
The RNA Template Channel of the RNA-Dependent RNA Polymerase as a Target for Development of Antiviral Therapy of Multiple Genera within a Virus Family
Virus replication relies on multiplication of viral genomes by viral polymerases. For enteroviruses, a large group of clinically important human pathogens for which no antiviral therapy is available, this function is performed by 3Dpol, the RNA-dependent RNA polymerase. 3Dpol is therefore an attractive target for novel antiviral strategies. Most polymerase inhibitors identified today are nucleoside analogs, a class of compounds that exert broad-spectrum activity but often suffer from off-target effects. Non-nucleoside inhibitors on the other hand, in general have a more narrow spectrum of activity and are more prone to resistance development because in most cases they bind the surface of the enzyme which is less conserved and structurally more flexible. In this study, we present the identification of GPC-N114 as a non-nucleoside inhibitor of 3Dpol with broad-spectrum antiviral activity against both enteroviruses and cardioviruses, which also belong to the picornavirus family. Remarkably, it acts by targeting the RNA template-primer binding site in the core of 3Dpol, making GPC-N114 the first anti-picornaviral compound with this mechanism of action. Thus, the characterization of GPC-N114 has led to the identification of a novel drug-binding pocket in 3Dpol that can serve as a starting point for antiviral drug design.
Published in the journal:
The RNA Template Channel of the RNA-Dependent RNA Polymerase as a Target for Development of Antiviral Therapy of Multiple Genera within a Virus Family. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004733
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004733
Summary
Virus replication relies on multiplication of viral genomes by viral polymerases. For enteroviruses, a large group of clinically important human pathogens for which no antiviral therapy is available, this function is performed by 3Dpol, the RNA-dependent RNA polymerase. 3Dpol is therefore an attractive target for novel antiviral strategies. Most polymerase inhibitors identified today are nucleoside analogs, a class of compounds that exert broad-spectrum activity but often suffer from off-target effects. Non-nucleoside inhibitors on the other hand, in general have a more narrow spectrum of activity and are more prone to resistance development because in most cases they bind the surface of the enzyme which is less conserved and structurally more flexible. In this study, we present the identification of GPC-N114 as a non-nucleoside inhibitor of 3Dpol with broad-spectrum antiviral activity against both enteroviruses and cardioviruses, which also belong to the picornavirus family. Remarkably, it acts by targeting the RNA template-primer binding site in the core of 3Dpol, making GPC-N114 the first anti-picornaviral compound with this mechanism of action. Thus, the characterization of GPC-N114 has led to the identification of a novel drug-binding pocket in 3Dpol that can serve as a starting point for antiviral drug design.
Introduction
The family Picornaviridae contains 12 genera, and includes many human and animal pathogens (reviewed in [1]). Among these is the genus Enterovirus which contains four human enterovirus species (HEV-A, -B, -C, -D), three human rhinovirus species (HRV-A, -B, -C), simian enterovirus, bovine enterovirus, and porcine enterovirus. The HEV species include poliovirus (PV), coxsackievirus (CV), echovirus, and several numbered enteroviruses (EV). PV is the cause of poliomyelitis, which can lead to acute flaccid paralysis. Enterovirus 71, a major cause of hand-foot-and-mouth disease, is also frequently associated with flaccid paralysis and is a growing concern due to major epidemics in Southeast Asia. Coxsackieviruses are the main cause of viral meningitis, conjunctivitis, herpangina, myocarditis, and pancreatitis. HRV infections manifest themselves in most cases as the relatively mild common cold, but can cause serious exacerbations in patients with asthma or chronic obstructive pulmonary disease (COPD). Other well-known picornavirus genera are Hepatovirus, which contains hepatitis A virus, Aphthovirus, which contains foot-and-mouth disease virus (FMDV), and Cardiovirus, which includes encephalomyocarditis virus (EMCV), Theiler's murine encephalomyelitis virus, and the recently discovered Saffold virus (SAFV), which, unlike the other cardioviruses, is a human-tropic virus.
Currently, the toolbox to control picornavirus infections consists solely of vaccines against PV, hepatitis A virus, and FMDV. Prevention of diseases caused by the non-polio enteroviruses through vaccination seems unachievable given the great number of serotypes, with about 30 coxsackieviruses, 30 echoviruses, 50 numbered enteroviruses, and more than 150 rhinoviruses [2]. With this in mind, current efforts are aimed at developing antiviral compounds with broad-spectrum activity, targeting a wide range of viruses within a genus or ideally even multiple genera. No specific antiviral drugs have yet been clinically approved for the treatment of enteroviruses or any other picornavirus.
Picornaviruses are non-enveloped RNA viruses with a single-stranded 7–8 kb RNA genome of positive polarity. Upon entry, the viral RNA is translated into a polyprotein which is proteolytically processed by viral proteases to release the structural proteins (VP1-4) and the non-structural proteins (2A-2B-2C-3A-3B-3C-3Dpol and in some genera L) as well as some stable precursors. The genome is replicated via a negative-sense RNA intermediate by 3Dpol, the viral RNA-dependent RNA polymerase (RdRP). For this, 3Dpol uses the viral protein 3B, in this context usually termed VPg (viral protein genome-linked), as a primer.
The structure of 3Dpol has been studied extensively in the past decades. Crystal structures have been reported of 3Dpol of the enteroviruses CVB3, PV, EV71, HRV1B, HRV14, and HRV16, and of the aphthovirus FMDV [3–10]. These enzymes adopt a classical right hand architecture, with fingers, palm and thumb subdomains providing the correct geometrical arrangement of substrate molecules and metal ions at the polymerase active site for catalysis [11]. In addition, in all picornavirus RdRPs, the conformation of the right hand is closed by the connection of fingers and thumb sub-domains through the N-terminal portion of the protein and several loops protruding from fingers, named the fingertips. This connection leads to the formation of a completely encircled active site and largely restricts the inter-domain mobility. Three well-defined channels have been identified in the RdRP structures that allow access to the active site of the incoming rNTPs and the RNA template, as well as the exit path of the newly synthesized dsRNA product [11]. Seven conserved structural elements, termed motifs A–G, have been defined. Motifs A–E, located in the palm subdomain, are involved in nucleotide and nucleic acid binding as well as catalysis. Motifs F and G, located in the fingers, play a critical role in the binding of nucleoside triphosphates and RNA templates [12].
Given its critical role in genome replication, 3Dpol is regarded as an attractive target for antiviral drugs. Polymerase inhibitors can be subdivided into two classes. Most inhibitors identified so far fall under the class of nucleoside/nucleotide analogs, which compete with endogenous nucleotides and may act as chain terminators and/or mutagens. One example in this class is ribavirin which induces lethal mutagenesis [13]. The second class of polymerase inhibitors are the non-nucleoside inhibitors (NNI) which can have a variety of mechanisms of action, e.g., stabilizing the enzyme such that necessary conformational changes cannot take place [14]. Frequently, these NNIs bind enzyme surfaces which are more variable between viruses and consequently NNIs are more likely to have a restricted spectrum of activity. Also, the barrier to resistance is lower due to the greater tolerance to amino acid substitutions. A few NNIs active against picornaviruses have been discovered, but for most of them, the mechanism of action remains poorly characterized [15–22]. In this study, we report a novel NNI, GPC-N114 that, unlike most NNIs, binds the core of 3Dpol and possesses broad-spectrum activity against enteroviruses and cardioviruses. GPC-N114 represents the first picornavirus 3Dpol inhibitor that targets the template-binding site and reveals a novel pocket in 3Dpol that is amenable for broad-spectrum inhibition.
Results
GPC-N114 is an inhibitor of enterovirus and cardiovirus replication
We recently described a series of 5-nitro-2-phenoxybenzonitriles that inhibit in vitro enterovirus replication [23]. Further optimization of this class of molecules led to the identification of 2,2'-[(4-chloro-1,2-phenylene)bis(oxy)]bis(5-nitro-benzonitrile), hereafter referred to as GPC-N114 (Fig. 1A), with potent and selective in vitro antiviral activity against CVB3. This small molecule inhibits CVB3 replication in multicycle CPE-reduction antiviral assay with a 50% effective concentration (EC50) of 0.15 ± 0.02 μM (Table 1).
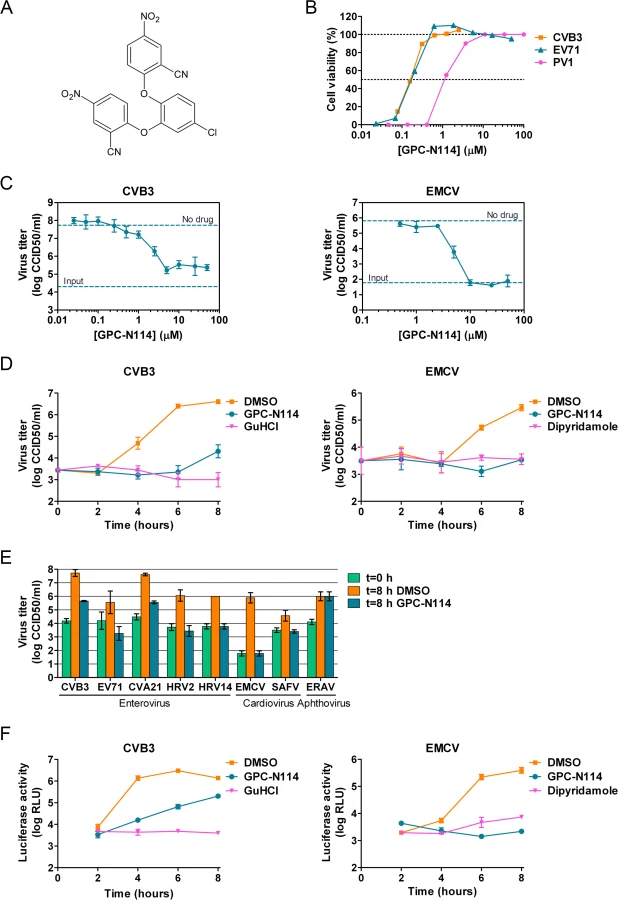
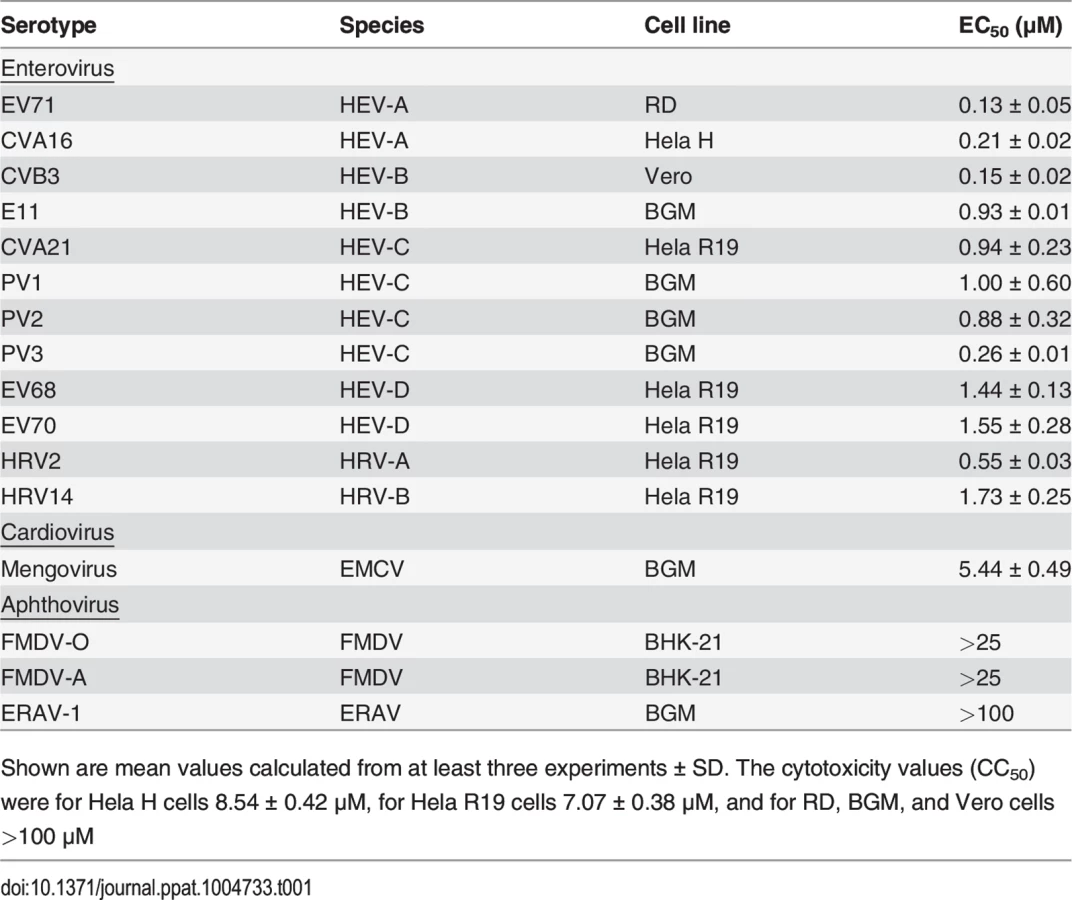
To study its spectrum of activity, GPC-N114 was evaluated in similar multicycle antiviral assays with representatives of the different HEV and HRV species, as well as several other picornaviruses. All enteroviruses and rhinoviruses included in this study were found to be sensitive to the inhibitory effect of GPC-N114, with EC50 values ranging from 0.1 μM to 1.7 μM (Fig. 1B and Table 1). GPC-N114 also inhibited the replication of EMCV (strain mengovirus), a member of the genus Cardiovirus (EC50 = 5.4 ± 0.49 μM). By contrast, FMDV and equine rhinitis A virus (ERAV), two members of the genus Aphthovirus, proved insensitive to its inhibitory effect (tested with concentrations up to 25 μM and 100 μM, respectively).
To evaluate the antiviral effect of GPC-N114 in a single round of replication, CVB3 and EMCV, as representatives of the enterovirus and cardiovirus genus, were incubated in the presence of different concentrations of the compound and virus titers were determined at 8 h post infection (p.i.). Cell viability assays performed in parallel showed that GPC-N114 was not toxic at these concentrations (S1A Fig.). For CVB3, the maximal inhibitory effect was obtained with concentrations of 3 μM and higher, although replication was not fully abrogated (Fig. 1C). Replication of EMCV was fully inhibited at concentrations of 10 μM or higher (Fig. 1C). Therefore, a concentration of 10 μM was selected for all further cellular assays. Detailed analysis of the kinetics of CVB3 and EMCV replication at this concentration showed that GPC-N114 strongly delayed virus replication (Fig. 1D and S1B Fig.). Comparable results were observed for a panel of other enteroviruses (EV71, CVA21, HRV2, and HRV14) and cardioviruses (EMCV and SAFV) (Fig. 1E and S1B Fig.). Again, no antiviral activity was observed against the aphthovirus ERAV (Fig. 1E).
Together, these results demonstrate that GPC-N114 exerts broad-spectrum in vitro antiviral activity, inhibiting the replication of a representative panel of viruses belonging to the enterovirus and cardiovirus genera from the picornavirus family.
GPC-N114 targets the RNA replication stage of the virus replication cycle
To identify the stage of the replication cycle that is targeted by GPC-N114, we used CVB3 and EMCV subgenomic replicons in which (a part of) the P1 coding region has been replaced with the firefly luciferase gene. Luciferase expression thus allows the quantification of translation and replication of the viral RNA. At 2 h post-transfection, a time point when no viral RNA replication has taken place yet [24], luciferase production from both replicons due to translation of incoming RNA was comparable in the presence and absence of GPC-N114 (Fig. 1F), suggesting that GPC-N114 does not affect translation. However, at later time points, luciferase levels were lower in GPC-N114-treated cells in comparison with mock-treated control cells for both the CVB3 and the EMCV replicon. In agreement with the single cycle assays, the EMCV replicon proved more sensitive to the inhibitory effect of GPC-N114 than the CVB3 replicon. These results indicate that GPC-N114 targets the viral RNA replication stage.
To determine whether the impaired replication in the presence of GPC-N114 is caused by a defect in the processing of the viral polyprotein, the viral proteins were visualized by metabolic labeling. CVB3-infected BGM cells were pulse-labeled with [35S]Met in the presence or absence of the compound between 5.5 and 6 h p.i., a time when translation of cellular, capped mRNAs is shut off by the virus [25]. The labeled proteins were analyzed by SDS-PAGE and autoradiography. The quantity and pattern of the viral proteins was similar for treated and untreated samples (S2 Fig.), demonstrating that GPC-N114 does not affect synthesis or processing of the viral polyprotein.
EMCV variants resistant to GPC-N114 carry mutations in 3Dpol
By serial passaging of virus in the presence of increasing concentrations of GPC-N114, we aimed to select for GPC-N114-resistant virus. For this, we used a protocol that previously allowed us to select virus variants resistant against a variety of compounds on different cell lines [26–28]. However, despite 32 passages on Vero cells and several attempts, no compound-resistant CVB3 isolates were obtained, indicating a high resistance barrier for this virus to GPC-N114. Also no resistant viruses were isolated on BGM cells. The possibility that the inability to obtain resistant CVB3 was due to the incomplete inhibition of replication by GPC-N114 (as indicated by single-cycle assays, though CPE was completely inhibited in multicycle assays), resulting in a too low selection pressure, seems unlikely because we also failed to isolate GPC-N114-resistant variants of PV1 (of which replication was completely blocked by GPC-N114 in a single-cycle assay, S3 Fig.) and E9. In contrast, three independent virus pools of EMCV showed phenotypic resistance after as few as three or four serial passages in the presence of suboptimal concentrations of the compound. Viral RNA was isolated from these virus pools and the regions coding for the non-structural proteins were sequenced. Interestingly, the three virus pools each contained a single missense mutation compared to wild-type (wt) virus. These mutations were located either at nucleotide 7115 (A7115G) or 7127 (A7127G) resulting in amino acid changes M300V and I304V in the viral RdRP, 3Dpol, respectively (Fig. 2A).
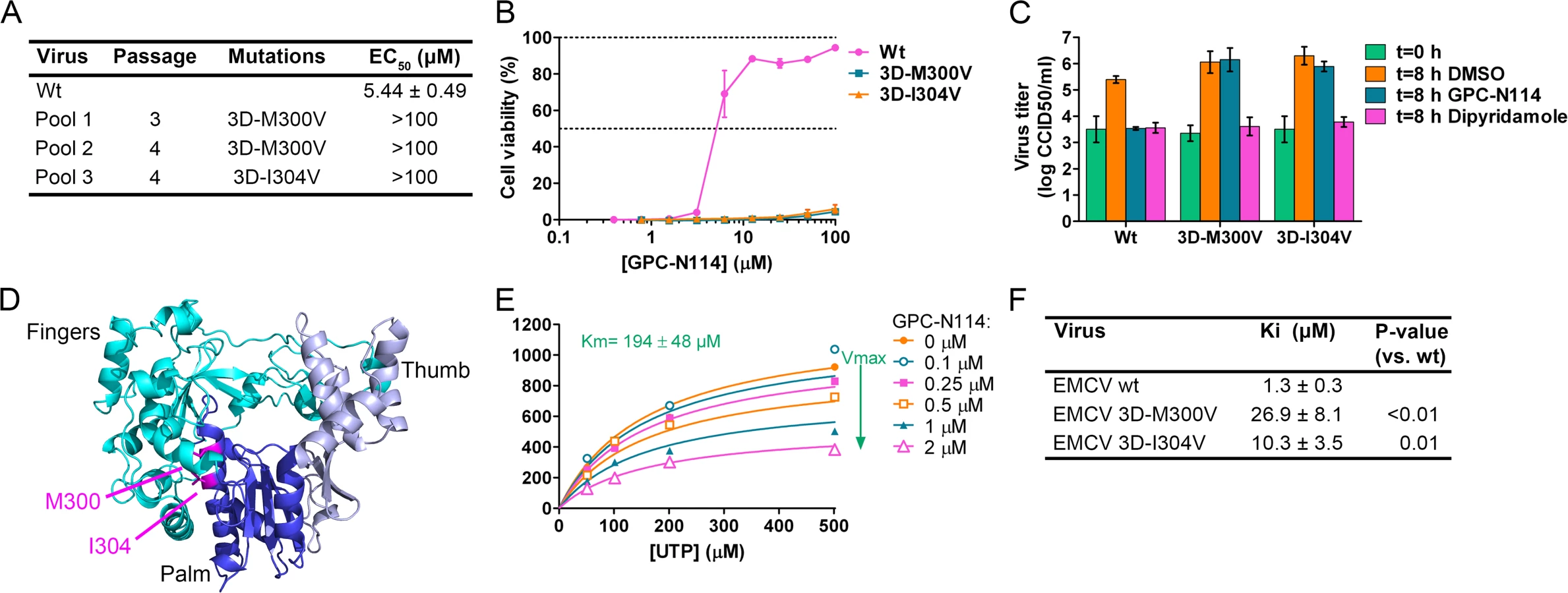
To verify the causal relationship between these mutations and the resistance phenotype, the mutations were introduced into the EMCV cDNA clone pM16.1 and mutant viruses were generated. In the multicycle CPE-reduction assay, the EC50 dramatically increased from 5.44 ± 0.49 μM for EMCV wt to >100 μM for EMCV 3D-M300V and 3D-I304V, indicating that the mutants were resistant to the compound (Fig. 2A and B). Accordingly, while the production of infectious virus particles by wt virus was strongly inhibited in a single cycle assay, virus titers of the mutant viruses after 8 h were unaffected by the presence of 10 μM GPC-N114 (Fig. 2C). As expected, the mutations did not confer resistance to the cardiovirus inhibitor dipyridamole. These results demonstrate that the mutations in 3Dpol were indeed responsible for the resistance phenotype of the isolated virus pools.
The X-ray structure of EMCV 3Dpol has been recently reported [29]. It shows that the amino acids M300 and I304 are both located in structural motif B both on the same face of helix α10 (which is part of the palm subdomain of the polymerase), close to the active site (Fig. 2D). Both residues are buried in the core of the protein and their side chains are involved in hydrophobic interactions that stabilize the local structure.
Having identified GPC-N114 resistance mutations in EMCV 3Dpol, we set out to test whether the corresponding mutations in CVB3 (i.e., 3D-I296V and 3D-M300V) would yield GPC-N114-resistant variants. Mutations were introduced in a CVB3 cDNA by site-directed mutagenesis and viruses were obtained by transfection of run-off transcripts in cells. The mutant viruses displayed similar increases in virus titer after 8 hours as CVB3 wt in the absence of compound, but did not show a compound-resistant phenotype (S4A Fig.). Also mutation S299T in CVB3 3Dpol, which was previously shown to provide resistance against the 3Dpol inhibitor amiloride [19,22], failed to confer resistance to GPC-N114 (S4B Fig.).
GPC-N114 inhibits in vitro polymerase activity
The location of the EMCV resistance mutations suggested that GPC-N114 targets the activity of 3Dpol. To verify this, biochemical assays were performed using purified EMCV 3Dpol in the presence or absence of GPC-N114. Briefly, elongation of a dT15 primer on a poly(rA) template by recombinant enzyme was determined by measurement of the rate of incorporation of [3H]UTP. Elongation activity of EMCV 3Dpol on the poly(rA) template was inhibited by GPC-N114 in a dose-dependent manner (Fig. 2E), which is in line with the resistance to the compound mapping to 3Dpol in EMCV. Using the same template-primer and metal ion (i.e., Mg2+), GPC-N114 did not affect the elongation activity of the polymerase domain of the dengue virus (DENV) NS5 RdRP (S5 Fig., IC50 > 100 μM), demonstrating that the inhibition of EMCV 3Dpol activity was not due to an unspecific interference with these components of the assay.
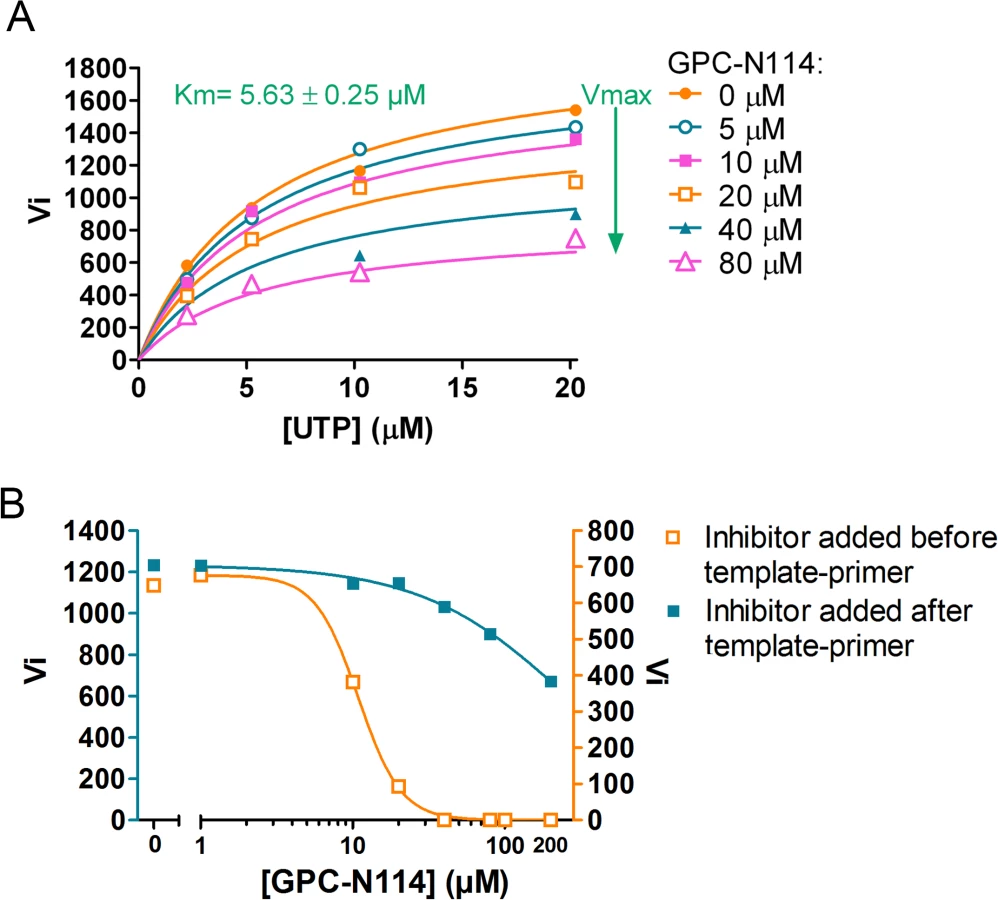
An inhibition constant (Ki) of 1.3 μM was calculated from the EMCV 3Dpol elongation activity measured at multiple UTP and inhibitor concentrations (Fig. 2F). Fig. 2E shows representative Michaelis-Menten plots of the results acquired with EMCV 3Dpol. The data were fitted by non-linear regression assuming a competitive, noncompetitive, uncompetitive or mixed mode of inhibition. The model of noncompetitive inhibition provided the best fit, with the maximum velocity (Vmax) of the reaction declining with increasing concentrations of the compound. This indicates a noncompetitive mode of inhibition with respect to UTP (and presumably NTPs in general). Markedly higher inhibition constants were observed for the two resistant mutants (M300V, 26.9 μM; I304V, 10.3 μM) as compared to that for the wt enzyme (1.3 μM) (Fig. 2F). Thus, GPC-N114 inhibited EMCV 3Dpol elongation activity, which could be counteracted by mutations M300V and I304V.
GPC-N114 binds 3Dpol at the site of the template acceptor nucleotide
Co-crystallization of 3Dpol in complex with GPC-N114 was attempted to study the binding of the compound to picornavirus polymerases. Co-crystals of EMCV 3Dpol with the inhibitor were not obtained, but the structure of the complex of CVB3 3Dpol with GPC-N114 was obtained by X-ray crystallography at 2.9 Å resolution (Table 2). The analysis of the difference electron density maps revealed the presence of extra densities to position the inhibitor within a pocket located at the bottom of the template channel, mimicking the position of the template acceptor nucleotide, the nucleotide which will base-pair with the incoming nucleotide (Fig. 3A-C and S6 Fig.). The GPC-N114-binding pocket is in close proximity to the location of the mutated residues in resistant EMCV 3Dpol (compare Fig. 2D and Fig. 3A). The compound bound in a conformation that perfectly fitted the shape of the pocket formed by residues L107, E108, L110, D111, T114 of motif G, R188, Y195, H199 of motif F, T294, S295, I296 of motif B and Y327 of motif A (Fig. 3A and 3B). Most of these residues are conserved in picornaviral polymerases (S7 Fig.). Among all contacts observed, the stacking interaction between Y195 and the central chlorophenyl ring of GPC-N114 appeared to be an important determinant of binding. This tyrosine is strictly conserved in enterovirus polymerases but not conserved in FMDV and EMCV. The first 2-cyano-4-nitrophenyl ring was oriented towards the entrance of the template channel, interacting with residues of motif G and B of CVB3 3Dpol, whereas the second 2-cyano-4-nitrophenyl ring appeared partially exposed to the solvent, occupying the position expected for the template acceptor base (Fig. 3C). The interacting residues appear to be in direct contact with the RNA templates in all picornaviral 3Dpol–RNA complexes determined so far [4,12,30,31]. Structural comparisons between unbound and GPC-N114-bound CVB3 3Dpol revealed that the polymerase did not undergo any major conformational change upon binding of the compound. This preservation of the template cavity conformation extended to the interacting side chains, which maintained their native conformation upon GPC-N114 binding.
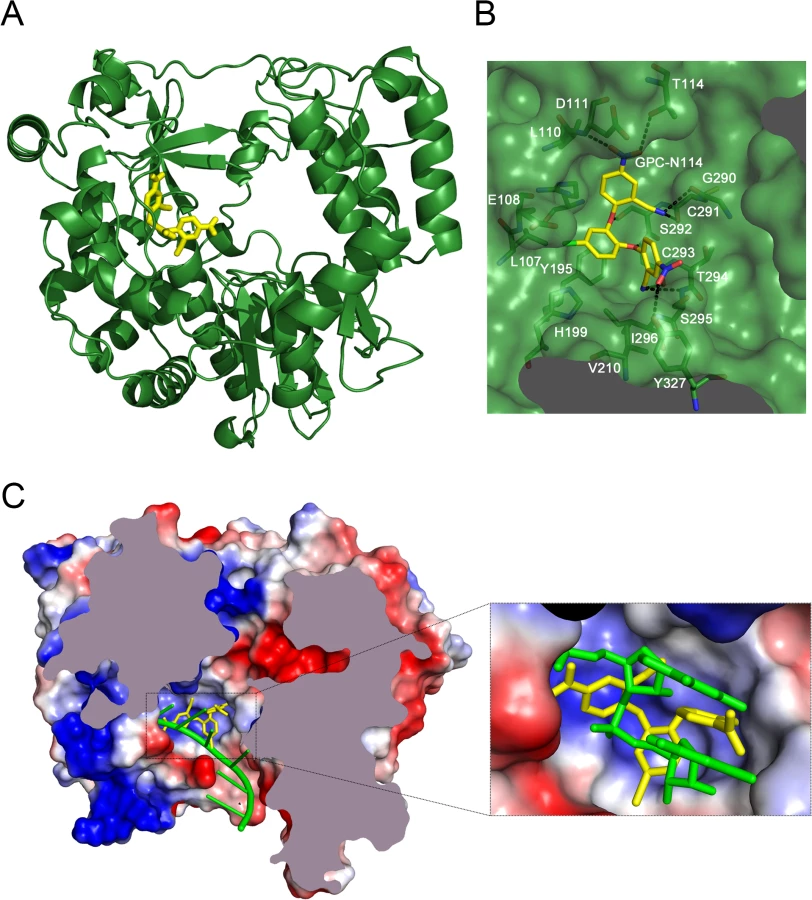
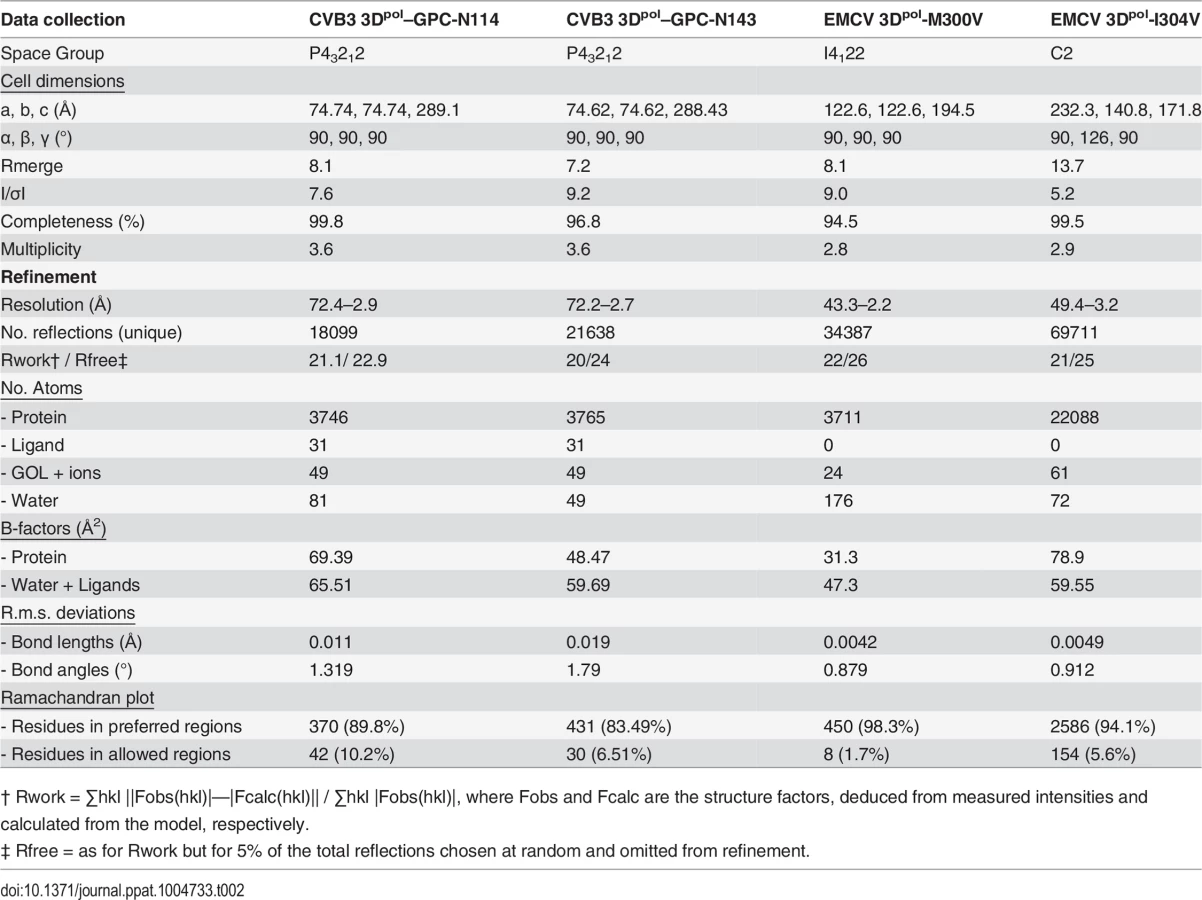
We also solved the X-ray structure of the complex of CVB3 3Dpol with a second inhibitor of the GPC series, GPC-N143, at 2.7 Å resolution (S8A Fig. and Table 2). This compound contains a central fluorophenyl ring instead of a chlorophenyl ring and displays similar antiviral activity as GPC-N114 (S8A and S8B Fig.). As expected, the GPC-N143 binding site is identical to that of GPC-N114, establishing almost equivalent interactions (Fig. 3B, S6 Fig. and S8D Fig.). Thus, the crystallographic data demonstrate that GPC-N114 and GPC-N143 specifically interact with CVB3 3Dpol at the template channel without having major effects on the structure of the enzyme.
Model of EMCV 3Dpol–GPC-N114 interactions and the structure of M300V and I304V mutants
All attempts to obtain the structure of EMCV 3Dpol–GPC-N114 complex were unsuccessful, however, the superposition of the CVB3 3Dpol–GPC-N114 complex onto the unbound EMCV 3Dpol structures allowed us to define the putative inhibitor binding site in the EMCV enzyme (Fig. 4A). The overall shape of this site is similar in the two enzymes, but some important contacts are lost in EMCV (e.g., the stacking interaction between the side chain Y195 and the central ring of GPC-N114 is absent due to the substitution of this residue by alanine in EMCV). The model also shows that the major polymerase–inhibitor interactions are mediated by residues of the motif B loop (β10-α10) (Fig. 4A and S7 Fig.). The resistance mutations at EMCV 3Dpol residues 300 and 304 of helix α10 are close to the putative GPC-N114 binding site. However, only the long lateral chain of the EMCV wt residue M300 is able to establish a direct contact with the compound.
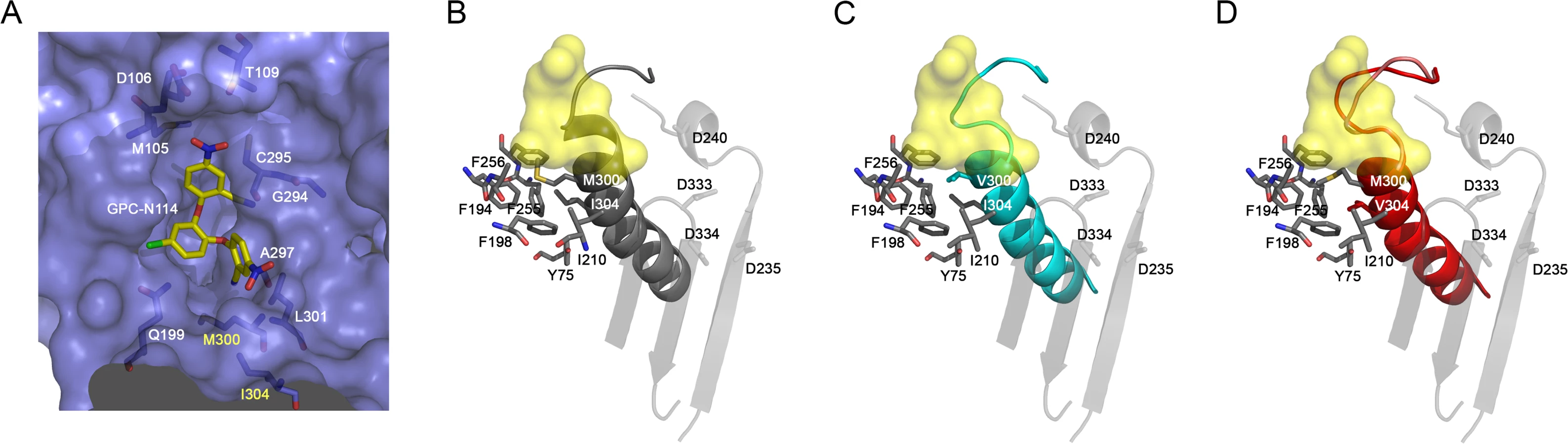
The apo structures of the EMCV M300V and I304V polymerase mutants were obtained using the same crystallization conditions as for the wt enzyme [29], yielding two different types of crystals for the mutants. The M300V mutant crystallized in the space group I4122 form with one 3Dpol molecule in the asymmetric unit (a.u.) and diffracted to 2.2 Å resolution. The 3Dpol I304V mutant crystallized in the space group C2 with six polymerase molecules per a.u. and diffracted to 3.2 Å resolution. Data collection and refinement statistics are summarized in Table 2. Structural comparisons showed that the mutant polymerases were almost identical to the wt enzyme. The main differences were concentrated in helix α10, which contains the mutated residues, and in the preceding loop B. The side chains of residues 300 and 304 participate in a large hydrophobic cluster linking α10 with helices α2 (Y75), α6 (F194, A195, F199), α7 (I210) and α8 (L241, F255 and F256), included in the inner fingers. The substitutions of the methionine and isoleucine side chains by the shorter valine in the mutants imply a weakening of these hydrophobic interactions. This could result in an increased flexibility of α10, including the disruption of the last helical turn, and of the B-loop, which appears to have a key role in GPC-N114 binding in EMCV 3Dpol (Fig. 4). In addition, the substitution with valine may disrupt the interaction of residue 300 with the compound.
GPC-N114 has a stronger inhibitory effect when added to 3Dpol before the RNA template-primer duplex
Prompted by the identification of the binding site of GPC-N114 on CVB3 3Dpol, we analyzed the effects of GPC-N114 on CVB3 3Dpol in more detail. Consistent with the data obtained for EMCV 3Dpol, GPC-N114 inhibited CVB3 3Dpol elongation activity of the homopolymeric substrate poly(rA)/dT15 (Fig. 5A). Again inhibition was noncompetitive with respect to UTP. Since the binding sites of GPC-N114 and the template on CVB3 3Dpol overlapped, we aimed to explore whether GPC-N114 competes with the RNA template-primer duplex for binding to 3Dpol. A possible method to test whether GPC-N114 precludes binding of the enzyme to RNA is by monitoring changes in fluorescence polarization signal [32] of a labeled symmetric heteropolymeric template-primer duplex (sym/sub-U) [33]. However, though GPC-N114 had an inhibitory effect in the elongation assays with the homopolymeric poly(rA)/dT15 template-primer (Fig. 5A), the compound failed to inhibit the incorporation of NTPs using the sym/sub-U template-primer, the latter being in contrast with what we observed in the elongation assays with the homopolymeric poly(rA)/dT15 template-primer (S9 Fig.). Therefore, an order-of addition assay was performed to compare the efficiency of GPC-N114 when added to the reaction mixture with 3Dpol before or after addition of poly(rA)/dT15. If GPC-N114 would affect productive binding of the template-primer to 3Dpol, the inhibitor should display an improved potency when added to the reaction mix with 3Dpol before the template-primer as compared to after. When enzyme was pre-incubated with the inhibitor before adding the template-primer on average a 3-fold (n = 3) reduction in the Vmax was observed (Fig. 5B), which is consistent with the reported inactivation of picornavirus polymerases when they are incubated in the absence of nucleic acid [34]. Importantly, the IC50 value for GPC-N114 on CVB3 3Dpol was dramatically reduced (on average 14-fold, n = 3) when the compound was added to the reaction mix before the template-primer (Fig. 5B). Unfortunately, this assay is not able to discriminate between direct competition and the formation of a nonproductive ternary complex consisting of GPC-N114–template-primer–3Dpol. Nevertheless, the observation that GPC-N114 has a stronger inhibitory effect on 3Dpol activity when added before the template-primer is compatible with the overlapping binding sites of the compound and the template-primer on 3Dpol.
Discussion
There is a great need for broad-range antiviral compounds to treat infections with enteroviruses, which includes important human pathogens such as poliovirus, coxsackievirus, enterovirus 71, and rhinovirus. Here, we report a novel small molecule, GPC-N114 that exerts broad-spectrum anti-enterovirus activity and also inhibits members of the genus Cardiovirus, such as EMCV. Analysis of its mechanism of action revealed that GPC-N114 inhibits virus replication at the stage of RNA replication. GPC-N114-resistant enterovirus variants could not be obtained, but compound-resistant EMCV variants were readily selected in the presence of suboptimal concentration of GPC-N114. These variants were found to carry mutations in the viral RdRP, 3Dpol. Consistently, the in vitro elongation activity of CVB3 and EMCV 3Dpol was inhibited by GPC-N114, and the mutations identified in compound-resistant EMCV 3Dpol rendered this polymerase less susceptible to inhibition. GPC-N114 did not compete with incoming NTPs, but interfered with productive binding of the template-primer to 3Dpol in a primer elongation assay. This is in agreement with the crystallographic studies of the CVB3 3Dpol–GPC-N114 complex which revealed that the binding site of the compound is located at the junction of the palm and the fingers domains, and overlaps partially with the binding site of the template.
A high barrier to resistance is a desirable feature for antiviral compounds since drug resistance represents a major problem in the treatment of viral infections. Notably, no GPC-N114-resistant enteroviruses (CVB3, PV1, E9) were obtained after several attempts and a large number of serial passages of these viruses in the presence of suboptimal concentrations of the compound. Attempts to generate compound-resistant CVB3 by introduction of the mutations that correspond to those identified in GPC-N114-resistant EMCV (i.e., 3D-I296V and 3D-M300V in CVB3), also did not result in a compound-resistant phenotype. The reason for the inability of enteroviruses to develop resistance against GPC-N114 remains to be established. A possible explanation is that mutations that would confer resistance to GPC-N114 also impair binding of the template-primer, thereby preventing replication. Although the exact reason remains to be determined, our data suggest that the compound-binding site in enterovirus 3Dpol lacks the conformational plasticity for resistance to develop, in contrast to most allosteric binding sites at the enzyme surface.
In contrast, EMCV 3Dpol is sufficiently plastic to allow for substitutions that result in compound-resistance. The amino acid changes are conservative with all three amino acids having similar properties. Based on the constructed model, only the long side chain of M300 (equivalent to I296 in CVB3 3Dpol, see Fig. 3B), would be in direct contact with the compound. In contrast, I304 seems to be located relatively far away of the GPC-N114 binding cavity. Both M300 and I304 are part of conserved structural motif B, which not only binds the RNA template in the active site but also participates in the mechanism of RNA translocation through fine movements of a loop located at the N-terminus of the motif, the B-loop [30,35]. Structural data from picornavirus polymerases [30,35] as well as other RdRPs [36] suggest that the B-loop is highly dynamic and that its dynamics can be regulated by external effectors [37]. The structures of the EMCV 3Dpol M300V and I304V mutants suggest that these mutations increase the flexibility of this loop, thereby affecting the binding site of GPC-N114 and probably losing a direct interaction with the shorter V300 side chain whilst retaining the ability to interact with the template-primer.
EMCV 3Dpol and CVB3 3Dpol display a high structural similarity, but there is a major difference in the main interactions of these polymerases with GPC-N114 that could possibly underlie the difference in resistance emergence. In CVB3 3Dpol, the main interaction with GPC-N114 is mediated by Y195. EMCV 3Dpol, however, possesses an alanine at this position which forms a weaker interaction (S10 Fig.). Due to this weaker interaction, a flexibility increase in the compound-binding area induced by the B loop resistance mutations may result in a decreased binding of GPC-N114 to EMCV 3Dpol. For CVB3 3Dpol, a possible increase in flexibility induced by B loop mutations may not, or to a lesser extent, affect the binding of GPC-N114 due to its strong interaction with Y195.
GPC-N114 proved to be not a very robust compound in biochemical assays, for which the reason might be that it interferes with the binding of a by far larger molecule, the RNA template-primer. Thus, despite our efforts, it remains to be established whether GPC-N114 inhibits 3Dpol elongation activity by competing with the template-primer for binding to 3Dpol or whether binding of the compound results in the formation of an unproductive conformation of the polymerase-template complex. Nevertheless, both the crystallographic studies as well as the order-of-addition experiment strongly suggest that GPC-N114 affects binding of the template-primer by 3Dpol. Compounds that interfere with binding of the template-primer have been described for polymerases of some viruses, including DENV and HIV [38,39], but not for any of the picornaviruses. The only known picornavirus polymerase inhibitors with a related mechanism of action are two compounds that target the incoming NTP binding site in the PV polymerase [15]. Clearly, the NTP binding site is close to, but distinct from, the GPC-N114 binding site. Hence, GPC-N114 is the first reported picornavirus polymerase inhibitor that binds the site of the template acceptor nucleotide.
Polymerase inhibitors have demonstrated their importance for the treatment of other viral infections such as those caused by HIV, herpesvirus, HBV and HCV. Unfortunately, it was not possible to evaluate the antiviral effect of GPC-N114 in an animal model, since—despite extensive attempts—we have not been able to formulate GPC-N114 (or GPC-N143) for administration. Notwithstanding this, the identification of a new druggable pocket in the polymerase through the characterization of the mechanism of action of GPC-N114 may be key in the (in silico) design of novel picornavirus polymerase NNIs.
Materials and Methods
Cells and reagents
Buffalo green monkey (BGM) kidney cells, HeLa R19 cells (obtained from G. Belov, University of Maryland and Virginia-Maryland Regional College of Veterinary Medicine, US), baby hamster kidney 21 (BHK-21) cells, and human rhabdomyosarcoma RD cells were grown at 37°C, 5% CO2 in Dulbecco’s Modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum and antibiotics [28,40]. The Hela H cells, BGM cells, and Vero cells used for the multicycle CPE-reduction assays were maintained in Minimal Essential Medium supplemented with 10% FBS, 1% bicarbonate, and 1% L-glutamine [28].
The synthesis of GPC-N114 will be published elsewhere. Guanidine hydrochloride (GuHCl) and dipyridamole were purchased from Sigma Aldrich. Dipyridamole was dissolved in ethanol and GuHCl in water. The other compounds were dissolved in DMSO. GPC-N114 was used in a concentration of 10 μM unless stated otherwise.
Viruses and infections
Coxsackievirus B3 (strain Nancy) was obtained by transfection of RNA transcripts of the full-length infectious clone p53CB3/T7 linearized with SalI into BGM cells [41]. EMCV, strain mengovirus, was obtained in a similar manner by transfection of RNA derived from cDNA clone pM16.1 into BHK-21 cells [42]. EV68, EV70, EV71 (BrCr), E11 (Gregory), CVA16 (G-10) and CVA21 (Coe) were obtained from the National Institute for Public Health and Environment (RIVM, the Netherlands). Poliovirus 1–3 Sabin reference strains were obtained from dr. J Martin (NIBSC, UK). Human rhinoviruses 2 and 14 were supplied by Joachim Seipelt (Medical University of Vienna, Austria). ERAV (NM11/67) was a gift from David Rowlands and Toby Tuthill (University of Leeds, UK). Saffold virus (type 3) was described previously [43]. The FMDV strains O1 Manisa and A22 Iraq 24/64 were used [44].
For virus infections, virus was added to subconfluent cell layers and allowed to adsorb for 30 minutes, after which virus was removed and fresh (drug-containing) medium was added to the cells. At the indicated times p.i. the cells were freeze-thawed three times to release intracellular virus particles. Virus titers were determined by endpoint titration according to the method of Reed and Muench and expressed as 50% cell culture infective doses (CCID50)/ml [45].
Multicycle CPE-reduction assays
The antiviral activity of compounds was determined as published previously [11,44]. In short, the medium was removed from cells grown in 96-well plates and a serial dilution of compound in medium with 2% FCS was added after which the cells were infected with virus at the lowest MOI sufficient for full CPE development in infected, untreated cultures after three days of incubation at 37°C. To quantify the level of CPE by MTS assay, the medium was then replaced with AQueous One Solution Cell Proliferation Reagent (Promega) diluted in medium and incubated at 37°C for 1–2 h. Subsequently, absorbance at 498 nm was measured. The measured values were used to calculate the cell viability ranging from 0% (full CPE with infected, untreated cultures) to 100% (uninfected, untreated cultures). The 50% effective concentration (EC50) was defined as the concentration of compound that resulted in a 50% reduction in virus-induced CPE and was calculated using logarithmic interpolation.
Subgenomic replicon assay
The subgenomic replicons of CVB3 (pRib-LUC-CB3/T7) and EMCV (pEMCV-FLuc) encode the cDNA clones of the corresponding virus except that the P1 area encoding the capsid proteins has been (partially) replaced by the code for firefly luciferase [24,46]. The plasmids were linearized with MluI or BamHI, respectively. RNA transcribed from the linearized plasmids was used to transfect BGM cells plated in 24-well plates using the DEAE-dextran method as described previously [24]. After transfection, cells were treated with DMSO or compound and incubated at 37°C. At 2, 4, 6, and 8 h post-transfection, cells were washed with PBS, lysed and luciferase levels were determined with the Luciferase Assay System (Promega) according to the manufacturer’s instructions.
A similar procedure was followed for the PV1 subgenomic replicon assay pXpA-RenR-PV except that luciferase activity was measured with the Renilla Luciferase Assay System (Promega), since this construct encodes the Renilla luciferase [47].
Metabolic labeling
For metabolic labeling of proteins, BGM cells were grown in 24-well plates to subconfluence and infected with CVB3 at an MOI of 50. At 5 h post-infection, the medium was replaced with 300 μl of methionine-free medium for 30 min. Subsequently, the cultures were pulse-labeled in methionine-free medium supplemented with [35S]Met in the absence or presence of 50 μM GPC-N114 for 30 min. At 6 h post-infection, cells were lysed with lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.05% SDS]. Laemmli sample buffer was added to the lysates, boiled for 5 min and analyzed on a 10% polyacrylamide gel containing SDS. The gels were fixed in 30% methanol-10% acetic acid, rinsed in DMSO, fluorographed with 20% 2,5-diphenyloxazole in DMSO, dried, and exposed to Kodak XAR film.
Generation of GPC-N114-resistant virus
To obtain drug-resistant virus, three independent virus pools of CVB3 and EMCV were serially passaged in the presence of a concentration series of GPC-N114 on subconfluent to confluent Vero cultures in 96-well plates. The amount of virus used for infection was selected so that full CPE was visible after 3 days of incubation in the absence of compound. The concentration series encompassed 8 different concentrations in 2-fold dilutions starting from 25–100 μM. After 3 to 4 days, the amount of CPE was assessed and supernatant collected from the culture with the highest concentration of compound that still exhibited full CPE was used to perform the next round of infection. This procedure was repeated 32 times (CVB3) or until there was a clear increase in the concentration required for inhibition of CPE (EMCV) compared to wt virus that was tested in parallel in each passage. Viral RNA was isolated from the resistant virus pools using the GenElute Mammalian Total RNA Miniprep Kit (Sigma Aldrich) and used for sequencing of the parts of the genome that encode the nonstructural proteins. The PCR product containing the mutations found in the virus pools generated in this procedure was digested with PinAI and NruI and cloned into pM16.1 to replace the corresponding wt sequence. The resulting infectious clones pM16.1-3D-M300V and pM16.1-3D-I304V were sequenced to confirm the presence of the mutations.
The analogous mutations in CVB3 (I296V and M300V) were introduced into the infectious cDNA p53CB3/T7CVB3 by site-directed mutagenesis. The mutant clones were sequenced to confirm the presence of the mutations. The construction of pRibCB3/T7 with the S299T mutation was described previously [48].
Recombinant EMCV and CVB3 was generated as described above and the presence of the mutations was again confirmed by sequence analysis.
Protein expression and purification for biochemical activity assays
Recombinant CVB3 3Dpol and the RdRP domain of DENV NS5 (NS5-RdRP) were obtained as previously described [49,50]. For expression and purification of EMCV 3Dpol, the coding sequence of 3Dpol (polyprotein domain from amino acid position 1834 to 2293) was amplified on EMCV cDNAs (pM16.1 wt, 3D-M300V, and 3D-I304V) and cloned into pETG20A (kindly provided by Dr. Arie Geerlof) by Gateway recombination (Life Technologies), downstream a cleavable Hexahistidine-Thioredoxin tag using a two-step PCR protocol. The protein was expressed in E. coli Rosetta (DE3) pLysS strain (Novagen) at 17°C in Tur*best Broth (Arcadia Biotech). The purification of the protein and the tag removal were performed in non-denaturing conditions as previously described [51]. The final size exclusion chromatography step was performed in 10 mM Hepes, 300 mM NaCl, 2mM dithiotreitol, pH 7.5.
Polymerase activity assays
The conditions for the polymerase elongation activity assays were adapted from conditions reported previously [10,50]. EMCV 3Dpol filter-binding polymerase activity assays were conducted in a mix of 50 μl containing 50 mM MOPS pH 7.0, 10 mM KCl, 4 mM MgCl2, 9% glycerol, 2 μM dT15 (Invitrogen) annealed to 350 nM poly(rA) (GE Healthcare, average size 519 nt) (i.e., a molar ratio dT15:poly(rA) of 5.7:1), 1 μM EMCV 3Dpol, 50/100/200 or 500 μM UTP and 2 μCi [3H]UTP (GE Healthcare, 35.5 Ci/mmol). The reaction mix excluding UTP was incubated with either DMSO or GPC-N114 with a constant concentration of 5% DMSO for 2 minutes after which the reaction was started by addition of labeled and unlabeled UTP. Samples of 10 μl were taken after 0, 10, 20 and 30 min of incubation at 30°C and added to 50 μl of 100 mM EDTA in 96-well sample plates to stop the reaction. Mixtures were then transferred onto glass fiber filter mats with DEAE active groups (DEAE filtermat, Wallac) using a Filtermat Harvester (Packard Instruments). Filtermats were washed three times with 0.3 M ammonium formate pH 8.0, twice with water, and once with ethanol after which they were dried and transferred into sample bags. Liquid scintillation fluid was added to the sample bags and incorporation of radiolabeled UTP was measured in counts per minute using a Wallac MicroBeta TriLux Liquid Scintillation Counter.
For the DENV NS5 elongation assay the reaction mixture contained 50 mM HEPES pH 8.0, 10 mM KCl, 10 mM DTT, 5 mM MgCl2, 2 μM dT15 annealed to 350 nM poly(rA), 0.4 μM DENV NS5-RdRP, 20 μM UTP and 2 μCi [3H]UTP. 10 μl samples were taken after 0, 30, 60, and 90 minutes. Subsequently, the same procedure was followed as for EMCV 3Dpol.
The CVB3 3Dpol reaction mixture contained 50 mM Tris pH 7.0, 10 mM KCl, 0.8 mM MgCl2, 9% glycerol, 2 μM dT15 annealed to 350 nM poly(rA), 20 nM CVB3 3Dpol, UTP (2, 5, 10 or 20 μM) and 0.5 μCi [3H]UTP. 10 μl samples were taken after 0, 5, 10, and 15 minutes. For the order-of-addition experiment with CVB3 3Dpol, a reaction premix was assembled as above but without poly(rA)/dT15, GPC-N114 and UTP. To this premix either a range of concentrations of GPC-N114 or 80 nM dT15 annealed to 14 nM poly(rA) were added. After an incubation of 1 min at RT, the other component was added to the mix followed by an incubation of several minutes at 30°C. The reaction was started by addition of UTP, and samples were taken after 0, 3, 6, and 9 minutes.
The data were fitted using non-linear regression using Graphpad Prism 5.0.3. Ki values averaged from at least three independent experiments were statistically compared between EMCV 3Dpol wt and the 3Dpol mutants using a Student’s t-test.
Protein expression and purification for crystallography
Recombinant CVB3 3Dpol was over-expressed essentially as previously described [49], except that the expression was carried out at 20°C. Stored pellets were resuspended in a cold lysis buffer containing 50 mM Tris HCl pH 9.0, 500 mM NaCl, 10 mM imidazole, 0.1% (v/v) Triton X-100, 5% glycerol, 1 mg/ml lysozyme and 10 μg/ml DNase, and cells were lysed by sonication. Cellular debris was pelleted by centrifugation and the soluble fraction was filtrated and loaded into a 5 ml HisTrap HP column (GE Healthcare). The protein was eluted using an imidazole gradient (10–500 mM) in 50 mM Tris pH 8.0 and 300 mM NaCl. Protein fractions were dialyzed against 20 mM Tris pH 9.0, 600 mM NaCl, 15% glycerol, 0.5 mM TCEP and further purified by size-exclusion chromatography on a Superdex 200 16/60 column (GE Healthcare). Purified protein was concentrated to 5 mg/ml in the same buffer, using a Vivaspin concentrator (10.000 MWCO PES, Sartorius Stedim Biotech). The quality of the protein obtained was checked by SDS-PAGE.
EMCV 3Dpol wt and mutants were expressed and purified as previously reported for EMCV 3Dpol wt [29]. Briefly, using the constructs described above, the polymerases were overexpressed in E. coli, and EMCV 3Dpol was purified by means of Ni-affinity (HisTrap HP, GE Healthcare). After that, the His-tag was removed by TEV digestion, and size exclusion chromatography (Superdex 200 16/60, GE Healthcare).
Crystallization and data collection
Crystals of isolated CVB3 3Dpol were obtained by the sitting-drop vapor-diffusion method at 16°C in 96-well plates (MRC, Swissci AG) using a Cartesian automated drop dispenser to mix 250 nl of 5 mg/ml protein solution with an equal volume of reservoir solution, containing 50 mM Tris pH 7.5, 24.5% (w/v) glycerol, 1.29 M ammonium sulfate. Highly reproducible crystals of 25x25x25 μm in size appeared between 2–3 days. Protein/inhibitor complexes were prepared by soaking CVB3 3Dpol crystals in the inhibitor solution for 72h. The inhibitor solutions contained 100 μM GPC-N114 or GPC-N143 in 2.5% DMSO, 0.65 M ammonium sulfate, 12.5% glycerol and 25 mM Tris pH 7.5. Crystals were then transferred to a cryo-protecting solution, containing 30% glycerol in the respective crystallization buffer prior to cooling by immersion in liquid nitrogen. The two datasets were collected at 100K by using synchrotron radiation at the European Synchrotron Radiation Facility (ESRF, Grenoble) on a Pilatus 6M S/N 60–104 detector at beamline ID 29, λ = 0.977Å. Diffraction images were processed with iMOSFLM [52] and internally scaled with SCALA (CCP4i [53]), achieving resolutions of 2.9Å and 2.7Å for the CVB3 3Dpol–GPC-N114 and CVB3 3Dpol–GPC-N143 crystals, respectively. Data collection statistics are given in Table 2. Coordinates have been deposited at the PDB with accession codes 4Y2A and 4Y34 for the complexes CVB3 3Dpol–GPC-N114 and CVB3 3Dpol–GPC-N143, respectively.
Crystals of EMCV 3Dpol wt and M300V and I304V, mutants were obtained as previously described [29]. The data sets were collected at the ALBA-CELLS synchrotron light facility (Barcelona) on a Pilatus 6M Dectris detector at beamline XALOC λ = 0.977Å. Diffraction images were processed with iMOSFLM and SCALA, achieving resolutions of 2.2Å and 3.2Å for the M300V and I304V respectively (Table 2). Coordinates have been deposited at the PDB with accession codes 4Y2C and 4Y3C for the structures of EMCV 3Dpol-M300V and EMCV 3Dpol-I304V, respectively.
Structure determination and refinement
The initial electron density maps of the CVB3 3Dpol–GPC-N114 and CVB3 3Dpol–GPC-N143 complexes were obtained after rigid-body fitting of the coordinates of the 3Dpol apoenzymes (PDB id. 3DDK) to the new unit cells (Table 2), using the program Refmac5 [54,55]. In both structures, the weighted 2|Fo|-|Fc| and |Fo|-|Fc| difference map showed the presence of extra densities that permitted the initial positioning of the inhibitor molecules. Inhibitor structure files were generated using “Online SMILES Translator and Structure File Generator” (http://cactus.nci.nih.gov/translate/) and its respective CIF dictionaries were created by Grade [56]. Several cycles of automatic refinement, performed with Refmac5, were alternated with manual model rebuilding using Coot [57]. The refinement statistics are summarized in Table 2.
The first electron density maps for both EMCV 3Dpol mutants were obtained after rigid-body fitting of the EMCV 3Dpol wt coordinates to the new unit cells. The PDB id 4NYZ, of space group I4122, was used as a model for the M300V structure and coordinates 4NZ0, of space group C2, were used as a model for the I304V mutant. Noncrystallographic symmetry (ncs) restraints were initially applied to the six independent molecules present in the asymmetric unit of the C2 crystals using Refmac5. However, the ncs restrained-refinement result was unstable because the presence of different conformations in many polymerase regions, especially in the surface-exposed loops, was affected by crystal packing interactions. Therefore, the variable regions were manually rebuilt using Coot, and the final cycles of automatic refinement with Refmac 5, were performed in absence of ncs restraints. The refinement statistics are given in Table 2.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Tapparel C, Siegrist F, Petty TJ, Kaiser L (2012) Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol.
2. Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, et al. (2012) Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier. pp. 855–880.
3. Appleby TC, Luecke H, Shim JH, Wu JZ, Cheney IW, Zhong W, Vogeley L, Hong Z, Yao N (2005) Crystal structure of complete rhinovirus RNA polymerase suggests front loading of protein primer. J Virol 79: 277–288. 15596823
4. Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N (2004) Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem 279: 47212–47221. 15294895
5. Hansen JL, Long AM, Schultz SC (1997) Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 5: 1109–1122. 9309225
6. Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA (2004) The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure 12: 1533–1544. 15296746
7. Thompson AA, Peersen OB (2004) Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J 23: 3462–3471. 15306852
8. Wu Y, Lou Z, Miao Y, Yu Y, Dong H, Peng W, Bartlam M, Li X, Rao Z (2010) Structures of EV71 RNA-dependent RNA polymerase in complex with substrate and analogue provide a drug target against the hand-foot-and-mouth disease pandemic in China. Protein Cell 1: 491–500. doi: 10.1007/s13238-010-0061-7 21203964
9. Campagnola G, Weygandt M, Scoggin K, Peersen O (2008) Crystal structure of coxsackievirus B3 3Dpol highlights the functional importance of residue 5 in picornavirus polymerases. J Virol 82: 9458–9464. doi: 10.1128/JVI.00647-08 18632862
10. Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B (2008) The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J Virol 82: 9577–9590. doi: 10.1128/JVI.00631-08 18632861
11. Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N (2006) A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol 16: 27–34. 16364629
12. Ferrer-Orta C, Agudo R, Domingo E, Verdaguer N (2009) Structural insights into replication initiation and elongation processes by the FMDV RNA-dependent RNA polymerase. Curr Opin Struct Biol 19: 752–758. doi: 10.1016/j.sbi.2009.10.016 19914060
13. Crotty S, Cameron CE, Andino R (2001) RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A 98: 6895–6900. 11371613
14. Hardy JA, Wells JA (2004) Searching for new allosteric sites in enzymes. Curr Opin Struct Biol 14: 706–715. 15582395
15. Campagnola G, Gong P, Peersen OB (2011) High-throughput screening identification of poliovirus RNA-dependent RNA polymerase inhibitors. Antiviral Res 91: 241–251. doi: 10.1016/j.antiviral.2011.06.006 21722674
16. Chen TC, Chang HY, Lin PF, Chern JH, Hsu JT, Chang CY, Shih SR (2009) Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob Agents Chemother 53: 2740–2747. doi: 10.1128/AAC.00101-09 19414569
17. Durk RC, Singh K, Cornelison CA, Rai DK, Matzek KB, Leslie MD, Schafer E, Marchand B, Adedeji A, Michailidis E, Dorst CA, Moran J, Pautler C, Rodriguez LL, McIntosh MA, Rieder E, Sarafianos SG (2010) Inhibitors of foot and mouth disease virus targeting a novel pocket of the RNA-dependent RNA polymerase. PLoS One 5: e15049. doi: 10.1371/journal.pone.0015049 21203539
18. Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD (2009) T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res 82: 95–102. doi: 10.1016/j.antiviral.2009.02.198 19428599
19. Harrison DN, Gazina EV, Purcell DF, Anderson DA, Petrou S (2008) Amiloride derivatives inhibit coxsackievirus B3 RNA replication. J Virol 82: 1465–1473. 18032495
20. Hung HC, Chen TC, Fang MY, Yen KJ, Shih SR, Hsu JT, Tseng CP (2010) Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J Antimicrob Chemother 65: 676–683. doi: 10.1093/jac/dkp502 20089540
21. Rodriguez PL, Carrasco L (1992) Gliotoxin: inhibitor of poliovirus RNA synthesis that blocks the viral RNA polymerase 3Dpol. J Virol 66: 1971–1976. 1372367
22. Gazina EV, Smidansky ED, Holien JK, Harrison DN, Cromer BA, Arnold JJ, Parker MW, Cameron CE, Petrou S (2011) Amiloride is a competitive inhibitor of coxsackievirus B3 RNA polymerase. J Virol 85: 10364–10374. doi: 10.1128/JVI.05022-11 21795353
23. Purstinger G, De Palma AM, Zimmerhofer G, Huber S, Ladurner S, Neyts J (2008) Synthesis and anti-CVB 3 evaluation of substituted 5-nitro-2-phenoxybenzonitriles. Bioorg Med Chem Lett 18: 5123–5125. doi: 10.1016/j.bmcl.2008.07.099 18710805
24. van Kuppeveld FJ, Galama JM, Zoll J, Melchers WJ (1995) Genetic analysis of a hydrophobic domain of coxsackie B3 virus protein 2B: a moderate degree of hydrophobicity is required for a cis-acting function in viral RNA synthesis. J Virol 69: 7782–7790. 7494289
25. Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW (1982) Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem 257: 14806–14810. 6294080
26. De Palma AM, Heggermont W, Lanke K, Coutard B, Bergmann M, Monforte AM, Canard B, De Clercq E, Chimirri A, Purstinger G, Rohayem J, van Kuppeveld F, Neyts J (2008) The thiazolobenzimidazole TBZE-029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C. J Virol 82: 4720–4730. doi: 10.1128/JVI.01338-07 18337578
27. De Palma AM, Thibaut HJ, van der Linden L, Lanke K, Heggermont W, Ireland S, Andrews R, Arimilli M, Al-Tel TH, De Clercq E, van Kuppeveld F, Neyts J (2009) Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob Agents Chemother 53: 1850–1857. doi: 10.1128/AAC.00934-08 19237651
28. Thibaut HJ, van der Linden L, Jiang P, Thys B, Canela MD, Aguado L, Rombaut B, Wimmer E, Paul A, Perez-Perez MJ, van Kuppeveld FJ, Neyts J (2014) Binding of glutathione to enterovirus capsids is essential for virion morphogenesis. PLoS Pathog 10: e1004039. doi: 10.1371/journal.ppat.1004039 24722756
29. Vives-Adrian L, Lujan C, Oliva B, van der LL, Selisko B, Coutard B, Canard B, van Kuppeveld FJ, Ferrer-Orta C, Verdaguer N (2014) The crystal structure of a cardiovirus RNA-dependent RNA polymerase reveals an unusual conformation of the polymerase active site. J Virol 88: 5595–5607. doi: 10.1128/JVI.03502-13 24600002
30. Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N (2007) Sequential structures provide insights into the fidelity of RNA replication. Proc Natl Acad Sci U S A 104: 9463–9468. 17517631
31. Kortus MG, Kempf BJ, Haworth KG, Barton DJ, Peersen OB (2012) A template RNA entry channel in the fingers domain of the poliovirus polymerase. J Mol Biol 417: 263–278. doi: 10.1016/j.jmb.2012.01.049 22321798
32. Mestas SP, Sholders AJ, Peersen OB (2007) A fluorescence polarization-based screening assay for nucleic acid polymerase elongation activity. Anal Biochem 365: 194–200. 17475199
33. Arnold JJ, Cameron CE (2000) Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J Biol Chem 275: 5329–5336. 10681506
34. Arias A, Arnold JJ, Sierra M, Smidansky ED, Domingo E, Cameron CE (2008) Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J Virol 82: 12346–12355. doi: 10.1128/JVI.01297-08 18829745
35. Gong P, Peersen OB (2010) Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 107: 22505–22510. doi: 10.1073/pnas.1007626107 21148772
36. Garriga D, Navarro A, Querol-Audi J, Abaitua F, Rodriguez JF, Verdaguer N (2007) Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 104: 20540–20545. 18077388
37. Garriga D, Ferrer-Orta C, Querol-Audi J, Oliva B, Verdaguer N (2013) Role of Motif B Loop in Allosteric Regulation of RNA-Dependent RNA Polymerization Activity. J Mol Biol.
38. Niyomrattanakit P, Chen YL, Dong H, Yin Z, Qing M, Glickman JF, Lin K, Mueller D, Voshol H, Lim JY, Nilar S, Keller TH, Shi PY (2010) Inhibition of dengue virus polymerase by blocking of the RNA tunnel. J Virol 84: 5678–5686. doi: 10.1128/JVI.02451-09 20237086
39. Wang LZ, Kenyon GL, Johnson KA (2004) Novel mechanism of inhibition of HIV-1 reverse transcriptase by a new non-nucleoside analog, KM-1. J Biol Chem 279: 38424–38432. 15231830
40. Zoll J, Galama JM, van Kuppeveld FJ, Melchers WJ (1996) Mengovirus leader is involved in the inhibition of host cell protein synthesis. J Virol 70: 4948–4952. 8763999
41. Wessels E, Duijsings D, Notebaart RA, Melchers WJ, van Kuppeveld FJ (2005) A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J Virol 79: 5163–5173. 15795300
42. Duke GM, Palmenberg AC (1989) Cloning and synthesis of infectious cardiovirus RNAs containing short, discrete poly(C) tracts. J Virol 63: 1822–1826. 2538661
43. Zoll J, Erkens HS, Lanke K, Verduyn LF, Melchers WJ, Schoondermark-van de Ven E, Roivainen M, Galama JM, van Kuppeveld FJ (2009) Saffold virus, a human Theiler's-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog 5: e1000416. doi: 10.1371/journal.ppat.1000416 19412527
44. Goris NE, Eble PL, de Jong MC, De Clercq K (2009) Quantification of foot-and-mouth disease virus transmission rates using published data. ALTEX 26: 52–54. 19326033
45. Reed LJ, Muench H (2012) A simple method of estimating the fifty per cent endpoints. Am J Hyg 27: 493–497.
46. Aminev AG, Amineva SP, Palmenberg AC (2003) Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res 95: 59–73. 12921996
47. Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E (2008) A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 4: e1000216. doi: 10.1371/journal.ppat.1000216 19023417
48. van Ooij MJM (2007) Biological characterization of a Coxsackie B3 virus revertant exhibiting a thermophilic phenotype. PhD thesis. Radboud University Nijmegen. Available: http://repository.ubn.ru.nl/bitstream/handle/2066/30016/30016.pdf?sequence=1.
49. Jabafi I, Selisko B, Coutard B, De Palma AM, Neyts J, Egloff MP, Grisel S, Dalle K, Campanacci V, Spinelli S, Cambillau C, Canard B, Gruez A (2007) Improved crystallization of the coxsackievirus B3 RNA-dependent RNA polymerase. Acta Crystallogr Sect F Struct Biol Cryst Commun 63: 495–498. 17554171
50. Selisko B, Dutartre H, Guillemot JC, Debarnot C, Benarroch D, Khromykh A, Despres P, Egloff MP, Canard B (2006) Comparative mechanistic studies of de novo RNA synthesis by flavivirus RNA-dependent RNA polymerases. Virology 351: 145–158. 16631221
51. Lantez V, Dalle K, Charrel R, Baronti C, Canard B, Coutard B (2011) Comparative production analysis of three phlebovirus nucleoproteins under denaturing or non-denaturing conditions for crystallographic studies. PLoS Negl Trop Dis 5: e936. doi: 10.1371/journal.pntd.0000936 21245924
52. Leslie AGW, Powell HR (2007) Processing Diffraction Data with Mosflm. In: Read R, Sussman JL, editors. Evolving Methods for Macromolecular Crystallography. Dordrecht, the Netherlands: Springer. pp. 41–51.
53. Potterton E, Briggs P, Turkenburg M, Dodson E (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr 59: 1131–1137. 12832755
54. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255. 15299926
55. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67: 355–367. doi: 10.1107/S0907444911001314 21460454
56. Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G (2011) Grade, version 1.1.0.
57. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. 15572765
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Is Antigenic Sin Always “Original?” Re-examining the Evidence Regarding Circulation of a Human H1 Influenza Virus Immediately Prior to the 1918 Spanish Flu
- An 18 kDa Scaffold Protein Is Critical for Biofilm Formation