightly restricted to the posterior of the oocyte, leads to a reorganization of the embryonic body plan and ectopic formation of germ cells [7,8]. Thus, proper deployment of Osk is critical.
The osk mRNA is present from the earliest stages of oogenesis, transcribed in nurse cells and rapidly transported into the oocyte. As oogenesis proceeds, osk mRNA persists in the oocyte, and is transiently enriched near the anterior at stage 8 before assuming its final position at the extreme posterior of the oocyte [9,10]. It is only at this point that substantial levels of Osk protein accumulate [11–13]. The absence of Osk protein at earlier stages, and from unlocalized osk mRNA, is due to translational repression. A key player in repression is Bruno (Bru; encoded by the aret gene), which binds to multiple sites in the osk 3′ UTR. Mutation of these sites leads to excess Osk activity and precocious Osk protein [11]. Translational repression by Bru has been recapitulated using in vitro assay systems from Drosophila tissues [14,15].
Two models have been proposed for the mechanism of repression by Bru. In one model, the events that occur at the mRNA 5′ cap, a structure bound by eIF4E, are targeted. During cap-dependent initiation, eIF4E binds to eIF4G, resulting eventually in assembly of a functioning ribosome. The Cup protein also binds to eIF4E, using the same site bound by eIF4G. Another interaction of Cup is with Bru, leading to the model: Cup is recruited to an osk mRNA by Bru, binds to the eIF4E bound to the 5′ cap of that mRNA, and thus blocks the required eIF4E/eIF4G interaction [16]. In the other model for Bru-dependent repression, Bru promotes osk mRNA oligomerization and formation of large silencing particles, which are proposed to be inaccessible to ribosomes [17]. In this model Cup is also involved, but not the ability of Cup to bind eIF4E. Oligomerization of osk mRNA is also promoted by direct RNA dimerization and formation of large RNP complexes by Polypyrimidine Tract Binding protein (PTB) [18,19].
After the osk mRNA has been localized to the posterior pole of the oocyte at stage 9, translational repression must be overcome [11–13]. This could be achieved in different ways, with or without the need for specific regulatory elements. At one extreme, and requiring no activation elements, is inactivation or degradation of the repressors. At the other extreme, the repressors would remain in place and functional, but independent forms of activation (mediated by further regulatory elements) would overcome repression by exerting more powerful positive influences on translation. Just as for repression, there appear to be multiple contributions to activation: a variety of proteins and regulatory sequences have been implicated, with no unifying model for how their actions collectively lead to activation [11–13,20–26].
Here we characterize the interactions of Bru, using in vitro assays to map protein-binding domains and sites of phosphorylation, and designing mutations that affect the interactions. A simplified in vivo system, focusing on Bru-mediated regulation in the absence of much of the complex regulation of osk mRNA, reveals one form of translational activation and provides concrete insights into its mechanism.
Results
Bru dimerizes via a domain that also mediates Cup binding
A GST pull-down assay was used to test for the ability of Bru to dimerize. Full-length Bru was expressed as a fusion to GST, and incubated with Bru bearing a His6 tag. Following affinity purification of GST::Bru with glutathione sepharose beads, copurification of His6::Bru was tested by Western blot analysis using the anti-His6 antibody. By this assay, Bru did dimerize while His6::Bru did not bind to GST alone (Fig. 1A).
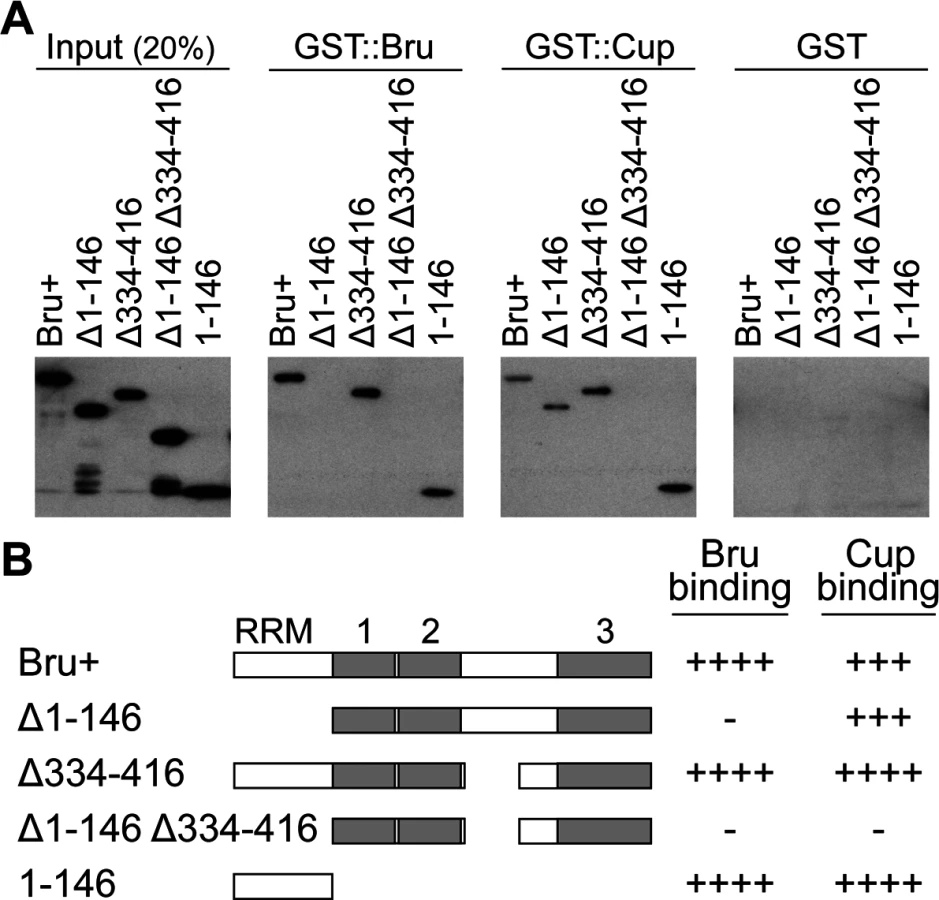
To map the domain of Bru responsible for dimerization, deletion derivatives of Bru (Fig. 1B) were tested in the GST::Bru pull-down assay. The three RRMs all function in RNA binding [27], so we focused on the other domains. Deletion of the Bru amino-terminal domain (aa1–146, Δ1–146) eliminated binding to GST::Bru, while deletion of most of the linker domain between RRMs 2 and 3 (aa334–416, Δ334–416) had no effect. The amino-terminal domain was not only required for dimerization with Bru, but was also sufficient: the isolated domain bound GST::Bru (Fig. 1A). The ability of Bru to dimerize provides an explanation for how Bru oligomerizes osk mRNA: a molecule of Bru bound to one osk mRNA could dimerize with a second molecule of Bru bound to a different osk mRNA. With the many Bru binding sites in the osk mRNA 3′ UTR [11,26], formation of large, highly interconnected protein-RNA assemblies is possible. This suggests that the proposed use of osk mRNA oligomerization as a mechanism of translational repression [17] would rely on Bru dimerization.
A second Bru interaction, with Cup, provides the basis for the other proposed mechanism of translational repression, in which Bru recruits Cup to the osk mRNA [16]. A GST::Cup pull-down assay was used to monitor interaction with Bru. As expected, full-length Bru (Bru+) bound GST::Cup. Deletion of either aa1–146 or aa334–416 of Bru had no dramatic effect on binding, but deletion of both domains eliminated binding. Just as for Bru dimerization, the isolated amino-terminal domain was sufficient for binding to GST::Cup (Fig. 1A,B).
The amino-terminal domain of Bru is required for translational repression
Our evidence that the amino-terminal domain of Bru is essential for dimerization and contributes to Cup binding suggested that this domain is likely to play an important role in repression. To test this prediction we established an in vivo tethering assay, in which translation of a GFP-MS2 reporter mRNA was monitored. The 3′ UTR of the GFP-MS2 mRNA includes multiple copies of the bacteriophage MS2 stem loop, a binding site for the MS2 coat protein (MCP [28]). Forms of Bru were expressed as fusions to MCP to direct binding to the reporter mRNA. Both the reporter mRNA and tethered Bru proteins were expressed in Drosophila ovaries using the UAS/GAL4 system.
The GFP-MS2 reporter by itself was expressed throughout the germline cells of the egg chamber (Fig. 2A). Coexpression of tethered Bru dramatically reduced the GFP level (10 fold; Fig. 2B,F). The strong reduction in GFP expression from tethered Bru was accompanied by a reduction in GFP-MS2 mRNA level (1.7 fold; Fig. 2G). Therefore, in this assay Bru both repressed translation and reduced mRNA stability, although repression was the stronger effect (compare Fig. 2F and 2G).

Testing Bru mutants in the tethering assay revealed that deletion of the amino-terminal domain led to a substantial increase in GFP, although not to the level in the absence of repression (Fig. 2C,F). Deletion of the linker domain had no strong effect (Fig. 2D,F). The combination of deleting both the amino-terminal domain and the linker domain was no stronger than deleting the amino-terminal domain alone (Fig. 2F,H), but the range of fluorescence intensities was greater (error bars in Fig. 2F) and some egg chambers had weaker repression (Fig. 2E). Both mutants with enhanced GFP also had slightly elevated GFP-MS2 mRNA (Fig. 2G), although the changes were strongest at the protein level (Fig. 2F,H). The differing activities of the mutant proteins were not due to differences in their expression (S1A Fig.). Thus, the mutants impaired both activities of Bru monitored in this assay: translational repression and destabilization of mRNA.
Bruno is a phosphoprotein
To allow translation of the osk mRNA once it has been localized to the posterior pole of the oocyte, there must be a release from repression. How this is accomplished is not known, but one possibility is that Bru is post-translationally modified to change its activity. To ask if Bru is phosphorylated, the conventional approach of testing for phosphatase-dependent changes in electrophoretic mobility of the protein was used. In untreated ovary extract, Bru appeared by Western blot analysis as a major band, with a faint lower-mobility band. Treatment with phosphatase eliminated the weak band. By contrast, addition of phosphatase inhibitors enhanced the minor band, consistent with the interpretation that this small fraction of Bru is phosphorylated (Fig. 3A left).
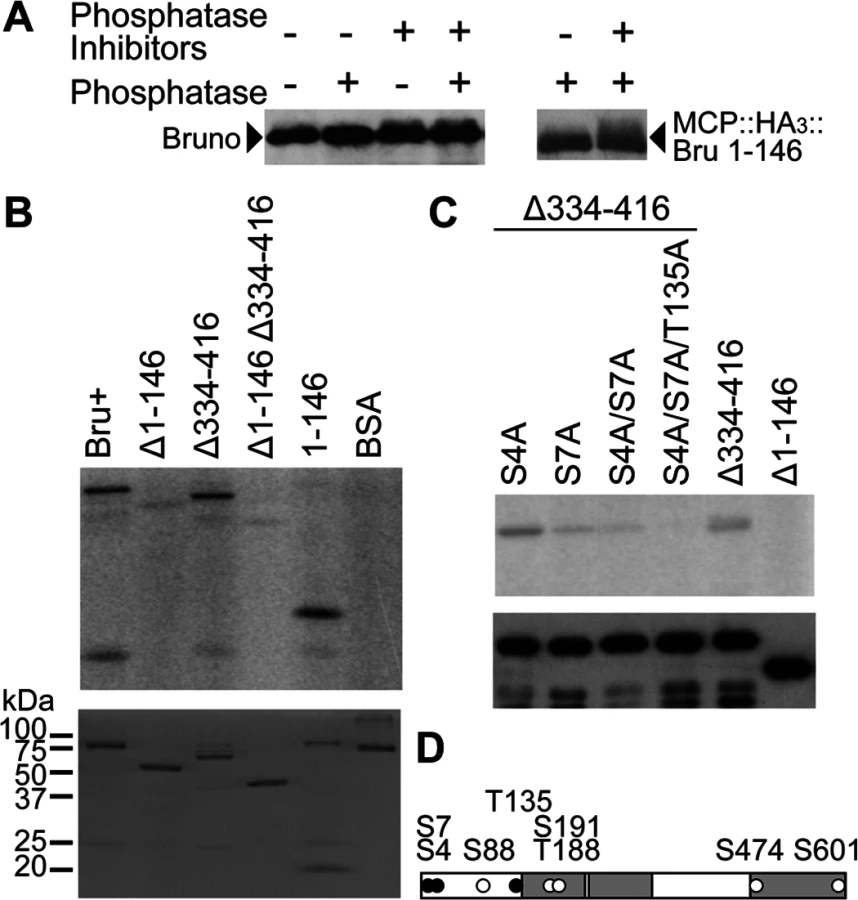
To make a more compelling case for phosphorylation, we also tested the MCP::HA3::Bru 1–146 protein from above, which at 32 kDa is substantially smaller than Bru (64 kDa) and thus might display a larger change in mobility from phosphorylation. This was indeed the case, and the difference between the major Bru band and the slower migrating fraction was more dramatic (Fig. 3A right). Bru phosphorylation was also analyzed by phosphate-affinity SDS-PAGE with the acrylamide-pendant Phos-tag, which separates different phosphoprotein isoforms [29]. Using this approach, multiple, different phosphorylated species could be detected (S2A Fig.).
Bruno is phosphorylated by PKA
The NetPhosK 1.0 and KinasePhos prediction programs were used to identify candidate phosphorylation sites in Bru. Both report multiple sites for many different kinases, although none of the candidate sites had scores suggesting a high probability of phosphorylation (S1 Table). Nevertheless Bru is phosphorylated, and so even the sites with modest scores remain as candidates. Several amino acids are predicted to be targets for Protein Kinase A (PKA), an interesting option since alteration of PKA activity affects osk expression pattern and embryonic body patterning [30].
To evaluate PKA, in vitro phosphorylation assays were performed using the PKA catalytic subunit and full-length Bru. Bru was strongly phosphorylated, while BSA (a negative control) was not (S3A Fig.). By contrast, neither Casein Kinase I (CK1) nor Calmodulin-dependent Protein Kinase II (CaMKII), which share a part of the recognition motif of PKA [31–33], supported detectable phosphorylation of full-length Bru (S3B Fig.).
To map the sites of phosphorylation, Bru deletion proteins from above (Fig. 1B) were used as substrates for PKA. Deletion of the amino-terminal domain greatly reduced phosphorylation, and the isolated domain was itself phosphorylated. Deletion of the linker region did not reduce phosphorylation, nor did it enhance the effect of deleting the amino-terminal domain (Fig. 3B). Thus, PKA phosphorylates the amino-terminal domain of Bru.
To identify sites of phosphorylation, we performed a tandem mass spectrometry (MS/MS) analysis of Bru phosphorylated in vitro. Although four candidate sites are predicted in the amino-terminal domain, only phosphoserine at position 7 (S7) was identified with high confidence and no ambiguity (Fig. 3D). S88 could not be tested since aa36–119 was undetectable due to a low coverage of MS/MS (see Materials and Methods), and a majority of peptides containing either S4 or T135 was detected as unphosphorylated. Nevertheless, there is still a possibility of weak phosphorylation below the limit of detection at either S4 or T135. Mutation of S7 to alanine (S7A) substantially reduced phosphorylation by PKA. Because the S7A mutant retained a low level of phosphorylation, we also tested mutations in the other predicted sites, either alone or in combinations. Of the mutants tested, S4A/S7A/T135A was most resistant to phosphorylation (Fig. 3C). The mutants, except for S4A, were also tested in the context of full-length Bru, and similar results were obtained (S2B Fig.).
Phosphomimetic mutations prevent Bru dimerization and impair Cup binding
Since the amino-terminal domain of Bru is essential for repression, the potential phosphorylation of one or more residues within this region might inhibit or enhance repression. We therefore asked if phosphomimetic mutations would interfere with Bru protein interactions mediated by the amino-terminal region and implicated in repression.
Pull-down assays were performed with GST::Bru and GST::Cup, using Bru mutants with phosphosilent alanine (A) or phosphomimetic glutamate (E) substitutions at one or more of the three residues that affect phosphorylation by PKA: S4, S7, and T135. None of the phosphosilent mutants showed reduced binding to GST::Bru (Fig. 4A) or GST::Cup (Fig. 4B), demonstrating that mutation of the affected residues did not inherently disrupt the protein interactions. By contrast, the phosphomimetic mutants altered interactions. S7E significantly reduced dimerization, and the S4E/S7E double mutant retained only a very low level of dimerization. Including the T135E mutation did not obviously further reduce dimerization by S7E (in S7E/T135E), but did reduce dimerization in the triple mutant (S4E/S7E/T135E) to below the level of detection (Fig. 4A and 4C).
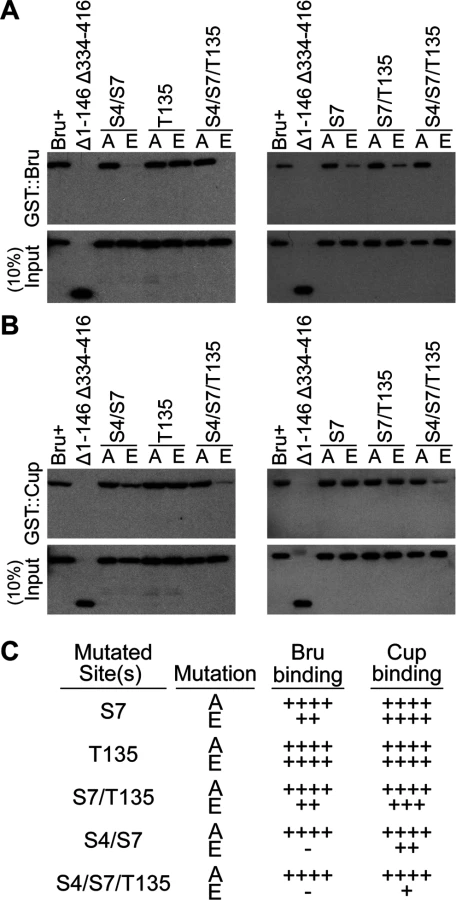
Bru binding to Cup was less sensitive to the phosphomimetic mutations. S7E did not reduce binding, and the double mutation combinations caused only modest defects. Even the S4E/S7E/T135E triple mutant retained detectable binding (Fig. 4B and 4C).
Because the S4E/S7E/T135E triple mutant eliminated detectable Bru dimerization, repression dependent on this interaction is expected to be disrupted. The prediction is less clear for Cup-dependent repression, given the residual Bru/Cup interaction.
Mutations that prevent Bru dimerization do not affect repression
Our analysis of PKA phosphorylation of Bru in vitro suggests that PKA may also modify the protein in vivo. Testing this prediction has proven to be challenging, with no conclusive answer. This is due in part to failure in making an antibody against the phospho-S7 peptide. Nevertheless, the Bru mutants defective in dimerization provide useful tools to test the importance of this interaction for Bru function. As one such test, we made use of the tethering assay to monitor translational repression. Notably, none of the mutants tested, including the S4E/S7E/T135E triple mutant that prevented Bru dimerization and impaired the Bru/Cup interaction, showed any substantial decrease in repression (Fig. 5). Likewise, there were no substantial changes in reporter mRNA levels (Fig. 5H).
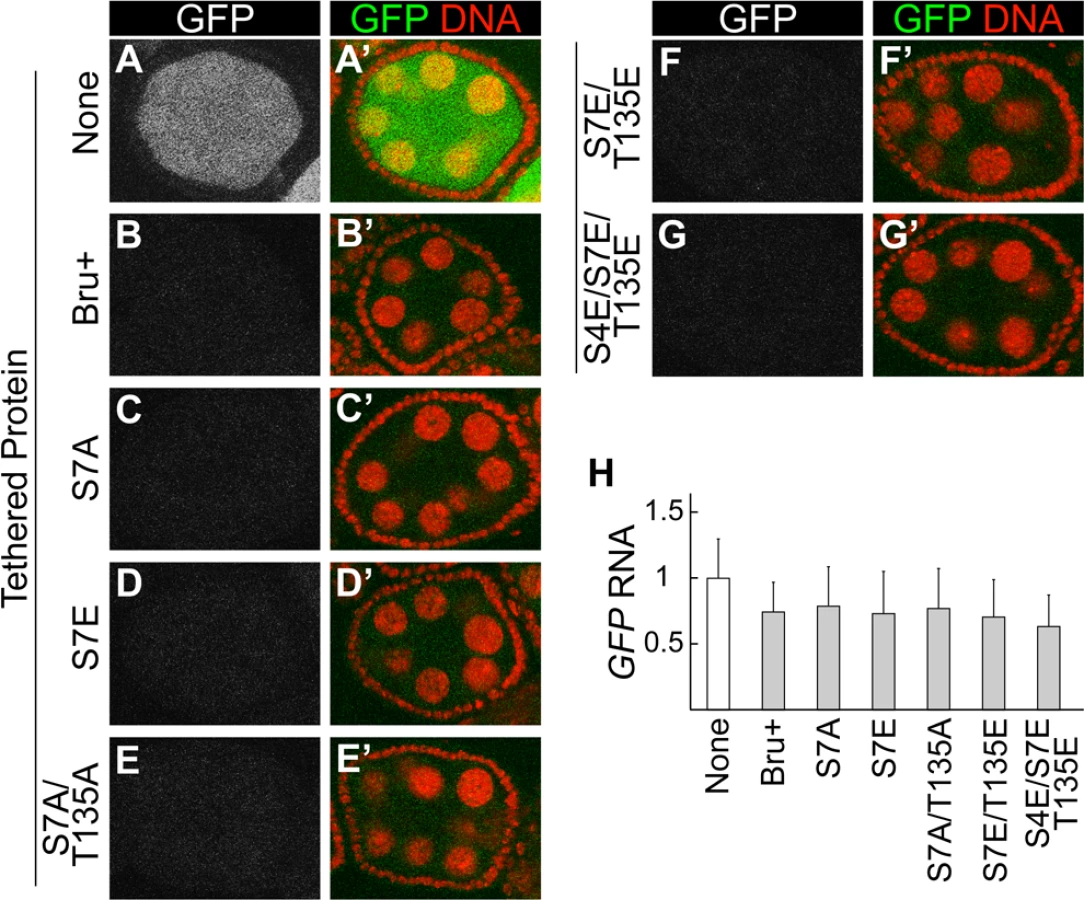
MCP is known to dimerize [34]. This property could substitute for Bru dimerization, and thus neutralize the effect of the dimerization-defective mutations. To address this possibility, we tested another tethering system which relies on binding of a bacteriophage lambda N peptide (which does not dimerize) to the boxB stem-loop RNA [35]. In this case, the reporter mRNA was GFP with 6 copies of the boxB sequence in the 3′ UTR (GFP-boxB), and the Bru proteins were expressed as fusions to the λN peptide. Just as with the other system, tethered Bru repressed translation of the reporter mRNA (compare S4A and S4B Fig.). Notably, the S4E/S7E/T135E triple mutant did not affect repression (S4C,D Fig.), confirming that dimerization is not required for translational repression.
Dimerization-defective Bru mutants have weakly impaired RNA-binding activity
Cooperative binding is a common strategy to enhance affinity for a substrate. Dimerization of Bru might facilitate cooperative binding to RNA, and if so, the mutations inhibiting dimerization are expected to impair RNA-binding activity of Bru. A UV-crosslinking assay was used to test Bru proteins for their ability to bind the osk 3′ UTR AB region RNA, which has multiple Bru binding sites [11]. The two mutants most strongly defective in dimerization, S4E/S7E and S4E/S7E/T135E, showed compromised RNA binding (S5A Fig.). After quantitation, normalization for protein levels and statistical analysis of three independent experiments, both mutants were considered to have a significant change in their RNA-binding ability when compared to their ala-mutant (dimerization-competent) counterparts (S5B Fig.). Although the reduction in RNA binding is modest, it is possible that this change could contribute to a reduction in Bru activity in vivo by weakening the interaction with target mRNAs, such as osk.
A reporter mRNA to detect the pattern of translation in vivo
The tethering assays were useful for monitoring, in isolation from much of the complex program of osk regulation, translational repression by Bru. However, these assays have limitations. First, any region-specific change in repressive activity might be missed, as the reporter protein is diffusible. Second, since the RNA-binding activity of Bru was not required in the assay, a possible disruption of repression by reduced RNA-binding affinity would not be detected.
As an alternate approach to address these limitations we wanted to maintain the use of a simplified system which focuses on repression by Bru, but allows detection of regional differences in translation under conditions where Bru binds directly to the mRNA. Our approach has two components. The first, described in the following paragraphs, is the development of an appropriate reporter mRNA. The second, described in the next section, is manipulation of the aret gene, which encodes Bru, to introduce the mutations that disrupt Bru dimerization.
A requirement for the reporter mRNA is that the encoded protein be anchored, such that its site of synthesis is revealed. The Osk protein is itself anchored to the oocyte cortex, with the amino-terminal domain of the Long Osk isoform required for this function [36]. To determine if this domain would anchor GFP, the UAS-osk1–534::GFP transgene (which includes nucleotides 1–534 of the osk mRNA, and thus the first 173 amino acids of Long Osk) was tested. While GFP alone appears diffuse throughout the germline cells (Fig. 6A), Osk::GFP is highly enriched at cortical regions of the oocyte and at cell boundaries in the nurse cells (Fig. 6B). Addition of the osk 3′ UTR to this transgene (in UAS-osk1–534::GFP-osk3′UTR) restricts expression to the posterior pole of the oocyte, where the mRNA is localized. Notably, the protein from osk1–534::GFP-osk3′UTR mRNA remains concentrated in a narrow crescent at the posterior pole, and does not diffuse extensively along the oocyte cortex (Fig. 6C). Therefore, the amino-terminal domain of Osk provides a useful anchoring domain to reveal the site of translation.
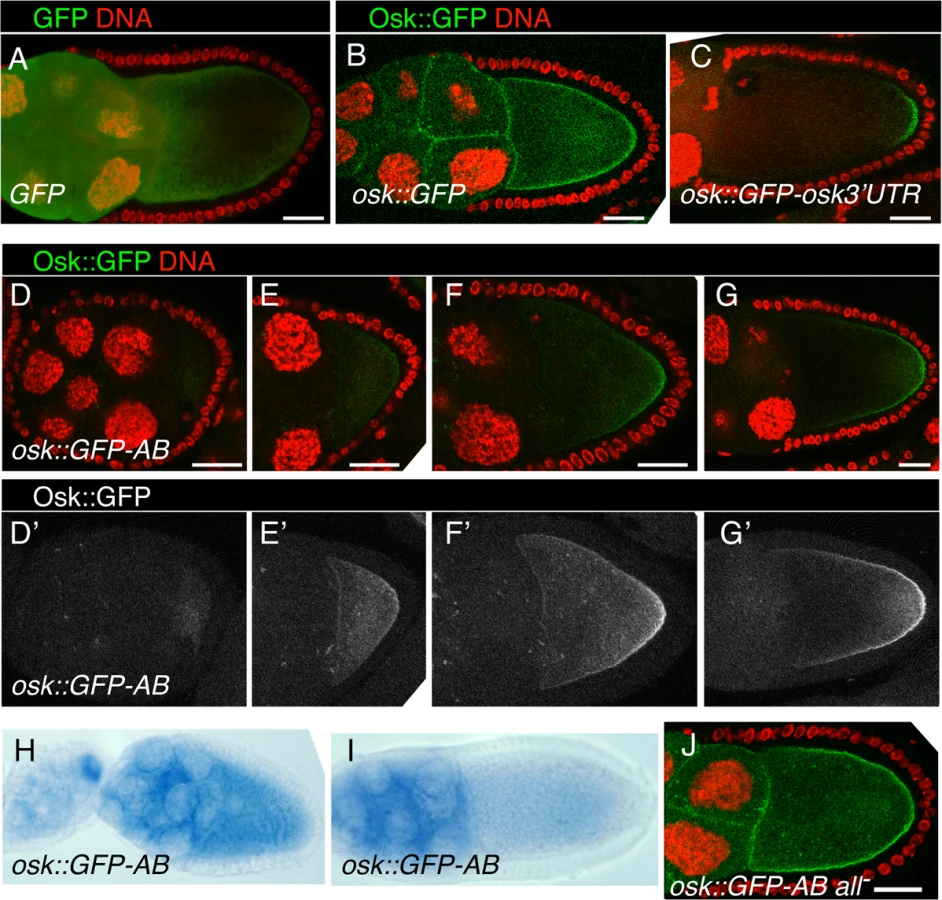
The reporter mRNA also needs to be subject to Bru-dependent repression, and with Bru binding directly to the mRNA. Just as in the tethering assay, we wanted to avoid the complexity of the full program of osk regulation. This can be achieved through the use of the osk 3′ UTR AB region, which contains multiple Bru binding sites [11,26]. Addition of this region to a GFP reporter confers efficient repression in the ovary [26], and as expected repression was dependent on the Bru binding sites (below).
Combining these two required features for the reporter mRNA, the UAS-osk1–534::GFP-AB transgene encodes an anchored GFP protein whose translation will be repressed by Bru.
Region-specific activation of osk mRNA translation
Characterization of the osk1–534::GFP-AB reporter mRNA confirmed the expected translational repression, but also revealed a novel pattern of region-specific translational activation.
During previtellogenic stages of oogenesis the osk1–534::GFP-AB mRNA was strongly repressed throughout the egg chamber, with no detectable GFP signal (Fig. 6D). Translation of osk1–534::GFP-AB mRNA continued to be strongly repressed in nurse cells at later stages of oogenesis. However, starting at stage 7/8 a faint Osk::GFP signal was detected in the oocyte (Fig. 6E). The intensity of the signal increased at later stages and, strikingly, Osk::GFP accumulated in a weak gradient extending from the posterior pole (Fig. 6F,G). This mRNA lacks localization signals required for posterior localization of osk mRNA [37,38], and, not surprisingly, we found no detectable posterior localization of the reporter mRNA (Fig. 6H,I). Therefore, translation of the reporter was activated independent of association with the mRNA localization machinery.
There are two options to explain the posterior gradient of the anchored Osk::GFP fusion protein. One is that activation of translation occurred in a posterior gradient essentially the same as that displayed by Osk::GFP. Alternatively, activation of translation could have occurred just where pole plasm assembles at the posterior pole of the oocyte (from the fraction of the mRNA located in that region by chance), followed by diffusion of Osk:GFP from that site to create the extended gradient we observe. However, as shown above, when we selectively expressed the same Osk::GFP fusion protein just at the posterior pole from a localized mRNA, it remained there and did not form a gradient (Fig. 6C). Thus, the gradient of Osk::GFP produced by the unlocalized transgene mRNA must have formed by translational activation in a broad and graded domain: highest at the posterior pole, extending with diminishing strength along almost the entire length of the oocyte, and virtually undetectable at the anterior margin of the oocyte.
These results reveal a spatially-restricted form of translational activation, which we call region-specific activation. This form of activation could inhibit Bru-mediated repression. Alternatively, activation could be independent, and simply superimposed on repression. To distinguish between these options we tested a version of the transgene, UAS-osk1–534::GFP-AB all-, with the Bru binding sites mutated. The mutations disrupted repression, allowing translation of the Osk::GFP protein, which appeared in nurse cells and oocytes (Fig. 6J). If activation inhibits repression by Bru, in the absence of repression there would be no detectable activation and the Osk::GFP protein should be present at similar levels along the anteroposterior axis of the oocyte. By contrast, if activation is independent of Bru, it should still occur when the Bru binding sites are mutated, leading to a higher level of Osk::GFP in the more posterior portion of the oocyte. We detected no posterior enhancement of Osk::GFP when repression was disrupted (Fig. 6J). Thus, region-specific activation of translation must act by inhibiting the repressive function of Bru.
Dimerization-defective Bru mutants have impaired region-specific activation of translation
For the assay system showing region-specific activation of translation, Bru is provided by the endogenous gene. To test dimerization-defective Bru in this assay, we used homologous recombination (HR) [39] to exchange exons that encode the amino-terminal region of Bru (Fig. 7A). The replacements were wild type (aret+), S4A/S7A/T135A (aret3ala), or S4E/S7E/T135E (aret3glu). Loss of aret function leads to an early arrest of oogenesis, with no oocyte specified [40]. Females in which the HR replacement alleles provided the only copy of aret all displayed normal progression through oogenesis, indicating that the mutations did not substantially alter aret function. Protein levels for the different alleles were similar (S6 Fig.). Each of the proteins showed the normal distribution of Bru (S6 Fig.), including the granular cytoplasmic appearance due to association with nuage and sponge bodies [41]. Embryos obtained from these females were tested for patterning defects. Although misregulation of osk expression—either too little or too much Osk activity—causes striking patterning defects [6–8], no such defects were found for any of the aret HR alleles. The only phenotype detected was for the aret3glu mutant, and was unrelated to any known osk defect: an increase in the proportion of embryos that fail to develop (Fig. 8J).


To ask if preventing dimerization of Bru influenced the regional activation of translation, expression of the osk1–534::GFP-AB mRNA was monitored in the engineered aret-mutant ovaries (Fig. 7B-G). Repression in nurse cells was similar for all genotypes, consistent with the absence of any repression defect in the tethering assays (Fig. 7B-D). Both aret3ala and aret3glu mutants reduced the degree of posterior activation, with the strongest effect from the Bru dimerization- defective aret3glu mutant: the fraction of oocytes with a strong posterior gradient was about half that of aret+, and over a third of the oocytes had no detectable posterior gradient (Fig. 7H). Thus, the aret3glu mutant substantially disrupted regional activation. Because this mutant, in effect, enhanced repression, these results are consistent with the failure to detect any loss of repression in the tethering assays.
The aret3ala and aret3glu mutants were also tested for an effect on Osk expression. Because defects in activation of osk mRNA translation can be most pronounced late in oogenesis [26], this analysis included the use of a genomic osk::GFP transgene for detection of Osk::GFP after deposition of the vitelline membrane (which is impermeable to antibodies), as well an epitope tagged osk transgene (oskHA) for detection at earlier stages. There was no defect in repression in stage 8 egg chambers (Fig. 8A-C), as expected from the tethering assay results. Likewise, we did not detect any difference in Osk expression pattern among the three replacement lines in stage 9 (Fig. 8D-F) or later (Fig. 8G-I) egg chambers, consistent with the absence of patterning defects (Fig. 8J) and suggesting redundancy in mechanisms of translational activation.
Discussion
To better understand translational regulation of osk mRNA by Bru, we characterized Bru protein interactions and developed in vivo assays to monitor Bru activity. Bru binds Cup, an interaction that is essential for at least one model of repression [16]. Here we have shown that Bru dimerizes, an interaction very likely to contribute to a second model of repression. In that model, the oligomerization of osk mRNA by Bru creates silencing particles, in which osk transcripts are made inaccessible to the translation machinery. The evidence for this model comes from an in vitro assay using purified Bru, which oligomerizes RNAs containing many Bru binding sites [17]. Because no other macromolecules are present in the assay, dimerization of Bru appears to be the only plausible option for oligomerization of the RNAs. The alternative is for the different Bru RNA-binding domains (there are three RRMs [27,42,43]) to bridge different RNAs, a kinetically unfavorable option: after the initial binding of one Bru domain to an RNA (a second order reaction), the subsequent binding of another Bru domain would strongly favor association with the same RNA (first order) as opposed to another RNA (second order). Furthermore, only one of the RRMs provides a high degree of binding specificity [43], and so specificity of oligomerization would be low (and inconsistent with the observed binding specificity of the intact protein [11,43]) if different domains bound to different RNAs.
We found that the amino-terminal domain of Bru is essential for dimerization. The same domain was sufficient for Cup binding, and deletion of both the amino-terminal and linker domains was required to eliminate Cup binding. These Bru/Cup interaction data using a GST pull-down assay contrast with those from the yeast two-hybrid assay, where the linker domain was shown to be both necessary and sufficient for Cup interaction, while the amino-terminal domain was neither necessary nor sufficient [16]. We do not know why there are differences. In both assays Bru is present as a protein fusion, and it may be that the different additional protein domains—GST in the one assay, and a transcriptional activation domain in the other assay—differentially constrain Bru binding.
To explore the role of dimerization on Bru activity in vivo, two types of simplified assays were used, both of which monitor the activity of Bru independent of much of the rest of the complex regulation of osk mRNA. In one type of assay, Bru was tethered to a reporter mRNA whose repression was monitored. For this assay, a variety of Bru mutants were used, both deletions and point mutations. In this assay the amino terminus of Bru was shown to be essential for repression, while the point mutants that prevent Bru dimerization and reduce Bru binding to Cup had no effect on repression. Therefore, Bru dimerization was not essential for repression. Notably, the tethering assay also revealed that Bru binding reduced stability of the reporter mRNA, in addition to translational repression. Regulation by Cup is known to alter mRNA stability in cultured cells [44], and our results are consistent with that property.
The second type of simplified in vivo assay made use of a reporter mRNA designed to display the pattern of translation within cells, not simply the total level of the encoded protein. Strikingly, Bru-dependent repression of the reporter mRNA was alleviated within the oocyte in a graded fashion, with peak translation at the posterior pole of the oocyte and extending with decreasing efficiency most of the distance to the anterior of the oocyte. Thus, this reporter revealed one form of translational activation—region-specific activation—for which concrete conclusions about mechanism can be drawn: activation is regional, but not narrowly restricted to the site at which osk mRNA is localized; activation is not mechanistically coupled to mRNA localization; and activation involves the inhibition of Bru repression.
To test the requirement for Bru dimerization in region-specific activation, homologous recombination was used to introduce point mutations into the aret locus. The new alleles can be used with no concerns about appropriateness of expression or artifacts from use of fusion proteins. Notably, the mutations preventing dimerization interfered with region-specific activation, suggesting that dimerization acts to disrupt repression, opposite of what might be expected for the silencing particles model of repression.
Region-specific activation disrupts or alters Bru activity, which could involve a direct effect on Bru, or be mediated by a regulatory element which could indirectly affect Bru. A direct effect on Bru could entail, for example, phosphorylation. We have shown that PKA phosphorylated Bru in vitro, and PKA plays a positive role in Osk expression [30]. PKA is involved in a posterior signal transduction event in the oocyte [45], and could thus act in the region where activation occurs (although there has been no demonstration of the site of PKA activity). However, phosphomimetic mutations of the sites at which PKA phosphorylates Bru in vitro did not promote activation, and instead diminished activation. Nevertheless, phosphorylation or another post-translational modification remains as a likely option for region-specific activation, as observed for other repressors of localized mRNAs in which this modification reduces RNA binding affinity [46–48]. The mutations mimicking phosphorylation of Bru also reduced RNA binding affinity, but by a modest degree and the significance of this effect is uncertain. It is noteworthy that region-specific activation was not tightly confined to the extreme posterior of the oocyte. This very strongly indicates that the factors involved are not restricted to the germ plasm that assembles in a crescent at the posterior pole.
If region-specific activation relies on a regulatory element, it must lie within the osk sequences of the osk1–534::GFP-AB mRNA, either the 5′ osk sequences or the 3′ UTR AB region. There has been no indication of a role for the AB region in translational activation. The 5′ osk sequences have been reported to include a translation activation element which mediates posterior-specific inhibition of Bru-mediated repression [21]. However, recent work shows that the 5′ element is not essential, and that the evidence for it functioning only at the posterior of the oocyte is based on an incorrect assumption (M Kanke and PMM, submitted).
Multiple factors and cis-acting elements have been implicated in translational activation of osk mRNA [11–13,20–26], and it is clear that the region-specific mechanism identified here is not the only form of activation. Indeed, the mutant aret alleles which disrupt regional activation of the osk1–534::GFP reporter mRNA have no detectable effect on regulation of endogenous osk mRNA. This is most likely due to redundancy, with the loss of one form of activation being obscured by persistence of other types of activation. Redundancy in activation mechanisms is not unexpected: there are multiple contributions to repression of osk mRNA translation [11,16,19,49–51], and the same might be expected for activation. Furthermore, different mechanisms could be required to overcome different forms of repression. Redundancy presents a substantial challenge, in part because it makes identification of factors and regulatory elements difficult. Each different type of activation may need to be characterized in isolation, before an understanding of the overall process can be obtained.
Another major challenge in understanding translational activation concerns the question of whether an activation mechanism serves as a prerequisite for translation, or is used in a spatially restricted manner. Addressing that question with in vitro systems would be extremely difficult, as spatial differences within the oocyte are lost in the process of preparing an extract. Our strategy of monitoring the pattern of translation in vivo provides a solution, and may be useful for analysis of other forms of activation.
Materials and Methods
Flies and transgenes
w1118 flies were used as the wild type. Transgenic fly stocks were established by standard methods. Expression of the UAS transgenes was driven by the nosGAL4VP16 [52] or matα4-GAL-VP16 driver [53], as indicated.
The P[UAS-GFP-MS218] and P[UAS-GFP-boxB6] transgenes are similar to UASp-GFP-312 [54], but with 18 copies of the MS2 binding sites or 6 copies of the boxB binding site instead of mi-312 targets. P[UAS-MCP::HA3::bru2–3-] shares the same synthetic 5′ UTR as UASp-GFP-312, fused to MCP, 3 copies of the HA epitope, and bru cDNA bearing point mutations in RRM2 (K239A, F241A) and RRM3 (N521A, F523A) [27]. Mutations in the bru sequences were introduced using restriction sites or PCR. P[oskT140::HA] and P[oskT140::GFP] are based on a genomic DNA fragment which provides complete osk function and regulation [10], and have either 3 copies of the HA epitope tag or mGFP6 [55] inserted after T140. To make P[UAS-osk1–534:GFP] (which encodes a fusion protein with the first 173 amino acids of Long Osk), P[UAS-GFP] [26] was modified by addition of osk sequences that include 20 nt of the 5′ flanking region and the first 534 nt of the mRNA (the 5′ UTR and the coding region for amino acids 1–173). Derivatives with the osk 3′ UTR AB region [11], a version of the AB region with Bru binding sites mutated (all-) [26], and with the entire osk 3′ UTR were made as described [26].
For homologous recombination of aret, a 2.1kb region (12,270,445–12,272,531; R5.48) including the first two protein-coding exons that encode the amino terminus of female Bru (up to aa193) was targeted. Details are provided in the Supplemental Materials.
Cloning, expression and purification of recombinant Bru and Cup proteins
GST::Bru was constructed by subcloning full-length bru cDNA into pGEX-2TK (GE Healthcare). GST::Cup577–947 was a gift from Robin Wharton [56]. Bru proteins for binding assays were tagged at the amino terminus with six histidine residues provided by the pET-15b (Novagen) vector and used for purification, and made use of the same mutations as in the UAS-MCP::HA::bru transgenes. GST fusion proteins were expressed in CodonPlus (Stratagene) E. coli, after induction with IPTG. Pelleted cells were resuspended in ice-cold GST lysis buffer (50mM Tris-Cl pH8.0, 150mM NaCl, 1mM EDTA, 1mM DTT, 2mg/ml lysozyme, and 0.1% IGEPAL-CA-630) supplemented with protease inhibitors (Complete, Mini, EDTA-free protease inhibitor cocktail tablet (Roche)), and extracts were prepared as previously described [27]. For His6 tagged proteins, extracts were prepared with His lysis buffer (50mM NaH2PO4H2O pH8.0, 300mM NaCl, 20mM imidazole, 0.01% β-Mercaptoethanol, and 2mg/ml lysozyme) supplemented with protease inhibitors. 250μl Ni-NTA Agarose (Quiagen) in 50% slurry was added per 1ml lysate, and the reaction was incubated for 1–2 hr at 4°C on a rotator. The lysate-Ni-NTA mixture was then loaded into a disposable column equilibrated with the His lysis buffer to remove flow-through and washed with increasing concentrations of imidazole in His lysis buffer (up to 40mM). The proteins were eluted with 250mM imidazole in His lysis buffer. Glycerol was added to the supernatant to 20% final volume and extracts were stored at -70°C.
GST pull-down assay
Equivalent amounts of GST::Bru, GST::Cup or GST was first immobilized on Glutathione Sepharose 4B (GE Healthcare) prepared in 50% slurry in binding buffer (50mM Tris-Cl pH7.5, 150mM NaCl, 10% glycerol, 1mM DTT, and 0.1% IGEPAL-CA-630) supplemented with protease inhibitors, by incubating with extract overnight at 4°C on a rotator. The beads were spun down, washed and resuspended in binding buffer to make 50% slurry. Then 20μl of this slurry was incubated with ~100ng of each of the N-terminally His6-tagged Bru proteins in 80μl reaction containing binding buffer for 2–3 hr at room temperature with rotation. The beads were spun down, washed with binding buffer, and boiled in 5μl 2X SDS loading buffer to elute the bound proteins. Eluates were separated by SDS-PAGE and analyzed by Western blotting. The mouse anti-His antibody (ABGENT) diluted at 1:2000 and alkaline phosphatase-conjugated goat anti-mouse antibody (Applied Biosystems) diluted at 1:5000 were used to detect Bru proteins.
In vitro phosphorylation assay
Phosphorylation reactions (20μl) contained ~250ng of purified substrate, 1–2 unit of recombinant PKA catalytic subunit, CK1 or CaMKII (all from NEB), and 0.2mM [γ-32P]ATP (adjusted to 250μCi/μmol, Perkin Elmer) in kinase buffer (buffer for PKA: 50mM Tris-Cl pH7.5, 100mM KCl, 5mM MgCl2, and 2.4mM DTT; buffers for CK1 or CaMKII provided by NEB) supplemented with protease inhibitors. Reactions were incubated at 30°C for 30 min and terminated by addition of 3X SDS loading buffer. Proteins were separated by SDS-PAGE, and gels were dried (Bio-Rad) and exposed to a Phosphor Screen (Molecular Dynamics) for 12 hr. The screen was then analyzed with a Typhoon laser scanner (GE Healthcare).
RNase protection assay
RNase protection assay was performed as previously described [26], except that quantitation was done using ImageJ.
Detection of phosphorylated Bru
Ovaries from young females, fed on yeast for 3–4 days, were dissected and extract was prepared as previously described [11] in ice-cold lysis buffer (50mM Hepes pH7.9, 150mM KCl, and 1% IGEPAL-CA-630) supplemented with protease inhibitors. Reactions (20μl) contained 10–15μg of ovary extract in phosphatase buffer (50mM Hepes pH7.9, 100mM NaCl, 10mM MgCl2, and 1mM DTT), and where indicated, one or more of the following components were added: 1–2 units of alkaline phosphatase (calf intestinal, NEB), 1.6M beta-glycero phosphate, and 40mM sodium vanadate. Reactions were incubated at 30°C for 1 hr and terminated by addition of 3X SDS loading buffer. Proteins were separated by SDS-PAGE and analyzed by Western blotting.
For phosphate-affinity SDS-PAGE using acrylamide-pendant Phos-tag (WAKO), 50μM Phos-tag and 200μM MnCl2 were added to both stacking and separating gels in solution.
Ovary imaging
Ovaries were processed for imaging as described [26], with an alternate protocol for detection of Bru [57]. Primary antibodies were used at the following dilutions: mouse anti-HA (Covance), 1:1000; rat anti-Bru, 1:500. Secondary antibodies, used at 1:800, were Alexa Fluor 488 goat anti-mouse (Invitrogen) and Cy5 goat anti-rat. DNA was stained with TO-PRO-3 Iodide (642/661, Invitrogen) diluted 1:1000. Samples were mounted in Vectashield (Vector Labs) and analyzed with a Leica TCS-SP laser scanning confocal microscope. Quantitation of GFP levels was done using the Macnification software from images obtained using a single plane of focus. The average green fluorescence was sampled from four different regions in the nurse cell cytoplasm of each of 10 to 11, stage 5 or 6 egg chambers.
Western blotting
Antibodies for western blotting were mouse anti-Bru (AN, unpublished) (1:8000), mouse anti-HA antibody (Covance) (1:1000), mouse anti-αTubulin (Sigma) (1:1000) and rat anti-Cup219 [58](1:2000). Alkaline phosphatase-conjugated secondary antibodies were used at 1:5000 (anti-mouse; Applied Biosystems) or 1:10000 (anti-rat; Sigma).
RNA binding assay
The osk 3’ UTR AB probe was transcribed using MAXIscript Kit (Ambion) and uniformly labeled with [α-32P]UTP (800Ci/mmol, Perkin Elmer). UV cross-linking assay was performed as described [11], except that 10X binding buffer consisted of 60mM Hepes pH7.9, 300mM KCl and 20mM MgCl2, and was supplemented with protease inhibitors (Roche). ~500ng of purified recombinant Bru proteins was used. After electrophoresis of cross-linked adducts, gels were dried (Bio-Rad) and exposed to a Phosphor Screen (Molecular Dynamics) for 12 hr. The screen was then analyzed with a Typhoon laser scanner (GE Healthcare).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Martin KC, Ephrussi A (2009) mRNA localization: gene expression in the spatial dimension. Cell 136: 719–730. doi: 10.1016/j.cell.2009.01.044 19239891
2. Holt CE, Schuman EM (2013) The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80: 648–657. doi: 10.1016/j.neuron.2013.10.036 24183017
3. Jung H, Gkogkas CG, Sonenberg N, Holt CE (2014) Remote control of gene function by local translation. Cell 157: 26–40. doi: 10.1016/j.cell.2014.03.005 24679524
4. Lasko P (2012) mRNA localization and translational control in Drosophila oogenesis. Cold Spring Harb Perspect Biol 4.
5. St Johnston D (1995) The intracellular localization of messenger RNAs. Cell 81: 161–170. 7736568
6. Lehmann R, Nüsslein-Volhard C (1986) Abdominal segmentation, pole cell formation, and embryonic polarity require the localized activity of oskar, a maternal gene in Drosophila. Cell 47: 141–152. 3093084
7. Ephrussi A, Lehmann R (1992) Induction of germ cell formation by oskar. Nature 358: 387–392. 1641021
8. Smith JL, Wilson JE, Macdonald PM (1992) Overexpression of oskar directs ectopic activation of nanos and presumptive pole cell formation in Drosophila embryos. Cell 70: 849–859. 1516136
9. Ephrussi A, Dickinson LK, Lehmann R (1991) oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell 66: 37–50. 2070417
10. Kim-Ha J, Smith JL, Macdonald PM (1991) oskar mRNA is localized to the posterior pole of the Drosophila ooctye. Cell 66: 23–35. 2070416
11. Kim-Ha J, Kerr K, Macdonald PM (1995) Translational regulation of oskar mRNA by bruno, an ovarian RNA-binding protein, is essential. Cell 81: 403–412. 7736592
12. Markussen F-H, Michon A-M, Breitwieser W, Ephrussi A (1995) Translational control of oskar generates Short OSK, the isoform that induces pole plasm assembly. Development 121: 3723–3732. 8582284
13. Rongo C, Gavis ER, Lehmann R (1995) Localization of oskar RNA regulates oskar translation and requires Oskar protein. Development 121: 2737–2746. 7555702
14. Lie Y, Macdonald PM (1999) Translational regulation of oskar mRNA occurs independent of the cap and poly(A) tail in Drosophila ovarian extracts. Development 126: 4989–4996. 10529417
15. Castagnetti S, Hentze MW, Ephrussi A, Gebauer F (2000) Control of oskar mRNA translation by Bruno in a novel cell-free system from Drosophila ovaries. Development 127: 1063–1068. 10662645
16. Nakamura A, Sato K, Hanyu-Nakamura K (2004) Drosophila Cup Is an eIF4E Binding Protein that Associates with Bruno and Regulates oskar mRNA Translation in Oogenesis. Dev Cell 6: 69–78. 14723848
17. Chekulaeva M, Hentze MW, Ephrussi A (2006) Bruno acts as a dual repressor of oskar translation, promoting mRNA oligomerization and formation of silencing particles. Cell 124: 521–533. 16469699
18. Jambor H, Brunel C, Ephrussi A (2011) Dimerization of oskar 3′ UTRs promotes hitchhiking for RNA localization in the Drosophila oocyte. RNA 17: 2049–2057. doi: 10.1261/rna.2686411 22028360
19. Besse F, Lopez de Quinto S, Marchand V, Trucco A, Ephrussi A (2009) Drosophila PTB promotes formation of high-order RNP particles and represses oskar translation. Genes Dev 23: 195–207. doi: 10.1101/gad.505709 19131435
20. Wilson JE, Connell JE, Macdonald PM (1996) aubergine enhances oskar translation in the Drosophila ovary. Development 122: 1631–1639. 8625849
21. Gunkel N, Yano T, Markussen FH, Olsen LC, Ephrussi A (1998) Localization-dependent translation requires a functional interaction between the 5′ and 3′ ends of oskar mRNA. Genes Dev 12: 1652–1664. 9620852
22. Chang JS, Tan L, Schedl P (1999) The Drosophila CPEB homolog, orb, is required for oskar protein expression in oocytes. Dev Biol 215: 91–106. 10525352
23. Micklem DR, Adams J, Grunert S, St Johnston D (2000) Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J 19: 1366–1377. 10716936
24. Castagnetti S, Ephrussi A (2003) Orb and a long poly(A) tail are required for efficient oskar translation at the posterior pole of the Drosophila oocyte. Development 130: 835–843. 12538512
25. Munro TP, Kwon S, Schnapp BJ, St Johnston D (2006) A repeated IMP-binding motif controls oskar mRNA translation and anchoring independently of Drosophila melanogaster IMP. J Cell Biol 172: 577–588. 16476777
26. Reveal B, Yan N, Snee MJ, Pai CI, Gim Y, Macdonald PM (2010) BREs mediate both repression and activation of oskar mRNA translation and act in trans. Dev Cell 18: 496–502. doi: 10.1016/j.devcel.2009.12.021 20230756
27. Snee M, Benz D, Jen J, Macdonald PM (2008) Two distinct domains of Bruno bind specifically to the oskar mRNA. RNA Biol 5: 49–57.
28. Romaniuk PJ, Lowary P, Wu H-N, Stormo G, Uhlenbeck OC (1987) RNA binding site of R17 coat protein. Biochem 26: 1563–1568.
29. Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757. 16340016
30. Yoshida S, Müller HA, Wodarz A, Ephrussi A (2004) PKA-R1 spatially restricts Oskar expression for Drosophila embryonic patterning. Development 131: 1401–1410. 14993189
31. Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ (1990) Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem 265: 14264–14269. 2117608
32. White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM (1998) Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem 273: 3166–3172. 9452427
33. Zetterqvist O, Ragnarsson U, Engstrom L (1990) Substrate specificity of cyclic AMP-dependent protein kinase. Peptides and protein phosphorylation: 171–187.
34. LeCuyer K, Behlen L, Uhlenbeck O (1995) Mutants of the bacteriophage MS2 coat protein that alter its cooperative binding to RNA. Biochemistry 22: 10600–10606.
35. De Gregorio E, Preiss T, Hentze MW (1999) Translation driven by an eIF4G core domain in vivo. EMBO J 18: 4865–4874. 10469664
36. Vanzo NF, Ephrussi A (2002) Oskar anchoring restricts pole plasm formation to the posterior of the Drosophila oocyte. Development 129: 3705–3714. 12117819
37. Kim-Ha J, Webster PJ, Smith JL, Macdonald PM (1993) Multiple RNA regulatory elements mediate distinct steps in localization of oskar mRNA. Development 119: 169–178. 8275853
38. Ghosh S, Marchand V, Gáspár I, Ephrussi A (2012) Control of RNP motility and localization by a splicing-dependent structure in oskar mRNA. Nat Struct Mol Biol 19: 441–449. doi: 10.1038/nsmb.2257 22426546
39. Huang J, Zhou W, Dong W, Watson AM, Hong Y (2009) Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc Natl Acad Sci U S A 106: 8284–8289. doi: 10.1073/pnas.0900641106 19429710
40. Schüpbach T, Wieschaus E (1989) Female sterile mutations on the second chromosome of Drosophila melanogaster. I. Maternal effect mutations. Genetics 121: 101–117. 2492966
41. Snee MJ, Macdonald PM (2009) Dynamic organization and plasticity of sponge bodies. Dev Dyn 238: 918–930. doi: 10.1002/dvdy.21914 19301391
42. Webster PJ, Liang L, Berg CA, Lasko P, Macdonald PM (1997) Translational repressor bruno plays multiple roles in development and is widely conserved. Genes Dev 11: 2510–2521. 9334316
43. Reveal B, Garcia C, Ellington A, Macdonald PM (2011) Multiple RNA binding domains of Bruno confer recognition of diverse binding sites for translational repression. RNA Biol 8: 1047–1060. doi: 10.4161/rna.8.6.17542 21955496
44. Igreja C, Izaurralde E (2011) CUP promotes deadenylation and inhibits decapping of mRNA targets. Genes Dev 25: 1955–1967. doi: 10.1101/gad.17136311 21937713
45. Lane ME, Kalderon D (1994) RNA localization along the anteroposterior axis of the Drosophila oocyte requires PKA-mediated signal transduction to direct normal microtubule organization. Genes Dev 8: 2986–2995. 7528157
46. Huttelmaier S, Zenklusen D, Lederer M, Dictenberg J, Lorenz M, et al (2005) Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438: 512–515. 16306994
47. Paquin N, Ménade M, Poirier G, Donato D, Drouet E, Chartrand P (2007) Local activation of yeast ASH1 mRNA translation through phosphorylation of Khd1p by the casein kinase Yck1p. Mol Cell 26: 795–809. 17588515
48. Deng Y, Singer RH, Gu W (2008) Translation of ASH1 mRNA is repressed by Puf6p-Fun12p/eIF5B interaction and released by CK2 phosphorylation. Genes Dev 22: 1037–1050. doi: 10.1101/gad.1611308 18413716
49. Saffman EE, Styhler S, Rother K, Li W, Richard S, Lasko P (1998) Premature translation of oskar in oocytes lacking the RNA-binding protein bicaudal-C. Mol Cell Biol 18: 4855–4862. 9671494
50. Nakamura A, Amikura R, Hanyu K, Kobayashi S (2001) Me31B silences translation of oocyte-localizing RNAs through the formation of cytoplasmic RNP complex during Drosophila oogenesis. Development 128: 3233–3242. 11546740
51. Wilhelm JE, Hilton M, Amos Q, Henzel WJ (2003) Cup is an eIF4E binding protein required for both the translational repression of oskar and the recruitment of Barentsz. J Cell Biol 163: 1197–1204. 14691132
52. Van Doren M, Williamson AL, Lehmann R (1998) Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr Biol 8: 243–246. 9501989
53. Martin SG, St Johnston D (2003) A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature 421: 379–384. 12540903
54. Reich J, Snee MJ, Macdonald PM (2009) miRNA-dependent translational repression in the Drosophila ovary. PLoS One 4: e4669. doi: 10.1371/journal.pone.0004669 19252745
55. Haseloff J (1999) GFP variants for multispectral imaging of living cells. Methods Cell Biol 58: 139–151. 9891379
56. Verrotti AC, Wharton RP (2000) Nanos interacts with cup in the female germline of Drosophila. Development 127: 5225–5232. 11060247
57. Snee MJ, Macdonald PM (2004) Live imaging of nuage and polar granules: evidence against a precursor-product relationship and a novel role for Oskar in stabilization of polar granule components. J Cell Sci 117: 2109–2120. 15090597
58. Nelson MR, Leidal AM, Smibert CA (2004) Drosophila Cup is an eIF4E-binding protein that functions in Smaug-mediated translational repression. EMBO J 23: 150–159. 14685270
59. Lyon A, Reveal B, Macdonald PM, Hoffman D (2009) Bruno protein contains an expanded RNA recognition motif. Biochemistry 48: 12202–12212. doi: 10.1021/bi900624j 19919093
60. Filardo P, Ephrussi A (2003) Bruno regulates gurken during Drosophila oogenesis. Mech Dev 120: 289–297. 12591598
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 2
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates