
A Unique Human Norovirus Lineage with a Distinct HBGA Binding Interface
Human norovirus (huNoV) has diverged into two major lineages (GI and GII) selected by the host histo-blood group antigens (HBGAs). Both lineages further diverge into various sub-lineages (genotypes) that recognize different ABH and Lewis antigens through a common HBGA binding interface shared among strains within each genogroup. In this study, through X-ray crystallography of the P domain of a GII.21 huNoV (OIF) we identified a unique lineage in GII consisting of GII.13 and GII.21 genotypes that recognize HBGAs through a binding interface distinct from the GII conventional binding interface. While the mechanism remains unknown, our finding raises an alert on future emergence of new lineages by the same way via developing new receptor binding interfaces, as well as further divergence of this new lineage into more sub-lineages recognizing different HBGAs, which may impact future epidemiology and strategies for disease control and prevention against huNoVs.
Published in the journal:
A Unique Human Norovirus Lineage with a Distinct HBGA Binding Interface. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1005025
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005025
Summary
Human norovirus (huNoV) has diverged into two major lineages (GI and GII) selected by the host histo-blood group antigens (HBGAs). Both lineages further diverge into various sub-lineages (genotypes) that recognize different ABH and Lewis antigens through a common HBGA binding interface shared among strains within each genogroup. In this study, through X-ray crystallography of the P domain of a GII.21 huNoV (OIF) we identified a unique lineage in GII consisting of GII.13 and GII.21 genotypes that recognize HBGAs through a binding interface distinct from the GII conventional binding interface. While the mechanism remains unknown, our finding raises an alert on future emergence of new lineages by the same way via developing new receptor binding interfaces, as well as further divergence of this new lineage into more sub-lineages recognizing different HBGAs, which may impact future epidemiology and strategies for disease control and prevention against huNoVs.
Introduction
Noroviruses (NoVs) are a group of non-enveloped, single stranded, positive-sense RNA viruses that constitute the Norovirus genus in the family Caliciviridae. NoVs are genetically diverse, containing six genogroups (GI to GVI) with over 35 genetic genotypes. NoVs exhibit wide host tropisms causing diseases in various mammalians including human. Human NoVs (huNoVs), consisting of mainly GI and GII NoVs, are the most important viral cause of epidemic acute gastroenteritis in humans [1], claiming over 200,000 lives each year [2]. NoVs are encapsulated by a protein capsid that is assembled by a single major structural protein, the capsid protein VP1. Each NoV capsid contains 180 copies of VP1, which are organized as a T = 3 icosahedron [3]. VP1 is divided into the N-terminal shell (S) and the C-terminal protruding (P) domain, forming the interior shell and the multiple protrusions of the capsid, respectively [3]. The P domain can be further divided into P1 and P2 subdomains, corresponding to the head as the outermost portion of NoVs, and the leg of the protrusion, respectively. Compared with S and the P1 domain, the P2 domain exhibits the most variable sequences, which are responsible for strain-specific virus-host interactions and immune responses of NoVs.
HuNoVs interact with histo-blood group antigens (HBGAs) in a strain-specific manner [4, 5]. HBGAs are fucose-containing glycans in specific sequences as determinants of human and animal blood types, including A/B/O, secretor (H), and Lewis (Le) or non-secretor (H negative) types. They are often present as parts of the carbohydrate moiety of cell surface glycoproteins and glycolipids with N - or O-linkage [6]. The biological roles of HBGAs in huNoV infection have been revealed by human volunteer challenge studies [7–9] and outbreak investigations [10, 11] of huNoVs, in which an association between the host susceptibility and the HBGA binding patterns of huNoVs has been established.
Despite the recent breakthroughs in culturing huNoVs in BJAB cells [12], the use of huNoV reverse genetics system [13], and the development of an immunocompromised mouse model [14] for huNoV propagation, an effective cell culture system or an animal model for huNoVs remains lacking. As a result, our understanding on huNoV-HBGA interactions relies mainly on data from in vitro studies using various recombinant subviral particles as models of huNoVs. Virus-like particles (VLPs), which are produced by expression full-length VP1 via a eukaryotic expression system, share similar structures and functions with the capsid of a native virion, and they have been used extensively as a huNoV surrogate. In addition, smaller P domain complexes that are self-assembled by expression of the huNoV P domain, including the P dimer [15], small P particle [16] and P particles [17, 18], are also used for the study of huNoV-HBGA interactions. Knowledge on the structures of the HBGA binding interfaces has been derived mainly from crystal structures of P domain dimers in complex with HBGA oligosaccharides [19–26], by taking advantage of its small size (~69 kDa) and easy production.
Structural analysis of known HBGA binding interfaces of huNoVs showed that GI and GII huNoVs recognize HBGAs through a conserved, genogroup-specific binding interface (reviewed in [27–29]), suggesting a strong selection of huNoV evolution by human HBGAs. On the other hand, the GI and GII HBGA-binding interfaces are distinct in the locations, structures, residue compositions, and HBGA binding modes [27–29], indicating a long separation of the two genetic lineages. In this study we report a new evolutionary lineage consisting of GII.13 and GII.21 genotypes within GII, which does not share the conventional GII HBGA binding interface (Fig 1), but remains binding ability to HBGAs [5, 30]. X-ray crystallography of the GII.21 OIF virus P domain complexed with a Lea antigen revealed a new HBGA binding interface that is distinct from the GII conventional binding interface. Sequence alignment further showed that the amino acid composition of the new binding interface of OIF is highly conserved among all members of both GII.21 and a closely related GII.13 genotype. These results indicate that the genetic branch consisting of GII.21 and GII.13 developed along a novel evolutionary path that split from the mainstream lineage of GII NoVs selected by HBGAs. While many questions on the cause and mechanisms behind the emergence of this new lineage remain unanswered, our data point towards a continual occurrence of new lineages, which may significantly impact future epidemiology and prevention strategies against huNoVs.

