
Accumulation of Glucosylceramide in the Absence of the Beta-Glucosidase GBA2 Alters Cytoskeletal Dynamics
During mammalian spermatogenesis, sperm with a head and a tail are formed from a round cell. This process is tightly regulated and involves the close interaction of somatic Sertoli cells and germ cells. Accumulation of the glycosphingolipid glucosylceramide in the absence of the beta-glucosidase GBA2 has been proposed to disturb sperm development, leading to morphological defects. However, the underlying mechanism is not known. Here, we demonstrate that accumulation of glucosylceramide in GBA2 knockout-mice controls the dynamics of the actin and microtubule cytoskeleton, which are crucial for sperm development. In particular, cytoskeletal structures at the interface between Sertoli and germ cells are disorganized, leading to malformation of the sperm head and a defect in acrosome formation. In summary, we provide mechanistic insights into how glucosylceramide controls cellular signaling and dysregulation of this essential glycosphingolipid leads to male infertility.
Published in the journal:
Accumulation of Glucosylceramide in the Absence of the Beta-Glucosidase GBA2 Alters Cytoskeletal Dynamics. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005063
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005063
Summary
During mammalian spermatogenesis, sperm with a head and a tail are formed from a round cell. This process is tightly regulated and involves the close interaction of somatic Sertoli cells and germ cells. Accumulation of the glycosphingolipid glucosylceramide in the absence of the beta-glucosidase GBA2 has been proposed to disturb sperm development, leading to morphological defects. However, the underlying mechanism is not known. Here, we demonstrate that accumulation of glucosylceramide in GBA2 knockout-mice controls the dynamics of the actin and microtubule cytoskeleton, which are crucial for sperm development. In particular, cytoskeletal structures at the interface between Sertoli and germ cells are disorganized, leading to malformation of the sperm head and a defect in acrosome formation. In summary, we provide mechanistic insights into how glucosylceramide controls cellular signaling and dysregulation of this essential glycosphingolipid leads to male infertility.
Introduction
Spermatogenesis occurs in the seminiferous tubules of the testis. Defects in sperm development often result in male infertility. The beta-glucosidase GBA2 plays an important role in sperm development [1]. GBA2 knockout-mice are subfertile, because sperm display severe morphological defects: heads are round rather than sickle-shaped, mitochondria are misaligned along the sperm flagellum, and the acrosome, needed to penetrate the egg coat, is lacking [1]. This phenotype is called globozoospermia [2]. GBA2 degrades the glycosphingolipid glucosylceramide (GlcCer) to glucose and ceramide. Accumulation of GlcCer in GBA2 knockout-mice has been proposed to underlie the defects in spermatogenesis leading to globozoospermia [1]. However, the underlying mechanism is not known.
Several knockout-mouse models display globozoospermia. In some models, vesicle fusion leading to acrosome formation is impaired [3–10]. The acrosome is a large, Golgi-derived vesicle that is tethered to the nuclear envelope [11]. The acrosome is formed in round and elongated spermatids [12,13] through budding of vesicles from the trans-Golgi network (TGN). These vesicles are transported to the nuclear envelope, where they fuse to form a single acrosomal vesicle [12,13]. However, other globozoospermia-related proteins are not involved in vesicular transport, but rather in acrosomal anchoring to the nuclear envelope or condensation of the sperm nucleus [14–16].
During spermiogenesis, spermatids undergo dramatic morphological changes, which occur while the cells are transported across the seminiferous epithelium into the lumen. The transport depends on the close interaction between developing sperm and Sertoli cells [17,18]. Actin bundles emanating from Sertoli cells into the ectoplasmic specialization (ES), a testis-specific adherens junction, undergo extensive re-organization while they break-down and reassemble to transport the developing sperm to the lumen [19,20]. A podosome-like structure, the so-called tubulobulbar complex, forms between spermatids and Sertoli cells; it internalizes intact junctions during sperm development and positions the developing sperm cell during the transit through the seminiferous tubules [21,22]. A bundle of filamentous actin (F-actin), which emanates from Sertoli cells, embraces each tubulobulbar complex; this interaction connects the endoplasmic reticulum (ER) of Sertoli cells to the tubulobulbar complex of spermatids [17]. Furthermore, the spermatid manchette, a microtubule-based structure that is transiently formed also contributes to shaping of the sperm head [23]. The manchette consists of a perinuclear microtubule ring. During spermatid elongation, this ring constricts to decrease the diameter of the elongating spermatid head [23].
Here, we demonstrate that cytoskeletal dynamics controlling sperm-head shaping and acrosome formation are affected by accumulation of GlcCer in GBA2 knockout-mice, which results in globozoospermia and, thereby, male infertility.
Results
GBA2 is expressed in Sertoli cells
To investigate the role of GBA2 during spermatogenesis, we analyzed GBA2 expression in the testis (Fig. 1A). Although the main defect in GBA2 knockout-mice occurs in sperm, GBA2 was only weakly if at all expressed in sperm (Fig. 1B). In fact, also mass spectrometry failed to detect peptides derived from GBA2 in mouse sperm. However, three lines of evidence demonstrate that GBA2 is expressed in Sertoli cells. First, immunofluorescent labeling of testis sections demonstrated co-localization of GBA2 and beta-tubulin III, a Sertoli cell marker (Fig. 1C). Second, Western blot-analysis of Sertoli cells, isolated from P7 old males, when the seminiferous epithelia consist exclusively of Sertoli cells and type A spermatogonia [24], showed that GBA2 is expressed in Sertoli cells (Fig. 1D). Finally, mass spectrometry analysis identified four and two unique GBA2 peptides in testis and Sertoli cells, respectively (S1 Fig). In summary, GBA2 is predominantly expressed in Sertoli cells.

In the mammalian testis, sperm are continuously produced throughout the reproductive period. Although spermatogenesis is precisely controlled, only the first spermatogenic wave is synchronized and, therefore, allows assigning the expression of a protein to the respective developmental stage. Thus, we analyzed the time course of GBA2 expression and activity during the first spermatogenic wave (P7: pre-puberty, P21: early puberty) and in adult testis (> 22 weeks). GBA2 followed the expression pattern of beta-tubulin III during the first spermatogenic wave (Fig. 1E). When normalizing the expression of GBA2 to beta-tubulin III, GBA2 expression increased from P7 to P21 and decreased again from P21 to adult (Fig. 1F, G). In parallel, GBA2 activity increased from P7 to P21 and decreased from P21 to adult (Fig. 1H), suggesting that GBA2 plays an important role during the first wave of spermatogenesis.
GlcCer accumulates in testis and sperm of GBA2 knockout mice
The fertility defect of GBA2 knockout-mice has been attributed to the accumulation of GlcCer in testis [1]. Germ and Sertoli cells contain a plethora of different glycosphingolipids (GSLs) [25]. Whereas germ cells contain glycosphingolipids (fucosylated GSLs, GSLs), ceramides (Cer), and sphingomyelings (Spm) with polyenoic very long-chain fatty acids, Sertoli cells predominantly harbor sphingolipids with saturated long-chain fatty acids [25]. We analyzed the amount of neutral sphingolipids in testis, Sertoli cells (P7), and sperm from wild-type and GBA2 knockout-mice by mass spectrometry and distinguished between sphingoid bases (long-chain bases, LCB; C18), ceramides (different chain length, saturated and unsaturated), hexosylceramides (HexCer; different chain length, saturated and unsaturated), and sphingomyelin (different chain length, saturated and unsaturated). In testis from GBA2 knockout-mice, total HexCer levels were dramatically increased, whereas total levels of LCB, Cer, and Spm were not affected (Fig. 2A). Using TLC (thin-layer chromatography), it has already been shown that in brain, only glucosylceramide, but not galactosylceramide accumulates in GBA2 knockout-mice [1]. Thus, the increase in HexCer levels can be attributed to an increase in GlcCer levels. Sertoli cells at P7 did not show a change in any of the sphingolipids (Fig. 2B), whereas in sperm, similar to testis, the GlcCer levels were increased (Fig. 2C). In both testis and sperm, C16 GlcCer levels were most dramatically increased, but also C18, C20, C22, and C24 GlcCer levels were elevated (Fig. 2D, E). In sperm, levels of long-chain GlcCer were similar between wild-type and GBA2 knockout-mice, whereas in testis, the C28 GlcCer levels were increased, indicating an accumulation in Sertoli cells (Fig. 2F). Thus, GlcCer accumulates in testis and sperm from adult GBA2 knockout-mice (˃ 22 weeks), but not in Sertoli cells at P7.

Loss of GBA2 results in cytoskeletal defects in the testis
Sperm from GBA2 knockout-mice display globozoospermia [1]: the heads are round rather than sickle-shaped and the acrosome is malformed (Fig. 3A). However, the underlying molecular mechanisms are not known. The cytoskeleton plays a major role during spermatogenesis, in particular in regulating the interaction between Sertoli and germ cells at the ES and in shaping the sperm head [20,23,26,27]. Using fluorescence microscopy, we investigated the distribution of the actin and microtubule cytoskeleton in the testis of adult wild-type and GBA2 knockout-mice (Fig. 3B): in wild-type testis, F-actin hoops in the ES displayed a sickle-shape alignment around sperm heads, whereas in GBA2 knockout-mice, F-actin accumulated in the ES and was misaligned around the round sperm heads (Fig. 3B). However, the microtubule network appeared rather normal (Fig. 3B).
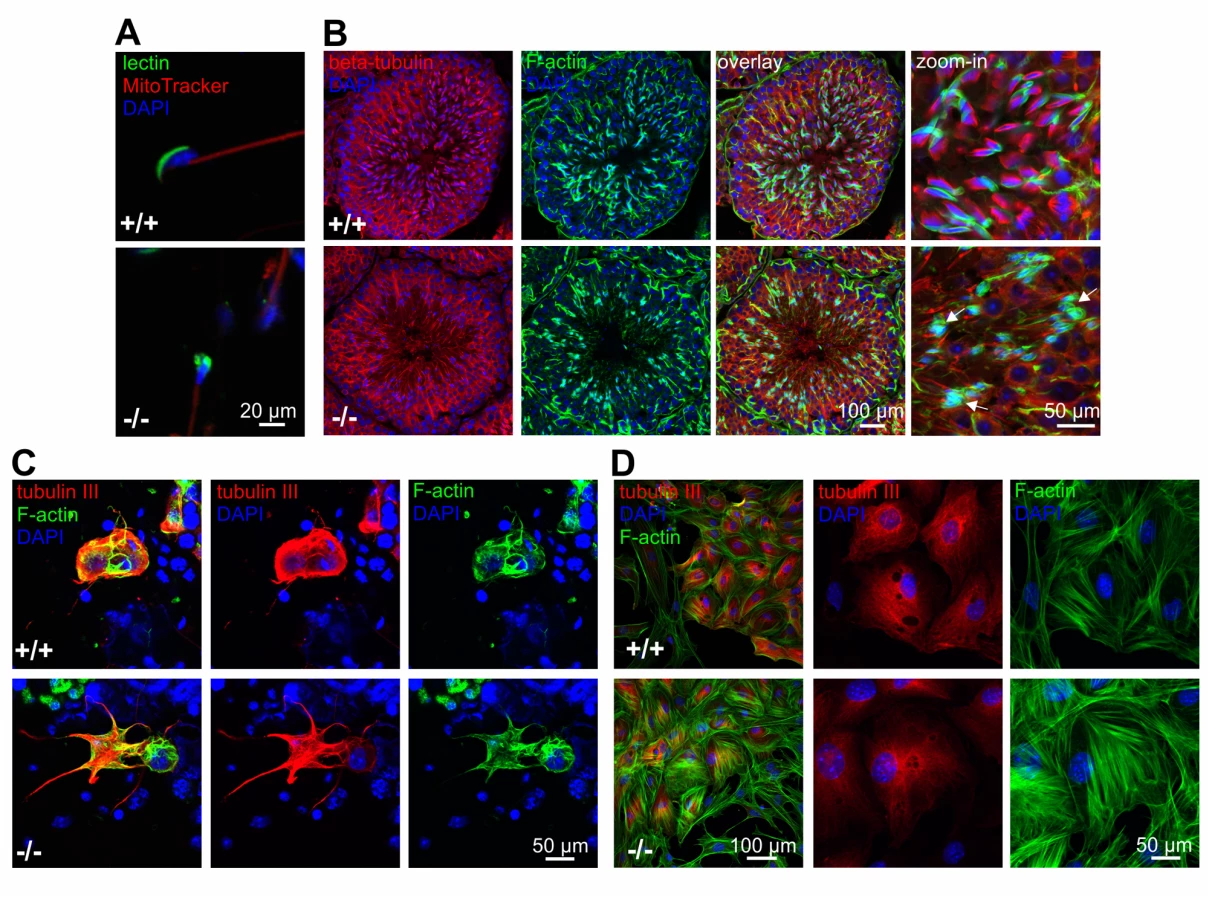
To analyze the cytoskeletal structures on a single cell level, we isolated Sertoli and germ cells from adult wild-type and GBA2 knockout-testis. In isolated cells, both the microtubule and F-actin network appeared more extensive in Sertoli cells from GBA2 knockout-mice with increased microtubule bundling and protrusive F-actin structures (Fig. 3C).
Together, our results show that cytoskeletal defects occur in particular in Sertoli cells, where GBA2 is predominantly expressed.
In testis, GBA2 expression and activity increased from P7 to P21. To correlate the expression and activity of GBA2 in wild-type mice with the cytoskeletal defects in GBA2 knockout-mice, we determined the developmental time course of the cytoskeletal defects. At P7, Sertoli cells from GBA2 knockout-mice did not show any obvious defect in the F-actin or microtubule structures (Fig. 3D), underlining the results from the lipidomics and indicating that P7 might be a too early time point during spermatogenesis for defects to occur. To follow the first spermatogenic wave in more detail, we analyzed the organization of the cytoskeleton in the testis at different stages until P34, when the first wave terminates. Until P21, no difference in the microtubule or actin cytoskeleton was observed between wild-type and GBA2 knockout-mice (Fig. 4A, B). However, at P23, the alignment of F-actin in GBA2 knockout-testis started to change and at P34, the F-actin structures around the sperm heads were misaligned and sperm organization inside the testis lumen was disturbed (Fig. 4C, D). Thus, the occurrence of cytoskeletal defects in GBA2 knockout-mice correlates with the time course of GBA2 expression and activity in wild-type mice.

Formation of the microtubule manchette is altered in GBA2 knockout-spermatids
Although GBA2 is predominantly expressed in Sertoli cells, GlcCer also accumulates in sperm from GBA2 knockout-mice (Fig. 2C). Thus, we hypothesized that the sperm cytoskeleton is also affected by accumulation of GlcCer. Abnormal sperm-head morphology has been linked to defects in the microtubule manchette [28–31]. The manchette is a microtubule structure that appears during the elongation process of spermatids [29,32,33], which first surrounds the proximal tip of the nucleus, gradually migrates towards the caudal end, and creates the force that is needed to shape the sperm head [34]. To analyze the formation of the microtubule manchette in wild-type and GBA2 knockout-spermatids, we monitored its formation in single, isolated germ cells (Fig. 4E). The microtubule manchette of wild-type spermatids was symmetric and conical-shaped, whereas the manchette of knockout spermatids was much more elongated. The manchette length of GBA2 knockout-spermatids was about twofold longer compared to wild-type spermatids (Fig. 4F;-/-: 10.9 ± 1.0 μm and +/+: 5.3 ± 0.5 μm, respectively). Thus, accumulation of GlcCer in the absence of GBA2 affects both the microtubule cytoskeleton of Sertoli cells and developing sperm.
Temporal control of acrosome formation in GBA2 knockout-sperm is disturbed
During sperm development, the cytoskeleton plays a crucial role in forming the acrosome [35]. Acrosome formation and the appearance of the sperm manchette start at the same time [34]. Immunofluorescent analysis of GBA2 knockout-sperm revealed that the formation of the acrosome is disrupted (Fig. 3A) [1]. We therefore analyzed the acrosome formation during the first spermatogenic wave (Fig. 5). The acrosome is derived from vesicles emanating from the TGN [12,13] and its formation can be divided into different phases: Golgi phase, cap phase, and acrosomal phase. In the Golgi phase, vesicles from the TGN fuse at one pole of the nucleus to form a single large vesicle that is attached to the nucleus. In the cap phase, the vesicle flattens over the nuclear membrane, forming a cap-like structure around the spermatid head. In the acrosome phase, the cap-like structures stretches out to form the final acrosome [36]. During the Golgi phase (P21), lectin staining revealed that vesicles in wild-type spermatids were completely fused and polarized, whereas in GBA2 knockout-spermatids, vesicles were still dispursed throughout the cell and not fused at one end of the cell (Fig. 5). This defect was even more prominent in the cap (P23) and the acrosome phase (P34, Fig. 5). The acrosomal cap at P23 was incomplete and at P34, only a few GBA2 knockout-spermatids showed an acrosomal-like structure. Furthermore, the nuclear morphology was altered, leaving sperm with round heads at P34 (Fig. 5). Thus, the cytoskeletal defects in developing sperm and Sertoli cells and the defect in acrosome formation occur in parallel in GBA2 knockout-mice.

Accumulation of GlcCer augments actin polymerization
Germ and Sertoli cells from adult testis are difficult to culture and to manipulate. In contrast, primary fibroblasts are easy to isolate and to maintain in culture. To gain a more mechanistic insight into how accumulation of GlcCer affects cytoskeletal dynamics, we analyzed dermal fibroblasts from adult wild-type and GBA2 knockout-mice as a model system. Wild-type fibroblasts express GBA2 (Fig. 6A) and GBA2 knockout-fibroblasts accumulate GlCer in the absence of GBA2 (Fig. 6B). Indeed, the F-actin and microtubule network appeared strikingly different in GBA2 knockout compared to wild-type fibroblasts, which resulted in a dramatic change in cell morphology—knockout cells were more filopodia-like with extensive protrusions emanating from the plasma membrane (Fig. 6C). However, when maintained in culture, dermal fibroblasts are morphologically heterogeneous, which makes it difficult to analyze cytoskeletal structures in detail. Thus, we seeded fibroblasts on chips that were coated with specific patterns (crossbow, disc, Y-shape, dumb-bell) to force the cells into a given shape. Wild-type fibroblasts fully adhered to the given fibronectin pattern, whereas GBA2 knockout-fibroblasts did not completely align with a given pattern and showed extensive F-actin structures protruding from the cell membrane (Fig. 6D). This was particularly evident for the crossbow and disc shape (Fig. 6D). Cells mainly contain three different F-actin structures: filopodia, lamellipodia, and stress fibers. We compared the different actin structures between wild-type and GBA2 knockout-fibroblasts on the crossbow shape and observed that GBA2 knockout-fibroblasts were more prone to develop filopodia and lamellipodia compared to wild-type cells, while stress fiber formation appeared normal (Fig. 6D, E). Furthermore, the average number of lamellipodia per cell was significantly increased in GBA2 knockout compared to wild-type fibroblasts (3.35 ± 0.58 vs 1.98 ± 0.29, respectively; Fig. 6E).

Actin dynamics are prominently regulated by three members of the Rho family of small GTPases [37]. The formation of filopodia is controlled by Cdc42, whereas lamellipodia formation depends on the activity of Rac1 and stress fiber formation on the activity of RhoA [38–42]. First, we analyzed the expression of Cdc42, Rac1, and RhoA on the mRNA level using qRT-PCR. The expression of RhoA was similar in wild-type and GBA2 knockout-fibroblasts, whereas the expression of both Rac1 and Cdc42 was slightly increased (1.22 ± 0.11 and 1.24 ± 0.15, respectively; Fig. 6F). However, this was not reflected on the protein level: neither the expression level of Rac1 nor of Cdc42 was different between wild-type and GBA2 knockout-fibroblasts (Fig. 6G, H). Thus, a change in the expression level of these key proteins controlling actin dynamics does not underlie the cytoskeletal defects observed in GBA2 knockout-fibroblasts.
Lamellipodia and filopodia formation is induced by actin polymerization [41]. To study the impact of GlcCer accumulation on actin polymerization, we determined the G- and F-actin content and calculated the F-actin/G-actin ratio as a read-out for actin polymerization. The F-actin/G-actin ratio was significantly higher in GBA2 knockout-fibroblasts compared to wild-type fibroblasts, indicating that actin polymerization was augmented in GBA2 knockout-fibroblasts (Fig. 6I). Although actin turnover in testis from adult GBA2 knockout-mice was similar to wild-type mice, we could clearly show that F-actin structures in the ES are augmented (Fig. 6J, 3B). This is probably due to the heterogeneity of cells in adult testis, which makes it difficult to detect a change in F-/G-actin in a particular subcellular structure like the ES.
Taken together, our results reveal that accumulation of GlcCer induces actin polymerization without changing the expression of RhoA, Rac1, or Cdc42.
Accumulation of GlcCer in dermal fibroblasts facilitates microtubule polymerization
An increase in the length of the manchette in GBA2 knockout-spermatids indicates that microtubules persist longer. Thus, we analyzed microtubule dynamics in wild-type and GBA2 knockout-fibroblasts. Cells were transfected with EB3-cherry, a fluorescent probe that binds to the plus-ends of growing microtubules and allows following microtubule assembly using live-cell imaging (Fig. 6K) [43]. Growing plus-ends appear as shooting comets, which allows determining the microtubule growth rate and persistence (Fig. 6L, M). Microtubule growth rate was not different between genotypes, but microtubules in GBA2 knockout-fibroblasts persisted significantly longer compared to wild-type fibroblasts (Fig. 6L, M). This is in line with the finding that microtubules in the manchette from GBA2 knockout-spermatids appear longer (Fig. 4E, F).
Accumulation of GlcCer accelerates cell migration
In fibroblasts, microtubule polymerization can induce actin polymerization and, together, promote cell migration [44]. Thus, we analyzed the migration of wild-type and GBA2 knockout-fibroblasts in a wound-healing assay. GBA2 knockout-fibroblasts migrated faster than wild-type cells, in particular 2 to 6 h after starting the assay (Fig. 7A, B). Thus, loss of GBA2 followed by an accumulation of GlcCer promotes microtubule and actin polymerization, which in turn changes cellular behavior.

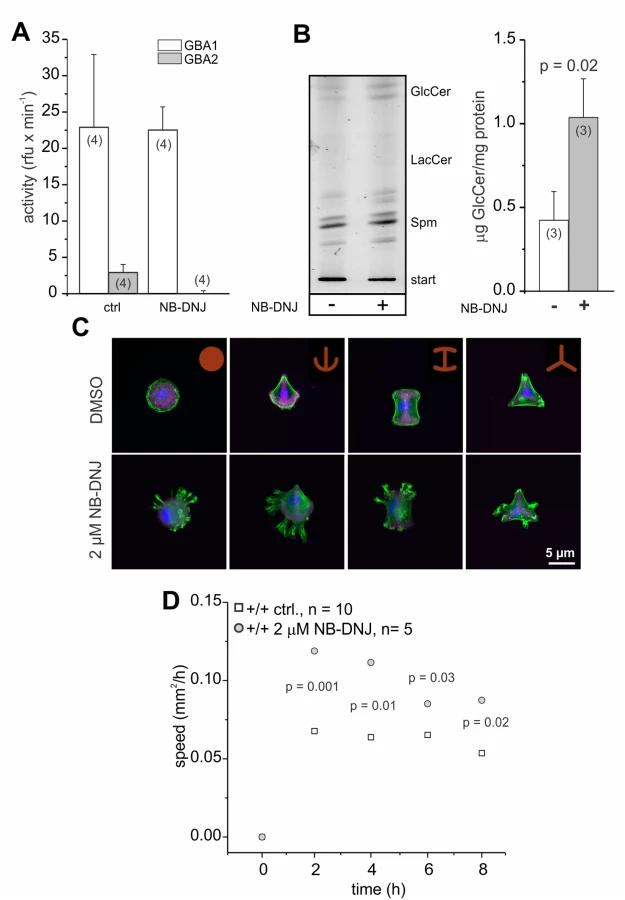
Inhibition of GBA2 with NB-DNJ also affects cytoskeletal dynamics and cell migration
To analyze whether the cellular defects observed in GBA2 knockout-fibroblasts are solely due to the loss of GBA2 and, thereby, accumulation of GlcCer, we independently assessed the contribution of GBA2 in controlling cytoskeletal dynamics by incubating wild-type fibroblasts with the GBA2 inhibitor NB-DNJ [45]. Treatment of fibroblasts for 48 h with 2 μM NB-DNJ abolished GBA2 activity (Fig. 8A). In turn, GlcCer accumulated (Fig. 8B). Similar to GBA2 knockout-fibroblasts, wild-type fibroblasts treated with NB-DNJ showed a striking difference in the organization of the F-actin cytoskeleton (Fig. 8C). We also analyzed the migration of cells treated with NB-DNJ. Similar to GBA2 knockout-fibroblasts, fibroblasts treated with NB-DNJ migrated faster than non-treated cells (Fig. 8D). Thus, inhibition of GBA2 has a similar effect on cytoskeletal dynamics and cellular behavior as genetically ablating GBA2.
Loss of GBA2/accumulation of GlcCer alters lipid packaging and membrane fluidity
The cell membrane is a lipid bilayer, in which proteins are incorporated. The lipid composition of the membrane determines its characteristics and determines the function of proteins at the membrane. GlcCer is incorporated into the inner leaflet of the plasma membrane [46]. We hypothesized that accumulation of GlcCer increases the amount of GlcCer in the plasma membrane and that this controls the function of proteins at the membrane. Thus, we analyzed lipid stacking in intact plasma membranes isolated from wild-type and GBA2 knockout-fibroblasts. Giant plasma-membrane vesicles (GPMV) were isolated from fibroblasts by chemical vesiculation [47] and analyzed using laurdan fluorescent labeling. Laurdan is a membrane dye that reports the extent of water penetration into the bilayer surface due to the dipolar relaxation effect [48]. Water penetration can be directly correlated with lipid packaging and membrane fluidity [49]. Thus, laurdan fluorescence reports lipid packaging in the membrane. For highly ordered membranes, laurdan emission peaks at around 440 nm, whereas it is shifted to longer wavelengths in relatively disordered membranes [47]. GPMVs from wild-type and GBA2 knockout-fibroblasts were loaded with laurdan and the emission spectrum was measured (Fig. 9). The emission peak for wild-type fibroblasts was centered at around 460 nm, whereas the emission peak for GBA2 knockout-fibroblasts was shifted to shorter wavelengths at around 440 nm. This demonstrates that lipid packaging in the plasma membrane form GBA2 knockout-fibroblasts is more ordered than in wild-type fibroblasts. To quantify the shift, we calculated the generalized polarization (GP) index. The GP index in GBA2 knockout-fibroblasts was significantly higher than in wild-type fibroblasts (0.15 ± 0.06 vs. 0.24 ± 0.03, respectively; Fig. 9). We performed similar experiments with wild-type fibroblasts treated with 2 μM NB-DNJ for 48 h. The emission peak for GPMVs isolated from treated cells was also shifted to shorter wavelength with a GP index of 0.26 ± 0.07 (Fig. 9).

In summary, our data indicate that accumulation of GlcCer in the absence of GBA2 leads to a more ordered lipid packaging in the plasma membrane. This in turn seems to control the activity of proteins incorporated in or associated with the plasma membrane, resulting in an increase in actin and microtubule polymerization. In vivo, this defect is most prominent in the testis, resulting in malformation of the acrosome and the sperm head and, thereby, in male infertility.
Discussion
Spermatogenesis is a complex process that relies on the interaction between germ and Sertoli cells. Our study reveals that the glycosphingolipid GlcCer regulates cytoskeletal dynamics in germ and Sertoli cells. Accumulation of GlcCer due to the lack of GBA2 activity induces actin polymerization and microtubule persistence, resulting in globozoospermia and male infertility.
The role of GlcCer in controlling cytoskeletal dynamics has not been established so far. However, it has been shown that ceramide, in particular C16 ceramide, promotes actin polymerization in mouse embryonic stem cells (ESCs) through PKC (protein kinase C) activation and FAK (focal adhesion kinase)/paxillin-dependent N-WASP/Cdc42/Arp2/3 complex formation [50]. This, in turn, increases cell migration. Ceramide serves as a building block for glycosphingolipids with GlcCer being one of them. Thus, the cellular changes observed after increasing C16 ceramide levels could also be due to an increase in GlcCer levels. Indeed, in GBA2 knockout-fibroblasts, we observed an increase in actin polymerization—similar to what has been described for ESCs treated with C16 ceramide [50]. Regulation of actin polymerization by lipids has also been shown for phospholipids using PIP2-containing vesicles [51]. In particular, PIP2 controls actin dynamics in highly polarized cells like T cells and neurons [52–54]. Here, PIP2 accumulates in membrane microdomains, where it directly regulates protein function and recruits proteins to form signaling complexes at the plasma membrane.
Not only phospholipids, but also glycosphingolipids accumulate in microdomains, where they act as signaling domains involved in recognition processes [55]. GlcCer is a structural element in the inner leaflet of the plasma membrane, but can also be incorporated into the outer leaflet of the plasma membrane. It has been suggested that GlcCer at the cytosolic surface of the plasma membrane forms signaling domains by recruiting specific proteins, thereby, controlling e.g. vesicle budding [46]. Using the environment-sensitive dye laurdan in GPMVs, we demonstrated that accumulation of GlcCer results in a more ordered lipid packaging of the plasma membrane. Future studies will reveal whether this occurs in a localized or uniform manner.
A change in lipid composition and/or lipid packaging of the membrane might also directly control protein function. For the EGF receptor (epidermal growth factor receptor), it has been demonstrated that the ganglioside GM3, surrounding the receptor in the membrane, regulates the allosteric transition from an inactive to an active EGF receptor dimer even in the absence of the ligand [56]. Furthermore, it has been shown that GlcCer controls the activity of a V-type ATPase in melanocytes [57]. Our results suggest that GlcCer accumulation in the plasma membrane controls the function of proteins like Cdc42, thereby, regulating cytoskeletal dynamics.
The role of GlcCer during spermatogenesis has been enigmatic so far. Our results suggest that GBA2 is predominantly expressed in Sertoli cells. However, more experiments are needed to clarify the physiological function of GBA2 during sperm development. In particular, Sertoli-cell specific knockout-mice will allow to unravel the role of GBA2 in Sertoli cells. In addition, analyzing the expression of GBA2 in different cell types during germ cell development will help to further understand GBA2 function in the testis.
Our results demonstrate that the GBA2 substrate GlcCer accumulates in both, Sertoli cells and sperm in GBA2 knockout-mice. Similar to our observations in dermal fibroblasts, cytoskeletal structures were also altered in Sertoli cells and sperm. Sertoli cells are polarized cells that are connected to germ cells via the ES at the apical end [20,27]. This allows to exchange proteins and lipids between these two cell types [58]. Thus, GlcCer could accumulate in germ cells even though GBA2 is not expressed there (at least not in detectable amounts). Furthermore, one function of Sertoli cells is to ensure a sufficient availability of lipids required for proliferation of millions of germ cells per day [58]. One way for the Sertoli cells to provide lipids is by recycling the lipid content of the residual body from spermatids, which is phagocytosed during spermatogenesis [58]. GBA2 could play a major role in this recycling process and accumulation of GlcCer in Sertoli cells would be further augmented. Accumulation of GlcCer in the testis in GBA2 knockout-mice occurs after P7. This is in line with our finding that GBA2 expression and activity in wild-type testis increases after P7 during sperm development. We hypothesize that GlcCer accumulates in particular at the junction between germ and Sertoli cells, similar to what has been shown for PIP2 in other polarized cells like T cells or neurons, where PIP2 forms microdomains at the immunological synapse or at the growth cone of the axon [52–54]. In GBA2 knockout-testis, F-actin hoops in the ES and the microtubule manchette in spermatids were majorly disrupted. F-actin hoops were misaligned around the developing sperm head and the microtubule manchette was more elongated. Both structures have been shown to be crucial for shaping the sperm head and forming the acrosome [35]. Interestingly, in GBA2 knockout-fibroblasts, actin polymerization was augmented and microtubule persistence was increased. These results indicate that lack of GBA2 and the concomitant accumulation of GlcCer affect similar signaling pathways in different cell types, suggesting that the regulation of cytoskeletal dynamics by GlcCer is a general mechanism.
Glycosphingolipids not only play a role in the plasma membrane, but also in membranes of organelles. In the Golgi and endosomes, glycosphingolipids have been shown to form microdomains that segregate membrane proteins and drive their sorting [59]. Furthermore, it has been proposed that GlcCer controls vesicle budding from the TGN [46]. The acrosome is formed by fusion of proacrosomal vesicles from the TGN. Thus, accumulation of GlcCer in sperm might disturb acrosome formation by inhibiting vesicle budding from the TGN and vesicle fusion. Indeed, vesicle fusion in GBA2 knockout-mice was disturbed during the cap and acrosome phase of acrosome formation, suggesting that GlcCer controls acrosome formation by regulating vesicle fusion.
In summary, our study reveals that GlcCer is a key regulator of cytoskeletal dynamics. This is particularly important during sperm development, opening up new avenues in understanding the molecular mechanisms underlying male infertility.
Material and Methods
Ethics statement
All animal experiments were in accordance with the relevant national and international guidelines and regulations. Animal procedures were approved by the local authorities (LANUV NRW). The generation of GBA2 knockout-mice has been described elsewhere [1].
Isolation of sperm and male germ cells
Sperm were isolated by incision of the cauda epididymis in modified TYH medium containing 138 mM NaCl, 4.8 mM KCl, 2 mM CaCl2, 1.2 mM KH2PO4, 1 mM MgSO4, 5.6 mM glucose, 0.5 mM sodium pyruvate, 10 mM L-lactate, pH 7.4. After 15 min swim out at 37°C and 5% CO2, sperm were counted. All subsequent experiments were performed at room temperature, unless otherwise stated.
For isolation of germ cells, testes were decapsulated and incubated in 1 ml Hank’s Balanced Salt Solution (HBSS) (20 mM HEPES, 137 mM NaCl, 5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 1.2 mM MgSO4, 1.3 mM CaCl2, 6.6 mM sodium pyruvate, 0.05% lactate, 5.6 mM glucose, pH 7.2) containing 0.5 mg/ml collagenase type IA (Sigma) for 30 min at 32°C. The dissociated interstitial cells were removed by two washing steps with HBSS. The seminiferous tubules were then incubated in 1 ml HBSS containing 0.5 mg/ml Trypsin type XIII (Sigma) and 1 μg/ml DNase I (Applichem) for 10 min at 32°C. Cell aggregates were sheared gently with a Pasteur pipette. The dispersed seminiferous cells were washed twice by centrifugation at 200 x g for 5 min at room temperature. The final cell pellet was re-suspended in HBSS and filtered through a Nylon mesh (40 μm mesh).
Isolation of P7 Sertoli cells
Seminiferous tubules were isolated from testes of 7 days old mice (P7) by removal of the tunica albuginea. The tubules were treated with 1 mg/ml collagenase (Sigma) at 37°C in a shaker for 8 min. The digestion was stopped by addition of DMEM/GlutaMax medium (Invitrogen) containing 10% FCS (Biochrom). The cell suspension was centrifuged at 400 x g for 8 min, the pellet was re-suspended in medium containing 0.5 mg/ml trypsin and 0.22 mg/ml EDTA (Sigma), and incubated in a shaker at 37°C for 5 min. The reaction was stopped by adding medium. The cell suspension was then treated with 1 μg/ml DNase I (Applichem) in a shaker at 37°C for 5 min. The cells were centrifuged at 600 x g for 10 min, and re-suspended in medium containing 70 U/ml penicillin, 70 μg/ml streptomycin, 100 mM sodium pyruvate, and 200 mM L-glutamine (all Life technologies). Cells were seeded at a density of 5 x 104 cells/5 cm cell culture plate (Greiner bio-one) and used on the 5th day for experiments. The purity of the cell population isolated using this protocol has been analyzed using immunocytochemistry using markers for Sertoli cells (beta-tubulin III, Sox9)—the preparation contains ca. 80% Sertoli cells.
Isolation of dermal fibroblasts
Dermal fibroblasts were isolated from mouse tails using collagenase digestion. Tail pieces were incubated in DMEM/GlutaMax containing 10% FCS, 100 mM sodium pyruvate, 200 mM L-glutamine, 70 I.U./ml penicillin, 70 μg/ml streptomycin, 0.1 mg/ml collagenase (Sigma) for 3 h, 37°C, and 5% CO2. After digestion, the supernatant was centrifuged for 5 min, 600 x g at room temperature. The cell pellet was re-suspended in DMEM medium, cells were plated on cell culture plates, and cultured at 37°C, 5% CO2. After 24 h, the medium was changed.
Antibodies and dyes
Primary antibodies: 4A12 (rat, ICC: 1:20), 2F8 (rat, WB: 1:50), pcGBA2 (rb, ICC/IHC: 1:2,000) [1,45], calnexin (Sigma #C4731, WB: 1:20,000), beta-tubulin-CY3 (Sigma #C4585, ICC: 1:200), beta-tubulin (Sigma T4026, WB: 1:1,000), HA (Roche #11867431001, WB: 1:10,000), beta-tubulin III (HISS Diagnostics, MMS-435P, ICC: 1:500, WB: 1:1,000).
Secondary antibodies: WB: IRDye680 and IRDye800 antibodies (LI-COR, 1:20,000); ICC: fluorescently-labeled antibodies (Dianova, 1:500).
Dyes: Alexa Fluor 488 Phalloidin (Molecular Probes, #A12379, ICC: 1:500), MitoTracker (Molecular Probes, M22426, ICC: 0.5 μM), peanut lectin (Sigma, #7381, ICC: 1:100), DAPI (Molecular Probes, D1306, ICC: 1:10,000).
Immunohistochemistry (IHC) and—cytochemistry (ICC)
Testes were fixed overnight with 4% paraformaldehyde/PBS, cryo-protected in 10 and 30% sucrose, and afterwards embedded in TissueTec (Sakura Finetek). Sperm were immobilized on microscope slides and fixed for 10 min. To block unspecific binding sites, frozen sections and sperm were incubated for 1 h with blocking buffer (0.5% Triton-X 100 and 5% ChemiBLOCKER (Millipore) in 0.1 M phosphate buffer, pH 7.4). Fibroblasts, Sertoli, and germ cells were fixed for 10 min. Primary antibodies were diluted in blocking buffer and incubated overnight. Fluorescent secondary antibodies were diluted in blocking buffer containing 0.5 μg/μl DAPI (Invitrogen) and pictures were taken on a confocal microscope (Olympus FV1000). For the analysis of cytoskeletal structures in dermal fibroblasts, cells were seeded on multi-pattern fibronectin-coated CYTOO chips (#10–900–13–06, CYTOO Cell Architects).
Protein preparation
All steps were performed at 4°C in the presence of mammalian protease inhibitor cocktail (Sigma Aldrich). Tissues or cells were homogenized in a 10-fold surplus (v/w) of hypotonic buffer (10 mM HEPES, 0.5 mM EDTA, pH 7.4) by using an Ultra-turrax (IKA) and three pulses (20 s each) of sonification (Branson sonifier). The suspension (total lysate) was centrifuged for 20 min at 1,000 × g. The supernatant (PNS, post-nuclear supernatant) was used for activity assays or Western-blot analysis.
Fluorescence-based GBA activity assays
The assay has been performed as described previously [45]. Briefly, cleavage of 4-methylumbelliferyl(MU)-beta-D-glucopyranoside (Sigma Aldrich) was monitored in real-time in a Fluostar Omega reader (BMG labtech) at 29°C using the filter pair 355 nm/460 nm for excitation and emission, respectively. The assays were performed in 384-well plates (Greiner) in the plate mode. Per well, 25 μl of lysate containing 20 μg of total protein were used. To discriminate between GBA1 and GBA2 activity, 30 μM CBE (Conduritol B epoxide, Sigma Aldrich), an inhibitor for GBA1, or 10 μM NB-DNJ (N-butyldeoxynojirimycin, Sigma Aldrich), an inhibitor for GBA2, were included. The pH of the protein lysates and the 4-MU-beta-D-glucopyranoside solution were adjusted by diluting with McIlvaine buffer. The assay was initiated by adding 5 μl of 4-MU-beta-D-glucopyranoside (10 mM) resulting in a final concentration of 1.67 mM. The hydrolysis of 4-MU-beta-D-glucopyranoside was monitored and recorded as a change of relative fluorescence units (rfu) per minute. Each analysis was performed as a quadruplicate in parallel. Per genotype, tissues or cells from three animals were analyzed if not otherwise stated.
Western-blot analysis
All samples were heated for 5 min at 95°C prior to separation on SDS-PAGE. For Western-blot analysis, proteins were transferred onto PVDF membranes and probed with antibodies by using the Odyssey Imaging System (LI-COR).
Transfection
1x106 mouse fibroblasts were re-suspended in 100 μl of transfection buffer (Neon transfection system, Life technologies) and 4 μg of plasmid DNA was added. Using a microporator mini (Digital Bio Technology, MP-100), 10 μl of the cell suspension were subjected to two pulses (20 ms each) of 1000 V and afterwards transferred to Poly-L-Lysine-coated glass-bottom dishes (Mat Tek, #P35G-1.5–20-C). A total of 30 μl of cells were electroporated. The cells were allowed to grow overnight at 37°C and 5% CO2 in medium.
Live-cell imaging
Cells were imaged 24 h after transfection using the DeltaVision Core microscope (Applied Precision, Inc.). Images were acquired every 3 s for 200–500 ms over 5 min and image analysis was done using Metamorph (version 7.0, Molecular Devices Corporation) using the track-points function. Microtubule tracks were followed from the first frame an EB3-labeled microtubule plus-tip appeared until the last frame (when the plus-tip was no longer visible). Data for velocity (microtubule growth-rate) and distance (microtubule persistence) were calculated. At least 10 microtubule tracks were followed in each cell, and at least 7 cells per cell line and genotype were analyzed.
Identification of mGBA2 by mass spectrometry
For GeLCMS, proteins of Sertoli cells, testis, and sperm were separated on SDS gels and stained with Coomassie. Per lane, 14–17 gel slices were excised, proteins were in-gel digested with trypsin (Promega), peptides were separated in a 90 or 180 min gradient by a nanoAcquity LC System equipped with a HSS T3 analytical column (1.8 μm particle, 75 μm x 150 mm) (Waters), and analyzed by ESI-LC-MS/MS using an LTQ Orbitrap Elite mass spectrometer (Thermo Scientific). All database searches were performed using SEQUEST as well as MS Amanda (Mechtler lab, Vienna, Austria) algorithm, embedded in Proteome Discoverer (Rev. 1.4, Thermo Electron 2008–2011), with a NCBI protein database (mouse, accession number NP_766280.2, accessed June 13, 2013). Only fully tryptic peptides with up to two missed cleavages were accepted. Oxidation of methionine was permitted as variable modification. The mass tolerance for precursor ions was set to 10 ppm; the mass tolerance for fragment ions was set to 0.4 amu. To filter the results, a peptide FDR threshold of 0.01 (q-value) according to Percolator was set in Proteome Discoverer; two peptides per protein and peptides with search result rank 1 were required.
Lipid analysis using thin-layer chromatography (TLC)
For lipid extraction, dermal fibroblasts from GBA2 wild-type (+/+) and knockout-mice (-/-) were grown until confluency, washed once in PBS, and harvested using trypsin/EDTA in medium. Cells were pelleted for 7 min at 700 x g and room temperature. Afterwards, cells were lysed in 1 ml bi-destilled water with three pulses (30 s each) of sonification (Branson sonifier). Lipids were extracted for 24 h at 37°C in chloroform/methanol/water (10/5/1, v/v/v). For a better analysis of glucosylceramide, glycerophospholipids were degraded by alkaline hydrolysis with 125 mM sodium hydroxide for 2 h at 37°C. After neutralization with acetic acid, lipid extracts were desalted by reversed-phase chromatography and separated into acidic and neutral glycosphingolipids as described previously [60,61].
For separation of neutral lipids by thin layer chromatography (TLC), lipids derived from an extract of 1–1.5 mg of total protein were applied to prewashed thin layer Silica Gel 60 (Merck, Darmstadt, Germany) and chromatograms were developed and quantified as described previously [61].
Extraction and quantification of sphingolipids using mass spectrometry
Sperm cells, testis, and Sertoli cells were frozen in liquid nitrogen and ground to a fine powder using the Precellys24 tissue homogenizer (PeqLab). Lipids were extracted and fractionated using solid-phase-extraction on silica columns [62]. Long chain bases (LCB), ceramides (Cer), and hexosylceramides (HexCer) were eluted with acetone/2-propanol (9/1, v/v) and sphingomyelin (Spm) was eluted with methanol. The purified sphingolipids were analyzed via direct infusion nanospray mass-spectrometry using an Agilent 6530 Accurate-Mass Q-TOF LC/MS device [62]. Sphingolipids were quantified after collision-induced dissociation by scanning for specific fragment ions: LCB, neutral loss of 18.0106; Cer and HexCer, precursor ion scanning for m/z 266.2842 (d18:0), m/z 264.2686 (d18:1) and m/z 262.2493 (d18:2); Spm, precursor ion scanning for m/z 184.0739. Internal standards were added for each sphingolipid class [63].
G-/F-Actin assay
The assay was performed according to the manufacturer’s protocol (#BK037, Cytoskeleton). In brief, cells were lysed in a detergent-based lysis buffer, which solubilizes G-actin but also stabilizes and maintains F-actin. As a control, protein lysates were treated with an F-actin polymerizing solution for 15 min at 37°C. Equal volumes of each samples were subjected to ultracentrifugation (100,000 x g, 1 h) to separate F-actin from G-actin. F-actin was maintained in the pellet fraction, whereas G-actin was maintained in the supernatant. The pellet was dissolved in F-actin depolymerizing buffer and incubated on ice for 1 h. Samples were run on a SDS-PAGE and analyzed by Western blot. Ratios of F-actin/G-actin for each genotype were calculated as percentage of the control.
Analysis of actin structures
Mouse fibroblasts were plated on Cytoo chips (Cytoo Cell Architects, # 10–900–13–06) placed in a 35 mm cell culture plate. Cells were fixed with 4% paraformaldehyde and labeled with Alexa Fluor 488 Phalloidin and DAPI. Images were taken using an Olympus FV1000 confocal microscope. Filopodia (slender actin-protrusions) and lamellipodia (wave-like actin extensions) structures were counted.
Wound-healing assay
Silicone cell culture-inserts (Ibidi) with a defined cell-free gap (width = 500 μm) were placed in 35 mm cell culture dishes. 4 x 104 cells were transferred into each of the culture inserts and incubated at 37°C, 5% CO2 for 2 h. Afterwards, inserts were removed and cells were washed with PBS. Fresh medium was added and a phase contrast image was taken (t = 0 h) using the Nikon eclipse (TE 2000-S) microscope. An image of the same region was taken every 2 hours (t = 2, 4, 6, 8 h). The area of the cell-free gap was measured using ImageJ (version 1.46m) and the speed of migration was calculated.
Isolation of GPMVs
Giant plasma-membrane vesicles (GPMVs) were isolated as described elsewhere [47]. In brief, dermal fibroblasts were incubated with GPMV buffer (10 mM HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7.4) containing 2 mM NEM for 1–2 h at 37°C, 5% CO2. The supernatant was centrifuged for 10 min at 2000 x g and room temperature to pellet cell debris and intact cells. The resulting supernatant was subjected to high-speed centrifugation for 1 h at 20,000 x g and 4°C to pellet the vesicles. The pellet was re-suspended in GPMV buffer.
Fluorescence spectroscopy
Measurements were performed in a quartz cuvette using the FluoroMax-4 Spectrofluorometer. The emission spectrum was recorded from 400 to 500 nm at 385 nm excitation to detect the lipid resonance-peak at 425 nm. All samples were normalized to the lipid resonance-peak for the GPMV buffer. GPMVs were labeled with 5 μM laurdan (6-Dodecanoyl-2-Dimethylaminonaphthalene, Molecular Probes, #D250) for 20 min at 23°C. Measurements were performed at 350 nm excitation and fluorescence emission was recorded from 400 to 600 nm. All measurements were done at 23°C. The GP value was calculated according to the following equation:

Supporting Information
Zdroje
1. Yildiz Y, Matern H, Thompson B, Allegood JC, Warren RL, et al. (2006) Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J Clin Invest 116: 2985–2994. 17080196
2. Dam AH, Feenstra I, Westphal JR, Ramos L, van Golde RJ, et al. (2007) Globozoospermia revisited. Hum Reprod Update 13: 63–75. 17008355
3. Funaki T, Kon S, Tanabe K, Natsume W, Sato S, et al. (2013) The Arf GAP SMAP2 is necessary for organized vesicle budding from the trans-Golgi network and subsequent acrosome formation in spermiogenesis. Mol Biol Cell 24: 2633–2644. doi: 10.1091/mbc.E13-05-0234 23864717
4. Heimann P, Laage S, Jockusch H (1991) Defect of sperm assembly in a neurological mutant of the mouse, wobbler (WR). Differentiation 47: 77–83. 1955109
5. Kang-Decker N, Mantchev GT, Juneja SC, McNiven MA, van Deursen JM (2001) Lack of acrosome formation in Hrb-deficient mice. Science 294: 1531–1533. 11711676
6. Moreno RD, Palomino J, Schatten G (2006) Assembly of spermatid acrosome depends on microtubule organization during mammalian spermiogenesis. Dev Biol 293: 218–227. 16540102
7. Paiardi C, Pasini ME, Gioria M, Berruti G (2011) Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis 1: 52–62. 21866276
8. Pierre V, Martinez G, Coutton C, Delaroche J, Yassine S, et al. (2012) Absence of Dpy19l2, a new inner nuclear membrane protein, causes globozoospermia in mice by preventing the anchoring of the acrosome to the nucleus. Development 139: 2955–2965. doi: 10.1242/dev.077982 22764053
9. Xiao N, Kam C, Shen C, Jin W, Wang J, et al. (2009) PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. J Clin Invest 119: 802–812. doi: 10.1172/JCI36230 19258705
10. Yao R, Ito C, Natsume Y, Sugitani Y, Yamanaka H, et al. (2002) Lack of acrosome formation in mice lacking a Golgi protein, GOPC. ProcNatlAcadSci USA 99: 11211–11216. 12149515
11. Kierszenbaum AL, Rivkin E, Tres LL (2003) Acroplaxome, an F-actin-keratin-containing plate, anchors the acrosome to the nucleus during shaping of the spermatid head. Mol Biol Cell 14: 4628–4640. 14551252
12. Abou-Haila A, Tulsiani DR (2000) Mammalian sperm acrosome: formation, contents, and function. Arch Biochem Biophys 379: 173–182. 10898932
13. Moreno RD, Alvarado CP (2006) The mammalian acrosome as a secretory lysosome: new and old evidence. Mol Reprod Dev 73: 1430–1434. 16894549
14. Escalier D, Silvius D, Xu X (2003) Spermatogenesis of mice lacking CK2alpha': failure of germ cell survival and characteristic modifications of the spermatid nucleus. Mol Reprod Dev 66: 190–201. 12950107
15. Fujihara Y, Satouh Y, Inoue N, Isotani A, Ikawa M, et al. (2012) SPACA1-deficient male mice are infertile with abnormally shaped sperm heads reminiscent of globozoospermia. Development 139: 3583–3589. doi: 10.1242/dev.081778 22949614
16. Xu X, Toselli PA, Russell LD, Seldin DC (1999) Globozoospermia in mice lacking the casein kinase II alpha' catalytic subunit. Nat Genet 23: 118–121. 10471512
17. Guttman JA, Takai Y, Vogl AW (2004) Evidence that tubulobulbar complexes in the seminiferous epithelium are involved with internalization of adhesion junctions. Biol Reprod 71: 548–559. 15084482
18. Russell L, Clermont Y (1976) Anchoring device between Sertoli cells and late spermatids in rat seminiferous tubules. Anat Rec 185: 259–278. 937734
19. Xiao X, Yang WX (2007) Actin-based dynamics during spermatogenesis and its significance. J Zhejiang Univ Sci B 8: 498–506. 17610330
20. Mruk DD, Cheng CY (2004) Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev 25: 747–806. 15466940
21. Young JS, Guttman JA, Vaid KS, Vogl AW (2009) Tubulobulbar complexes are intercellular podosome-like structures that internalize intact intercellular junctions during epithelial remodeling events in the rat testis. Biol Reprod 80: 162–174. doi: 10.1095/biolreprod.108.070623 18799754
22. Russell LD (1979) Spermatid-Sertoli tubulobulbar complexes as devices for elimination of cytoplasm from the head region late spermatids of the rat. Anat Rec 194: 233–246. 464324
23. Kierszenbaum AL, Rivkin E, Tres LL (2007) Molecular biology of sperm head shaping. Soc Reprod Fertil Suppl 65: 33–43. 17644953
24. Bellve AR, Millette CF, Bhatnagar YM, O'Brien DA (1977) Dissociation of the mouse testis and characterization of isolated spermatogenic cells. J Histochem Cytochem 25: 480–494. 893996
25. Rabionet M, van der Spoel AC, Chuang CC, von Tumpling-Radosta B, Litjens M, et al. (2008) Male germ cells require polyenoic sphingolipids with complex glycosylation for completion of meiosis: a link to ceramide synthase-3. J Biol Chem 283: 13357–13369. doi: 10.1074/jbc.M800870200 18308723
26. Kierszenbaum AL (2004) Polycystins: what polycystic kidney disease tells us about sperm. Mol Reprod Dev 67: 385–388. 14991728
27. Mruk DD, Cheng CY (2004) Cell-cell interactions at the ectoplasmic specialization in the testis. Trends Endocrinol Metab 15: 439–447. 15519891
28. Meistrich ML, Trostle-Weige PK, Russell LD (1990) Abnormal manchette development in spermatids of azh/azh mutant mice. Am J Anat 188: 74–86. 2346121
29. Russell LD, Russell JA, MacGregor GR, Meistrich ML (1991) Linkage of manchette microtubules to the nuclear envelope and observations of the role of the manchette in nuclear shaping during spermiogenesis in rodents. Am J Anat 192: 97–120. 1759685
30. Nayernia K, Vauti F, Meinhardt A, Cadenas C, Schweyer S, et al. (2003) Inactivation of a testis-specific Lis1 transcript in mice prevents spermatid differentiation and causes male infertility. J Biol Chem 278: 48377–48385. 13129914
31. Akhmanova A, Mausset-Bonnefont AL, van Cappellen W, Keijzer N, Hoogenraad CC, et al. (2005) The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev 19: 2501–2515. 16230537
32. Kierszenbaum AL (2001) Spermatid manchette: plugging proteins to zero into the sperm tail. Mol Reprod Dev 59: 347–349. 11468770
33. Wolosewick JJ, Bryan JH (1977) Ultrastructural characterization of the manchette microtubules in the seminiferous epithelium of the mouse. Am J Anat 150: 301–331. 920632
34. Tarsounas M, Pearlman RE, Moens PB (2001) CLIP-50 immunolocalization during mouse spermiogenesis suggests a role in shaping the sperm nucleus. Dev Biol 236: 400–410. 11476580
35. Sperry AO (2012) The dynamic cytoskeleton of the developing male germ cell. Biol Cell 104: 297–305. doi: 10.1111/boc.201100102 22276751
36. Moreno RD, Ramalho-Santos J, Sutovsky P, Chan EK, Schatten G (2000) Vesicular traffic and golgi apparatus dynamics during mammalian spermatogenesis: implications for acrosome architecture. Biol Reprod 63: 89–98. 10859246
37. Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701. doi: 10.1038/nrm2476 18719708
38. Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509–514. 9438836
39. Machesky LM, Hall A (1996) Rho: a connection between membrane receptor signalling and the cytoskeleton. Trends Cell Biol 6: 304–310. 15157438
40. Nobes CD, Hall A (1995) Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans 23: 456–459. 8566347
41. Ridley AJ, Hall A (1992) The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389–399. 1643657
42. Van Aelst L, D'Souza-Schorey C (1997) Rho GTPases and signaling networks. Genes Dev 11: 2295–2322. 9308960
43. Efimov A, Schiefermeier N, Grigoriev I, Ohi R, Brown MC, et al. (2008) Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J Cell Sci 121: 196–204. doi: 10.1242/jcs.012666 18187451
44. Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED (1999) Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol 1: 45–50. 10559863
45. Korschen HG, Yildiz Y, Raju DN, Schonauer S, Bonigk W, et al. (2013) The non-lysosomal beta-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J Biol Chem 288: 3381–3393. doi: 10.1074/jbc.M112.414714 23250757
46. van Meer G, Wolthoorn J, Degroote S (2003) The fate and function of glycosphingolipid glucosylceramide. Philos Trans R Soc Lond B Biol Sci 358: 869–873. 12803919
47. Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, et al. (2012) Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat Protoc 7: 1042–1051. doi: 10.1038/nprot.2012.059 22555243
48. Weber G, Farris FJ (1979) Synthesis and spectral properties of a hydrophobic fluorescent probe: 6-propionyl-2-(dimethylamino)naphthalene. Biochemistry 18: 3075–3078. 465454
49. Parasassi T, Gratton E (1995) Membrane lipid domains and dynamics as detected by Laurdan fluorescence. J Fluoresc 5: 59–69. doi: 10.1007/BF00718783 24226612
50. Park SS, Kim MO, Yun SP, Ryu JM, Park JH, et al. (2013) C(16)-Ceramide-induced F-actin regulation stimulates mouse embryonic stem cell migration: involvement of N-WASP/Cdc42/Arp2/3 complex and cofilin-1/alpha-actinin. Biochim Biophys Acta 1831: 350–360. doi: 10.1016/j.bbalip.2012.09.005 22989773
51. Groves JT, Kuriyan J (2010) Molecular mechanisms in signal transduction at the membrane. Nat Struct Mol Biol 17: 659–665. doi: 10.1038/nsmb.1844 20495561
52. Meiri KF (2004) Membrane/cytoskeleton communication. Subcell Biochem 37: 247–282. 15376624
53. Fooksman DR, Shaikh SR, Boyle S, Edidin M (2009) Cutting edge: phosphatidylinositol 4,5-bisphosphate concentration at the APC side of the immunological synapse is required for effector T cell function. J Immunol 182: 5179–5182. doi: 10.4049/jimmunol.0801797 19380760
54. Khuong TM, Habets RL, Slabbaert JR, Verstreken P (2010) WASP is activated by phosphatidylinositol-4,5-bisphosphate to restrict synapse growth in a pathway parallel to bone morphogenetic protein signaling. Proc Natl Acad Sci U S A 107: 17379–17384. doi: 10.1073/pnas.1001794107 20844206
55. Hakomori S, Handa K, Iwabuchi K, Yamamura S, Prinetti A (1998) New insights in glycosphingolipid function: "glycosignaling domain," a cell surface assembly of glycosphingolipids with signal transducer molecules,involved in cell adhesion coupled with signaling. Glycobiology 8: xi–xix. 9840984
56. Coskun U, Grzybek M, Drechsel D, Simons K (2011) Regulation of human EGF receptor by lipids. Proc Natl Acad Sci U S A 108: 9044–9048. doi: 10.1073/pnas.1105666108 21571640
57. van der Poel S, Wolthoorn J, van den Heuvel D, Egmond M, Groux-Degroote S, et al. (2011) Hyperacidification of trans-Golgi network and endo/lysosomes in melanocytes by glucosylceramide-dependent V-ATPase activity. Traffic 12: 1634–1647. doi: 10.1111/j.1600-0854.2011.01263.x 21810155
58. Gillot I, Jehl-Pietri C, Gounon P, Luquet S, Rassoulzadegan M, et al. (2005) Germ cells and fatty acids induce translocation of CD36 scavenger receptor to the plasma membrane of Sertoli cells. J Cell Sci 118: 3027–3035. 15972317
59. van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124. doi: 10.1038/nrm2330 18216768
60. Reichelt J, Breiden B, Sandhoff K, Magin TM (2004) Loss of keratin 10 is accompanied by increased sebocyte proliferation and differentiation. Eur J Cell Biol 83: 747–759. 15679119
61. Yildiz Y, Hoffmann P, Vom Dahl S, Breiden B, Sandhoff R, et al. (2013) Functional and genetic characterization of the non-lysosomal glucosylceramidase 2 as a modifier for Gaucher disease. Orphanet J Rare Dis 8: 151. doi: 10.1186/1750-1172-8-151 24070122
62. Wewer V, Dombrink I, vom Dorp K, Dormann P (2011) Quantification of sterol lipids in plants by quadrupole time-of-flight mass spectrometry. J Lipid Res 52: 1039–1054. doi: 10.1194/jlr.D013987 21382968
63. Ginkel C, Hartmann D, vom Dorp K, Zlomuzica A, Farwanah H, et al. (2012) Ablation of neuronal ceramide synthase 1 in mice decreases ganglioside levels and expression of myelin-associated glycoprotein in oligodendrocytes. J Biol Chem 287: 41888–41902. doi: 10.1074/jbc.M112.413500 23074226
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 3
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs