
Accurately Assessing the Risk of Schizophrenia Conferred by Rare Copy-Number Variation Affecting Genes with Brain Function
Investigators have linked rare copy number variation (CNVs) to neuropsychiatric diseases, such as schizophrenia. One hypothesis is that CNV events cause disease by affecting genes with specific brain functions. Under these circumstances, we expect that CNV events in cases should impact brain-function genes more frequently than those events in controls. Previous publications have applied “pathway” analyses to genes within neuropsychiatric case CNVs to show enrichment for brain-functions. While such analyses have been suggestive, they often have not rigorously compared the rates of CNVs impacting genes with brain function in cases to controls, and therefore do not address important confounders such as the large size of brain genes and overall differences in rates and sizes of CNVs. To demonstrate the potential impact of confounders, we genotyped rare CNV events in 2,415 unaffected controls with Affymetrix 6.0; we then applied standard pathway analyses using four sets of brain-function genes and observed an apparently highly significant enrichment for each set. The enrichment is simply driven by the large size of brain-function genes. Instead, we propose a case-control statistical test, cnv-enrichment-test, to compare the rate of CNVs impacting specific gene sets in cases versus controls. With simulations, we demonstrate that cnv-enrichment-test is robust to case-control differences in CNV size, CNV rate, and systematic differences in gene size. Finally, we apply cnv-enrichment-test to rare CNV events published by the International Schizophrenia Consortium (ISC). This approach reveals nominal evidence of case-association in neuronal-activity and the learning gene sets, but not the other two examined gene sets. The neuronal-activity genes have been associated in a separate set of schizophrenia cases and controls; however, testing in independent samples is necessary to definitively confirm this association. Our method is implemented in the PLINK software package.
Published in the journal:
Accurately Assessing the Risk of Schizophrenia Conferred by Rare Copy-Number Variation Affecting Genes with Brain Function. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001097
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001097
Summary
Investigators have linked rare copy number variation (CNVs) to neuropsychiatric diseases, such as schizophrenia. One hypothesis is that CNV events cause disease by affecting genes with specific brain functions. Under these circumstances, we expect that CNV events in cases should impact brain-function genes more frequently than those events in controls. Previous publications have applied “pathway” analyses to genes within neuropsychiatric case CNVs to show enrichment for brain-functions. While such analyses have been suggestive, they often have not rigorously compared the rates of CNVs impacting genes with brain function in cases to controls, and therefore do not address important confounders such as the large size of brain genes and overall differences in rates and sizes of CNVs. To demonstrate the potential impact of confounders, we genotyped rare CNV events in 2,415 unaffected controls with Affymetrix 6.0; we then applied standard pathway analyses using four sets of brain-function genes and observed an apparently highly significant enrichment for each set. The enrichment is simply driven by the large size of brain-function genes. Instead, we propose a case-control statistical test, cnv-enrichment-test, to compare the rate of CNVs impacting specific gene sets in cases versus controls. With simulations, we demonstrate that cnv-enrichment-test is robust to case-control differences in CNV size, CNV rate, and systematic differences in gene size. Finally, we apply cnv-enrichment-test to rare CNV events published by the International Schizophrenia Consortium (ISC). This approach reveals nominal evidence of case-association in neuronal-activity and the learning gene sets, but not the other two examined gene sets. The neuronal-activity genes have been associated in a separate set of schizophrenia cases and controls; however, testing in independent samples is necessary to definitively confirm this association. Our method is implemented in the PLINK software package.
Introduction
Multiple recent studies have demonstrated a convincing and statistically significant excess of rare CNVs in individuals affected by schizophrenia compared to unaffected individuals [1]–[4]. Similar observations have now been made separately in autism [5]–[7] and bipolar disorder [8]. However, it is typically not readily evident which individual CNV events are pathogenic since (1) many rare events are seen in the general population and the excess in cases is relatively modest and (2) individual events are too rare to demonstrate definitive association in realistically sized patient collections. Hence, it is challenging to translate these rare CNV events into a clear understanding of disease pathology. To identify candidate genes for follow-up, investigators have employed statistical tests of gene set enrichment, originally developed as an effective approach to interpret gene expression data [9]. Practically, these analyses identify functional gene sets or ‘pathways’ that are over-represented among those genes affected by case CNVs compared to unaffected genes [1], [8], [10], [11], often relying on online resources such as Panther [12], Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com), and Gene Ontology [13].
For example, gene set enrichment analyses by Walsh et al. suggested that rare CNVs in schizophrenia preferentially disrupt those genes with neuro-developmental functions [1]; Zhang et al. similarly reported that rare CNVs in bipolar disorder preferentially overlap genes involved in behavior and learning [8]. More recently Glessner et al. reported that genes affected by rare and common CNVs in autism are also involved in brain function [11]. While these initial results are highly promising, the gene set enrichment statistical framework as applied to copy number variation is critically limited and potentially confounded.
The key analytical question in this setting is whether an event that impacts a set of genes or a pathway, increases disease risk compared to events that do not impact that pathway. Under the hypothesis that events affecting a specific brain-function pathway are pathogenic, the rate of those events affecting the brain-function pathway should indeed be greater in cases than in controls – ideally fully explaining the observed genome-wide differences in case and control event rates. An alternative possibility is that the increased rate and size of CNVs in cases represents a mutational syndrome or genomic instability, and that they are not in themselves pathogenic. Under that possibility, case events should not preferentially impact any particular gene set; however, differences is size and rate might be observed.
The commonly used gene set enrichment analytical approach used to address this question falls short on two accounts: (1) they do not rigorously compare case event rates to control event rates and (2) since they examine affected genes rather than events, they do not accurately account for the fact that multiple genes might be contributed from a single event or that single genes may be affected by multiple events. Here we propose a straight forward statistical test to explicitly compare the rate of CNVs impacting a specific gene set in cases to the rate in controls that carefully accounts for background differences in CNV rate and size.
A possible consequence of not rigorously comparing event rates in cases to controls is that sets consisting of genes that are more frequently affected by CNVs might spuriously appear to be highly enriched in cases, but also will be highly enriched in controls. Examples of such genes include large genes spanning a massive portion of the genome or those whose functions are highly redundant or non-essential. This is a particularly important issue for neuropsychiatric disease considering the reportedly large size of genes with brain function. Multiple studies of CNVs in the general population have reported enrichment for neuro-physiological genes [14], [15] – suggesting that brain-function genes may be susceptible to CNV events in general, possibly due to their large size or other factors. In fact, published events in neuropsychiatric disease studies often implicate large genes (see Table S1). Others have already noted that gene size itself can bias pathway enrichment analyses in other contexts, such as annotating non-coding elements for function [16], [17]. In particular, Taher et al noted that randomly selected positions in the genome are enriched for proximity to genes involved in “development”, “cell-adhesion”, and “nervous system development” [17]. Some of the published disease studies attempted to address this issue indirectly by applying similar analyses to control events and note the lack of statistical evidence of enrichment for brain function gene sets [1], [8]; however, control events are typically fewer and smaller, implicating many fewer genes, and therefore simply comparing the statistical significance of gene set enrichment results in cases and controls is not adequate.
One possible consequence of examining genes rather than the events they occur in is that individual large events contribute many genes and may skew the analysis much more than smaller events, and cause spurious findings. This is of particular concern since genes with common function can often cluster together on the genome and a single event in one individual affecting a cluster of related genes can naively appear to implicate an entire pathway [18], [19]. One interesting example is the reported enrichment of psychological disorder genes in the Zhang et al data set (see Table S1); 11 of the 16 deleted psychological disorder genes are in the 22q11.21 region observed in two individuals in the data set [8]. These genes are possibly annotated as psychological disorder genes since rare deletions in 22q11.21 have long been observed among schizophrenia cases [2]. Removal of the two individuals with 22q11.21 events eliminates any enrichment for the psychological disorder gene set – suggesting that there is little evidence that this particular set is necessarily relevant to disease outside of the 22q11.21 region. Of course at least one gene in this region is pathogenic, but it is unlikely that >10 in this region are and that in aggregate define a key pathogenic set.
A second possible consequence of examining genes and not the events they occur in is that genes affected in both cases and controls, but at different rates, are not properly accounted for. For instance, a critical gene affected by many pathogenic events contributes equally to a gene set enrichment analyses as a gene sporadically affected by a single event. One interesting example is the NRXN1 gene, a large gene that plays an important role in synaptic development [20]. Since CNV events affecting NRXN1 have been observed in both schizophrenic cases and controls, they would contribute equally to a pathway analysis of case events as they would to one of control events. However, the rate of functional events observed in cases is significantly more than in controls; pathway-based approaches could be bolstered if methods explicitly take into account these differences between cases and controls event rates for genes of interest.
Here, we describe a case-control statistical test, cnv-enrichment-test, to explicitly compare the rate of CNVs impacting specific genes sets in cases to controls. We show how cnv-enrichment-test is robust to even extreme biases in gene size and case-control differences in CNV rate and size. We also demonstrate how standard gene set enrichment approaches is often confounded under realistic scenarios, by gene size and other gene structural features; we demonstrate these confounders in a set of 2,415 controls genotyped for rare single-event deletions. We finally apply the cnv-enrichment-test to examine genes with brain function within a large dataset of CNVs identified in schizophrenia cases and controls published by the International Schizophrenia Consortium (ISC) [2] and demonstrate nominal evidence of association for previously described gene sets.
Results and Discussion
Standard enrichment analysis to test gene sets affected by rare CNVs
Set enrichment is the standard approach to test whether genes impacted by CNVs in cases affect specific pathways. Specifically, the overlap between the set of genes affected by CNVs is compared to the set of genes with a particular function.
Genes affected by a CNV might be defined as disrupted genes or overlapped genes. Disrupted genes are those genes that have a CNV boundary that falls within the boundaries of its transcript [1]. Overlapped genes are a superset of those genes whose transcripts are either disrupted by a CNV or are fully contained by a CNV [8]. Since genes rarely overlap each other, a single CNV event might contribute up to two disrupted genes but many overlapped genes. Both have been previously examined in the literature. Unless otherwise specified, this study emphasizes overlapped genes.
After identifying the genes affected (overlapped or disrupted) by a CNV, we then identify genes with a specific process or within a specific pathway. We apply a two-tailed Fisher's test to assess whether the number of affected pathway genes is statistically significantly different than might be affected by chance. The critical assumption in gene set-based analyses is that there is a single independent observation per gene, not connected to the gene's size or structural features.
A case-control framework to test gene sets affected by rare CNVs
As an alternative, we propose a simple case-control strategy to test gene sets or pathways for association to disease: the “cnv-enrichment-test”. This strategy is consistent with the case-control association framework used in CNV and SNP disease association studies [21], [22]. A direct case-control comparison avoids any ascertainment bias that might be the consequence of structural features of genes within a set, since the same biases will apply equally to both cases and controls.
We are careful to control for case-control differences in CNV rate and size, since those differences can artificially induce a pathway association. For example, if the rare CNV rate in cases is more frequent or larger than in controls, then on average all genes will be impacted more often in cases, and any arbitrary gene set might appear to be affected more commonly in cases than in controls. Also, if rare CNVs are smaller but more frequent in cases than in controls, then sets of larger genes might appear to be impacted more often in cases than in controls.
To assess whether CNV events specifically overlapping genes in the pathway of interest are enriched in cases compared to controls, we propose the following logistic model:where pi is the probability that individual i is affected, ci is an integer that indicates the number of rare CNVs that an individual i has, si is the average size of those events, gi is the count of gene within a pre-specified gene set affected by a cnv, and e is an error term. The terms θ, γ, β0, and β1 are logistic regression parameters that are optimally determined to maximize the likelihood of the data. The θ term (the intercept) represents the background log likelihood for each individual, γ is the increase in log-likelihood per affected gene within the gene set, β0 is the increase in log-likelihood per rare CNV, and β1 is the increase in log likelihood per kilobasepair of average rare CNV size. The cnv-enrichment-test simply tests if γ is significantly different from 0.
In principle, previous studies in schizophrenia that have shown excess CNVs in affected individuals corresponding to a positive β0. It has also been demonstrated that individuals with neuropsychiatric disease often have larger events, consistent with a positive β1 term. On the other hand, if there is a “causal” gene set g, then adding it to the model should attenuate the magnitude of both β0 and β1 and result in a convincingly positive γ. An independent odds ratio estimate, eγ, can be calculated for the additional increased risk of disease if an event affects a gene in set g.
This approach is not confounded by functionally related genes that cluster on the genome. Since risk is estimated on a per individual basis, a single spurious observation will not dramatically impact the statistical significance of any of the parameter estimates. So, a rare single event, which happens to overlap multiple related genes within the gene set that is being tested, will not contribute substantially to the significance of γ - even though potentially many genes from that pathway are implicated. Of course, if many such events are observed, with a proclivity towards either cases or controls, then estimates for γ might appropriately be more significant.
The approach can be extended to do a meta-analysis if patient data is aggregated, and indicator variables are included to denote the dataset that the patient sample was derived from. Indicator variables would potentially account for specific differences across data sets, such as the proportion of individuals that are cases and also underlying biases in case severity.
This approach can be facilely applied to gene-sets ranging widely in size. It can equally be applied to a single gene, for example to identify whether a gene such as NRXN1 has more case-events than control-events after controlling for genome-wide differences in CNV size and rate. It can also be easily applied to the set of all genes in the human genome to test if genes in general are more often affected in cases than controls. We caution that in data sets with too few individuals, association to smaller gene sets might be difficult to detect given power limitations; furthermore the asymptotic p-value might be inaccurate. In cases where too few events have been genotyped the asymptotic p-value can be replaced by a p-value based on robust permutation testing instead.
We have implemented this test in the publicly available genetic data analysis software, PLINK [23].
CNV-enrichment-test is robust to skewed gene size, even if there are case-control differences between the size and rate of CNVs
To demonstrate that the cnv-enrichment-test does not detect spurious associations due to gene features that predispose key gene sets towards CNVs, we carefully considered gene size. We created an extreme hypothetical scenario (S0, see Table S2). Here, every fifth gene was designated as a hypothetical “brain gene”; brain genes were set to be considerably larger than other genes (50 kb versus 10 kb). For a single hypothetical chromosome, 250 Mb in length, we placed 2000 evenly spaced, non-overlapping genes. In all scenarios we simulated CNV data for 2000 cases and 2000 controls, specifying the mean CNV size at 100 kb (range 10 kb to 150 kb, standard deviation 30 kb) and the CNV rate per individual at 0.25. Reassuringly, in this simulation cnv-enrichment-test for “brain-genes” demonstrated p<0.05 association in 4.1% of 10,000 simulated datasets, suggesting that it estimates the type I error rate accurately (see Figure 1).

However, in practice, differences between the size and rate of CNVs might be present due to true genetic differences between cases and controls, as demonstrated in neuropsychiatric disease, or technical differences in array intensity or genotyping platform.
Our method must be robust to these differences and must not spuriously identify pathways with large genes as a consequence of these differences. To test for this we created four extreme scenarios (S1–S4, see Table S2). Under S1, we dramatically reduced the control rate of CNVs to 0.05/individual while retaining the same rate in cases (0.25/individual). Under S2, we fixed the rate at 0.25/individual in both cases and controls, but reduced the mean CNV size in cases (60 kb) compared to controls (100 kb). Under S3, we assigned cases the greater rate and mean size (0.25/individual and 100 kb) compared to controls (0.05/individual and 60 kb); this scenario is analogous to schizophrenia where events are larger and more frequent in cases. Under S4, we assigned cases had a greater rate, but smaller mean size (0.25/individual and 60 kb) compared to controls (0.05/individual and 100 kb); this scenario might occur if higher quality genotyping is applied in cases only resulting in better ability to detect smaller CNVs than in controls. We found that the proposed method that controlled for both CNV rate and average CNV size was robust under each of these extreme scenarios and for 10,000 simulated datasets demonstrated appropriate type I error rate at p<0.05 under all scenarios (see Figure 1).
To illustrate the importance of controlling for CNV rate and size in this setting where a pathway consists of systematically larger genes, we examined more limited models that do not control for either or both the CNV rate and size. All of these models caused inappropriately high type I error rates under at least one of the above scenarios (see Figure 1) and would demonstrate spurious association to “brain genes”. A simple association test (M0) that does not account for either for CNV rate or size at all demonstrates higher rates of false associations under all simulated scenarios where there are case-control differences in size and rate of CNVs (S1–S4). Similarly, controlling for differences in rate only (M1) demonstrates higher rates of false associations under almost all simulated scenarios, except for S0 and S1. Controlling for differences in size only (M2) demonstrates higher rates of false associations under almost all simulated scenarios, except for S0 and S3. Finally, controlling for differences in total CNV burden (M3) demonstrates higher rates of false associations under S3 and S4 all simulated scenarios.
Four plausible sets of genes with brain function
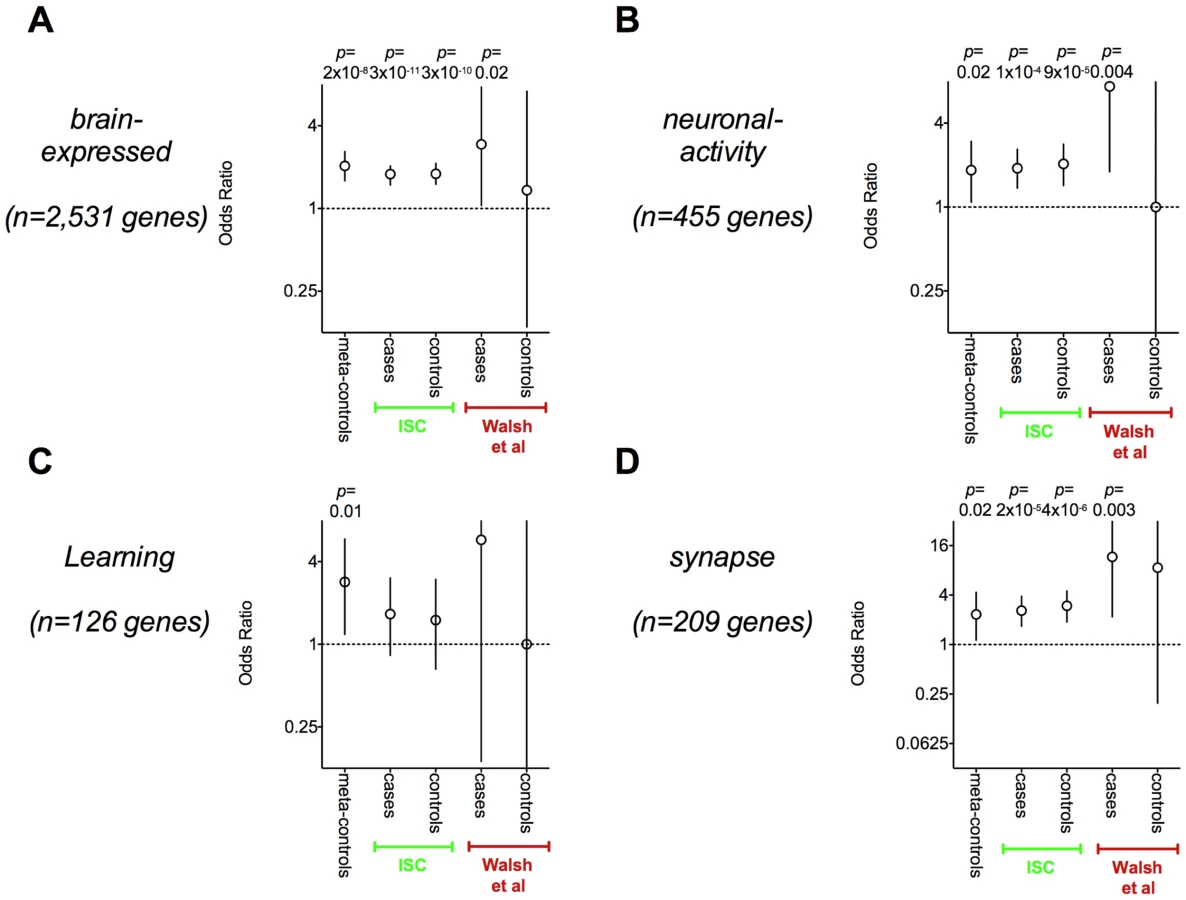
To broadly define genes that control brain function, we used a gene expression tissue atlas to define a broad set of 2,531 preferentially brain-expressed genes (see Materials and Methods). For secondary analyses, we compiled three more sets of general interest to neuropsychiatric disease: (1) 455 neuronal-activity genes defined by Panther and highlighted previously in schizophrenia by Walsh et al [1], (2) 126 learning genes defined by Ingenutiy and highlighted by Zhang et al in bipolar disease [8], and (3) 209 synapse genes defined by Gene Ontology. The gene sets overlap; 12 genes are in all four sets.
Application of set enrichment to rare CNVs in controls demonstrates that brain function genes are enriched
To demonstrate some of the limitations associated with standard set enrichment tests to assess critical gene functions examined the aforementioned gene sets in rare CNVs from controls recruited from the general populations. We used Affymetrix 6.0 chips in conjunction with stringent and uniform quality control to genotype 2,415 unaffected individuals (see Table S3 and Table S4) from four separate studies [8], [24]–[26]; hereafter referred to as ‘meta-controls’. We identified 1,054 single event deletions ranging from 20 kb to 1.9 Mb in size. To obtain the most confident calls possible, we focused only on deletions (see Materials and Methods) – though including duplications does not substantially impact our results.
Strikingly, many of the genes that are disrupted (and therefore also overlapped) by rare deletions within the meta-controls have been proposed as candidate genes for neuro-developmental diseases including: GRM5, GRM8, FHIT, OPCML, PTPRD, NRXN3, NRG3, CNTNAP2, AUTS2, CTNNA3, DLG2, ERBB4, PTPRM, and NRXN1. All of these genes are among the largest in the human genome, with transcripts extending from 550 kb to 2.2 Mb of genome. Except for GRM5 and PTPRM, they are all greater than 1 Mb in length. In particular DLG2, ERBB4, PTPRM, and NRXN1 were disrupted by 12 individual events in our study; Walsh et al. highlighted these four genes as potentially pathogenic based on pathway analysis [1].
As previously observed by Redon et al [14] and Yim et al [15], genes affected by rare CNVs are involved disproportionately in brain function in this control population. The set of genes disrupted by deletions within the meta-controls are enriched for brain-expressed genes (OR = 2.0, p = 2×10−8) and other brain function gene sets as well (see Figure 2). The enrichment is present, though somewhat less pronounced, if all genes overlapping deletions are included (OR = 1.63, p = 4×10−6, see Figure 2).
Gene size confounds gene set enrichment approaches
To explain this enrichment of rare CNVs affecting brain-function genes in controls, we conjectured that the gene set enrichment approach is confounded by gene size. Three observations support this possibility. First, the transcripts of brain-expressed genes are significantly larger than of other human genes (p = 9×10−82 by non-parametric rank-sum test, see Figure 3A). The median length of all human gene transcripts is 28.2 kb; in contrast the median length of brain expressed gene transcripts is 47.2 kb (1.7 fold longer). In fact of the genes longer than 1 Mb, 32 out of 48 (67%) are brain-expressed. Genes in the three other gene sets are also significantly longer (1.2–3.1 fold). Second, we note that the genes affected by CNVs are also large. Genes disrupted by events in these meta-controls, as well as previously published data sets by Zhang et al, Walsh et al, and the ISC were large (p<2×10−10, see Figure 3B). The bias towards large genes is still present, though mitigated, if the analysis is expanded to include all overlapping genes (p<0.01, see Figure 3B). Smaller genes overlapping a CNV are much more likely to be fully contained by that CNV while larger genes are more likely to extend beyond the boundaries of the CNV and hence be disrupted by that CNV. Third, almost all gene ontology [13] (GO) categories consisting of genes with an average size >200 kb are preferentially affected by rare deletions within the meta-controls (see Table S5). These codes implicate functions such as cell adhesion and recognition, neuron recognition, and synaptic pathways.

Random genomic segments also demonstrate enrichment for brain genes
To quantify the extent to which observed enrichment for these gene sets was simply a consequence of their large size, we tested whether randomly placed genomic segments affect genes with brain function genes preferentially also (see Table 1). We created 1,000 sets of 1,054 randomly positioned non-overlapping segments of equal size and probe density as those rare deletions observed in the meta-controls (see Materials and Methods). Brain-expressed genes were enriched among overlapping genes [OR = 1.67 (1.17–2.26)] and disrupted genes [OR = 2.08 (1.71–2.49)]; the enrichment for brain expressed and other brain genes sets was comparable to the enrichment in observed data.

However, there are two key differences in the results of real rare CNVs and simulated CNVs. Observed rare deletions overlap 35% fewer genes than random segments – suggesting unsurprisingly that deletions overlapping genes are selected against. Possibly, events affecting potentially critical genes that, if affected, disrupt normal human development are selected against. But, on the other hand, the pattern for the largest genes is strikingly different – the observed rare deletions actually overlap 26% more of those genes >1 Mb in length than random segments. This suggests a predilection for large genes that cannot be accounted for simply by their larger genomic footprint.
Gene size, structure, and genic density all independently predict whether a gene is independently affected by a CNV
To explain the discrepancy between the size and number of genes affected in real CNVs and simulated segments, we speculated that while rare events affecting genes are negatively selected against, those that affect large genes might be less strongly selected against. Possibly, large genes have certain structural features that tend to make them relatively preferred targets of rare CNVs above and beyond their simple large size. For example, a CNV within a long gene might be more likely to fall within a large intron and not disrupt the coding sequence, and therefore have less-clear relevance to gene function. Furthermore, since genes tend not to overlap, a CNV of a particular size that overlaps larger genes may affect fewer genes than one that overlaps many smaller genes, and may therefore be less likely to impact some nearby essential gene.
To test whether these factors might play a role we tabulated three relevant structural features for each gene (see Materials and Methods): (1) transcript length, (2) a gene neighborhood density score, representing the expected number of additional nearby genes that a randomly placed CNV affects, and (3) a gene structure score, that represents the expectation that a randomly placed overlapping deletion is fully intronic. The first parameter simply accounts for the size of the ‘target’. The other two parameters account for the possibility that CNVs overlapping certain genes might be more likely to be functionally consequential. We found that all of these variables individually correlated with the likelihood that a gene is overlapped by deletions in meta-controls (see Figure S1). We then conducted a conditional analysis and found that even though they are inter-correlated, they each independently predict the probability that a gene is deleted in the meta-controls – removal of any single parameter significantly affects a logistic regression model's predictive ability (see Table 2). The additional factors of gene density and structure could account for the reduced number of affected genes overall and the increased proportion of larger genes compared to random segments in the genome.
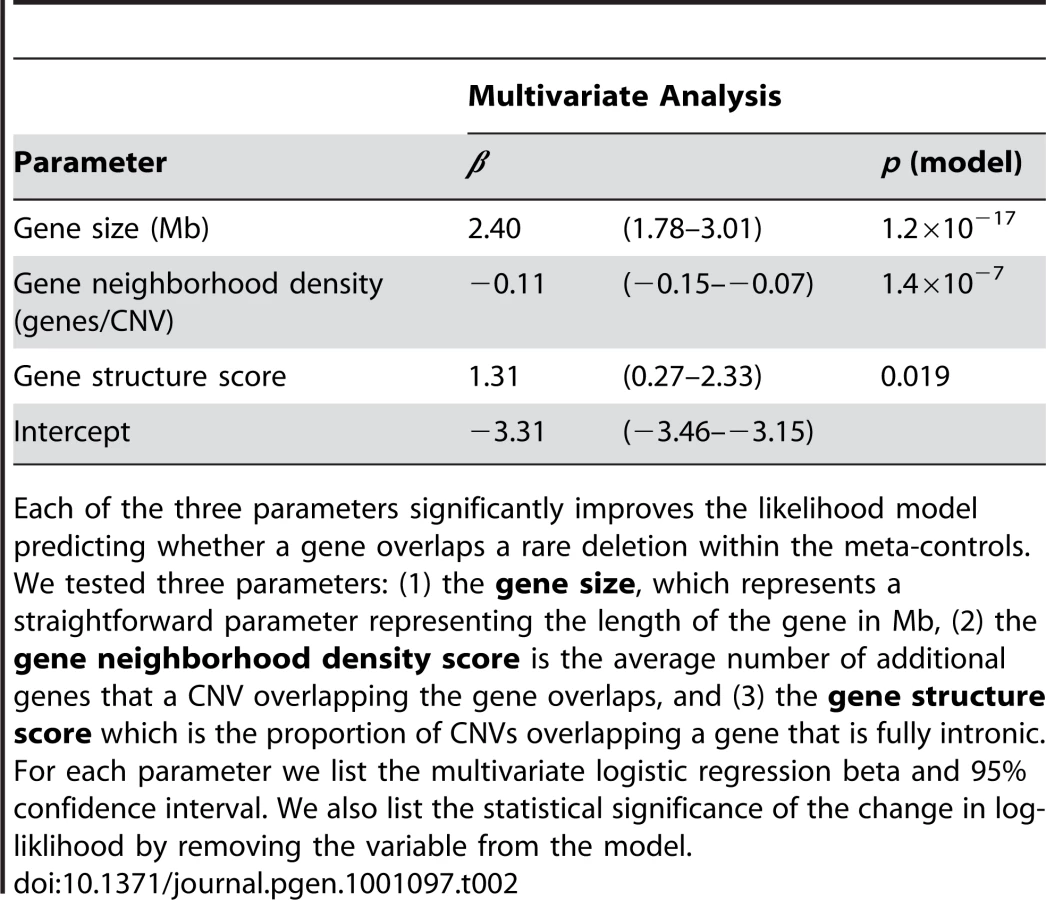
One possible strategy to correct gene set based analyses is to devise a score that encapsulates the structural features of genes, and their predicted propensity to be affected by a CNV. This provides a robust approach to assess pathway enrichment in he suboptimal situation when controls are not available (e.g. when evaluating a collection of de novo case-only Autism deletions). We present such a CNV-propensity gene score (CNVprop) that represents an empirical estimate of the log-likelihood that a gene is overlapped by a CNV based on gene structural features based on the parameters from Table 2. CNVprop can be used as a covariate within a logistic regression framework in assessing enrichment of a gene set. We provide the CNVprop scores of genes in Table S6. While other methods to correct for gene size have been proposed in the literature, they do not specifically account for additional effects from gene density and intron structure, which are likely specific to CNV events. This approach, however, is still not ideal since it fails to account for multiple genes contributed by a single event, or genes being affected multiple times by an individual CNV event.
Gene enrichment of brain function genes in schizophrenia case and control CNVs are equivalently significant
To further demonstrate application of gene set analysis and its potential pitfalls, we used a large data set published by the ISC with many rare (<1% frequency) deletions and duplications identified from 3,391 affected by schizophrenia and 3,181 unaffected individuals (see Table S3).
In order to replicate the analysis published by Walsh et al [1], we conducted set-based analyses of genes disrupted by CNV events within the ISC cases. We observed enrichment of brain-expressed genes (p = 3×10−11, two-tailed Fisher's exact, Figure 2). However, when we examined genes disrupted within controls in the ISC, we observed similar evidence for brain-expressed genes (p = 3×10−10, see Figure 2). Critically, the odds ratios (ORs) for enrichment of brain-expressed genes among genes disrupted in affected individuals and unaffected individuals were difficult to distinguish in this analysis. We observed similar trends towards enrichment for brain-expressed genes overlapped by CNVs (OR = 1.1, p = 0.06 for cases, OR = 1.08, p = 0.20 for controls, data not shown) and overlapped by very rare single event CNVs (OR = 1.5, p = 0.004 for cases, OR = 1.6, p = 0.01 for controls, data not-shown). Furthermore, with the exception of the learning genes, all brain function gene sets demonstrated significant enrichment within ISC cases and controls (see Figure 2).
We applied the same analysis to a data set with a small number of CNVs published by Walsh et al and demonstrated similar effects (see Figure 2). Cases tended to be more statistically significant for all of the gene sets than controls, since they were better powered with more affected genes. However, confidence intervals were wide in this analysis, and it was unclear it there were true case-control differences.
In both data sets – while statistically significant enrichment for brain function genes is observed in cases, it is not clear that the effect size is any different than in controls.
Application of cnv-enrichment-test to previously published schizophrenia data sets
We applied the case-control cnv-enrichment-test to check CNVs published by the ISC and by Walsh et al to test whether case events were enriched for genes with brain function relative to controls. In the ISC data, we had already reported elsewhere increased genome-wide rates and sizes for case CNVs [2]. Walsh et al had demonstrated genome-wide enrichment separately.
We applied the cnv-enrichment-test to the four gene sets (brain-expressed, neuronal-activity, learning and synapse as described above). The results in Table 3 report the empirical 1-tailed p-values for a test of enrichment of the genes in the set relative to the genome-wide baseline rates of all CNVs; for the smaller gene sets standard asymptotic tests yielded unreliable estimates, due to the sparse nature of the data (for example 7 case events, 0 control events for neuronal genes in Walsh et al). In this context, the empirical significance values obtained via permutation will be robust to these sparse cell counts. Of course, for the larger gene sets, and all of the gene sets in the larger ISC data set, analytical p-values corresponded closely to permuted p-values.

There was no evidence of enrichment among case-CNVs compared to control CNVs for brain-expressed and synapse genes (p>0.12, one-tailed analysis, see Table 3). This is in marked contrast to the observed enrichment of these same brain gene sets in the case-only analyses presented in Figure 2 that did not account for gene size.
However, the neuronal-function gene set demonstrated evidence of association to schizophrenia cases for both Walsh et al (p = 0.00045) and the ISC data (p = 0.04). There was also evidence of association of the learning gene set within the ISC data (p = 0.009) but not in the Walsh et al data (p = 0.35).
We want to emphasize that these results are not adjusted for multiple hypotheses testing – and the plausible number of independent gene sets. In this study alone we have tested four separate gene sets. Ultimately, convincing associations will require larger data sets. As additional samples are genotyped for CNVs, the relevance of the neuronal-function genes might be more clearly established.
Of note, considering only deletions within the ISC data, the effect of neuronal-function gene set enrichment is stronger (p = 0.0067, with higher rates in cases). Similarly, considering only deletions within the ISC data, the effect of the learning gene set enrichment is also stronger (p = 0.002, with higher rates in cases).
In both cases neuronal-function and learning gene sets, the effect sizes associated with an event affecting a gene is modest ranging from 1.2–1.7. This suggests that even if the set associations are ultimately validated, that rare CNV events affecting genes within these sets certainly do not fully explain the pathogenicity of rare CNVs.
Conclusions
The cnv-enrichment-test is an extremely versatile test to identify whether a gene set of interest is associated with case-control status. We have shown that it is robust to confounders, such as case-control differences in CNV rate and gene size, while standard gene set enrichment approaches are not.
Since the cnv-enrichment-test can be applied easily to a wide range of gene sets, there may be the temptation to examine data sets by testing a compendium of gene-sets. Generally, we discourage this approach, and urge investigators to look at specific sets of interest. Assessing the significance of association statistics when testing a large compendium of gene-sets is complex since there is a large number of highly overlapping sets; correcting for the large burden of multiple hypotheses testing appropriately can be challenging. However, should one decide to test such a compendium of gene-sets, it is important that investigators permute the case-control status within their own data set, and apply the same battery of tests to make sure that the actual data set is obtaining levels of significance that are beyond that of the permuted data sets.
We have also shown that pathway analyses with standard gene set enrichment approaches are confounded by gene size and structure. This issue is of particular importance when considering genes with brain function – since those genes are significantly larger than other human genes. We have demonstrated how a large set of brain-expressed genes seem to be impacted by CNVs in both case and control populations when using gene set enrichment approaches, and how this effect is largely the consequence of the size of these genes. The brain-expressed genes were selected for having significantly greater expression in neuronal tissues as opposed to non-neuronal tissues. Certainly genes with important brain functions that are ubiquitously expressed in all tissues might be missed by such a strategy, as might genes with very low expression levels overall. However, we observed very similar results for three other separately curated sets of genes with brain function; this suggests that gene size and spurious pathway associations may be of particular importance for brain function genes.
The approach we describe here can be applied more broadly than within the context of CNVs; the cnv-enrichment-test can be applied to any situation where disease-associated genomic segments are defined. For example, linkage disequilibrium blocks around associated SNPs can be defined as disease-associated genomic-segments. The potential for gene size and structure confounding pathway analyses extends beyond CNV studies, and applies equally to pathway analyses within other types of genetic studies, including SNP association studies, as noted by Wang et al [27] and exon re-sequencing studies. For example, in a study looking at classes of genes that are disproportionately affected by rare exonic mutations, the total length of the coding sequence will be a key confounding variable. Similarly, studies looking at classes of genes that contain a single SNP nominally associated to disease, confounding variables might include the number of independent SNPs examined, the physical size of the gene, and the recombination hotspots across the length of the gene. In any case, careful case-control comparisons are essential to avoid these confounders.
Many of the genes involved in brain function are compelling candidate genes for neurological and psychiatric diseases – and indeed they may be the most vulnerable to CNVs. The purpose of this manuscript is not to question the results of the original publications, but to rather set up a rigorous statistical approach that allows investigators to accurately estimate effect sizes of events impacting specific gene sets of interest and also to precisely replicate reported results.
Materials and Methods
Compiling Gene Sets
Brain-Expressed
To identify genes with specific expression in the brain, we obtained a large publicly available human tissue expression microarray panel (GEO accession: GSE7307) [28]. We analyzed the data using the robust multi-array (RMA) method for background correction, normalization and polishing [29]. We filtered the data excluding probesets with either 100% ‘absent’ calls (MAS5.0 algorithm) across tissues, expression values <20 in all samples, or an expression range <100 across all tissues. To represent each gene, we selected the corresponding probeset with the greatest intensity across all samples. We included expression profiles from some 96 healthy tissues and excluded disease tissues and treated cell lines. We averaged expression values from replicated tissues averaged into a single value. To assess whether genes had differential expression for CNS tissues, we compared the 27 tissue profiles that represented brain or spinal cord to the remaining 69 tissue profiles with a one-tailed Mann-Whitney rank-sum test. We identified those genes obtaining p<0.01 as preferentially expressed.
Synapse
We downloaded Gene Ontology [30] structure and annotations on December 2006. Since it was available, we used a previous version of Gene Ontology to ensure independence from the results of recent genetic scans. We expanded human gene annotations to include annotations from orthologous genes, identified through Homologene [30] from model organisms. We identified those genes that were annotated with the ‘Synaptic Junction’ code (GO:0045202), or descendents of that code.
Neuronal Activity
We downloaded the list of genes within the category ‘Neuronal Activities’ (BP00166) listed in the Panther database [12].
Learning
We downloaded the list of genes within the category ‘Behavior-Learning’ listed in the Ingenuity Application.
To avoid spurious results and focus on a consistent set of genes across all studies, we included in our analysis only autosomal genes that (1) had at least one annotations in GO and (2) passed quality control criteria in the data set used to identify brain-expressed genes. The resulting set consisted of 14,565 annotated genes.
Obtaining Walsh CNV data
We obtained rare event deletions and duplications from Table 2 in the original publication of the data [1].
Obtaining ISC CNV data
Rare (<1% frequency) event deletions and duplications were provided directly by request from the International Schizophrenia Consortium.
Identifying Rare CNVs in the Meta-controls
We obtained data from unaffected individuals with informed consent from four Institutional Review Board approved studies: macular degeneration [26], myocardial infarction [25], bipolar disease [8], and multiple sclerosis [24]. We obtained Affymetrix 6.0 raw intensity data for all samples and ran the Birdsuite software on each plate individually [31]; CNV calls were based on Birdseye output. We then analyzed healthy unaffected individuals from each of four studies separately. First we filtered individuals on SNP data, removing individuals with >5% missing data. Second, in situations where Birdseye called two nearby segments (<10 kb) with identical copy number and there was a low confident segment in between (LOD<3), we merged those segments. Third, we exclude all CNVs that (1) overlap CNVs from a map of common variation [32], or (2) failed stringent quality control criteria (<20 kb in length or <10 LOD or <10 probes). Fourth we removed those individuals in each study that were outliers in either excessive number of CNVs, or in excessive aggregate length of CNVs – we defined outlier as the median plus the 1.5 times the inter-quartile range. We then combined all CNVs into a single data set, and identified single-events (i.e. non-overlapping) deletions.
Placing Genomic Segments
We produced 1000 sets of non-overlapping segments throughout the genome. Each set consisted of segments matched for size and probe-denisty (+/−10%) to each observed single-event deletions in meta-controls. Since we were simulating rare events, random events were not allowed to overlap regions with known copy number variation [32] or in regions where we observed an overlapping event (i.e. not a singleton) in the meta-controls.
Defining Gene Parameters
For each gene we defined three parameters (1) gene length, (2) gene neighborhood density score, and (3) gene structure score.
Gene length was simply the length of the gene transcript in mega-basepairs.
To calculate a neighborhood density score for a gene, we consider a CNV overlapping a gene. The neighborhood density score is then the expected number of additional nearby genes overlapped by the same CNV. To empirically estimate the distribution of sizes of rare CNVs, we utilized the sizes, s, of observed single event deletions in the meta-controls. Then to calculate the gene neighborhood density score, gdi, for gene i, we used the following formula:where p is a genomic position, overlapi(p, s) is an indicator function that is 1 if a segment of length s starting at position p overlaps gene i, or is otherwise 0. Similarly overlapall(p, s) is the number of genes that a segment of length s starting at position p overlaps. In the numerator we subtract one off, since we want to exclude gene i itself
To calculate a gene structure score, we calculated the expected proportion of overlapping CNVs that would not affect the coding sequence of the gene (i.e. be fully intronic). To empirically estimate the distribution of sizes of rare CNVs, we again used the sizes, s, of observed single event deletions in the meta-controls. Then to calculate the gene structure score, gsi, for gene i, we used the following formula:where p is a genomic position, overlapi(p, s) is an indicator function that is 1 if a segment of length s starting at position p overlaps gene i, or is otherwise 0. Similarly intronici(p, s) is an indicator variable that is 1 if a segment of length s starting at position p does not overlap a coding sequence, or otherwise is 0.
Statistical Models to Assess Gene Set Enrichment Across CNVs
In order to produce a framework to test gene-sets and their association to disease, we used a linear/logistic regression framework in which phenotype is regressed on the number of genes intersected (or disrupted) by one or more CNVs and covariates. We considered five different models to test for enrichment of CNVs in a pathway of interest, and tested them with simulated datasets.
For a disease outcome, a standard model, M0, is as follows:where for individual i, gi is the number of genes in a pathway of interest that intersected/disrupted by a CNV. The θ term is the logistic regression intercept and represents the background log likelihood for each individual, while γ is the logistic regression parameter for gi.
Model M1 controls for potential genome-wide differences in CNV burden between cases and controls:where ci is the total number of CNVs in a given individual i. The β0 term is the logistic regression parameter for ci.
Model M2 alternatively controls for CNV size:where si is the individual's mean CNV size in kb. If for a particular individual ci = 0 (i.e. they do not have any CNVs) then si is set to the sample mean of s rather than 0 or missing. (Otherwise, if many individuals have no CNVs, a strong correlation will be induced between the rate and average size of CNVs.) The β1 term is the logistic regression parameter for si.
Model M3 alternatively controls for an individual's total CNV burden expressed in terms of total kb deleted or duplicated, written here as the product of ci and si:The β2 term is the logistic regression parameter for (ci·si).
Finally, the cnv-enrichment-test model controls explicitly for potential case/control differences in both the number and size distributions of CNVs:This is the model introduced in the main text labeled as the “cnv-enrichment-test”. As above, if for a particular individual ci = 0 (they do not have any CNVs) then si is set to the mean size of all CNVs in the sample as opposed to zero.
Under all circumstances, the null hypothesis for the 2-sided test of enrichment is H0: γ = 0.
Testing Models in a Simulated Case-Control Framework
We conducted simulations to understand the performance characteristics of these different analytic approaches (M0–M3 and cnv-enrichment-test) to test for enrichment of case CNVs in a set of genes. We explicitly adopt extreme conditions in these simulations, to best illustrate the robustness of each approach under the broadest range of conditions.
For each individual, we simulated data for a single hypothetical chromosome, 250 Mb in length. We placed 2000 evenly-spaced, non-overlapping genes on the hypothetical chromosome, where every fifth gene was designated as a “brain gene”. We assigned brain genes to be considerably larger than other genes (50 kb versus 10 kb). In all scenarios we simulated CNV data for 2000 cases and 2000 controls. For cases and controls, the mean CNV size was either 60 kb or 100 kb, as detailed in Table S2 (range 10 kb to 150 kb, standard deviation 30 kb). Under all scenarios, individuals had either 0 or 1 CNV, with rates given in Table S2.
All datasets were simulated under the null hypothesis of no enrichment for brain genes; that is, CNVs were randomly placed on the hypothetical chromosome, similarly for both causal and controls. Under five scenarios, S0 to S4, we altered the mean CNV rate and CNV size for cases and controls independently, in order to induce enrichment of CNVs in brain genes arising solely as a consequence of CNV rate and size. Under the first scenario, S0, there were no differences between cases and controls in the rate and size of CNVs: we therefore expected all methods to give appropriate type I error rates here. Under S1, the rate of CNVs was higher in cases. Under S2, the average CNV size was smaller in cases. Under S3, cases had a greater number, and larger, CNVs than controls. Under S4, cases had a greater number, but smaller, CNVs than controls.
For each scenario, we simulated 10,000 datasets to calculate the type I error rate for the enrichment test, for a nominal rate of 0.05.
Implementation
This test is implemented in PLINK v1.07 (–cnv-enrichment-test). It is appropriate for either continuous or disease traits and allows for the inclusion of multiple other covariates and for empirical significance tests.
The following examples illustrate basic usage. If the file genes.dat contains the locations of all genes (i.e. as available from the resources section of the PLINK website, glist-hg18) and the file pathway.txt is a file of gene names forming the pathway to be tested for enrichment and the CNV data are in the files mycnv.cnv, mycnv.cnv.map and mycnv.fam (see website CNV page for details), then one can ask whether a) genes are enriched for CNVs, b) a subset of genes are enriched, relative to the whole genome, c) a subset of genes are enriched, relative to all genes. The latter form of the enrichment test might be desirable, for example, to determine whether any enrichment is general to all genes, or specific to a subset of genes.
a) Enrichment of genic CNVs
./plink ––cfile mycnv
––cnv-count genes.dat
––cnv-enrichment-test
b) Enrichment of pathway genes CNVs, relative to all CNVs
./plink ––cfile mycnv
––cnv-count genes.dat
––cnv-subset pathway.txt
––cnv-enrichment-test
c) Enrichment of pathway genes CNVs, relative to all genic CNVs
./plink ––cfile mycnv
––cnv-intersect genes.dat
––cnv-write my-genic-cnv
./plink ––cfile my-genic-cnv
––cnv-count genes.dat
––cnv-subset pathway.txt
––cnv-enrichment-test
The usual modifiers (to define intersection differently, allow for a certain kb border around each gene, filter on CNV size, type or frequency, etc) are all available. Under all circumstances, 2-sided asymptotic p-values are returned. Alternatively, permutation testing can be applied and 1-sided empirical p-values are returned (positive enrichment in cases, based on estimated regression coefficient).
For additional information consult the PLINK website (http://pngu.mgh.harvard.edu/purcell/plink/), the resources subsection (gene list) (http://pngu.mgh.harvard.edu/purcell/plink/res.shtml), or the CNV file format subsection (http://pngu.mgh.harvard.edu/purcell/plink/cnv.shtml).
Supporting Information
{kind=link}
Zdroje
1. WalshT
McClellanJM
McCarthySE
AddingtonAM
PierceSB
2008 Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320 539 543
2. International Schizophrenia Consortium 2008 Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455 237 241
3. StefanssonH
RujescuD
CichonS
PietilainenOP
IngasonA
2008 Large recurrent microdeletions associated with schizophrenia. Nature 455 232 236
4. XuB
RoosJL
LevyS
van RensburgEJ
GogosJA
2008 Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 40 880 885
5. MarshallCR
NoorA
VincentJB
LionelAC
FeukL
2008 Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82 477 488
6. SebatJ
LakshmiB
MalhotraD
TrogeJ
Lese-MartinC
2007 Strong association of de novo copy number mutations with autism. Science 316 445 449
7. WeissLA
ShenY
KornJM
ArkingDE
MillerDT
2008 Association between Microdeletion and Microduplication at 16p11.2 and Autism. N Engl J Med 358 667 675
8. ZhangD
ChengL
QianY
Alliey-RodriguezN
KelsoeJR
2009 Singleton deletions throughout the genome increase risk of bipolar disorder. Mol Psychiatry 14 376 380
9. MoothaVK
LindgrenCM
ErikssonKF
SubramanianA
SihagS
2003 PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34 267 273
10. EliaJ
GaiX
XieHM
PerinJC
GeigerE
2010 Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 5 637 646
11. GlessnerJT
WangK
CaiG
KorvatskaO
KimCE
2009 Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459 569 573
12. MiH
Lazareva-UlitskyB
LooR
KejariwalA
VandergriffJ
2005 The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 33 D284 288
13. AshburnerM
BallCA
BlakeJA
BotsteinD
ButlerH
2000 Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 25 29
14. RedonR
IshikawaS
FitchKR
FeukL
PerryGH
2006 Global variation in copy number in the human genome. Nature 444 444 454
15. YimSH
KimTM
HuHJ
KimJH
KimBJ
2010 Copy number variations in East-Asian population and their evolutionary and functional implications. Hum Mol Genet 19 1001 1008
16. StanleySM
BaileyTL
MattickJS
2006 GONOME: measuring correlations between GO terms and genomic positions. BMC Bioinformatics 7 94
17. TaherL
OvcharenkoI
2009 Variable locus length in the human genome leads to ascertainment bias in functional inference for non-coding elements. Bioinformatics 25 578 584
18. KanehisaM
GotoS
2000 KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28 27 30
19. CohenBA
MitraRD
HughesJD
ChurchGM
2000 A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet 26 183 186
20. RujescuD
IngasonA
CichonS
PietilainenOP
BarnesMR
2009 Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet 18 988 996
21. The Wellcome Trust 2007 Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 661 678
22. BarnesC
PlagnolV
FitzgeraldT
RedonR
MarchiniJ
2008 A robust statistical method for case-control association testing with copy number variation. Nat Genet 40 1245 1252
23. PurcellS
NealeB
Todd-BrownK
ThomasL
FerreiraMA
2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
24. De JagerPL
JiaX
WangJ
de BakkerPI
OttoboniL
2009 Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 41 776 782
25. KathiresanS
VoightBF
PurcellS
MusunuruK
ArdissinoD
2009 Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 41 334 341
26. NealeBM
FagernessJ
ReynoldsR
SobrinL
ParkerM
2010 Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U S A 107 7395 7400
27. WangK
ZhangH
KugathasanS
AnneseV
BradfieldJP
2009 Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am J Hum Genet 84 399 405
28. RothRB
HeveziP
LeeJ
WillhiteD
LechnerSM
2006 Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 7 67 80
29. IrizarryRA
BolstadBM
CollinF
CopeLM
HobbsB
2003 Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31 e15
30. WheelerDL
BarrettT
BensonDA
BryantSH
CaneseK
2008 Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36 D13 21
31. KornJM
KuruvillaFG
McCarrollSA
WysokerA
NemeshJ
2008 Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 40 1253 1260
32. McCarrollSA
KuruvillaFG
KornJM
CawleyS
NemeshJ
2008 Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40 1253 1260
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2010 Číslo 9
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data