Results
Crystal structure of the native OIF P domain protein
The crystal of the native P domain protein belongs to the P21 space group, with two monomers forming a dimer in an asymmetric unit. The final refined structure of native P domain includes residues 223 to 527, with the exception of a loop region comprising residues 340~342 due to the lack of recognizable electron density. While the P domain of OIF virus shares only ~50% sequence homology with other GII P domains of known crystal structures, it shows an arrangement of overall and secondary structures similar to those huNoV P proteins, including GII.4 VA387 [20], GII.10 Vietnam 026 [23], GII.9 VA207 [21], and GII.12 Hiro [23] with Cα atoms r.m.s.d. = 0.85 Å, 0.83 Å, 0.82 Å, and 0.62 Å, respectively. Like other huNoVs, the OIF P domain has two moieties (Fig 2), with the inner portion or P1 subdomain (residues 223 to 272, and 416 to 527) constituting the leg, and the outer moiety or P2 subdomain (residues 273 to 415) forming the protruding head of the P dimer.

The two monomers of the OIF P dimer are related by a non-crystallographic two-fold axis, which forms the biologically active protrusion of the NoV capsid. The P dimer has a dimension of 55 Å×64 Å×70 Å with an extensively buried interface of 3,500 Å2 between two protomers, including hydrophobic and hydrophilic interacting residues from both P1 and P2 subdomains (Fig 2A). These extensive inter-molecular interactions contribute to the stability of the P dimer. Although the OIF P dimer shares similar global structures with the previously reported P dimers of other huNoVs, significant differences on the top surface are clearly seen, mainly due to the differences in the sequences, lengths, and conformations of several surface loops (Figs 3 and 4) (see below).


Top surface structure of the P dimer
The P-loop of the OIF P dimer exhibits a unique conformation, taking a ~90° flip (Fig 3), which results in its extruding out of the flat surface and approaching the opposite protomer. This leads to greater exposure of the P-loop on the top surface of the P dimer compared to that in other GII huNoVs (Fig 4). As a result, the majority of the S-loop is covered in the OIF virus. Such structural rearrangements destroy the structural integrity of the conventional GII HBGA binding interfaces that are formed mainly by P - and S-loops (Figs 2 and 4). On the other hand, the B - and the T-loops also exhibit unique conformations (Fig 3), approaching to each other in closer proximity with the N-loop (Fig 4A). This special structural feature allows the formation of new HBGA binding interface (see below), formed by the B-, T - and N-loops, a scenario that is absent in all other known GII huNoV structures (Fig 4). The protrusions of the P - and A-loops directed towards the opposite protomer restructure the boundary between two P monomers into a saw tooth-like structure, in contrast to the straight line-like pattern in other GII NoVs (Fig 4, compare G and H). These different surface structures and conformations of OIF P dimer might also contribute to the difference in antigenic and immunological properties of GII.21 compared to other huNoV genotypes.
The HBGA binding interface
As shown in previous studies [5, 30], the OIF virus that does not share the conventional GII HBGA binding interface only recognizes Lea antigen. To understand their binding mechanism, we co-crystallized the OIF P domain with Lea trisaccharide (Lea-tri). The P domain-Lea-tri complex was crystallized in P212121 space group, with one homodimer in each asymmetric unit. The Lea-tri is clearly visible in the 2Fo-Fc difference electron density map, with all three rings of the Lea-tri well fitted into the map (Fig 5A). Two symmetric Lea binding interfaces are identified on the top surface of the P dimer (Fig 5B), each of which is formed by eleven residues from the P2 domain of a single protomer. Specifically, residues W296 from the B-loop, S354 and S357 from the N-loop, N392 and T395 from the T-loop form the depressed region at the bottom of the binding pocket (Fig 5C–5E). We noticed that, although S357 does not interact directly with the Lea-tri, it forms a strong hydrogen bond (2.6Å) with the D297 to stabilize the side chain of the latter, ensuring the structural integrity of the binding pocket (Fig 5E and 5F). The surrounding wall of the binding pocket is built by D294 and Y295 from B-loop, T356 and E358 from N-loop, and N394 from T-loop (Fig 5C–5E). None of these residue compositions are conserved with those of the known GII HBGA binding interfaces that is contributed by residues from P-, S-, and A-loops (Figs 2 and 4). The OIF HBGA binding interface also differs completely from those of the GI huNoVs (Fig 4F). Thus, this OIF binding interface represents a previously unrecognized HBGA binding interface.

The β-Gal binding site
The HBGA binding pocket interacts with Lea antigen through the β-galactose (β-Gal), one of the two saccharides of the precursor disaccharide, as the major binding saccharide (MaBS). The β-Gal is firmly held inside the binding pocket by nine direct hydrogen bonds formed by the side chains of S354, T356, N392, N394 and T395, in which the side chains of S354 from the bottom and N394 from the wall of the pocket form three strong hydrogen bonds (2.7 Å or 2.6 Å) with β-Gal (Fig 5E and 5F). It is noteworthy that the side chain of W296 is oriented nearly in parallel with the β-Gal ring, which leads to a hydrophobic interaction/stacking with the latter to further support the interaction between β-Gal and the binding pocket. In addition, the side chains of D294, D297 and E358 form water-bridged hydrogen bonds with β-Gal via a common water molecule, contributing to the binding outcomes between the OIF P domain and the Lea antigen. Together, these amino acids form the β-Gal binding site that is the major component of the HBGA binding interface of OIF virus.
Le Fuc binding site
As a result of the strong interactions between the β-Gal and the binding pocket, the Lewis fucose (Le-Fuc, α-1,4-fucose) is orientated between the walls of N394 and E358. Only a single direct hydrogen bond (2.7 Å) forms between E358 and the Le-Fuc (Fig 5E and 5F) and therefore, the Le-Fuc is a minor binding saccharide (MiBS). As a result of the β-Gal/Le-Fuc-binding pocket interactions, the N-acetyl-β-glucosamine (β-GlcNAc), the other saccharide of the precursor disaccharide, is pointing away from the surface of the binding interface. No hydrogen bond was observed between β-GlcNAc and the binding interface (Fig 5C–5F).
Validation of the HBGA binding interface
We further investigated the roles of the eleven residues that form the HBGA binding interface of the OIF virus using single-point mutagenesis (Figs 5 and 6). Compared with the wild type P particle of OIF virus that binds strongly to Lea oligosaccharide, but not other oligosaccharides representing different HBGA types, including precursors (Fig 6A), mutant P particles with a single mutation at one of those amino acids to an alanine lost the binding to Lea antigen completely (Fig 6C–6L) or nearly completely (Fig 6B). These results confirmed the importance of these residues for the structural and functional integrity of the HBGA binding interface. In addition, except for a slight increase in binding to the A antigen of the E358A mutant, no changes in binding to other HBGA types were observed for these interface mutants.

Glycerol binds the HBGA binding pocket of the OIF P domain
Analysis of the native OIF P domain structure revealed that a glycerol molecule occupies the HBGA binding pocket, which was clearly visible from the 2Fo-Fc omit map (Fig 7A). Originally, glycerol was part of the protease solution acting as a stabilizer during the cleavage of the GST-P domain fusion protein. The glycerol molecule is held firmly inside the HBGA binding pocket by eight direct hydrogen bonds between the hydroxyl groups of the glycerol and the side chains of N392, N394 and T395 from the T-loop, T356 and the main chain of S357 from the N-loop (Fig 7B and 7C). Interestingly, the glycerol molecule resembles partial structures (C2, C3, and C4 with their hydroxyl groups) of the β-Gal that contributes the vast majority of the interactions with the binding pocket (Compare Fig 5F with Fig 7C), thus explaining the observed interactions.
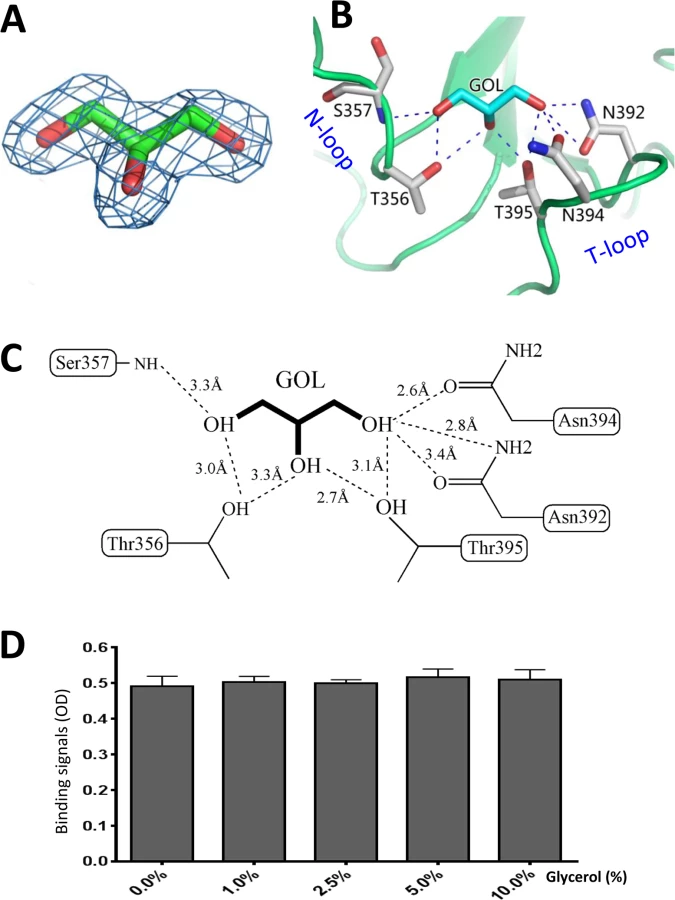
Glycerol blocks the interaction of OIF P domain with the Lea antigen
We found that inclusion of glycerol in any process of purification or crystallization would prevent the binding of the Lea antigen to the same binding pocket. The later success in identification of HBGA binding interface (Fig 5) of OIF virus using the P protein without glycerol proved our assumption that glycerol occupies the HBGA binding pocket and blocks further interaction of the binding pocket with the Lea antigen. Co-crystallization followed by structural determination using the P protein in the presence or absence of glycerol confirmed our observation. We then performed ELISA binding assays to test if the monomeric glycerol (1–10%) can block binding of polyvalent Lea-tri-PAA (polyacrylamide) conjugates (2 μg/mL) to OIF P particles (4 μg/mL). We did not observe any detectable blocking effects (Fig 7D), indicating that the free glycerol can block binding of free, monomeric Lea-tri, but cannot inhibit the binding of polyvalent Lea-tri molecules to the HBGA binding pocket.
Conservation of the HBGA binding pocket of the GII.13/GII.21 lineage
The unique HBGA binding interface of the GII.21 OIF virus prompted us to examine whether this is a common feature of this specific huNoV lineage. Representative P domain sequences of GII.13/21 huNoVs were aligned and compared with those of GII.17, which is genetically closest to the GII.13/21 lineage (Fig 1A), as well as with that of GII.4, which is the most prevalent genotype. We focused on the surface loops that form the OIF HBGA binding pocket (B-, T, and N-loops) and the conventional GII binding interface (P-, S - and A-loops) (Fig 8). We found that most members of the GII.13/21 lineage share the major residues that form the novel HBGA binding pocket, indicating a new evolutionary selection has occurred among members of this genetic lineage. In contrast, GII.17 retained the conventional GII HBGA binding interfaces, indicating that the occurrence of the OIF-like binding pocket in the GII.13/21 lineage was after the evolutionary divergence with the GII.17 genotype.

Discussion
GI and GII NoVs constitute the vast majority of huNoVs, and they were shown previously to interact with HBGAs through two distinct, genogroup-specific HBGA binding interfaces [31]. The two HBGA binding interfaces differ in their locations on the top of the protruding dimer, their residue composition as well as their interacting modes with HBGAs, but are highly conserved among genotypes within each of the two genogroups (reviewed in [27–29, 31]). It is therefore suggested that GI and GII NoVs must split from their common ancestor a long time ago during evolution into two independent evolutionary lineages, from which the two genogroup specific HBGA binding interfaces evolved individually. It is noteworthy that both binding interfaces recognize most, if not all, of the same repertoire of the polymorphic human HBGAs despite their distinct features, indicating that HBGA is an important selection factor in the evolution of huNoVs.
The presence of the two different HBGA binding interfaces in huNoVs raises the question as to whether such an event may occur again during evolution, especially within the same genogroup. We noted earlier on that a genetic branch that consists of GII.13 and GII.21 genotypes does not share the residue composition of the HBGA binding interface of the mainstream GII NoVs, but maintains HBGA binding function [5, 30], suggesting that this GII.13/21 lineage may interact with HBGAs through an unknown binding interface. By performing X-ray crystallography of the native P domain of the GII.21 OIF virus and its complex with Lea trisaccharide (Lea-tri), we found that GII.21 huNoVs recognize HBGAs via a previously unrecognized binding interface. The newly identified HBGA binding interface is spatially, structurally, and compositionally distinct from the well-studied conventional GII and GI HBGA binding interface.
The conventional GII HBGA binding interface is formed by three surface loops, i.e. the P - and A-loops from the P2 subdomain and the S-loop from the P1 subdomain. The binding interface is located directly on the boundary between the two P protomers, which makes each binding interface bivalent contributed by residues from both P protomers. In the GII.21 OIF virus, however, the conventional HBGA binding interface has been clearly abolished by the rearrangement of the three loops. Through a 90 degree flip, the P-loop extends to the opposite protomer, covering the vast majority of the S-loop and moving away from the A-loop. Nevertheless, this dramatic structural rearrangement of the OIF P dimer also helps to reconstruct a new HBGA binding interface, formed by other three surface loops, i.e. the B-, N - and T loops from the P2 subdomain of a single P protomer. We also noted that the binding interface corresponding to that of OIF virus does not exist in the mainstream GII NoVs, because the B - and T-loops are far away from each other in those GII NoVs.
In addition to the location, residue composition, and structure, the two GII HBGA binding interfaces are also different in their binding modes to HBGAs. The general binding modes of huNoVs have recently been summarized [28, 29]. GI NoVs bind HBGAs via a Gal (theα - or the β-Gal), while GII NoVs bind via a Fuc (the α-1, 2/3/4 Fuc) as the major binding saccharide (MaBS). The OIF virus interacts with the Lea antigen via the β-Gal as the MaBS, distinct from the convention GII, but similar to the GI binding interfaces. In addition, the OIF-Lea interaction relies mainly on the β-Gal that contributes 11 out of total 12 hydrogen bonds, including 9 direct ones. The α-Fuc (Le-Fuc) contributes only one hydrogen bond, while the β-GlcNAc does not participate directly in binding. This “upright” binding mode is also different from the “flat” mode observed in the conventional GII binding interfaces, which often includes two saccharides as the minor binding saccharides (MiBS), which usually contribute three or more interacting bonds. Again, the binding mode of OIF virus to HBGA appears to be more similar to that of GI NoV than to GII NoV [28, 29].
Our crystal structures of the HBGA binding interface of OIF virus complexed with Lea-tri reveal why OIF virus does not bind H, A, and B antigens. The H epitope (α-1,2 Fuc) and the A/B epitope (α-GalNAc/Gal) are linked to the 2’ and 3’ hydroxyl groups of β-Gal, respectively [29]. However, both the 2’ and 3’ hydroxyl groups of the β-Gal are located at the bottom of the binding pocket (Fig 5). Any addition of an extra saccharide at one of these positions would cause serious steric hindrance, and thus prevent the binding of H, A, and B antigens to the binding pocket of the OIF virus. This binding mode may also explain the absence of changes of the binding specificity through the eleven single residue mutations at the amino acids forming the OIF binding interfaces (Fig 6). This HBGA binding interface and its binding mode appear not to provide much flexibility to different HBGAs. Thus, this scenario of OIF virus differs clearly from observed flexibility of the conventional GII binding interface that is capable to bind more than one HBGA type and that can change binding specificity through a single residue mutation in or around the binding interface [32–36].
The formation of the new HBGA binding interface in the GII 13/21 lineage also raises questions as to how the conventional GII HBGA binding interface has been abandoned, and how a new interface was formed. The huge structural variation of the conventional GII binding interface in OIF virus may represent the accumulation of multiple gradual changes over time, following the acquisition of the novel binding interface and loss of binding function of the old one and HBGA selection. The movements of the B-, T - and N-loops to form the new HBGA binding interface can be monitored through the huNoVs with known crystal structures (Fig 4). The three loops are far away in GI.1 (Norwalk virus), GII.4 (VA387), GII.9 (VA207), GII.12 (Hiro), but are closer sterically in GII.10 (Vietnam026). Thus, movements of these loops are the structural basis for the formation of the functional HBGA binding interface found in OIF virus, although how such a functional binding interface was created in detail remains to be elucidated.
How the GII.13/21 lineage with the new HBGA binding interface split from the GII.17 genotype with the conventional one remains unclear. The capsid proteins (VP1s) of the GII.13/21 lineage share generally ~75% sequence identify with that of the GII.17 NoVs and these differences concentrate in their P domains that shows only ~62% sequence identity. This great sequence difference indicates that the two lineages have split from each other for a long time, which makes an attempt to reconstruct their common ancestor difficult. We speculate that the GII.17 P domain must have similar surface topology and loop organization to those of the mainstream GII NoVs due to the fact that the GII.17 NoV retains the conventional binding interface. In contrast, the GII.21 OIF shows very different surface topology and loop arrangements compared with the other GII NoVs, leading to the formation of the new HBGA binding interface. Unfortunately, the lack of the crystal structures of a GII.17 P domain prevents our further understanding on the evolutionary scenarios of the separation between the GII.13/GII.21 lineage and the GII.17 genotype. Thus, future study to solve the structures of a GII.17 P domain is of significance.
While the conservation of the conventional GI and GII HBGA binding interfaces indicates that there was selection pressure imposed by HBGAs functioning as attachment factors or receptors, the conservation of the HBGA binding interface of the GII.13/21 lineage (Fig 8) also indicates that new selection pressure of HBGA for the new binding interface has been established. Thus, it is plausible to expect that the conserved binding interface of the GII.13/21 lineage will continue to be maintained, just like the GI and GII binding interfaces. It would be important to show whether different strains of the GII.13/21 lineage can also recognize different HBGAs, just as GI and GII NoVs do. It was noted that Y295 in the binding pocket of GII.21 OIF is replaced by an asparagine (N) in GII.13 (Fig 8) that is highly conserved among most known GII.13 isolates [37]. It would be of significance to assess how this mutation could affect the HBGA binding profiles of GII.13 NoVs. In addition, N392 is another important residue for the structural and functional integrity of the GII.21 OIF binding interface, which was confirmed by the loss of the binding function through a N392A mutation (Fig 6J). This residue is also conserved among GII.13 NoVs (N395, Fig 8). However, we noted an exception, a GII.13 isolate called Kashiwa 47 (BAC05515) has an N395D mutation [37] and the recombinant VLPs of this isolate did not bind human saliva samples and tested synthetic HBGAs [38], consistent with our result of N392A mutation in GII.21 OIF.
Members of the GII.13/21 lineage revealed increased prevalence in the recent years. Surveillance indicates that the GII.13 and GII.21 huNoVs continue to cause outbreaks in the USA (CaliciNet http://www.cdc.gov/norovirus/reporting/calicinet/data.html), Europe (NoroNet, http://www.rivm.nl/dsresource?objectid=rivmp:248062&type=org&disposition=inline), and other countries [37, 39, 40], indicating that the GII.13/21 lineage is of clinical importance. It was also noted that, after being silent for the past decades, GII.17 NoVs became dominant in southern China in the past winter [41], overwhelmed GII.4 NoVs. Thus, continual surveillance of the epidemiological trends of the GII.13/17/21 lineage will be necessary in the future.
The fact that a new functional carbohydrate binding interface can be created through evolution raises the possibility of zoonotic transmission of NoVs. A new binding interface could recognize different host factors or receptors among human populations or animal species. The currently known host tropisms of different human and animal NoVs may be examples of such possibility. The genetic branch of porcine NoVs, consisting of the GII.11, GII.18 and GII.19 genotypes may be another example. Since these porcine NoVs share the conventional GII binding interface, even though they do not infect humans, it would be important to determine what host factor that porcine NoVs may recognize and understand how such molecule(s) interact with the conserved GII binding interface of porcine NoVs.
Another important finding of this study is the strong interaction of the HBGA binding pocket with a glycerol molecule, a fact mirrored by its inhibition of OIF P domain interaction with Lea antigen. Thus, glycerol, a small, low cost compound, may be a promising antiviral candidate against infection of the GII.13/21 NoVs. The glycerol molecule shares the structure of the β-Gal that is involved in the interaction to the binding pocket. A similar scenario was also observed in a previous study showing that a citrate molecule occupies the binding pocket of a GII.10 NoV (Vietnam026) [42] and importantly, does so with similar binding affinity observed between HBGAs and GII.10 P domains. However, our results showed that the free, monomeric glycerol cannot inhibit the interaction of the OIF P particle with multivalent Lea-tri-PAA conjugates, most likely due to avidity effects. Therefore, further investigations will be necessary to develop polymers that contain multivalent structures of glycerol molecules as antiviral candidates. Most importantly, our study should provide a solid structural basis and a model for future studies.
In summary, we have identified and proved the genetic branch of GII.13/21 as new evolutionary lineage with a novel binding interface for HBGAs. We have elucidated the structural basis for the abolishment of the conventional GII HBGA binding interface and the development of the new binding site. We also showed that the new HBGA binding interface is conserved among members of the GII.13/21 lineage, indicating that a new selection pressure has been asserted through the interaction with HBGAs. Finally, we have identified glycerol as a potential low-cost compound to become an antiviral candidate for future development. In conclusion, our data provide new insights into the complex interactions between the diverse huNoV and the polymorphic HBGAs.
Materials and Methods
Protein expression and purification
OIF (Operation Iraqi Freedom) virus is a GII.21 huNoV that was isolated from a huNoV outbreak in a US military deployed to Iraq in 2003. The cDNA sequences encoding the P domain of the OIF virus (accession number: AY675554; residues 220 to 527) was cloned into pGEX-6P-1 expression vector between the EcoR I and Xho I sites of the MCS. The OIF P domain was expressed as a GST fusion protein in Escherichia Coli BL 21 (DE 3) upon induction with 0.5 mM Isopropyl β-D-1-Thiogalactopyranoside (IPTG) at 16°C for 18 hours, at an OD600 nm of 0.6–0.8. The recombinant protein was purified using glutathione-sepharose 4B (GE Healthcare Life Sciences) following the manufacturer’s guidelines. The GST-P domain fusion protein was cleaved on the resin by Prescission Protease (GE Healthcare Life Sciences) at 4 °C overnight, and the P proteins were further purified by Resource Q anion ion exchange (GE), using a buffer containing 20 mM HEPES (pH 7.5), with P domain protein eluted at approx. 100 mM NaCl.
Crystallization of native P domain protein and its complex with Lea trisaccharide
The purified native P domain protein was dialyzed against buffer containing 20 mM HEPES (pH 7.5), 150mM NaCl and 5% (v/v) glycerol, before it was concentrated to 13 mg/mL for crystallization. Native P domain crystals were grown by hanging-drop vapor diffusion method, with the crystallization droplet containing 1 μL protein and 1 μL reservoir solution containing 0.1 M MES (pH 6.5), 0.25 M (NH4)2SO4, 18% (w/v) polyethylene glycol (PEG) 3350. The crystals were grown at 16°C and harvested after approx. one week.
While performing the crystallization experiments, we observed that the presence of glycerol inhibited the co-crystallization of the OIF P domain-Lea complex. The P protein used for co-crystallization with Lea-trisaccharide was purified with identical solutions (see above), but in the absence of glycerol. The Lea trisaccharide [β-Gal-(1,3)-(α-Fuc-(1,4)) - GlcNAc] (J&K, China) was dissolved in double distilled water and prepared as 20 mM solution, and then mixed with an equal volume of native P domain protein (26 mg/mL), and incubated at 4°C for 1 hour before crystallization. The final reservoir for the growth of complex crystals contained 0.25 M (NH4)2SO4, 18% (w/v) PEG 3350. Micro-seeding technique was used to aid the growth of complex crystal with native crystal seeds 16 hours after the setting of crystallization droplets. OIF P domain-Lea complex crystals were harvested in three days.
Data collection and processing, structure determination and refinement
The cryo-protectants of crystals of unliganded P domain and P domain-Lea complex were the corresponding reservoir solutions complemented with 15% (v/v) PEG 400. Crystals were briefly soaked in the cryo-protectant for 5 seconds before being mounted for diffraction test. The diffraction data for native crystal were collected at the beamline 41U of SPRING8 (Japan) at a wavelength of 1.0000 Å, while the Lea complex data were collected at rotating-anode X-ray source MicroMax-007/Satun 944 HG/Varimax HF (Institute of Biophysics, CAS, Beijing) at the wavelength of 1.5418 Å. Diffraction data were processed, scaled, and merged using the HKL-2000 program package [43]. Data collection statistics are summarized in Table 1.

The native crystal structures were solved by molecular replacement method using Phaser of CCP4 suite [44] and the GII.9 NoV VA207 P domain (PDB ID:3PUN) structure as the initial search model. Automatic structure building and refinement were carried out using Phenix program [45] and manual adjustment was done using the program COOT [46] with guidance of (2Fo-Fc) and (Fo-Fc) electron density maps. Water molecules were added at the final round of structure optimization at (Fo-Fc) electron density map peaks (>2.5 σ) where they can form stable hydrogen bonding with nearby amino acid residues. The phases and structures of the P domain-Lea complex were solved using the final refined structure of native P domain protein as model. Structure refinement statistics are summarized in Table 2. The final structure validation was done with the PROCHECK [47], with no residue found at disallowed region of the Ramachandran plot. Structural analysis was performed using EdPDB [48] and Pymol [49].

Production of OIF P particles containing single mutations
Single residue mutations were introduced to the HBGA binding site of the OIF P domain by site-directed mutagenesis using the expression plasmid of the wild type OIF P domain as template. Mutagenesis was carried out using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technology, CA) and corresponding primer pairs containing the mutation sites. After confirmation of mutations, the P proteins were expressed as P particles and purified using an E. coli system (BL21) as described elsewhere [31, 50, 51]. The GST-P domain fusion proteins were cleaved by thrombin (GE Healthcare Life Sciences) to allow the P proteins to self-assemble into P particles. P particle formation was monitored by gel filtration through a size-exclusion column (Superdex 200, GE Healthcare Life Sciences) powered by an AKTA-FPLC system (model 920, GE Healthcare Life Sciences, Piscataway, NJ) followed by SDS-PAGE electrophoresis. The P particles showed a molecular weight of 830 kDa. None of the mutations in this study affected the formation of P particles, as confirmed by FPLC (data not shown). All mutant P particles can be well detected by the hyperimmune serum against norovirus VLPs that was used in the HBGA binding and blocking assays (below).
HBGA binding and blocking assays
HBGA assays were carried out as described previously [5]. Briefly, a panel of synthetic oligosaccharides (GlycoTech, Gaithersburg, MD) representing types A, B, H1, H2, H3, Lea, Leb, Lex, Ley, sialyl Lea, and sialyl Lex as well as the disaccharides representing the type 1 and type 2 precursors of HBGAs at 2 μg/mL were coated on microtiter plates at 4°C overnight. After blocking with 5% (w/v) non-fat milk, the coated oligosaccharides were incubated with the affinity-column purified OIF P particles (10 ng/μL) for 60 minutes at 37°C. The bound OIF P particles were detected as described previously, using an in-house hyperimmune rabbit serum against various huNoVs [5, 30]. For glycerol blocking assays, the OIF P particles (4 μg/mL) were mixed with the monomeric glycerol (1–10%) and incubated with the coated polyvalent Lea-tri-PAA (polyacrylamide) conjugates (2 μg/mL). The bound OIF P particles were detected as described above.
Accession numbers
The cDNA sequences encoding the P domain of the OIF virus has been submitted to GenBank previously with an accession number of AY675554. Coordinates and structure factors of the native OIF P protein and the complex with Lea trisaccharide have been deposited in the Protein Data Bank (www.pdb.org) with the pdb accession codes of 4RLZ and 4RM0, respectively. The GenBank accession numbers of the sequences that were used in Figs 1 and 8 were shown in their figure legends.
Zdroje
1. Payne DC, Vinje J, Szilagyi PG, Edwards KM, Staat MA, Weinberg GA, et al. Norovirus and medically attended gastroenteritis in U.S. children. The New England journal of medicine. 2013;368(12):1121–30. doi: 10.1056/NEJMsa1206589 23514289.
2. Patel MM, Widdowson MA, Glass RI, Akazawa K, Vinje J, Parashar UD. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg Infect Dis. 2008;14(8):1224–31. Epub 2008/08/06. 18680645. doi: 10.3201/eid1408.071114
3. Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. X-ray crystallographic structure of the Norwalk virus capsid. Science. 1999;286(5438):287–90. 10514371.
4. Huang P, Farkas T, Marionneau S, Zhong W, Ruvoen-Clouet N, Morrow AL, et al. Noroviruses Bind to Human ABO, Lewis, and Secretor Histo-Blood Group Antigens: Identification of 4 Distinct Strain-Specific Patterns. J Infect Dis. 2003;188(1):19–31. 12825167.
5. Huang P, Farkas T, Zhong W, Tan M, Thornton S, Morrow AL, et al. Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J Virol. 2005;79(11):6714–22. Epub 2005/05/14. doi: 79/11/6714 [pii] doi: 10.1128/JVI.79.11.6714–6722.2005 15890909; PubMed Central PMCID: PMC1112114.
6. Ravn V, Dabelsteen E. Tissue distribution of histo‐blood group antigens. Apmis. 2000;108(1):1–28. 10698081
7. Frenck R, Bernstein DI, Xia M, Huang P, Zhong W, Parker S, et al. Predicting Susceptibility to Norovirus GII.4 by Use of a Challenge Model Involving Humans. J Infect Dis. 2012;206(9):1386–93. doi: 10.1093/infdis/jis514 22927452.
8. Hutson AM, Atmar RL, Graham DY, Estes MK. Norwalk virus infection and disease is associated with ABO histo-blood group type. J Infect Dis. 2002;185(9):1335–7. 12001052.
9. Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, Lindblad L, et al. Human susceptibility and resistance to Norwalk virus infection. Nat Med. 2003;9(5):548–53. 12692541.
10. Tan M, Jin M, Xie H, Duan Z, Jiang X, Fang Z. Outbreak studies of a GII-3 and a GII-4 norovirus revealed an association between HBGA phenotypes and viral infection. Journal of medical virology. 2008;80(7):1296–301. 18461617. doi: 10.1002/jmv.21200
11. Nordgren J, Nitiema LW, Ouermi D, Simpore J, Svensson L. Host genetic factors affect susceptibility to norovirus infections in Burkina Faso. PLoS One. 2013;8(7):e69557. doi: 10.1371/journal.pone.0069557 23894502; PubMed Central PMCID: PMC3716642.
12. Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346(6210):755–9. doi: 10.1126/science.1257147 25378626.
13. Katayama K, Murakami K, Sharp TM, Guix S, Oka T, Takai-Todaka R, et al. Plasmid-based human norovirus reverse genetics system produces reporter-tagged progeny virus containing infectious genomic RNA. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(38):E4043–52. doi: 10.1073/pnas.1415096111 25192933; PubMed Central PMCID: PMC4183323.
14. Taube S, Kolawole AO, Hohne M, Wilkinson JE, Handley SA, Perry JW, et al. A mouse model for human norovirus. mBio. 2013;4(4):e00450–13. doi: 10.1128/mBio.00450-13 23860770; PubMed Central PMCID: PMC3735125.
15. Tan M, Hegde RS, Jiang X. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol. 2004;78(12):6233–42. 15163716.
16. Tan M, Fang PA, Xia M, Chachiyo T, Jiang W, Jiang X. Terminal modifications of norovirus P domain resulted in a new type of subviral particles, the small P particles. Virology. 2011;410(2):345–52. Epub 2010/12/28. doi: S0042-6822(10)00735-X [pii] doi: 10.1016/j.virol.2010.11.017 21185050; PubMed Central PMCID: PMC3064930.
17. Tan M, Fang P, Chachiyo T, Xia M, Huang P, Fang Z, et al. Noroviral P particle: structure, function and applications in virus-host interaction. Virology. 2008;382(1):115–23. 18926552. doi: 10.1016/j.virol.2008.08.047
18. Tan M, Jiang X. The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. J Virol. 2005;79(22):14017–30. 16254337.
19. Bu W, Mamedova A, Tan M, Xia M, Jiang X, Hegde RS. Structural basis for the receptor binding specificity of Norwalk virus. J Virol. 2008;82(11):5340–7. Epub 2008/04/04. doi: JVI.00135-08 [pii] doi: 10.1128/JVI.00135-08 18385236.
20. Cao S, Lou Z, Tan M, Chen Y, Liu Y, Zhang Z, et al. Structural basis for the recognition of blood group trisaccharides by norovirus. J Virol. 2007;81(11):5949–57. Epub 2007/03/30. doi: JVI.00219-07 [pii] doi: 10.1128/JVI.00219-07 17392366; PubMed Central PMCID: PMC1900264.
21. Chen Y, Tan M, Xia M, Hao N, Zhang XC, Huang P, et al. Crystallography of a lewis-binding norovirus, elucidation of strain-specificity to the polymorphic human histo-blood group antigens. PLoS pathogens. 2011;7(7):e1002152. Epub 2011/08/04. doi: 10.1371/journal.ppat.1002152 PPATHOGENS-D-10-00601 [pii]. 21811409; PubMed Central PMCID: PMC3141052.
22. Choi JM, Hutson AM, Estes MK, Prasad BV. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(27):9175–80. Epub 2008/07/05. doi: 0803275105 [pii] doi: 10.1073/pnas.0803275105 18599458.
23. Hansman GS, Biertumpfel C, Georgiev I, McLellan JS, Chen L, Zhou T, et al. Crystal Structures of GII.10 and GII.12 Norovirus Protruding Domains in Complex with Histo-Blood Group Antigens Reveal Details for a Potential Site of Vulnerability. J Virol. 2011;85(13):6687–701. Epub 2011/04/29. doi: JVI.00246-11 [pii] doi: 10.1128/JVI.00246-11 21525337.
24. Kubota T, Kumagai A, Ito H, Furukawa S, Someya Y, Takeda N, et al. Structural basis for the recognition of Lewis antigens by genogroup I norovirus. J Virol. 2012;86(20):11138–50. doi: 10.1128/JVI.00278-12 22855491; PubMed Central PMCID: PMC3457155.
25. Shanker S, Czako R, Sankaran B, Atmar RL, Estes MK, Prasad BV. Structural analysis of determinants of histo-blood group antigen binding specificity in genogroup I noroviruses. J Virol. 2014;88(11):6168–80. doi: 10.1128/JVI.00201-14 24648450; PubMed Central PMCID: PMC4093872.
26. Hao H, Chen Y, Xia M, Tan M, Liu W, Guan X, et al. Crystal structures of GI.8 Boxer P dimers in complex with HBGAs, a novel evolutionary path selected by the Lewis epitope. Protein and Cell. 2014;in press.
27. Tan M, Jiang X. Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLoS pathogens. 2010;6(8):e1000983. Epub 2010/09/25. doi: 10.1371/journal.ppat.1000983 20865168; PubMed Central PMCID: PMC2928792.
28. Tan M, Jiang X. Norovirus-host interaction: Multi-selections by human histo-blood group antigens. Trends in microbiology. 2011;19(8):382–8. Epub 2011/06/28. doi: S0966-842X(11)00108-9 [pii] doi: 10.1016/j.tim.2011.05.007 21705222; PubMed Central PMCID: PMC3149758.
29. Tan M, Jiang X. Histo-blood group antigens: a common niche for norovirus and rotavirus. Expert Rev Mol Med. 2014;16:e5. doi: 10.1017/erm.2014.2 24606759.
30. Tan M, Zhong W, Song D, Thornton S, Jiang X. E. coli-expressed recombinant norovirus capsid proteins maintain authentic antigenicity and receptor binding capability. Journal of medical virology. 2004;74(4):641–9. 15484274.
31. Tan M, Xia M, Chen Y, Bu W, Hegde RS, Meller J, et al. Conservation of carbohydrate binding interfaces: evidence of human HBGA selection in norovirus evolution. PLoS One. 2009;4(4):e5058. Epub 2009/04/02. doi: 10.1371/journal.pone.0005058 19337380; PubMed Central PMCID: PMC2660415.
32. Chen YT, Tan M, Xia M, Hao N, Zhang XJC, Huang PW, et al. Crystallography of a Lewis-Binding Norovirus, Elucidation of Strain-Specificity to the Polymorphic Human Histo-Blood Group Antigens. Plos Pathogens. 2011;7(7). doi: e100215210.1371/journal.ppat.1002152. WOS:000293339300040.
33. Tan M, Xia M, Cao S, Huang P, Farkas T, Meller J, et al. Elucidation of strain-specific interaction of a GII-4 norovirus with HBGA receptors by site-directed mutagenesis study. Virology. 2008;379(2):324–34. doi: 10.1016/j.virol.2008.06.041 18692213
34. de Rougemont A, Ruvoen-Clouet N, Simon B, Estienney M, Elie-Caille C, Aho S, et al. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J Virol. 2011;85(9):4057–70. Epub 2011/02/25. doi: JVI.02077-10 [pii] doi: 10.1128/JVI.02077-10 21345963; PubMed Central PMCID: PMC3126233.
35. Lindesmith LC, Donaldson EF, Lobue AD, Cannon JL, Zheng DP, Vinje J, et al. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008;5(2):e31. 18271619. doi: 10.1371/journal.pmed.0050031
36. Jin M, Tan M, Xia M, Wei C, Huang P, Wang L, et al. Strain-specific interaction of a GII.10 Norovirus with HBGAs. Virology. 2015;476 : 386–94. doi: 10.1016/j.virol.2014.12.039 25591173.
37. Hoa-Tran TN, Nakagomi T, Sano D, Sherchand JB, Pandey BD, Cunliffe NA, et al. Molecular epidemiology of noroviruses detected in Nepalese children with acute diarrhea between 2005 and 2011: increase and predominance of minor genotype GII.13. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2015;30 : 27–36. doi: 10.1016/j.meegid.2014.12.003 25497351.
38. Shirato H, Ogawa S, Ito H, Sato T, Kameyama A, Narimatsu H, et al. Noroviruses Distinguish between Type 1 and Type 2 Histo-Blood Group Antigens for Binding. J Virol. 2008;82(21):10756–67. doi: 10.1128/JVI.00802-08 18701592
39. Yahiro T, Wangchuk S, Wada T, Dorji C, Matsumoto T, Mynak ML, et al. Norovirus GII.21 in Children with Diarrhea, Bhutan. Emerg Infect Dis. 2015;21(4):732–4. doi: 10.3201/eid2104.141856 25811105; PubMed Central PMCID: PMC4378497.
40. Bruggink LD, Dunbar NL, Marshall JA. Norovirus genotype diversity associated with gastroenteritis outbreaks in aged-care facilities. Epidemiology and infection. 2015 : 1–5. doi: 10.1017/S095026881500031X 25721658.
41. Jing L, Limei S, Lin F, Feng Y, Yanling M, Jiaqian L, et al. Gastroenteritis Outbreaks Caused by Norovirus GII.17, Guangdong Province, China, 2014–2015. Emerging Infectious Disease journal. 2015;21(7). doi: 10.3201/eid2107.150226
42. Hansman GS, Shahzad-Ul-Hussan S, McLellan JS, Chuang GY, Georgiev I, Shimoike T, et al. Structural basis for norovirus inhibition and fucose mimicry by citrate. J Virol. 2012;86(1):284–92. doi: 10.1128/JVI.05909-11 22031945; PubMed Central PMCID: PMC3255897.
43. Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276 : 307–26. doi: 10.1016/S0076-6879(97)76066-X WOS:A1997BH42P00020.
44. Bailey S. The Ccp4 Suite—Programs for Protein Crystallography. Acta Crystallogr D. 1994;50 : 760–3. WOS:A1994PK56800011.
45. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58 : 1948–54. doi: 10.1107/S0907444902016657 WOS:000178745000009.
46. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60 : 2126–32. doi: 10.1107/S0907444904019158 WOS:000225360500002.
47. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26(2):283–91.
48. Zhang XJ, Matthews BW. EDPDB: a multifunctional tool for protein structure analysis. Journal of Applied Crystallography. 1995;28(5):624–30.
49. DeLano WL. The PyMOL molecular graphics system. 2002.
50. Tan M, Fang P, Chachiyo T, Xia M, Huang P, Fang Z, et al. Noroviral P particle: Structure, function and applications in virus-host interaction. Virology. 2008;382 : 115–23. Epub 2008/10/18. doi: S0042-6822(08)00579-5 [pii] doi: 10.1016/j.virol.2008.08.047 18926552.
51. Tan M, Jiang X. The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. Journal of Virology. 2005;79(22):14017–30. 16254337.
52. Kroneman A, Vega E, Vennema H, Vinje J, White PA, Hansman G, et al. Proposal for a unified norovirus nomenclature and genotyping. Archives of virology. 2013;158(10):2059–68. doi: 10.1007/s00705-013-1708-5 23615870.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2015 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein