
Identification of Driving Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer
Little is known about the molecular biology of medullary thyroid cancer (MTC), which is a rare disease. Genomics are increasingly being used to improve our knowledge about disease biology and to identify therapeutic targets in many cancers. Here, we report the largest genomic results of MTC to date. MTC tissue frequently included several mutations. For the first time, anaplastic lymphoma kinase (ALK) rearrangements were detected in MTC: one case with a glutamine:fructose-6-phosphate transaminase 1 (GFPT1)-ALK fusion, and another case with an echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion. The fusion mechanism of the novel GFPT1-ALK fusion was successfully investigated using molecular biology techniques. In addition, an inhibitor of ALK (crizotinib) dramatically decreased the number of metastatic MTC lesions harboring the EML4-ALK fusion, thus verifying the fusion as a promising target in MTC. Our findings suggest that using rapidly improving sequencing techniques and accumulated genomic data to comprehensively perform genetic analyses on rare tumors, such as MTC, will help to improve the poor prognosis of orphan diseases.
Published in the journal:
Identification of Driving Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005467
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005467
Summary
Little is known about the molecular biology of medullary thyroid cancer (MTC), which is a rare disease. Genomics are increasingly being used to improve our knowledge about disease biology and to identify therapeutic targets in many cancers. Here, we report the largest genomic results of MTC to date. MTC tissue frequently included several mutations. For the first time, anaplastic lymphoma kinase (ALK) rearrangements were detected in MTC: one case with a glutamine:fructose-6-phosphate transaminase 1 (GFPT1)-ALK fusion, and another case with an echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion. The fusion mechanism of the novel GFPT1-ALK fusion was successfully investigated using molecular biology techniques. In addition, an inhibitor of ALK (crizotinib) dramatically decreased the number of metastatic MTC lesions harboring the EML4-ALK fusion, thus verifying the fusion as a promising target in MTC. Our findings suggest that using rapidly improving sequencing techniques and accumulated genomic data to comprehensively perform genetic analyses on rare tumors, such as MTC, will help to improve the poor prognosis of orphan diseases.
Introduction
Many cancer gene profiling studies have recently been published, describing genetic trends that are not limited to specific cancers. Next-generation sequencing (NGS) is an important tool for detecting genetic alterations in many kinds of cancers, as it allows for millions of nucleic acid sequences to be simultaneously sequenced within a short period of time and is more cost-effective than older methods. Thus, many researchers and physicians anticipate that NGS will bring the concept of personalized cancer therapy to fruition.
Medullary thyroid cancer (MTC) is a rare malignancy that accounts for up to 3–5% of thyroid cancers. It is derived from calcitonin-secreting para-follicular C cells and can arise in a familial (25%) or sporadic (75%) pattern. Genetic and epigenetic alterations play important roles in the progression and prognosis of MTC [1–3]. Genes encoding the ret proto-oncogene (RET) and Ras (RAS) are commonly mutated in MTC [4, 5]. The RET mutation is believed to be a causative event in both familial and sporadic MTC [6, 7]. In the Mitogen-activated protein kinase (MAPK) pathway, the RAS mutation is another genetic rearrangement that is prevalent in sporadic MTC and other types of thyroid cancer [2] but the prevalence and significance of other genetic mutations including BRAF in MTC remain unclear.
MTC has a different response to treatment than that of well-differentiated thyroid cancers. Because radioactive iodine does not accumulate in MTC, few therapeutic options are available for advanced MTC. Inhibitors of RET, such as cabozantinib and vandetanib, have recently been shown to be effective in advanced MTC [8, 9]. However, whether the RET mutation is a predictive factor for the success of these drugs is unclear [9].
Recently, the rearrangement of anaplastic lymphoma kinase (ALK) was detected in a small but significant proportion of patients with non-small cell lung cancer (NSCLC) [10]. Several ALK inhibitors, including crizotinib, have achieved dramatic responses in cases of NSCLC harboring ALK rearrangements [11–13]. Although ALK rearrangement has also been episodically observed in a small set of other cancer types, little is known about ALK rearrangements in MTC [14, 15].
In this study, we used targeted NGS and various methods to examine the genetic profiles of MTC and detect ALK rearrangements.
Results
Basal characteristics and prevalence of gene mutations that are detected by AmpliSeq
Eighty-four samples (11 hereditary, 41 sporadic and 32 unknown) from patients with MTC (mean age of 48.5 years) and 36 paired normal thyroid tissue samples were successfully sequenced. The normal thyroid tissue samples in the MTC patients were used as matched control samples. Of the cases, 32 were male and 52 were female. Detailed demographic, clinic-pathological and genetic characteristics are listed in Table 1 and S2 Table. Hereditary MTCs were defined as cases having either positive germ-line RET mutations in blood tests or possession of a strong family history with MTC in at least four family members [16]. The unknown group was composed of MTC cases with no blood RET test and no family history of MTC/MEN. The mean value of variant coverage was 593 reads, and the variant coverage ranged from 19 to 1,482 reads. Overall, 101 mutations were observed in the MTC samples. Most mutations (N = 96, 95.0%) were single-nucleotide variants; 5 were deletions. The most common mutation occurred in RET, which was observed in 47 cases, followed by mutations in genes encoding Harvey rat sarcoma viral oncogene homolog (N = 14), serine/threonine kinase 11 (N = 11), v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (N = 6), mutL homolog 1 (N = 4), Kiesten rat sarcoma viral oncogene homolog (N = 3), MET proto-oncogene (N = 3), ATM serine/threonine kinase (N = 2), kinase insert domain receptor (N = 2), adenomatous polyposis coli (APC; N = 2), B-raf proto-oncogene (N = 1), cadherin 1 (N = 1), epidermal growth factor receptor (N = 1), cyclin-dependent kinase inhibitor 2A (CDKN2A, N = 1), Janus kinase 3 (N = 1), protein tyrosine phosphatase, non-receptor type 1 (N = 1), SMAD family member 4 (N = 1) and von Hippel-Lundau tumor suppressor (N = 1). We did not detect any dominant gene mutations in 20 MTC samples, which all exhibited wild-type RET, HRAS and KRAS. These are listed in S1 Table and shown in Fig 1.


Specific types of gene mutations
The commonly observed RET mutations occurred in exons 10, 11, 15, and 16. Previous studies have shown that M918T is the most common RET mutation in MTC [2, 10]. Similarly, M918T (N = 19) was the most common RET mutation in our samples, followed by C634Y (N = 7), C634W (N = 4), C634G (N = 4), C630R (N = 4), D631Y (N = 2), and others (N = 7). All HRAS mutations occurred in exon 3. The mutant amino acid sequence in each of the HRAS mutant cases was Q61K (N = 13). KRAS mutations were observed in three cases (Q61R, 2 and G48R, 1), and BRAF mutation was found in only one case. The dominant amino acid sequence in STK11 was F354L (N = 7). Other mutated genes are shown in S1 Table.
Comparative analysis between MTC tissue and matched normal thyroid tissue
We compared the genetic landscapes between 36 MTC tissues and their matched normal thyroid tissues: this group was composed of 16 sporadic, 5 hereditary and 15 cases with unknown information about heredity (Fig 2). In the hereditary MTC cases, RET mutations were observed in MTC and their matched normal thyroid tissues: these RET mutation types included C634Y, D631Y, and C634W, which are well known to be associated with the MEN2A [17, 18]. One case, which had been classified as an unknown subgroup based on blood test or family history, was found to have C634W mutation in both MTC and normal tissue, leading us to suspect that this case might be hereditary MTC. In 16 sporadic MTC group, several RET mutation types (M918T, C630R, C618S and deletion) were detected in MTC tissues, but not in the matched normal thyroid tissues. The M918T RET mutations and Q61K HRAS mutations were observed only in the MTCs of the sporadic or unknown subgroups, suggesting that these mutations are pathognomonic somatic mutation in MTC. In addition, two KRAS (G48R, Q61R) and one MET (A986T) mutations were also observed only in MTC tissues. However, STK11 (F354L), MLH1, KIT, and KDR mutations were observed in both MTC and normal thyroid tissues, which leads their pathognomonic natures unresolved in MTC.

Screening for ALK rearrangement and identification of ALK fusion
In parallel with targeted sequencing using AmpliSeq, we screened for ALK rearrangements. Ninety-eight cases were screened using immunohistochemistry (IHC), and 83 of these cases were also evaluated using AmpliSeq. Nine ALK-positive cases were found with IHC scores of 1+ (N = 7), 2+ (N = 1), and 3+ (N = 1). We also performed ALK fluorescence in situ hybridization (FISH) testing on ALK-positive samples that were identified via IHC. The two samples with 2+ and 3+ IHC scores exhibited ALK break-apart rearrangements (Fig 3).
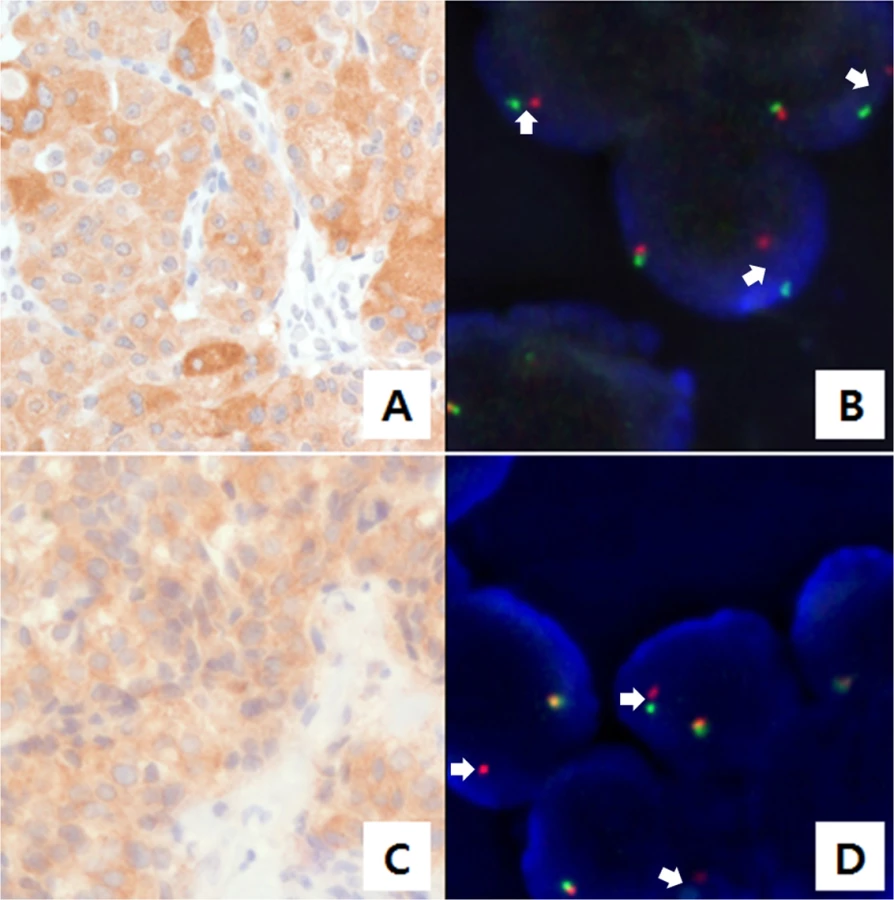
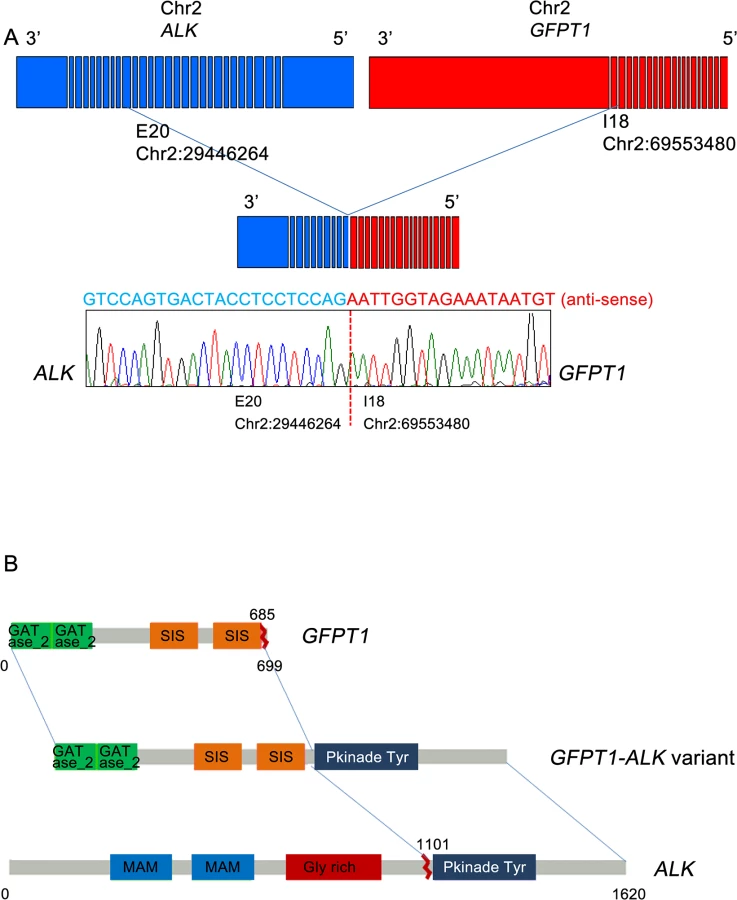
For the two cases harboring ALK break-apart rearrangements, targeted cancer panel sequencing (HiSeq 2500, Illumina, USA) was performed to detect the breakpoints and 5’ fusion partner genes of ALK. This process revealed two distinct ALK fusions. For the first case, a novel fusion gene was detected: 5’ glutamine:fructose-6-phosphate transaminase 1 (GFPT1; located in 2p13) was fused to 3’ ALK (located in 2p23) with preservation of the ALK kinase domain (Fig 4A and 4B). The breakpoints in GFPT1 and ALK were in intron 18 and exon 20, respectively. Based on the gene direction and location, the structural variation was presumed to be intra-chromosomal translocation or deletion. To confirm the fusion, we amplified the genomic fusion point between GFPT1 and ALK using genomic DNA of the MTC tissue. PCR analysis and Sanger sequencing revealed the same results as that of the customized targeted cancer panel (Figs 4B and S1). For the second case, the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion was detected. The breakpoints were located in intron 13 of EML4 and intron 19 of ALK, which indicates that this fusion is the most common variant (E13; A20) in NSCLC [19, 20]. This case exhibited metastatic lesions after thyroidectomy and was enrolled in a Phase I crizotinib trial (NCT01121588). After crizotinib therapy, the tumor lesions in the lung, liver, and bone shrank remarkably, and plasma calcitonin levels decreased. The final results will be disclosed with the full clinical study.
Discussion
We identified two types of ALK fusion genes in MTC by sequencing via IHC, FISH, and NGS analyses. Of the two fusion types, the EML4-ALK fusion was the same as the most commonly detected variant in NSCLC, [19] where the EML4-ALK fusion is a strong predictive factor for the efficacy of ALK inhibitors [13, 21, 22]. In the current study, the patient with metastatic MTC harboring the EML4-ALK fusion showed a dramatic response to crizotinib. We are the first to report an MTC case with a targetable EML4-ALK fusion gene. Previously, Kelly et al. used the Illumina HiSeq sequencing system to identify one papillary thyroid cancer case with an EML4-ALK fusion [15]. However, they also tested 22 medullary carcinoma cases and did not find any cases with the EML4-ALK fusion, as evaluated by reverse transcription-PCR. Their failure to detect the ALK rearrangement in MTC is understandable, given that our prevalence rate of ALK fusions in the current study was only 2% (2 out of 98 cases). This suggests that more efficient strategies are needed to detect the ALK rearrangement. Results from the current study suggest that IHC-based screening, along with FISH-based confirmation and targeted NGS, may be a cost-effective and reliable method to detect ALK rearrangements.
Most importantly, we detected a novel GFPT1-ALK fusion that has not been reported in any type of cancer. GFPT1 is a key enzyme in the biosynthesis of N-acetylglucosamine and is required for critical events in neuromuscular transmission [23]. Until now, several fusion partners of ALK have been reported in various cancers [24–28]. Among them, huntingdon-interacting protein (HIP1)-ALK and RAN-binding protein 2 (RANBP2)-ALK, which have been reported to exist in NSCLC and inflammatory myofibroblastic tumors, respectively, show clinical responses to crizotinib [25, 26]. In the current study, the MTC case harboring the GFPT1-ALK fusion showed strong ALK protein expression and did not exhibit co-existing genetic mutations; both of these factors may support an important role for this fusion gene in the pathogenesis of this MTC case. However, we were unable to validate whether GFPT1-ALK was a driving oncogene or a therapeutically targetable gene. Whether GFPT1-ALK is also a predictor for ALK inhibitors is unclear.
Currently, vandetanib and cabozantinib are approved for the treatment of MTC by the U.S. Food and Drug Administration. However, the prognosis of patients with metastatic MTC is still poor, due to the inherent resistance to radioiodine therapy and aggressive nature of this disease. Furthermore, the rarity of MTC makes it hard to perform prospective studies to find new agents. Therefore, the comprehensive genetic analysis of MTC can help to identify effective ways to improve its prognosis. Despite the low frequency of ALK rearrangements in MTC, our techniques can be used to detect target genes in other rare diseases.
In addition, our sequencing analysis of MTC is the largest to date. Previously, Agrawal et al. published the largest genomic analysis of MTC [5], where they performed whole-exome sequencing of 17 sporadic MTCs and 40 additional MTCs (hereditary or sporadic) for validation. RET was the dominant mutation (43/57) in that study. We used a larger sample size and accurate verification by comparing 36 pairs of MTC with matched normal thyroid tissues that were acquired from the same person.
In the comparison analyses, all five hereditary cases were observed to have germ-line RET mutations in both MTC and control tissues. However, M918T RET (N = 10), Q61K HRAS (N = 7), KRAS (N = 2), and MET (N = 1) mutations were harbored dominantly in MTCs. Simbolo et al. identified RET, HRAS, KRAS and STK11 mutations as significant somatic mutations in MTCs, whereas TP53, KDR, KIT, MET, PIK3CA and ATM mutations were classified as nonpathogenic germ-line variants [29]. Our current data are compatible with that report. Interestingly, however, the F354L STK11 mutation, regarded as significant somatic mutation by Simbolo et al., was observed in both MTCs and control tissues of our seven cases. Therefore, we presume that the F354L STK11 mutation is a germ-line mutation in MTC.
In conclusion, we report that the EML4-ALK fusion, which was found for the first time in MTC, could be an effective molecular target of crizotinib. Furthermore, our results also suggest that the novel GFPT1-ALK fusion can be a potential candidate for molecular target therapy. This study included the largest set of molecular profile data in MTC to date, which was achieved by using high-depth NGS panel sequencing, and also presented the genetic landscape of MTC. Further translational research is needed to determine the oncogenic roles of these mutations in MTC.
Materials and Methods
Ethics statement
Written informed consent was obtained from all participants, and this study was approved by the Institutional Review Board of Samsung Medical Center. (SMC 2013-02-010).
Searching for genetic mutation profiles by Ampliseq
We collected data on patients who were histologically diagnosed with MTC without the coexistence of tumors on the parathyroid and adrenal gland. All patients received surgical treatment at Samsung Medical Center between June 2000 and January 2013. Among 101 MTC specimens, 17 were excluded based on quality control (N = 5), preparation failure (N = 11), and sequencing failure (N = 1). The remaining 84 MTC samples were sequenced using an Ion Torrent Personal Genome Machine (IT-PGM, Life Technologies, Grand Island, NY, USA), which takes real-time measurements of hydrogen ions that are produced during DNA replication and allows for rapid sequencing. Eight normal thyroid tissues were obtained by thyroidectomy and sequenced. Mutation profiles between MTC and normal thyroid tissues from eight individuals were compared.
We constructed libraries using the Ion AmpliSeq Panels, Ion AmpliSeq Library Kit, and Ion Xpress Barcodes, as well as 10 ng of DNA sample per pool (Life Technologies). The amplicons were ligated to Ion Adapters and purified. For barcoded library preparations, barcoded adapters from the Ion Xpress Barcode Adapters 1–96 Kit were substituted for the non-barcoded adapter mix in the Ion AmpliSeq Library Kit. Next, the multiplexed barcoded libraries were enriched by clonal amplification using emulsion polymerase chain reaction (PCR) on Ion Sphere Particles (Ion PGM Template 200 Kit) and loaded on an Ion 316 Chip. Massively parallel sequencing was carried out on an Ion PGM using the Ion PGM Sequencing 200 Kit v2. The Ion AmpliSeq Cancer Hotspot Panel v2 covered hotspot regions of 50 oncogenes and tumor suppressor genes.
The primary filtering process was performed with the Torrent Suite v4.0.0 and Ion Torrent Variant Caller v4.0 software and included signal processing, base calling, assigning quality scores, adapter trimming, PCR duplicate removal, read alignment (to human genome reference 19), mapping quality control, coverage analyzing, and variant calling [30]. To detect variants, a minimum coverage of 100 reads was achieved with a cutoff value of at least 5% in the variant calling rate (frequency). Variant calls were further analyzed by using ANNOVAR variant filtering and COSMIC database (dbSNP build 137) annotating, and these analyses were based on changes in the amino acid sequence.
ALK immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH)
The ALK IHC assay used a mouse monoclonal ALK antibody (5A4, Novocastra, Newcastle, United Kingdom) and the antibody for ALK was diluted to 1:30, treated, and incubated at 42°C for 2 hours. ALK IHC scores were assigned as follows: 0, no staining; 1+, faint or weak staining intensity with more than 5% tumor cells or any staining intensity with ≤5% tumor cells; 2+, moderate cytoplasmic reactivity with more than 5% tumor cells; and 3+, granular cytoplasmic reactivity of strong intensity in more than 5% of tumor cells [31]. Cases that showed ALK-positive staining with a score of 1+ or greater were analyzed by FISH with the Vysis ALK Break-Apart FISH Probe Kit (Abbott Laboratories, Abbott Park, IL). Samples were considered positive for ALK FISH if more than 15% of cells were positive or an isolated red signal (IRS) in tumor cells.
Customized targeted cancer panel sequencing for ALK fusion genes
Genomic DNA extraction was performed using the QIAamp DNA mini kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. The Nanodrop 8000 UV-Vis spectrometer (Thermo Scientific Inc., DE, USA), Qubit 2.0 Fluorometer (Life Technologies), and 2200 TapeStation Instrument (Agilent Technologies, Santa Clara, CA, USA) were used to check the concentration, purity, and degradation of extracted genomic DNA. For the next step, samples that passed our quality control thresholds were used.
Genomic DNA (250 ng) from the tissues was sheared by the Covaris S220 (Covaris, Woburn, MA, USA) and used for the construction of the library using customized RNA baits and the SureSelect XT reagent kit, HSQ (Agilent Technologies), according to the manufacturer’s protocol. The customized RNA baits covered whole exons and flanking intronic sequences of the 83 genes. After enriched exome libraries were multiplexed, the libraries were sequenced on the HiSeq 2500 sequencing platform (Illumina, USA), as described previously [32]. Briefly, a paired-end DNA sequencing library was prepared through the following processes: genomic DNA shearing, end-repair, A-tailing, paired-end adaptor ligation, and amplification. After the library was hybridized with bait sequences for 16 hours, the captured library was purified and amplified with an index barcode tag. Then, the quality and quantity of the captured library were measured. Sequencing of the exome library was carried out using the 100-bp, paired-end mode of the TruSeq Rapid PE Cluster kit and TruSeq Rapid SBS kit (Illumina, San Diego, CA, USA).
PCR for ALK fusion genes
The newly identified glutamine:fructose-6-phosphate transaminase 1 (GFPT1)-ALK fusion gene was detected by targeted cancer panel sequencing, and its respective genomic rearrangement was confirmed by genomic PCR analysis, followed by Sanger sequencing. Genomic DNA was isolated from formalin-fixed, paraffin-embedded (FFPE) tumor samples using a ReliaPrep FFPE genomic DNA extraction kit (Promega, Madison, WI, USA). The PCR products were indicative of fusion points within intron 18 of GFPT1 and exon 20 of ALK, based on target sequencing results. PCR analysis of genomic DNA for GFPT1-ALK was performed with a pair of primers flanking the putative fusion point: GFPT1 F (5’-TCTGTGTGAACTGGCACCTT-3’) and ALK R (5’-ATTCAGCCCCTACACTGCAC-3’). PCR products were then separated on a 2% E-Gel SizeSelect agarose gel (Invitrogen, Carlsbad, CA, USA). For genomic PCR controls, we used DNA from the same FFPE tumor samples with glyceraldehyde-3-phosphate dehydrogenase PCR primers. In reactions that produced a PCR product of the expected size, the amplicons underwent gel purification and sequencing using a 3130 XL ABI Prism sequencer (Applied Biosystems, Foster City, CA, USA) with Bigdye Terminator v3.1 Cycle sequencing kits, according to the manufacturer’s instructions.
Supporting Information
{kind=link}
Zdroje
1. Cerrato A, De Falco V, Santoro M. Molecular genetics of medullary thyroid carcinoma: the quest for novel therapeutic targets. J Mol Endocrinol. 2009;43: 143–155. doi: 10.1677/JME-09-0024 19383830
2. Goutas N, Vlachodimitropoulos D, Bouka M, Lazaris AC, Nasioulas G, Gazouli M. BRAF and K-RAS mutation in a Greek papillary and medullary thyroid carcinoma cohort. Anticancer Res. 2008;28: 305–308. 18383861
3. Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93: 682–687. 18073307
4. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16: 441–467. 15982921
5. Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013;98: E364–369. doi: 10.1210/jc.2012-2703 23264394
6. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2: 851–856. 8103403
7. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363: 458–460. 8099202
8. Wells SA Jr., Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28: 767–772. doi: 10.1200/JCO.2009.23.6604 20065189
9. Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31: 3639–3646. doi: 10.1200/JCO.2012.48.4659 24002501
10. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27: 4247–4253. doi: 10.1200/JCO.2009.22.6993 19667264
11. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363: 1693–1703. doi: 10.1056/NEJMoa1006448 20979469
12. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368: 2385–2394. doi: 10.1056/NEJMoa1214886 23724913
13. Shaw AT, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370: 2537–2539.
14. Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363: 1727–1733. doi: 10.1056/NEJMoa1007056 20979472
15. Kelly LM, Barila G, Liu P, Evdokimova VN, Trivedi S, Panebianco F, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A. 2014;111: 4233–4238. doi: 10.1073/pnas.1321937111 24613930
16. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276: 1575–1579. 8918855
17. Beldjord C, Desclaux-Arramond F, Raffin-Sanson M, Corvol JC, De Keyzer Y, Luton JP, et al. The RET protooncogene in sporadic pheochromocytomas: frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab. 1995;80: 2063–2068. 7608256
18. Sanso GE, Domene HM, Garcia R, Pusiol E, de M, Roque M, et al. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer. 2002;94: 323–330. 11900218
19. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448: 561–566. 17625570
20. Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46: 1773–1780. doi: 10.1016/j.ejca.2010.04.002 20418096
21. Girard N. Crizotinib in ALK-positive lung cancer. Lancet Oncol. 2012;13: 962–963. doi: 10.1016/S1470-2045(12)70375-3 22954506
22. Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15: 1119–1128. doi: 10.1016/S1470-2045(14)70362-6 25153538
23. Senderek J, Muller JS, Dusl M, Strom TM, Guergueltcheva V, Diepolder I, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88: 162–172. doi: 10.1016/j.ajhg.2011.01.008 21310273
24. Wong DW, Leung EL, Wong SK, Tin VP, Sihoe AD, Cheng LC, et al. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer. 2011;117: 2709–2718. doi: 10.1002/cncr.25843 21656749
25. Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, et al. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J Thorac Oncol. 2014;9: 1821–1825. doi: 10.1097/JTO.0000000000000368 25393796
26. Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70: 10038–10043. doi: 10.1158/0008-5472.CAN-10-2956 21030459
27. Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1995;267: 316–317.
28. Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood. 2000;95: 2144–2149. 10706887
29. Simbolo M, Mian C, Barollo S, Fassan M, Mafficini A, Neves D, et al. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014;465: 73–78. doi: 10.1007/s00428-014-1589-3 24828033
30. Roy S, Durso MB, Wald A, Nikiforov YE, Nikiforova MN. SeqReporter: automating next-generation sequencing result interpretation and reporting workflow in a clinical laboratory. J Mol Diagn. 2014;16: 11–22. doi: 10.1016/j.jmoldx.2013.08.005 24220144
31. Paik JH, Choe G, Kim H, Choe JY, Lee HJ, Lee CT, et al. Screening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer: correlation with fluorescence in situ hybridization. J Thorac Oncol. 2011;6: 466–472. doi: 10.1097/JTO.0b013e31820b82e8 21258247
32. Lee YS, Cho YS, Lee GK, Lee S, Kim YW, Jho S, et al. Genomic profile analysis of diffuse-type gastric cancers. Genome Biol. 2014;15: R55. doi: 10.1186/gb-2014-15-4-r55 24690483
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 8
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- Molecular Basis of Gene-Gene Interaction: Cyclic Cross-Regulation of Gene Expression and Post-GWAS Gene-Gene Interaction Involved in Atrial Fibrillation