
The White-Nose Syndrome Transcriptome: Activation of Anti-fungal Host Responses in Wing Tissue of Hibernating Little Brown Myotis
White-nose syndrome is the most devastating epizootic wildlife disease of mammals in history, having killed millions of hibernating bats in North America since 2007. We have used next-generation RNA sequencing to provide a survey of the gene expression changes that accompany this disease in the skin of bats infected with the causative fungus. We identified possible new mechanisms that may either provide protection or contribute to mortality, including inflammatory immune responses. Contrary to expectations that hibernation represents a period of dormancy, we found that gene expression pathways were responsive to the environment. We also examined which genes were expressed in the pathogen and identified several classes of genes that could contribute to the virulence of this disease. Gene expression changes in the host were associated with local inflammation despite the fact that the bats were hibernating. However, we found that hibernating bats with white-nose syndrome lack some of the responses known to defend other mammals from fungal infection. We propose that bats could be protected from white-nose syndrome if these responses could be established prior to hibernation or if treatments could block the virulence factors expressed by the pathogen.
Published in the journal:
The White-Nose Syndrome Transcriptome: Activation of Anti-fungal Host Responses in Wing Tissue of Hibernating Little Brown Myotis. PLoS Pathog 11(10): e32767. doi:10.1371/journal.ppat.1005168
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005168
Summary
White-nose syndrome is the most devastating epizootic wildlife disease of mammals in history, having killed millions of hibernating bats in North America since 2007. We have used next-generation RNA sequencing to provide a survey of the gene expression changes that accompany this disease in the skin of bats infected with the causative fungus. We identified possible new mechanisms that may either provide protection or contribute to mortality, including inflammatory immune responses. Contrary to expectations that hibernation represents a period of dormancy, we found that gene expression pathways were responsive to the environment. We also examined which genes were expressed in the pathogen and identified several classes of genes that could contribute to the virulence of this disease. Gene expression changes in the host were associated with local inflammation despite the fact that the bats were hibernating. However, we found that hibernating bats with white-nose syndrome lack some of the responses known to defend other mammals from fungal infection. We propose that bats could be protected from white-nose syndrome if these responses could be established prior to hibernation or if treatments could block the virulence factors expressed by the pathogen.
Introduction
White-nose syndrome (WNS) is an epizootic disease that has killed millions of bats in North America [1, 2]. WNS is caused by the psychrophile Pseudogymnoascus destructans (Pd) (formerly Geomyces destructans), an ascomycete fungal pathogen [3–5] that affects bats during hibernation. Pd grows at temperatures between 2 and 18°C and can infect bats while they hibernate [4, 6]. Pd is invasive and damages the cutaneous tissues of bats, including the wing [7], forming characteristic cupping erosions that are diagnostic of Pd infection [8]. Mortality rates due to WNS vary by species. In the little brown myotis, Myotis lucifugus, the mortality rate is up to 91% in affected caves [9, 10] whereas WNS resistance has been reported in the big brown bat, Eptesicus fuscus [11]. Bats in Europe are exposed to endemic Pd, but do not exhibit WNS mortality and appear to be resistant to the disease [12], despite cutaneous invasion by Pd [13].
Cutaneous infection by Pd causes some species of bats to arouse more frequently from torpor [5, 14, 15]. Although hibernating mammals spend less than 1% of their time euthermic [16], they use up to 90% of their stored energy during these periods [17, 18]. Because each arousal in little brown myotis utilizes an estimated 108 mg of stored fat [18], the increase in arousal frequency caused by WNS explains 58% of the morbidity rate associated with Pd infection [14]. Other factors that are also associated WNS pathology include effects of Pd infection on the integrity of wing tissue [7, 19], electrolyte balance and hydration [7, 20, 21], chronic respiratory acidosis [22], oxidative stress [23], and immune function [24]. The relative importance of each of these mechanisms in causing death in WNS is not clear, and the most likely model that has emerged is a multi-stage progression of WNS with contributions of several of these factors [22]. Differences in susceptibility to WNS between species in North America may be explained in part by different responses to Pd infection such as changes in thermoregulatory behavior. Understanding host responses to Pd infection may provide insight that could be useful for improving survival of affected species.
Cutaneous fungal infections in mammals are first recognized by components of the innate immune system, including C-type lectin receptors and Toll-like receptors [25]. Conserved components of the fungal cell wall activate pattern recognition receptors on phagocytes such as neutrophils, macrophages, and dendritic cells, and on epithelial cells [26]. Activation of these cells can lead to induction of the inflammasome, the production of inflammatory cytokines, and generation of reactive oxygen species that can mediate fungal cell killing [25]. The importance of the innate immune response to the initial recognition of fungal infections is demonstrated by the observation that deficiencies in these signaling pathways can lead to chronic fungal infections in humans [27, 28]. In the absence of invasion, colonization by commensal fungi can be maintained through tolerance mechanisms mediated by interactions with dendritic cells and epithelial cells in the skin [29]. Local activation of innate immune pathways can slow the growth of invasive pathogenic fungi and promote tolerance, possibly leading to a commensal relationship with the fungus [30], but is not usually sufficient to clear infections. Clearance of infections typically requires T helper (Th) cells, as demonstrated by the susceptibility of patients with acquired immune deficiency syndrome, immunosuppressant therapy, or chemotherapy to fungal infections [31]. These T cell responses can be mediated by Th17 cells [32, 33] or, in some cases, Th1 cells [34], with Th2 responses typically associated with greater susceptibility [35]. Th17 responses can contribute to clearance of invasive fungal infections through the actions of IL-17A and IL-22 [36] and the further recruitment and activation of neutrophils [37]. These T cell subsets have not been well characterized in bats, but those T-cell mediated immune mechanisms that have been studied appear to be conserved between bats and other mammals [38–41].
Fungal infections in animals are typically life-threatening only upon suppression of adaptive immune responses in the host, such as when chytrid fungus (Batrachochytrium dendrobatidis) blocks lymphocyte-mediated inflammatory responses [42]. Hibernation produces a natural suppression of some immune responses in mammal species where it has been studied. During hibernation, when the conservation of energy is critical, certain immunological mechanisms are downregulated while others remain unaffected [43–51]. Changes during hibernation can include depressed antibody responses [44, 52], decreased ability of T and B lymphocytes to proliferate in response to challenge [53, 54], and reduced complement activity [47]. Hibernation does not affect all immune responses equally, as shown in thirteen-lined ground squirrels (Ictidomys tridecemlineatus) that have a suppressed T-independent antibody response but are capable of mounting a T cell-dependent response during hibernation [44]. Studies of transcriptome-wide changes during hibernation in squirrels [55–59] have shown expression changes in genes involved in metabolism, oxidative stress, protein folding, ischemia/hypoxia, and other processes, but these studies were not examining an active immune response. Hibernation is also known to affect the distribution of leukocytes [45, 60] and platelets [61]. However, we have an incomplete understanding of how hibernation affects the suppression, or subsequent recovery, of immune responses [43], or how immune physiology in bats during hibernation may differ from that of rodents.
The cost of immune suppression during torpor is presumably outweighed by the benefits of energy conservation because most pathogens are not capable of proliferating at the low body temperatures of hibernating animals. However, the psychrophilic nature of Pd allows it to infect bats within hibernacula [2, 4]. The brief euthermic bouts of hibernating bats are shorter than most other hibernating mammalian species [14, 62] and it may not be possible for a bat naïve to a pathogen to mount a primary immune response in the few hours that it is euthermic throughout the hibernation season. We have observed antibody responses to Pd in bats, but these responses are strongest in active bats exposed to Pd after emergence from hibernation [63]. Therefore, hibernating bats may keep pathogens in check by relying on hypothermia, innate immune responses, and/or memory immune responses. The psychrophilic nature of Pd overcomes the first of these barriers to infection and the difficulty in fighting fungal pathogens with innate mechanisms alone may allow Pd to proliferate and invade the cutaneous tissues of bats.
The WNS panzootic has created an urgent need to understand if North American bat populations can persist in the presence of the fungal pathogen [1, 10]. Understanding the complete array of host responses mounted by bats afflicted with WNS may help illuminate sources of variation in survival within and among bat species. To determine which host responses are activated by Pd infection, we measured transcriptome-wide gene expression levels in bat wing tissue from hibernating bats affected by WNS. Gene expression was compared to bats that were hibernated in captivity in the absence of Pd exposure. We hypothesized that Pd infection would cause changes in gene expression that would reveal physiological responses during WNS that might be either protective or pathological. By using next-generation RNA sequencing to examine transcriptome-wide gene expression changes we expected to discover consistent patterns of host responses that occur in Pd-infected tissues. Combined with changes in gene expression within the Pd pathogen, these results have provided a survey of the host and pathogen interactions occurring during WNS.
Results
Gene Expression Changes Revealed by Next Generation RNA Sequencing
To determine the host response mounted by little brown myotis to Pd during hibernation, we measured changes in gene expression at the whole transcriptome level. Wing tissue samples were obtained from hibernating little brown myotis with no known exposure to Pd and bats exhibiting physical signs of WNS, as shown in Table 1. Histopathology [8] and quantitative PCR (qPCR) for Pd [64] were used to confirm the WNS status of each bat (Table 1). Cupping erosions diagnostic of WNS were found on all 6 bats captured in Kentucky, but on none of the 5 bats from states negative for WNS at the time of capture. Low levels of neutrophilic inflammation were found in all 11 wing samples (Table 1; Infl), although this inflammation was not associated with sites of Pd infection. All 6 WNS-affected bats tested positive for Pd by qPCR, although the fungal load measured on wing swabs (Table 1; qPCR) did not correlate with the number of cupping erosions found by histology (Table 1; WNS). As previously shown [5, 14, 15], WNS-affected bats had significantly lower body condition (Table 1; SMI; p = 0.017, t = 2.9255, df = 9).

Next generation RNA sequencing (RNA-Seq) was performed using poly-A selected RNA isolated from each RNAlater-preserved wing tissue sample (S1 Table). Using expression levels of Pd-derived transcripts, we confirmed that all 6 WNS-affected bats had abundant expression of Pd genes. The Pd-derived transcripts were not present at significant levels in any of the 5 samples from unaffected bats (S2 Table; p = 2.2x10-6, t = 21.5, df = 5.33), including the MN090 sample that had tested positive for Pd by qPCR in one of the two replicates (Table 1). Because high levels of differential expression of Pd transcripts would make it more difficult to detect significant changes in host gene expression, the assembly was filtered [65] to remove Pd-derived sequences. Comparison of the filtered assembly with the original revealed that removing the Pd sequences did not significantly decrease the completeness of the assembly (S3 Table) as determined by BUSCO [66]. This filtered assembly (S1 Dataset) was used to calculate differential expression in host genes between the unaffected and WNS-affected samples.
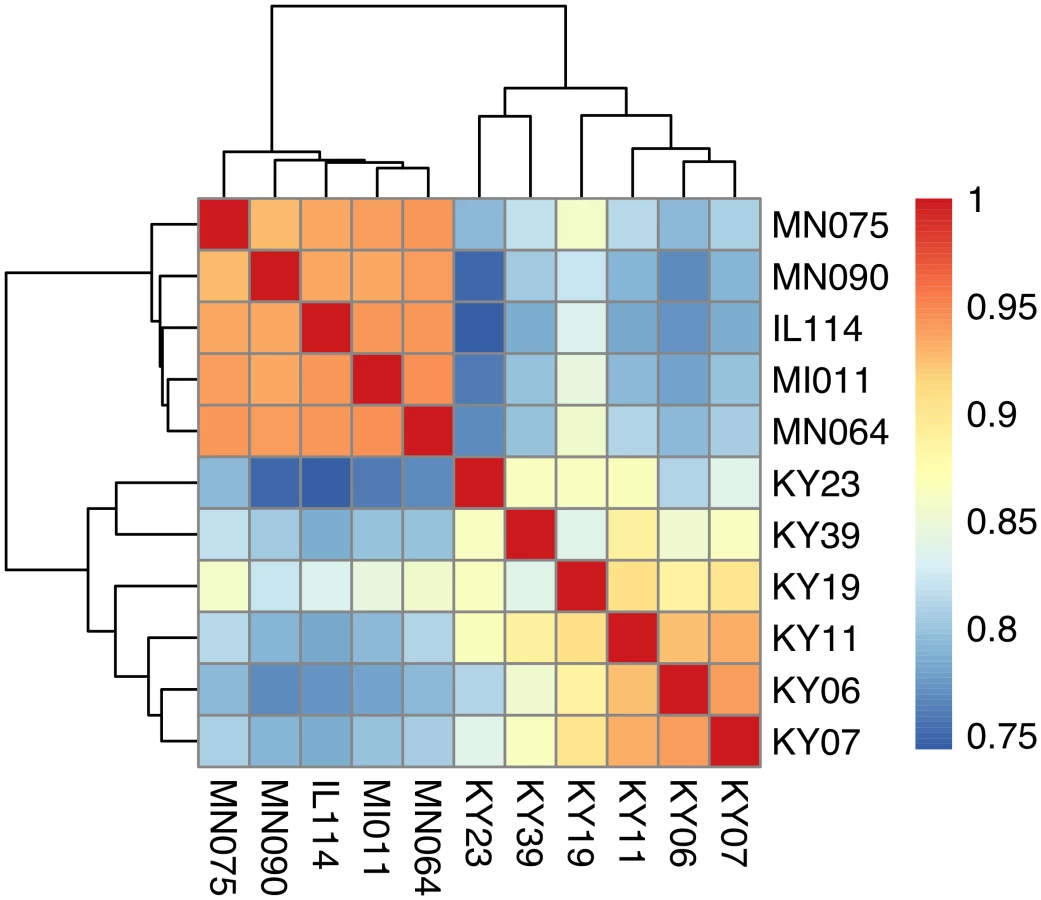
We compared host gene expression across all samples (S2 Dataset) using DESeq2 [67] to identify transcript clusters that were expressed at a minimum of 2-fold difference and significant at a false discovery rate (FDR) of 0.05 (S1 Fig). We found 1804 transcript clusters that were expressed at higher levels, and 1925 transcript clusters expressed at lower levels, in WNS-affected bat tissues (S4 Table). Hierarchical clustering (Fig 1) revealed that expression of these transcripts from all 5 bats without WNS was similar to each other. Gene expression in wing tissue from WNS-affected bats was different from unaffected bats and more similar to each other, as predicted. The normalized expression levels of the 3729 identified transcript clusters differentially expressed are listed in S4 Table.
Differential expression of individual gene isoforms was further analyzed using EBSeq [68], an empirical Bayesian approach to modeling gene expression. For each transcript cluster identified as differentially expressed by DESeq2, we used EBSeq to determine if any of the individual transcripts were differentially expressed at a posterior probability greater than 0.99 (S4 Table). Of the 3729 differentially expressed transcript clusters identified by DESeq2, EBSeq identified at least one differentially expressed transcript for 1427 (38% of total, 43% of upregulated genes and 33% of downregulated genes). These results indicate that differences in gene expression are likely due to alternative splicing or other isoform differences for many of the differentially expressed genes.
To annotate the functions of these genes and identify those likely to be involved with host responses to Pd infection, we used the Trinotate pipeline. BLAST was used to identify 1365 upregulated transcripts and 325 downregulated transcripts in WNS-affected tissues with significant homology to known genes from vertebrates in the Swissprot database. Of the 2295 remaining transcripts, 13 were mapped to genes from non-vertebrates in the Swissprot database, presumably due to environmental contamination or incomplete removal of Pd transcript sequences. Of the 2842 trinity transcript clusters without a BLASTx match in Swissprot, 2731 (96%) were found to align to sequences (e-value < 0.0001) in the little brown myotis genome. Of the aligned transcripts, 204 (7.4%) were found to correspond to previously identified non-coding RNA sequences. Of the 111 transcript clusters without a transcript that aligned to the little brown myotis genome or Swissprot, BLAST was used to align their transcripts to the UniRef90 database. We found that 7 genes aligned to vertebrate homologs, 9 aligned to fungal homologs, and 15 aligned to other metagenomic sequences. We were unable to identify homologous sequences for any transcripts from 80 (2.1%) of the transcript clusters that were differentially expressed.
Expression levels for the Swissprot-identified transcript clusters with the 100 lowest adjusted p values are shown in Fig 2 (see S4 Table for all results). Some of the differentially expressed genes with putative functions that were predicted to associate with host responses to a fungal pathogen are listed in Table 2. WNS caused dramatic changes in expression of genes involved in inflammation, immune responses, wound healing, metabolism, and oxidative stress, even though the bats were hibernating during the Pd infection. Most of these genes were upregulated in WNS-affected tissues, while a much smaller number of identified genes with putative functions in these categories were downregulated (Tables 2 and S4).


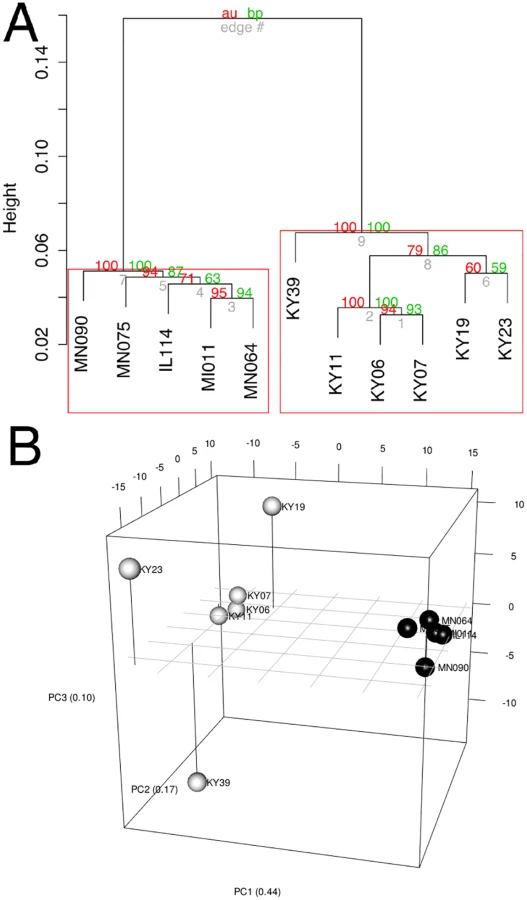
To determine if all 6 little brown myotis with WNS exhibit similar changes in gene expression, we performed clustering analysis of the differentially expressed transcripts (Fig 1). To confirm the significance of these patterns of gene expression, bootstrap analysis of clustering was performed [69]. The clustering of the unaffected samples together and the clustering of the WNS-affected samples together was verified with a confidence of 99% (Fig 3A). Principal component analysis was performed to better understand the relationships between the transcripts expressed in the 11 samples (Fig 3B). All 5 samples from unaffected bats were very similar based on the first three principal components identified, which account for 71% of the variance in these transcripts. The WNS-affected bat samples have more diverse gene expression (S5 Table) and PC1 (accounting for 44% of the variance) differentiates all 6 from the unaffected bat samples. The genes represented by PC1 include those that are more highly expressed in unaffected than WNS-affected wing tissue (Fig 2). PC2 (17% of the variance) and PC3 (10% of the variance) distinguish the KY19, KY23, and KY39 samples from the other two WNS-affected samples and from the unaffected samples. The rotation values of principal component analysis (S5 Table) reveal that inflammatory genes made the greatest contribution to PC2. Clustering analysis revealed diverse host responses among the bats infected with Pd.
Metabolic and Inflammatory Immune Pathways Associated with WNS
We next examined the functional pathways that were most affected in little brown myotis infected with Pd. For this gene ontology analysis, DESeq2 results on transcript isoforms were used with a higher FDR threshold of 0.1, as is typical for this type of analysis. From WNS-affected bat tissue, 3104 upregulated transcripts were aligned with BLAST to the human Uniprot database. Homologs for these transcripts were identified and a list of 1937 unique Ensembl IDs associated with upregulated genes was generated (S6 Table). GOrilla [70] was used to determine significantly upregulated gene ontology categories from the Uniprot GO ID database (Table 3 and S7 Table) and REVIGO [71] was used to visualize biological processes that were significantly overrepresented in the WNS-affected transcriptome (Fig 4). The functional analysis revealed that Pd infection increases expression of genes involved in metabolism, defense responses, and other pathways (Table 3).


For the transcripts that showed lower expression in WNS-affected tissue (Fig 2), the same analysis was performed. Of the 1152 identified transcripts that were downregulated at an FDR of 0.1, 694 homologous human genes were identified by BLAST and mapped to Ensembl gene IDs (S6 Table). GOrilla did not identify any Biological Process categories that were significantly downregulated in the WNS-affected bat tissue.
Host-Pathogen Interactions during WNS
To examine the gene expression of the Pd pathogen using a dual RNA-Seq approach [72], we separately generated a genome-guided Trinity assembly (S3 Dataset) with the Broad Institute G. destructans genome. The reads from each of the WNS-affected tissues were mapped onto this assembly with Bowtie and gene expression estimated using RSEM (S4 Dataset). Expression levels for the Pd genes with the greatest variance are shown in Fig 5. Hierarchical clustering (Fig 6) and principal component analysis (S2 Fig) of the differentially expressed transcripts indicated that Pd gene expression was most similar in the wing tissues from bats obtained from the same hibernaculum (Table 1). The expression patterns of Pd genes were more similar for KY06, KY07, and KY11, which corresponds to bats captured in Cave 1 in Kentucky, and for KY19 and KY23, which were captured from Cave 2.


The possible functions of the Pd genes expressed among the WNS-affected samples were analyzed by sequence homology. We first examined the expression levels of a family of secreted proteases that have been proposed to be involved in Pd virulence [73, 74] and found that these alkaline proteases were expressed by Pd in all 6 wing samples (Table 4). Destructin-2 was the most highly expressed isoform in all WNS-affected bat Pd samples.

We next examined the Pd transcript clusters for additional factors that could affect virulence. Alignment by BLAST to the Swissprot and Uniprot90 databases identified 12 056 transcripts with significant homology to known fungal genes (S8 Table). For the remaining 67 Pd transcript clusters, Trinotate was not able to identify known functional domains or signal peptides present in these previously uncharacterized Pd transcripts. The results from the BLAST alignment were examined for genes known to be involved in processes that could affect Pd virulence, such as secreted proteases [73–75], metal binding proteins [76], fungal cell wall remodeling [76, 77], and other virulence factors [75, 77, 78]. This analysis identified 46 Pd genes that could be involved in pathogenesis (Table 5), including additional secreted proteases that could be involved in tissue invasion.

Because the tissue samples were collected from bats from 6 different hibernacula for this study, it is possible that differences in host or pathogen gene expression reflect differences in the environmental conditions present in each location, including the microbiome. In addition, the housing of the unaffected bats in captivity for 13 weeks prior to analysis could also have affected the microbiome. To examine the differences in the skin microbiome between the bats, we used MG-RAST to identify the lowest common ancestor of metagenomic sequences present (S8 Table). Although there were some differences observed in the bacterial microbiomes present on the wings of the 11 bats, there were no significant changes between the WNS-affected and unaffected samples when bacteria were identified at the class level. Several strains of Pseudomonas fluorescens isolated from bat tissues have been identified with Pd growth inhibiting properties [79]. MG-RAST analysis showed that Pseudomonas species are present in all 11 samples (S8 Table). P. fluorescens transcripts represented 2.8±0.6% of transcripts identified from gammaproteobacteria and 0.40±0.05% of all bacteria on the wings of unaffected bats and 0.37±0.07% of all bacteria on WNS-affected bats. P. fluorescens was present on all little brown myotis sampled, but was rare and relative abundance was not statistically different between WNS-affected and unaffected bats (p = 0.49, t = -0.71, df = 9).
Discussion
The comparison of host gene expression between WNS-affected and unaffected little brown myotis clearly demonstrates that Pd infection causes physiological responses in wing tissue, where substantial fungal invasion of the skin occurs in WNS-affected bats [8]. The changes in transcript levels that we have observed indicate that host responses to fungal infection remain intact during hibernation and are similar to those observed during the initial stages of fungal infection in euthermic mammals [32]. These host responses include acute inflammation, wound healing, and metabolic changes. Pathogen gene expression varies among bats with WNS, suggesting host-pathogen interactions that mediate pathogenesis. Together, these results lay a foundation to determine which host and pathogen responses contribute to WNS resistance and susceptibility and identify targets to increase survival.
Host Response to Pd Infection
The gene expression changes we observed in the wing tissue of WNS-affected bats are similar to those observed in other cutaneous fungal infections [80]. Cutaneous Candida albicans infections in humans and mice typically initiate an immune response by activating pattern recognition receptors of the C-type lectin family [81–83] and the toll-like receptor family, both of which we found upregulated in WNS-affected bat wing tissue (S4 Table). These included C-type lectin domain (CLEC) family members CLEC4D (MCL), CLEC4E (MINCLE), CLEC7A (Dectin-1), CLEC6A (Dectin-2), and Toll-like receptor 9. In mice and humans, protective host responses to C. albicans are usually characterized by many of the same cytokines and chemokines [29] that we have found upgregulated in WNS-affected wing tissue, including the cytokines IL-1β, IL-6, G-CSF, IL-23A, and IL-17C. Little brown myotis infected with Pd are increasing transcription of the key genes necessary for initiating a host response that provides protection from fungal infection. This clearly demonstrates that hibernation does not prevent innate immune responses in bats infected with Pd and that, although they are not closely related to rodents and primates [41], bats respond to fungal infections similarly to these other mammals.
The responses to Pd infection within bat wing tissue may be mediated by keratinocytes in the epithelial tissue. Activation of pattern recognition receptors by fungal ligands is expected to induce keratinocytes to produce many of the cytokines that we have found upregulated at the transcript level in WNS-affected bat wing tissue [84]. In addition to the cytokines typically involved in C. albicans responses described above, keratinocytes are also known to express the chemokine Ccl2 and the cytokines IL-20 and IL-24 in response to pattern recognition receptor activation [85]. Keratinocytes and fibroblasts are also known to exhibit a paracrine loop of IL-1 and IL-6 activation [86] that enhances wound healing and host defense to microbial infection and we found evidence of IL-1 and IL-6 receptor activation in the increased RNA levels for transcription factor p65, NFκB, and P-selectin glycoprotein ligand 1 (S4 Table). Another important cytokine produced by epithelial cells in response to infection is IL-17C [87]. This is an atypical IL-17 family member that is expressed by epithelial cells and causes autocrine responses in the epithelial cells that also express the IL-17RA and IL-17RE heterodimeric IL-17 receptor [87]. The wing tissue transcriptomes from WNS-affected and unaffected bats show similar expression levels of both IL-17RA and IL-17RE (S2 Dataset) and would, therefore, be expected to be responsive to IL-17C. The gene ontology analysis also found evidence for functional enrichment of genes involved in keratinocyte differentiation, presumably due to wound healing responses. Keratinocytes or other epithelial cells in bat wing tissue appear to have responded to the invasion of the epidermis by fungal hyphae.
Genes for pro-inflammatory mediators characterized the innate immune response that we observed in the wing tissue of Pd infected bats. Under euthermic conditions this would be expected to provide protection by the recruitment of monocytes and neutrophils, mediated by G-CSF, IL-23A, Ccl2, IL-17C and IL-6 [88], and the initiation of an adaptive Th17 or Th1 response. However, under the constraints of hibernation, responses that require leukocyte migration do not appear to occur in Pd-infected bats. We do not find strong evidence of increased expression for genes characteristic of either innate or adaptive leukocytes, except for L-selectin, which is expressed on T cells, and CD177, which is expressed on neutrophils. Lower than expected levels of monocyte, neutrophil, Th1, and Th17 cell recruitment may be related to the sequestration of leukocytes during hibernation [45]. However, we have observed neutrophil recruitment in hibernating little brown myotis in response to another fungal infection (Table 1). In the histological examination of the current samples, we found neutrophilic inflammation in both WNS-affected and unaffected wing tissue (Table 1). However, this inflammation did not occur at the sites of Pd infection. Curiously, we found a significant increase in WNS-affected tissue for transcripts for CD3γ and CD45 that could be expressed by gamma-delta T cells or other innate lymphocytes that reside in the skin [89]. It is possible that Pd is specifically suppressing neutrophil and/or T cell recruitment by interfering with chemotactic signals, similar to the suppression of inflammatory immune responses during chytridiomycosis in amphibians [42]. However, analysis of tissue levels of the cytokines and chemokines is necessary to confirm the secretion of these proteins. Because neutrophils and T cells do not appear to be recruited to sites of Pd infection during hibernation, only local inflammatory mediators may be available and they appear to be unable to control the infection in little brown myotis.
In addition to immune responses, hibernating bats also respond to Pd infection in other ways. We found transcripts for proteins from many pathways involved in metabolism, signaling, gene expression, transport, migration, and differentiation that were altered in WNS-affected bats (Fig 4). We cannot exclude the possibility that some of these differences were due to the different hibernation conditions of the two groups of bats. However, the differential expression of the genes in these pathways demonstrates that they are subject to regulation during hibernation and can respond to infection, tissue damage, and/or environmental changes.
Host responses to fungal infection can be influenced by changes in the pathogen, including gene expression changes in the colonizing fungus, such as C. albicans [29]. We found significant variability in the gene expression by Pd, which is particularly interesting because all Pd in North America is presumed to be a clone of the same mating type [90]. The pathogen has adopted different gene expression profiles in the 6 bat tissues (Fig 5), perhaps in response to differences in the host environments. Correspondingly, host gene expression patterns also show differences between the WNS-affected tissue samples. Of particular interest is the observation that the cytokine and chemokine genes found in principal component 2 of our PCA analysis (Fig 3B and S5 Table) are expressed at very different levels in the 6 Pd-infected samples. From this study we cannot determine whether the differences in pathogen gene expression are driven by differences in the host environment or vice versa. Although all 6 WNS-affected bats had visible signs of WNS, had similar Pd burdens, and similar histopathology, it is possible that the differences in host or pathogen gene expression that we observed may have affected progression of WNS and survival.
Responses that May Contribute to WNS Mortality
Because the increased frequency of arousals from torpor appears to be a primary cause of WNS mortality [5, 14, 22], we considered possible mechanisms that could affect torpor bout length. The increased gene expression of IL-1, IL-6, and other pro-inflammatory cytokines mediates a local acute inflammatory response to Pd. These cytokines also have systemic effects that modify behavior and thermoregulation [91]. In addition to cytokine and chemokine transcript increases, we also found increased transcripts for the enzyme cyclooxygenase-2 (prostaglandin G/H synthase 2) and both secreted and cytosolic phospholipase A2 that form critical inflammatory lipid mediators such as prostaglandin H2. The eicosanoids generated by these enzymes, along with the actions of the upregulated genes kallikrein-6 and cathepsin S, are expected to generate pain and itching by locally activating neuronal nociceptors [92, 93]. This, in turn, could affect torpor bout length and/or behavior during periodic arousals. Indeed, we have documented significantly more grooming in WNS-affected bats infected in the wild [94], although a different study on laboratory-infected bats did not find similar behavior changes [95]. Together, the upregulated genes will likely generate an inflammatory microenvironment within the wing that may contribute to the robust wound healing response that we observe in WNS-affected bats. However, inflammation can also play a detrimental role in some diseases [96]. Further tissue damage and subsequent wound healing occurs in surviving bats upon emergence from hibernation [19]. These local affects of inflammation (pain and itching) as well as systemic effects are likely to play a key role in WNS pathology.
In addition to the gene expression changes that may contribute to acute inflammation locally within the epithelial tissues invaded by Pd, the systemic release of febrile cytokines such as IL-6 could affect the signals that control hibernation arousal. However, an exogenous pyrogen, lipopolysaccharide, is not able to provoke arousals in hibernating golden-mantled ground squirrels [97], so it may be unlikely that inflammation or febrile cytokines can directly trigger arousal in WNS-affected bats. Intracerebroventricular injection of prostaglandin E2 in golden-mantled ground squirrels induces arousal from torpor and a febrile response during an extended periodic arousal [97]. Our observation of increased expression of the enzyme that generates prostaglandin H2 may provide a mechanism that explains the shortened torpor bouts in WNS-affected bats, if it can be shown that this enzyme is active in the tissue and produces enough prostaglandin H2 to act systemically.
In addition to the changes in expression of genes involved in immune responses and wound healing, we also found significant changes in metabolic genes. We found evidence of gene expression changes consistent with increased fat metabolism, including changes in transcripts for apolipoproteins, lipid transport proteins, protein metabolism, and carbohydrate metabolism. Of particular interest, we found increases in the expression of hydroxycarboxylic acid receptors 2 and 3 that are known to mediate adiponectin secretion [98]. This suggests that infection with Pd may directly trigger changes in lipid and carbohydrate metabolism that contribute to WNS pathology. These changes stand in contrast to the changes that have been seen in the brain transcriptome of hibernating horseshoe bats (Rhinolophus ferrumequinum) [99] and the brain proteome of hibernating Rickett’s big-footed bats (Myotis pilosus) [100], which show decreased fat metabolism during torpor. By leading to premature depletion of fat stores, these gene expression changes could contribute to WNS mortality.
The other changes in host gene expression that we observed are consistent with a multi-stage progression model of WNS [22]. We also found support for changes in genes involved in oxidative stress [23] and body fluid levels, which may contribute to WNS progression. Together, the pattern of gene expression changes that we find in little brown myotis with WNS suggests that a combination of maladaptive responses may contribute to mortality. However, the number of upregulated genes involved in the acute inflammatory response suggests that excessive inflammation may also be a factor contributing to pathology even prior to emergence from hibernation when it is suspected to contribute to wing damage [101].
Implications for Future Studies
The changes in host transcript levels that we have found are presumably caused by physiological responses of the host to infection. However, caution must be used when extending these transcriptional responses to functional mechanisms because the current study does not measure protein or metabolite levels directly. Future studies will be necessary to determine which of the gene expression changes observed affect which host response mechanisms.
The little brown myotis chosen for the WNS-affected samples were exhibiting WNS pathology and appeared unlikely to survive at the time of sample collection. For this reason, it is presently uncertain which of the gene expression changes that we have observed are contributing to protection and which are pathological. Another factor that likely contributes to the variation in gene expression that we observed among the samples collected from free-ranging bats is the time since the most recent arousal from torpor. Prior to collection of each wing tissue sample, bats were artificially aroused for 30 to 120 minutes. This period of arousal is similar in duration to the natural arousals during hibernation for little brown myotis [14], and presumably of sufficient duration for some innate immune responses to occur and for transcript levels to be altered. One reason for this procedure was to avoid disparities between the elapsed time from the most recent arousal bout until tissue collection. For the WNS-affected bats we could not determine when the most recent natural arousal would have occurred, but it would have likely been more recently than in unaffected animals, as affected animals arouse from torpor more frequently [14]. In the current study we cannot resolve whether the changes in gene expression that we observed occurred during the most recent arousal, during previous periodic arousals, or during torpor. Future studies will be needed to determine which of the changes in gene expression that we observed during WNS in bats in the wild also vary in controlled captive hibernation conditions when prior arousal patterns are known. Further studies are also needed to compare the physiological responses in bats exhibiting WNS morbidity to responses in less susceptible bats, such as European species, North American species that are less susceptible like the big brown bat [11], and the remnant populations of little brown myotis that appear to have developed tolerance or resistance to Pd [1]. Such studies should point to a path forward for bats in North America to persist in a landscape where Pd is endemic.
Conclusions
Little brown myotis mount a host response to Pd infection during hibernation. Which components of this response are protective or contribute to WNS pathology remains to be resolved. The innate immune response we have observed would be expected to promote a Th17-directed adaptive immune response that could clear the infection. However, the energetic constraints of hibernation may prevent little brown myotis from execution of the Th17- and neutrophil-mediated phases of the immune response. This may lead to excessive inflammatory responses, either during hibernation or upon emergence. The changes in host gene expression that we observed demonstrate that during Pd infection, little brown myotis also alter other defense responses, metabolic pathways, and transcription. Numerous Pd genes that may contribute to virulence were identified and these represent potential pathogen responses to host defense. Hibernation does not prevent a host response to infection and a better understanding of the differences between host and pathogen responses in bats susceptible to WNS and those resistant may lead to ways for increasing survival.
Materials and Methods
Ethics Statement
This study was carried out on bats from non-endangered species in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All methods were approved by the Institutional Animal Care and Use Committee at Bucknell University (protocol DMR-016). Animals were humanely euthanized by isoflurane anesthesia overdose followed by decapitation. In Illinois, animal collection was conducted by state wildlife officials and a numbered permit was not required. Scientific collector’s permits were obtained in Michigan (SC1448), Minnesota (201174), and Kentucky (SC1411147).
Samples
We collected hibernating little brown myotis from cave or mine walls at the locations listed in Table 1. Bats collected from all locations are expected to be from the same genetic population of eastern little brown myotis [102]. For bats unaffected by WNS, little brown myotis were first swabbed on the left forearm for quantitative PCR analysis. After measurements were taken, bats were individually placed in cloth bags and hung in constant temperature thermoelectric coolers (Koolatron PC-3) maintained at ~7°C. Water-saturated sponges were placed in the bottom of each cooler to maintain humidity during transportation to Bucknell University. Bats were housed for 13 weeks in a Percival (model I36VLC8) environmental chamber with conditions set to 4°C and 95% relative humidity. Bats were provided water throughout hibernation. Bats were aroused from hibernation for 30–120 minutes prior to euthanasia. For WNS-affected bats, little brown myotis were collected in the field, measured, swabbed for quantitative PCR, and humanely euthanized after being aroused from hibernation for 60–120 minutes. Scaled mass index (SMI) was calculated using the formula (mass(in g))*(38.01/(forearm length(in mm))^1.406 [103]. Wing tissue was placed in formalin for histology and placed in RNAlater (Sigma-Aldrich) for gene expression analysis. RNAlater samples were stored at ambient temperature for up to 24 hours before long-term storage at -80°C. RNA was purified from 50 mg of wing tissue using a QIAGEN RNeasy Mini Kit. All samples used for RNA sequencing had RNA integrity values greater than 7.0 using an Agilent Bioanalyzer.
Verification of WNS Status
Wing skin tissue was removed from the bones of the arm and digits and rolled onto 2 cm paraffin wax logs. The logs were then fixed in 10% neutral buffered formalin for at least 24 hours. Each log was cut into 3 pieces that were processed into paraffin blocks overnight in a Tissue-Tek VIP processor (Sakura Finetek). The pieces were embedded in paraffin blocks, sectioned at 3 microns, and stained with periodic acid Schiff with a hematoxylin counterstain [8]. WNS lesions (Table 1; WNS) were identified as cupping erosions with fungal hyphae and conida present. Inflammatory foci (Table 1; Infl) were identified as clusters of infiltrating neutrophils and were not associated with the asymmetrical curved conidia of Pd.
To determine presence or absence of Pd on bats unaffected by WNS, each swab was tested twice by quantitative PCR [64] by Jeffrey T. Foster at University of New Hampshire. A cycle-threshold less than 40 was used as a positive result. One of the 5 unaffected bats had one positive and one negative test (Table 1), but histology (Table 1) and subsequent RNA sequencing determined this to most likely be a false positive (S2 Table; p = 2.2x10-6). For bats affected by WNS, we performed quantitative PCR to measure the Pd load, in genomic equivalents normalized to swabs spiked with 10 000 Pd conidia, that were detected on each bat [15].
Next Generation RNA Sequencing
The Genome Sequencing and Analysis Facility at the University of Texas at Austin performed all library preparation and quality control procedures. Directional RNA libraries were prepared with poly-A mRNA enrichment, dUTP/UDG strand-specific labeling, fragmentation, and 200 base pair size selection. RNA-Seq was performed in two lanes of an Illumina HiSeq 2500 with 101 base pair length reads obtained.
Transcriptome Assemblies
The paired reads from all samples were preprocessed by removing adapters and using trimmomatic PE [104] with settings of Illumina clip:2:30:10, seed mismatches:2, palindrome threshold:30, clip threshold:10, leading:5, trailing:5, minlength:36. The remaining paired reads were then combined and Trinity (v2.0.4) was used in strand-specific mode (RF) to construct a de novo assembly [105]. K-mer in silico read normalization with maximum coverage of 50 resulted in 22 482 456 read pairs that were used for assembly out of 177 755 004 total. The assembly was then filtered to remove Pd sequences using the program Deconseq [65] with the Broad Institute Geomyces destructans genome 20631–21 used to identify pathogen sequences and with the little brown myotis genome (Myoluc2.0) used to retain host sequences. Bowtie 1.0.1 [106] was used to determine the number of reads that mapped to each transcript in the assembly.
Differential Expression
The script align_and_estimate_abundance.pl included in the Trinity v2.0.6 distribution [105] was used to estimate expression levels for each transcript. Bowtie 1.0.1 [106] was used to map reads (including unpaired reads after quality trimming) from each sample onto the assembly. RSEM v1.2.20 [107] was used to apply an expectation maximization algorithm to predict gene expression counts for each transcript. Expression levels are presented after trimmed mean of M-values (TMM) normalization in fragments per kilobase of transcript per million mapped reads (FPKM). DESeq2 v1.8.1 [67] was used to determine the probability of differential expression for each Trinity transcript cluster that had a minimum RSEM-estimated count, before normalization, of 5 across all samples. For DESeq2 analysis, the default values for removing outliers and filtering lowly expressed transcripts were used. An alpha value of 0.05 was used instead of the default of 0.1 to decrease the number of differentially expressed genes identified. Posterior probabilities of differential expression for individual transcript isoforms were estimated using a Bayesian approach with EBSeq v1.8.0 [68]. False discovery rate [108] was used to control for multiple comparisons. NCBI BLAST v2.2.29+ [109] was used to identify the highest-ranking match for each isoform in the UniProt Swissprot database (downloaded on Sep 17, 2014) with an e-value cutoff of 1x10-5.
Hierarchical clustering of samples and genes was performed within R 3.1.2 using the hclust function with the complete linkage method. Bootstrap analysis of clustering was performed using the pvclust 1.3–2 package and 1000 replications [69]. Principal component analysis was performed using the prcomp function and visualized with the rgl 0.93.1098 package.
Gene Ontology
NCBI BLAST v2.2.29+ [109] was used with an e-value cutoff of 1x10-5 to identify homologs in the Uniprot Swissprot human protein database (downloaded on Nov 25, 2014) for transcripts significantly upregulated in WNS-affected bat wing tissue with an FDR of less than 0.1 (in order to increase the number of genes prior to subsequent analysis with higher stringency FDR). Unique Ensembl gene IDs were identified for 1144 of the 1922 upregulated transcripts and 481 of the 1356 downregulated transcripts. GOrilla [70] was used with a p value cutoff of 0.001 to identify upregulated or downregulated biological processes by comparison to the background list of 12 828 human genes identified by BLAST in the Trinity assembly. Multiple testing correction [108] was used with an FDR cutoff of 0.01. Results were visualized as a treemap with REVIGO [71].
Pd Gene Analysis
Trinity v2.0.4 was used to generate a Pd assembly in genome-guided mode with jaccard clipping and using the Broad Institute G. destructans genome 20631–21. This assembly was used to assess pathogen gene expression in the samples from WNS-affected bats using RSEM v1.2.20 [107]. Trinotate v2 was used to annotate the Pd transcripts by using NCBI BLAST v2.2.29+ [109] and both the Swissprot and Uniref90 databases (downloaded on Sep 17, 2014).
Metagenome Analysis
Reads for each sample were analyzed using MG-RAST v.3.5 [110] to identify metagenomic sequences after filtering against the B. taurus genome (the taxonomically closest genome available for filtering). For assignment of organism abundance, the best hit classification was used with the M5NR database, maximum e-value cutoff of 1x10-5, minimum identity cutoff of 60%, and minimum alignment length cutoff of 15.
Supporting Information
{kind=link}
Zdroje
1. Coleman JT, Reichard JD. (2014) Bat White-Nose Syndrome in 2014: A Brief Assessment Seven Years After Discovery of a Virulent Fungal Pathogen in North America. Outlooks on Pest Management 25: 374–377.
2. Blehert DS. (2012) Fungal disease and the developing story of bat white-nose syndrome. PLoS Pathog 8: e1002779. doi: 10.1371/journal.ppat.1002779 22829763
3. Blehert DS, Hicks AC, Behr M, Meteyer CU, Berlowski-Zier BM, et al. (2009) Bat white-nose syndrome: an emerging fungal pathogen? Science 323: 227–227. doi: 10.1126/science.1163874 18974316
4. Lorch JM, Meteyer CU, Behr MJ, Boyles JG, Cryan PM, et al. (2011) Experimental infection of bats with Geomyces destructans causes white-nose syndrome. Nature 480: 376–378. doi: 10.1038/nature10590 22031324
5. Warnecke L, Turner JM, Bollinger TK, Lorch JM, Misra V, et al. (2012) Inoculation of bats with European Geomyces destructans supports the novel pathogen hypothesis for the origin of white-nose syndrome. Proc Natl Acad Sci U S A 109: 6999–7003. doi: 10.1073/pnas.1200374109 22493237
6. Verant ML, Boyles JG, Waldrep Jr W, Wibbelt G, Blehert DS. (2012) Temperature-dependent growth of Geomyces destructans, the fungus that causes bat white-nose syndrome.
7. Cryan PM, Meteyer CU, Boyles JG, Blehert DS. (2010) Wing pathology of white-nose syndrome in bats suggests life-threatening disruption of physiology. BMC Biol 8: 135-7007-8-135.
8. Meteyer CU, Buckles EL, Blehert DS, Hicks AC, Green DE, et al. (2009) Histopathologic criteria to confirm white-nose syndrome in bats. Journal of Veterinary Diagnostic Investigation 21: 411–414. 19564488
9. Turner GG, Reeder DM, Coleman JTH. (2011) A five year assessment of mortality and geographic spread of white-nose syndrome in North American bats and a look to the future. Bat Research News 52: 13–27.
10. Frick WF, Puechmaille SJ, Hoyt JR, Nickel BA, Langwig KE, et al. (2015) Disease alters macroecological patterns of North American bats. Global Ecol Biogeogr 24: 741–749.
11. Frank CL, Michalski A, McDonough AA, Rahimian M, Rudd RJ, et al. (2014) The Resistance of a North American Bat Species (Eptesicus fuscus) to White-Nose Syndrome (WNS). PloS One 9: e113958. doi: 10.1371/journal.pone.0113958 25437448
12. Wibbelt G, Puechmaille SJ, Ohlendorf B, Mühldorfer K, Bosch T, et al. (2013) Skin Lesions in European Hibernating Bats Associated with Geomyces destructans, the Etiologic Agent of White-Nose Syndrome. PloS One 8: e74105. doi: 10.1371/journal.pone.0074105 24023927
13. Bandouchova H, Bartonicka T, Berkova H, Brichta J, Cerny J, et al. (2015) Pseudogymnoascus destructans: evidence of virulent skin invasion for bats under natural conditions, Europe. Transboundary and Emerging Diseases 62: 1–5. doi: 10.1111/tbed.12282 25268034
14. Reeder DM, Frank CL, Turner GG, Meteyer CU, Kurta A, et al. (2012) Frequent arousal from hibernation linked to severity of infection and mortality in bats with white-nose syndrome. PLoS One 7: e38920. doi: 10.1371/journal.pone.0038920 22745688
15. Johnson JS, Reeder DM, McMichael JW III, Meierhofer MB, Stern DW, et al. (2014) Host, pathogen, and environmental characteristics predict white-nose syndrome mortality in captive little brown myotis (Myotis lucifugus). PLoS ONE 9: e112502. doi: 10.1371/journal.pone.0112502 25409028
16. Geiser F. (2004) Metabolic rate and body temperature reduction during hibernation and daily torpor. Annu Rev Physiol 66: 239–274. 14977403
17. Thomas DW, Cloutier D. (1992) Evaporative water loss by hibernating little brown bats, Myotis lucifugus. Physiol Zool: 443–456.
18. Thomas DW, Dorais M, Bergeron J. (1990) Winter energy budgets and cost of arousals for hibernating little brown bats, Myotis lucifugus. J Mammal 71: 475–479.
19. Fuller NW, Reichard JD, Nabhan ML, Fellows SR, Pepin LC, et al. (2011) Free-ranging little brown myotis (Myotis lucifugus) heal from wing damage associated with white-nose syndrome. Ecohealth 8: 154–162. doi: 10.1007/s10393-011-0705-y 21922344
20. Cryan PM, Meteyer CU, Blehert DS, Lorch JM, Reeder DM, et al. (2013) Electrolyte depletion in white-nose syndrome bats. J Wildl Dis 49: 398–402. doi: 10.7589/2012-04-121 23568916
21. Willis CK, Menzies AK, Boyles JG, Wojciechowski MS. (2011) Evaporative water loss is a plausible explanation for mortality of bats from white-nose syndrome. Integr Comp Biol 51: 364–373. doi: 10.1093/icb/icr076 21742778
22. Verant ML, Meteyer CU, Speakman JR, Cryan PM, Lorch JM, et al. (2014) White-nose syndrome initiates a cascade of physiologic disturbances in the hibernating bat host. BMC Physiol 14: 10. doi: 10.1186/s12899-014-0010-4 25487871
23. Moore MS, Reichard JD, Murtha TD, Nabhan ML, Pian RE, et al. (2013) Hibernating little brown myotis (Myotis lucifugus) show variable immunological responses to white-nose syndrome. PloS One 8: e58976. doi: 10.1371/journal.pone.0058976 23527062
24. Moore MS, Reichard JD, Murtha TD, Zahedi B, Fallier RM, et al. (2011) Specific alterations in complement protein activity of little brown myotis (Myotis lucifugus) hibernating in white-nose syndrome affected sites. PLoS One 6: e27430. doi: 10.1371/journal.pone.0027430 22140440
25. LeibundGut-Landmann S, Wüthrich M, Hohl TM. (2012) Immunity to fungi. Curr Opin Immunol 24: 449–458. doi: 10.1016/j.coi.2012.04.007 22613091
26. Brown GD. (2011) Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol 29: 1. doi: 10.1146/annurev-immunol-030409-101229 20936972
27. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, et al. (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357: 1608–1619. 17881745
28. Liu L, Okada S, Kong X, Kreins AY, Cypowyj S, et al. (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208: 1635–1648. doi: 10.1084/jem.20110958 21727188
29. Gow NA, van de Veerdonk, Frank L, Brown AJ, Netea MG. (2012) Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nature Reviews Microbiology 10: 112–122.
30. Netea MG, Brown GD, Kullberg BJ, Gow NA. (2008) An integrated model of the recognition of Candida albicans by the innate immune system. Nature Reviews Microbiology 6: 67–78. 18079743
31. Romani L. (2011) Immunity to fungal infections. Nat Rev Immunol 11: 275–288. doi: 10.1038/nri2939 21394104
32. Hernández-Santos N, Gaffen SL. (2012) Th17 cells in immunity to Candida albicans. Cell Host & Microbe 11: 425–435.
33. Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. (2010) IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol 185: 5453–5462. doi: 10.4049/jimmunol.1001153 20921529
34. Smeekens SP, Ng A, Kumar V, Johnson MD, Plantinga TS, et al. (2013) Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nature Communications 4: 1342. doi: 10.1038/ncomms2343 23299892
35. Blanco JL, Garcia ME. (2008) Immune response to fungal infections. Vet Immunol Immunopathol 125: 47–70. doi: 10.1016/j.vetimm.2008.04.020 18565595
36. De Luca A, Zelante T, D'Angelo C, Zagarella S, Fallarino F, et al. (2010) IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunology.
37. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, et al. (2009) Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206: 299–311. doi: 10.1084/jem.20081463 19204111
38. Brook CE, Dobson AP. (2015) Bats as ‘special’reservoirs for emerging zoonotic pathogens. Trends Microbiol 23: 172–180. doi: 10.1016/j.tim.2014.12.004 25572882
39. Cogswell-Hawkinson AC, McGlaughlin ME, Calisher CH, Adams R, Schountz T. (2011) Molecular and phylogenetic characterization of cytokine genes from Seba’s short-tailed bat (Carollia perspicillata). Open Immunology Journal 4: 31–39.
40. Iha K, Omatsu T, Watanabe S, Ueda N, Taniguchi S, et al. (2009) Molecular cloning and sequencing of the cDNAs encoding the bat interleukin (IL)-2, IL-4, IL-6, IL-10, IL-12p40, and tumor necrosis factor-alpha. Journal of Veterinary Medical Science 71: 1691–1695. 20046044
41. Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, et al. (2013) Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339: 456–460. doi: 10.1126/science.1230835 23258410
42. Fites JS, Ramsey JP, Holden WM, Collier SP, Sutherland DM, et al. (2013) The invasive chytrid fungus of amphibians paralyzes lymphocyte responses. Science 342: 366–369. doi: 10.1126/science.1243316 24136969
43. Bouma HR, Carey HV, Kroese FG. (2010a) Hibernation: the immune system at rest? J Leukoc Biol 88: 619–624.
44. Bouma HR, Henning RH, Kroese FG, Carey HV. (2012) Hibernation is associated with depression of T-cell independent humoral immune responses in the 13-lined ground squirrel. Dev Comp Immunol.
45. Bouma HR, Strijkstra AM, Boerema AS, Deelman LE, Epema AH, et al. (2010b) Blood cell dynamics during hibernation in the European Ground Squirrel. Vet Immunol Immunopathol 136: 319–323.
46. Kurtz CC, Carey HV. (2007) Seasonal changes in the intestinal immune system of hibernating ground squirrels. Dev Comp Immunol 31: 415–428. 16930701
47. Maniero GD. (2002) Classical pathway serum complement activity throughout various stages of the annual cycle of a mammalian hibernator, the golden-mantled ground squirrel, Spermophilus lateralis. Dev Comp Immunol 26: 563–574. 12031416
48. Maniero GD. (2005) Ground squirrel splenic macrophages bind lipopolysaccharide over a wide range of temperatures at all phases of their annual hibernation cycle. Comp Immunol Microbiol Infect Dis 28: 297–309. 16182368
49. Jaroslow BN, Serrell BA. (1972) Differential sensitivity to hibernation of early and late events in development of the immune response. J Exp Zool 181: 111–116. 4556623
50. Larsen B. (1971) Antibody formation in hedgehogs. II. Hibernating animals. Comp Biochem Physiol A Comp Physiol 38: 571–580. 4396827
51. Manasek FJ, Adelstein SJ, Lyman CP. (1965) The effects of hibernation on the in vitro synthesis of DNA by hamster lymphoid tissue. J Cell Physiol 65: 319–324. 5891143
52. Cahill JE, Lewert RM, Jaroslow BN. (1967) Effect of hibernation on course of infection and immune response in Citellus tridecemlineatus infected with Nippostrongylus brasiliensis. J Parasitol 53: 110–115. 6017221
53. Sieckmann DG, Jaffe H, Golech S, Cai D, Hallenbeck JM, et al. (2014) Anti-lymphoproliferative activity of alpha-2-macroglobulin in the plasma of hibernating 13-lined ground squirrels and woodchucks. Vet Immunol Immunopathol 161: 1–11. doi: 10.1016/j.vetimm.2014.05.006 25113962
54. Maniero GD. (2000) The influence of temperature and season on mitogen-induced proliferation of ground squirrel lymphocytes. In: Heldmaier G, Klingenspor M, editors. Life in the Cold. New York: Springer. pp. 493–503.
55. Hampton M, Melvin RG, Kendall AH, Kirkpatrick BR, Peterson N, et al. (2011) Deep sequencing the transcriptome reveals seasonal adaptive mechanisms in a hibernating mammal. PloS One 6: e27021. doi: 10.1371/journal.pone.0027021 22046435
56. Hampton M, Melvin RG, Andrews MT. (2013) Transcriptomic analysis of brown adipose tissue across the physiological extremes of natural hibernation. PloS One 8: e85157. doi: 10.1371/journal.pone.0085157 24386461
57. Yan J, Burman A, Nichols C, Alila L, Showe LC, et al. (2006) Detection of differential gene expression in brown adipose tissue of hibernating arctic ground squirrels with mouse microarrays. Physiol Genomics 25: 346–353. 16464973
58. Schwartz C, Hampton M, Andrews MT. (2013) Seasonal and regional differences in gene expression in the brain of a hibernating mammal. PloS One 8: e58427. doi: 10.1371/journal.pone.0058427 23526982
59. Williams DR, Epperson LE, Li W, Hughes MA, Taylor R, et al. (2005) Seasonally hibernating phenotype assessed through transcript screening. Physiol Genomics 24: 13–22. 16249311
60. Bouma HR, Kroese FG, Kok JW, Talaei F, Boerema AS, et al. (2011) Low body temperature governs the decline of circulating lymphocytes during hibernation through sphingosine-1-phosphate. Proc Natl Acad Sci U S A 108: 2052–2057. doi: 10.1073/pnas.1008823108 21245336
61. de Vrij EL, Vogelaar PC, Goris M, Houwertjes MC, Herwig A, et al. (2014) Platelet dynamics during natural and pharmacologically induced torpor and forced hypothermia. PloS One 9: e93218. doi: 10.1371/journal.pone.0093218 24722364
62. Melvin RG, Andrews MT. (2009) Torpor induction in mammals: recent discoveries fueling new ideas. Trends in Endocrinology & Metabolism 20: 490–498.
63. Johnson JS, Reeder DM, Lilley TM, Czirják GÁ, Voigt CC, et al. (2015) Antibodies to Pseudogymnoascus destructans are not sufficient for protection against white-nose syndrome. Ecology and Evolution 5: 2203–2214. doi: 10.1002/ece3.1502 26078857
64. Muller LK, Lorch JM, Lindner DL, O’Connor M, Gargas A, et al. (2013) Bat white-nose syndrome: a real-time TaqMan polymerase chain reaction test targeting the intergenic spacer region of Geomyces destructans. Mycologia 105: 253–259. doi: 10.3852/12-242 22962349
65. Schmieder R, Edwards R. (2011) Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PloS One 6: e17288. doi: 10.1371/journal.pone.0017288 21408061
66. Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics.
67. Love MI, Huber W, Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 25516281
68. Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, et al. (2013) EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29: 1035–1043. doi: 10.1093/bioinformatics/btt087 23428641
69. Suzuki R, Shimodaira H. (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22: 1540–1542. 16595560
70. Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. doi: 10.1186/1471-2105-10-48 19192299
71. Supek F, Bošnjak M, Škunca N, Šmuc T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PloS One 6: e21800. doi: 10.1371/journal.pone.0021800 21789182
72. Westermann AJ, Gorski SA, Vogel J. (2012) Dual RNA-seq of pathogen and host. Nature Reviews Microbiology 10: 618–630. doi: 10.1038/nrmicro2852 22890146
73. Pannkuk EL, Risch TS, Savary BJ. (2015) Isolation and identification of an extracellular subtilisin-like serine protease secreted by the bat pathogen Pseudogymnoascus destructans. PLoS One 10: e0120508. doi: 10.1371/journal.pone.0120508 25785714
74. O'Donoghue AJ, Knudsen GM, Beekman C, Perry JA, Johnson AD, et al. (2015) Destructin-1 is a collagen-degrading endopeptidase secreted by Pseudogymnoascus destructans, the causative agent of white-nose syndrome. Proc Natl Acad Sci U S A 112: 7478–7483. doi: 10.1073/pnas.1507082112 25944934
75. Smyth C, Schlesinger S, Overton B, Butchkoski C. (2013) The alternative host hypothesis and potential virulence genes in Geomyces destructans. Bat Res News 54: 17–24.
76. Whittington A, Gow NA, Hube B. (2014) From commensal to pathogen: Candida albicans. In: Anonymous Human Fungal Pathogens.: Springer. pp. 3–18.
77. Casadevall A, Steenbergen JN, Nosanchuk JD. (2003) ‘Ready made’virulence and ‘dual use’virulence factors in pathogenic environmental fungi—the Cryptococcus neoformans paradigm. Curr Opin Microbiol 6: 332–337. 12941400
78. Swidergall M, Ernst JF. (2014) Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot Cell 13: 950–957. doi: 10.1128/EC.00093-14 24951441
79. Hoyt JR, Cheng TL, Langwig KE, Hee MM, Frick WF, et al. (2015) Bacteria isolated from bats inhibit the growth of Pseudogymnoascus destructans, the causative agent of white-nose syndrome. PLoS ONE 10: e0121329. doi: 10.1371/journal.pone.0121329 25853558
80. Rizzetto L, Kuka M, De Filippo C, Cambi A, Netea MG, et al. (2010) Differential IL-17 production and mannan recognition contribute to fungal pathogenicity and commensalism. J Immunol 184: 4258–4268. doi: 10.4049/jimmunol.0902972 20228201
81. Reid DM, Gow NA, Brown GD. (2009) Pattern recognition: recent insights from Dectin-1. Curr Opin Immunol 21: 30–37. doi: 10.1016/j.coi.2009.01.003 19223162
82. Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, et al. (2009) C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci U S A 106: 1897–1902. doi: 10.1073/pnas.0805177106 19171887
83. Zhu L, Zhao X, Jiang C, You Y, Chen X, et al. (2013) C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39: 324–334. doi: 10.1016/j.immuni.2013.05.017 23911656
84. Bernard F, Morel F, Camus M, Pedretti N, Barrault C, et al. (2012) Keratinocytes under fire of proinflammatory cytokines: bona fide innate immune cells involved in the physiopathology of chronic atopic dermatitis and psoriasis. J Allergy 2012.
85. Kunz S, Wolk K, Witte E, Witte K, Doecke W, et al. (2006) Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp Dermatol 15: 991–1004. 17083366
86. Werner S, Krieg T, Smola H. (2007) Keratinocyte–fibroblast interactions in wound healing. J Invest Dermatol 127: 998–1008. 17435785
87. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, et al. (2011) IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12: 1159–1166. doi: 10.1038/ni.2156 21993848
88. Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, et al. (2014) Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, ROR [gamma] t and dectin-2. Nat Immunol 15: 143–151. doi: 10.1038/ni.2797 24362892
89. Heath WR, Carbone FR. (2013) The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 14: 978–985. doi: 10.1038/ni.2680 24048119
90. Palmer JM, Kubatova A, Novakova A, Minnis AM, Kolarik M, et al. (2014) Molecular characterization of a heterothallic mating system in Pseudogymnoascus destructans, the fungus causing white-nose syndrome of bats. G3 (Bethesda) 4: 1755–1763.
91. Harden LM, du Plessis I, Poole S, Laburn HP. (2008) Interleukin (IL)-6 and IL-1β act synergistically within the brain to induce sickness behavior and fever in rats. Brain Behav Immun 22: 838–849. doi: 10.1016/j.bbi.2007.12.006 18255258
92. Moalem G, Tracey DJ. (2006) Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev 51: 240–264. 16388853
93. Couture R, Harrisson M, Vianna RM, Cloutier F. (2001) Kinin receptors in pain and inflammation. Eur J Pharmacol 429: 161–176. 11698039
94. Brownlee-Bouboulis SA, Reeder DM. (2013) White-nose syndrome-affected little brown myotis (Myotis lucifugus) increase grooming and other active behaviors during arousals from hibernation. J Wildl Dis 49: 850–859. doi: 10.7589/2012-10-242 24502712
95. Wilcox A, Warnecke L, Turner JM, McGuire LP, Jameson JW, et al. (2014) Behaviour of hibernating little brown bats experimentally inoculated with the pathogen that causes white-nose syndrome. Anim Behav 88: 157–164.
96. Tobin DM, Ramakrishnan L. (2013) TB: the Yin and Yang of lipid mediators. Current Opinion in Pharmacology 13: 641–645. doi: 10.1016/j.coph.2013.06.007 23849093
97. Prendergast BJ, Freeman DA, Zucker I, Nelson RJ. (2002) Periodic arousal from hibernation is necessary for initiation of immune responses in ground squirrels. Am J Physiol Regul Integr Comp Physiol 282: R1054–62. 11893609
98. Plaisance EP, Lukasova M, Offermanns S, Zhang Y, Cao G, et al. (2009) Niacin stimulates adiponectin secretion through the GPR109A receptor. Am J Physiol Endocrinol Metab 296: E549–58. doi: 10.1152/ajpendo.91004.2008 19141678
99. Lei M, Dong D, Mu S, Pan Y, Zhang S. (2014) Comparison of brain transcriptome of the greater horseshoe bats (Rhinolophus ferrumequinum) in active and torpid episodes. PloS One 9: e107746. doi: 10.1371/journal.pone.0107746 25251558
100. Zhang Y, Pan Y, Yin Q, Yang T, Dong D, et al. (2014) Critical roles of mitochondria in brain activities of torpid Myotis ricketti bats revealed by a proteomic approach. Journal of Proteomics 105: 266–284. doi: 10.1016/j.jprot.2014.01.006 24434588
101. Meteyer CU, Barber D, Mandl JN. (2012) Pathology in euthermic bats with white nose syndrome suggests a natural manifestation of immune reconstitution inflammatory syndrome. Virulence 3.
102. Vonhof MJ, Russell AL, Miller-Butterworth CM. (2015) Range-Wide Genetic Analysis of Little Brown Bat (Myotis lucifugus) Populations: Estimating the Risk of Spread of White-Nose Syndrome. PloS One 10: e0128713. doi: 10.1371/journal.pone.0128713 26154307
103. Peig J, Green AJ. (2009) New perspectives for estimating body condition from mass/length data: the scaled mass index as an alternative method. Oikos 118: 1883–1891.
104. Bolger AM, Lohse M, Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. doi: 10.1093/bioinformatics/btu170 24695404
105. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8: 1494–1512. doi: 10.1038/nprot.2013.084 23845962
106. Langmead B, Trapnell C, Pop M, Salzberg SL. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. doi: 10.1186/gb-2009-10-3-r25 19261174
107. Li B, Dewey CN. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323-2105-12-323.
108. Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society.Series B (Methodological): 289–300.
109. McGinnis S, Madden TL. (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32: W20–5. 15215342
110. Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, et al. (2008) The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386-2105-9-386.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2015 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Chronobiomics: The Biological Clock as a New Principle in Host–Microbial Interactions
- Interferon-γ: The Jekyll and Hyde of Malaria
- Crosslinking of a Peritrophic Matrix Protein Protects Gut Epithelia from Bacterial Exotoxins
- Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis