
Male-Biased Aganglionic Megacolon in the TashT Mouse Line Due to Perturbation of Silencer Elements in a Large Gene Desert of Chromosome 10
Hirschsprung’s disease (also known as aganglionic megacolon) is a severe congenital defect of the enteric nervous system (ENS) resulting in complete failure to pass stools. It is characterized by the absence of neural ganglia (aganglionosis) in the distal gut due to incomplete colonization of the embryonic intestines by neural crest cells (NCC), the ENS precursors. Hirschsprung’s disease has an incidence of 1 in 5000 newborns and a 4:1 male sex bias. Although many genes have been associated with this complex genetic disease, most of its heritability as well as its male sex bias remain unexplained. Here, we describe an insertional mutant mouse line (“TashT”) in which virtually all homozygotes display colonic aganglionosis due to defective migration of enteric NCC, but in which only a subset of homozygotes develops megacolon. Surprisingly, this group is almost exclusively male. The TashT ENS defect stems, at least in part, from the disruption of long-range interactions between evolutionarily conserved elements with silencer activity and Fam162b, resulting in NCC-specific upregulation of this uncharacterized protein coding gene. Global analysis of gene expression further revealed that several hundreds of genes are significantly deregulated in TashT enteric NCC. Interestingly, this dataset includes multiple X-linked candidate genes potentially underlying the male sex bias. Taken together, our data pave the way for a clearer understanding of the intriguing male sex bias of Hirschsprung’s disease.
Published in the journal:
Male-Biased Aganglionic Megacolon in the TashT Mouse Line Due to Perturbation of Silencer Elements in a Large Gene Desert of Chromosome 10. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005093
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005093
Summary
Hirschsprung’s disease (also known as aganglionic megacolon) is a severe congenital defect of the enteric nervous system (ENS) resulting in complete failure to pass stools. It is characterized by the absence of neural ganglia (aganglionosis) in the distal gut due to incomplete colonization of the embryonic intestines by neural crest cells (NCC), the ENS precursors. Hirschsprung’s disease has an incidence of 1 in 5000 newborns and a 4:1 male sex bias. Although many genes have been associated with this complex genetic disease, most of its heritability as well as its male sex bias remain unexplained. Here, we describe an insertional mutant mouse line (“TashT”) in which virtually all homozygotes display colonic aganglionosis due to defective migration of enteric NCC, but in which only a subset of homozygotes develops megacolon. Surprisingly, this group is almost exclusively male. The TashT ENS defect stems, at least in part, from the disruption of long-range interactions between evolutionarily conserved elements with silencer activity and Fam162b, resulting in NCC-specific upregulation of this uncharacterized protein coding gene. Global analysis of gene expression further revealed that several hundreds of genes are significantly deregulated in TashT enteric NCC. Interestingly, this dataset includes multiple X-linked candidate genes potentially underlying the male sex bias. Taken together, our data pave the way for a clearer understanding of the intriguing male sex bias of Hirschsprung’s disease.
Introduction
The enteric nervous system (ENS) is the intrinsic neural network of the gastrointestinal tract. One of its essential roles is to regulate intestinal motility. The ENS is made up of interconnected neural ganglia, themselves composed of neurons and supporting glial cells, forming two main parallel networks: the submucosal plexus and the myenteric plexus. The muscles of the bowel wall that ensure peristaltic movements are controlled by the myenteric plexus of the ENS.
The ENS is constructed during embryo development by derivatives of migrating neural crest cells (NCC) [1]. These multipotent cells originate from the dorsal part of the neural tube, undergo an epithelial-mesenchymal transition, and migrate extensively to contribute to numerous embryonic structures. Among several different cell types, NCC generate melanocytes as well as all enteric neurons and glia. The developing bowel is mainly colonized by NCC derivatives originating from the vagal region of the neural tube. Such colonization proceeds as a rostro-caudal wave lasting more than 5 days in the mouse (from embryonic day (e) 9.0 to 14.5), with NCC derivatives first entering the foregut, passing through the midgut (prospective small intestine) and finally populating the hindgut (prospective colon) either by migrating through the intestinal mesenchyme [2] or by taking a shortcut via the mesentery [3]. The hindgut is the last part of the intestines to be colonized and, therefore, the most susceptible to enteric NCC (eNCC) developmental defects. Sacral NCC also contribute to the ENS, but this later contribution is minor and cannot compensate for a lack of vagal NCC [4].
Defects in hindgut colonization by eNCC result in a lack of neural ganglia in the colon, leading to intestinal blockage due to absence of peristalsis. This phenotype is generally described as “aganglionic megacolon” because of the subsequent massive accumulation of fecal material and severe distention of the colon. In humans, this condition is called Hirschsprung's disease (HSCR) and, depending on the length of aganglionosis, is clinically subdivided in short-segment (i.e. restricted to the rectosigmoid colon) and long-segment forms. Short-segment HSCR represents the vast majority of cases and is more common in males than females, with an overall ratio of ~4:1 [5]. In patients displaying longer segments of aganglionosis, the sex bias is much less pronounced or absent altogether. Although mutations in at least 15 genes have been implicated in HSCR, heritability is unexplained for the majority of cases [6]. HSCR is thus a classic example of a complex disease involving multiple genes, incomplete penetrance, variable expressivity and an intriguing male bias.
Most known HSCR-associated genes encode players from two signaling pathways: the GDNF ligand/ RET receptor and EDN3 ligand/ EDNRB receptor pathways. In fact, RET is the main gene associated with HSCR [5]. For both pathways, the receptor is found at the surface of eNCC while the ligand is dynamically secreted from the surrounding mesenchyme during the colonization phase. The role of GDNF/RET and EDN3/EDNRB signaling in ENS formation has been well conserved evolutionarily and studies in animal models have revealed that both pathways profoundly influence every key aspect of eNCC development such as proliferation, survival, differentiation and, most especially, migration [7]. Mouse models have been particularly informative in this regard and multiple lines bearing mutation—either spontaneous or targeted—of genes encoding members of Gdnf/Ret and Edn3/Ednrb pathways have been studied [8–13]. However, the incomplete penetrance and, above all, the male bias observed in human HSCR have been poorly replicated in current animal models [14].
Here, we report the creation of a new insertional mutant mouse model for HSCR that displays, for the first time, incomplete penetrance of the aganglionic megacolon phenotype with a very strong male bias. Extensive characterization of this mouse line and independent validation via transient transgenesis indicate that this outcome is, at least in part, initiated by the specific upregulation of Fam162b in NCC.
Results
Phenotypic overview of the TashT mouse line
The TashT mouse line was obtained from an insertional mutagenesis screen for genes involved in NCC development. This screen was based on the random insertion of a Tyrosinase (Tyr) minigene in the FVB/n genetic background. Owing to its specific expression in melanocytes, the Tyr minigene rescues the albino phenotype of FVB/n mice and thus provides a visible—and generally uniform—pigmentation marker for transgenesis [15]. Since melanocytes are derived from NCC, this genetic tool also proved to be a potent indicator of abnormal NCC development via identification of non-uniform pigmentation patterns. This approach yielded several transgenic mutant lines (to be described elsewhere) among which TashT (Tachetée, in French) was identified due to its variegated pigmentation (Fig. 1a).
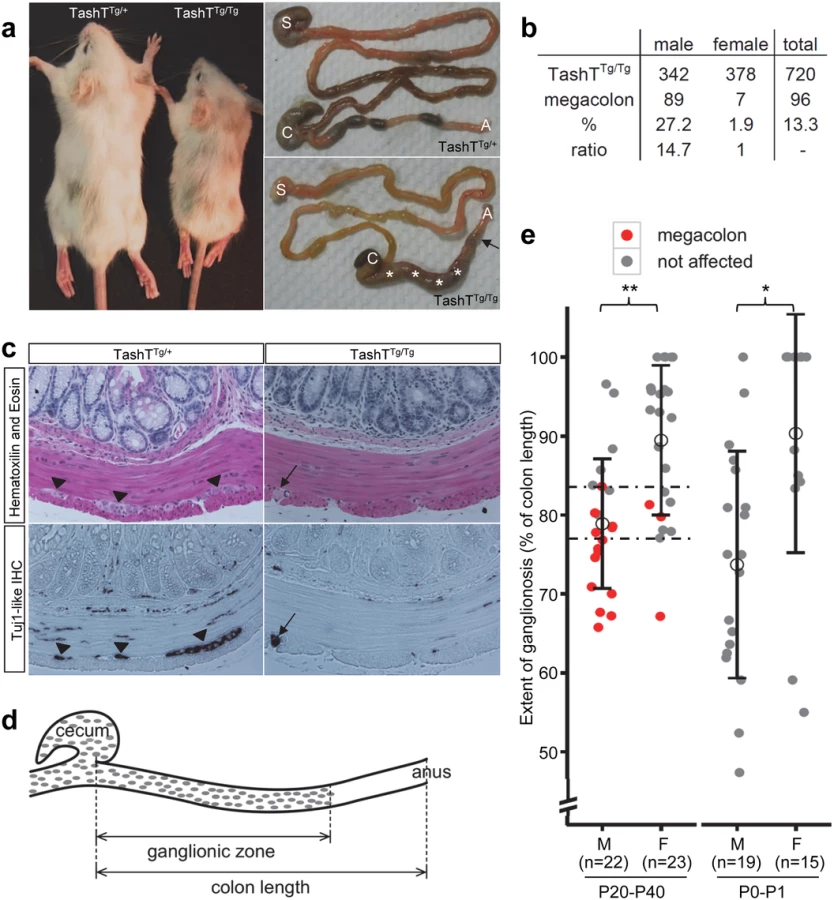
In addition to the pigmentary anomalies that are similar in both heterozygous and homozygous mutants, a subset of TashTTg/Tg animals suffer from aganglionic megacolon around weaning age. These animals are smaller than unaffected TashTTg/Tg siblings (reaching about 74% of littermate weight) and exhibit bowel obstruction concomitant with lack of myenteric ganglia in the distal colon (Fig. 1a,c). The most striking and interesting feature of this lethal phenotype is the fact that the vast majority of affected animals are male (Fig. 1b). In addition, we found via histological analyses that colonic aganglionosis can not only be detected in megacolon-suffering but also in non-affected TashTTg/Tg animals of both sexes (Fig. 1c). To determine whether megacolon expressivity could be correlated with extent of aganglionosis, we undertook a systematic analysis of the length of the colonic ENS for a random group of TashTTg/Tg animals of weaning age via staining of acetylcholinesterase activity (Fig. 1d). In accordance with such correlation, quantification results first revealed that—regardless of the sex of the animal—a minimal length of the colon (~ 80%; critical region in Fig. 1e) has to be properly innervated in FVB/n mice in order to avoid blockage. No intermediate phenotype was noted in the course of these analyses as none of the non-affected animals showed signs of abnormal accumulation of feces in the colon. Furthermore, these results confirmed that most TashTTg/Tg animals exhibit aganglionosis in the distal colon and that males (ganglionated on average over 79% of total colon length) are more affected than females (ganglionated on average over 89% of total colon length) (Fig. 1e). Importantly, a similar statistically significant difference between male and female animals (74% vs 90%) was also observed in neonatal colons, thus confirming the developmental origin of this male-biased defect.
Detailed analysis of TashTTg/Tg eNCC reveals a cell migration defect
To analyze ENS formation in TashT embryos, we took advantage of the fact that this line bears a second co-injected transgene (pSRYp[1.6kb]-YFP) that labels migrating NCC derivatives (including eNCC of vagal and sacral origin) with YFP fluorescence [16] (see also S1 Fig). Whole-mount detection of fluorescence in dissected stage-matched embryonic intestines revealed that, in comparison to TashTTg/+ or G4-GFP control embryos [17] (S2 Fig), a colonization delay by eNCC of vagal origin is clearly observed for TashTTg/Tg embryos starting around e11.0 (Fig. 2a). Although ectopic fluorescence in the cecum and proximal hindgut regions impeded precise visualization of the migration front between e11.5 and e14.5, we found that this delay persists through the time at which normal colonization of the digestive tract is overtly completed (e15.5). The presence of scattered fluorescent cells beyond the chains of vagal-derived eNCC at this stage also suggests that the contribution of sacral-derived eNCC to the distal hindgut is not abrogated in TashTTg/Tg embryos.
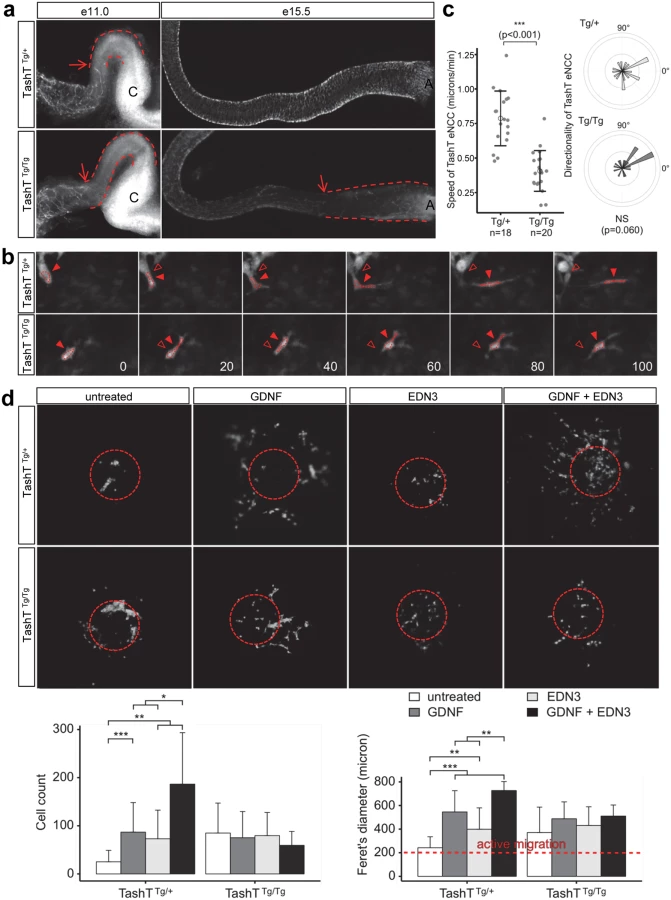
Closer inspection of the migration front at e11.0 showed that cell protrusions in the form of filopodia were not overtly affected in TashTTg/Tg eNCC (S3 Fig), suggesting that cells could still investigate, and had the capacity to respond to, their environment. To further characterize the colonization defect, leader cells at the tip of eNCC chains were then visualized during several hours in ex vivo cultures of e11.0 intestines from littermate control (TashTTg/+) and mutant (TashTTg/Tg) embryos (Figs. 2b, S4 and S1–S2 Videos). It is noteworthy that TashTTg/Tg intestines were selected for these analyses on the basis of the severity of their colonization defect in order to increase the odds of detecting differences between control and mutant eNCC. Using these conditions, we found that the average migration speed, and therefore travel distance, is almost halved in TashTTg/Tg leader eNCC while directionality of migration is not noticeably affected (Fig. 2c). Given the selection bias towards more affected embryos, it is important to bear in mind that this severe effect is most likely not representative of the true average migration speed of mutant eNCC. It should also be noted that the small difference in eNCC location along the intestines—before cecum (TashTTg/Tg) vs entry of cecum (TashTTg/+)—cannot account for the observed difference in speed since the eNCC migration front normally displays a fairly stable net speed of ~35 micron/hour (~0.58 micron/min) between e10.5 and e12.5 [18].
Migration of eNCC is dependent on multiple signaling pathways among which GDNF/RET and EDN3/EDNRB are recognized as the most critical regulators [19–22]. To evaluate the status of these signaling pathways in TashT embryos, we made use of a recently described quantitative migration assay using e12.5 midgut explants [23]. With control TashTTg/+ or G4-GFP tissues, collagen gels containing either GDNF or EDN3 increased the number of cells coming out of the explants (Fig. 2d). Interestingly, when these ligands were used in combination, a synergistic increase in eNCC numbers invading the collagen was observed. However, little reaction to these extracellular ligands was detected in eNCC derived from TashTTg/Tg embryos (Fig. 2d). In fact, eNCC from homozygous embryos migrated out of intestinal explants even in the absence of exogenous ligands, suggesting they lost some sensitivity to their endogenous microenvironment and distinguished poorly between intestinal tissue and collagen gel.
Premature differentiation or a scarcity of progenitor cells can disturb eNCC colonization and lead to incomplete ENS formation [19,22,24,25]. To verify whether these processes might contribute to the TashT phenotype, we performed a detailed marker analysis of embryonic intestines in order to quantify neuronal and glial differentiation as well as proliferation and cell death of eNCC. Quantification of proliferation and cell death in e12.5 stage-matched bowel tissues failed to reveal any significant difference between TashTTg/Tg and control TashTTg/+ embryos (S5 Fig). Assessment of neuronal differentiation at the same stage also failed to reveal any significant difference (S6a-S6b Fig). On the other hand, glial differentiation at e15.5 was found to be less prevalent in TashTTg/Tg distal bowel tissues, to the benefit of undifferentiated progenitors (S6c-S6d Fig). This, however, is most likely a consequence of the delay in rostro-caudal colonization, and goes contrary to the idea that premature differentiation is the cause of the TashT migration defect. Taken together, these results thus highlight the eNCC migration defect (concomitant with insensitivity towards GDNF and EDN3) as the principal cause of the TashT aganglionosis phenotype. Moreover, since no clear sex bias was observed in these analyses, the relatively modest male bias in phenotype severity is most likely the result of an accumulation of subtle differences during the whole ENS developmental time window.
The TashT transgenic insertion disrupts evolutionarily conserved regions possessing silencer activity
Breeding of the TashT line revealed systematic co-segregation of pigmentation with YFP fluorescence, meaning co-integration of both transgenes into a single autosomal locus which is frequent when an equimolar mixture of each transgene is micro-injected [15]. FISH analysis first allowed a rough estimate of the localization of the transgene insertion site on chromosome 10 at bands B2–B3 (S7a Fig). To obtain a more precise localization, we sequenced the whole genome of a TashTTg/Tg mouse. Mapping of high-throughput paired sequencing reads allowed us to localize the transgenic insertion around the middle of a 3.3Mb gene desert between Hace1 and Grik2 (Fig. 3a,b). Twice as many reads were observed in a 26kb non-coding region of chromosome 10B2, indicating a duplication. Flanking this duplicated region were paired reads with one end mapping to chromosome 10 and the other end mapping to sequences corresponding to either one or the other transgene (Fig. 3a). A schematic representation of the inferred organization of the TashT transgene insertion site is shown at the bottom of Fig. 3a. The number of transgene copies was estimated from the mapping data and the total size of the insertion calculated to be about 700kb.
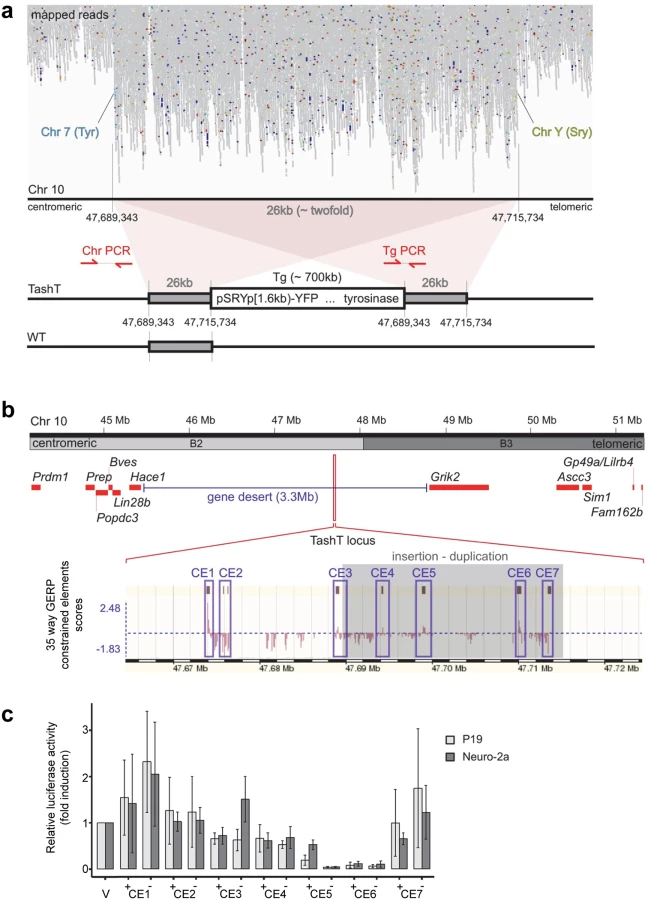
The TashT locus contains several blocks of evolutionary conserved non-coding sequences (Fig. 3b). To evaluate their regulatory potential, we cloned seven ~1kb fragments containing most of these constrained elements (CE)—named CE1 to CE7—in a luciferase expression vector bearing a minimal thymidine kinase promoter. Transcriptional activity was then assessed in various cell lines (Neuro-2a, P19, Cos7 and NIH 3T3) via luciferase assays (Figs. 3c and S9). Overall, this analysis revealed very strong repression activity in a cell type-independent manner for two of the cloned regions (CE5 and 6) (Figs. 3c and S9). These luciferase assays thus suggest that the TashT transgenic insertion has disrupted at least one important long-range regulatory element that normally represses expression of a surrounding gene. However, expression of the two most proximal neighboring genes on each side of the gene desert (Lin28b and Hace1 as well as Grik2 and Ascc3) (Fig. 3b) was found to be similar in TashTTg/Tg and control G4-GFP e12.5 eNCC recovered by FACS (S8 Fig).
The transcriptional signature of TashTTg/Tg eNCC
To cast a wider net and detect transcript variation in an unbiased manner, we sequenced the rRNA-depleted transcriptome of FACS-recovered eNCC from anterior intestinal tissues of stage-matched control (G4-GFP) and TashTTg/Tg e12.5 embryos. This stage was chosen because it combines ease of intestine dissection with clear presence of the eNCC colonization defect in TashTTg/Tg tissues. It is also important to note that this analysis was restricted to anterior intestinal tissues only (prospective oesophagus, stomach and small intestine) because endogenous YFP labelling in the TashT line is strictly specific to eNCC in these regions (S1b Fig). As mentioned above (see Fig. 2a), the TashT line exhibit ectopic YFP fluorescence in non-NCC derivatives in more posterior regions (prospective cecum and colon) and, therefore, these regions were excluded from both control and mutant cell preparations.
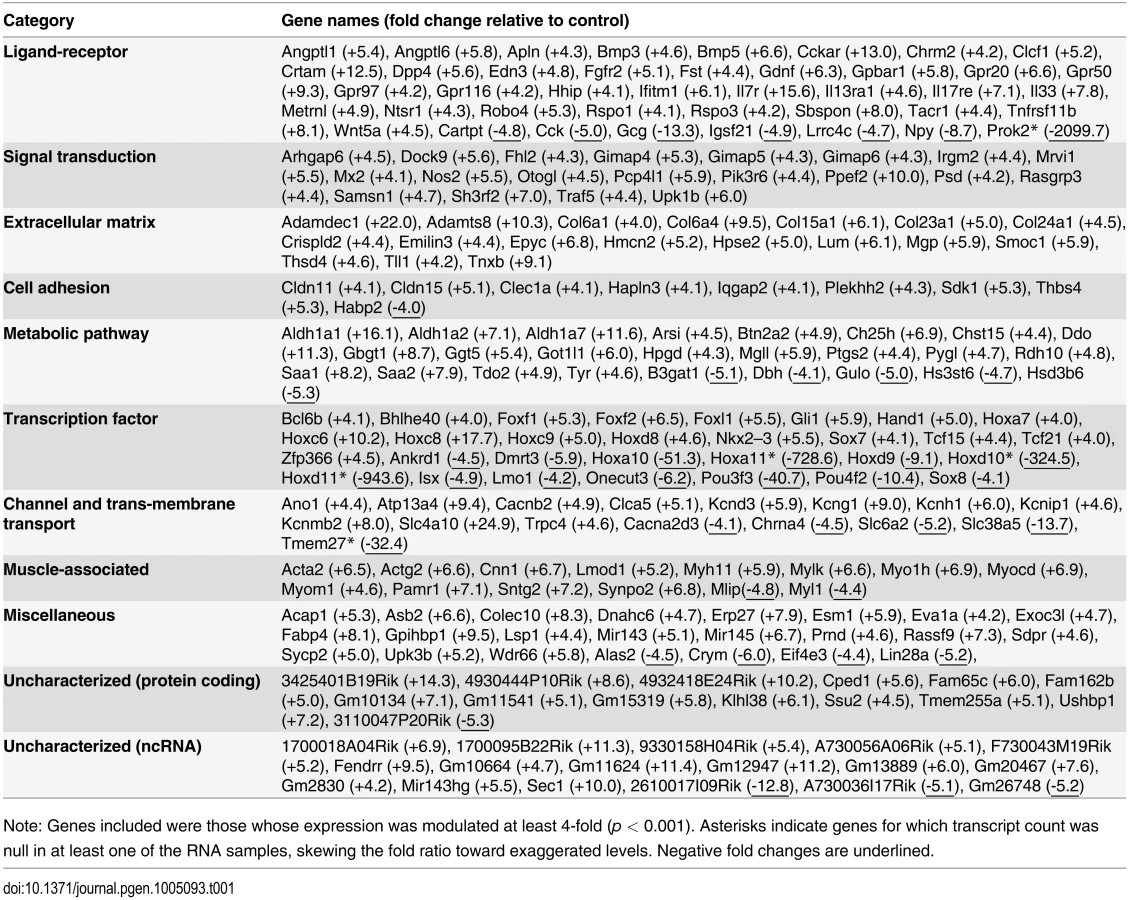
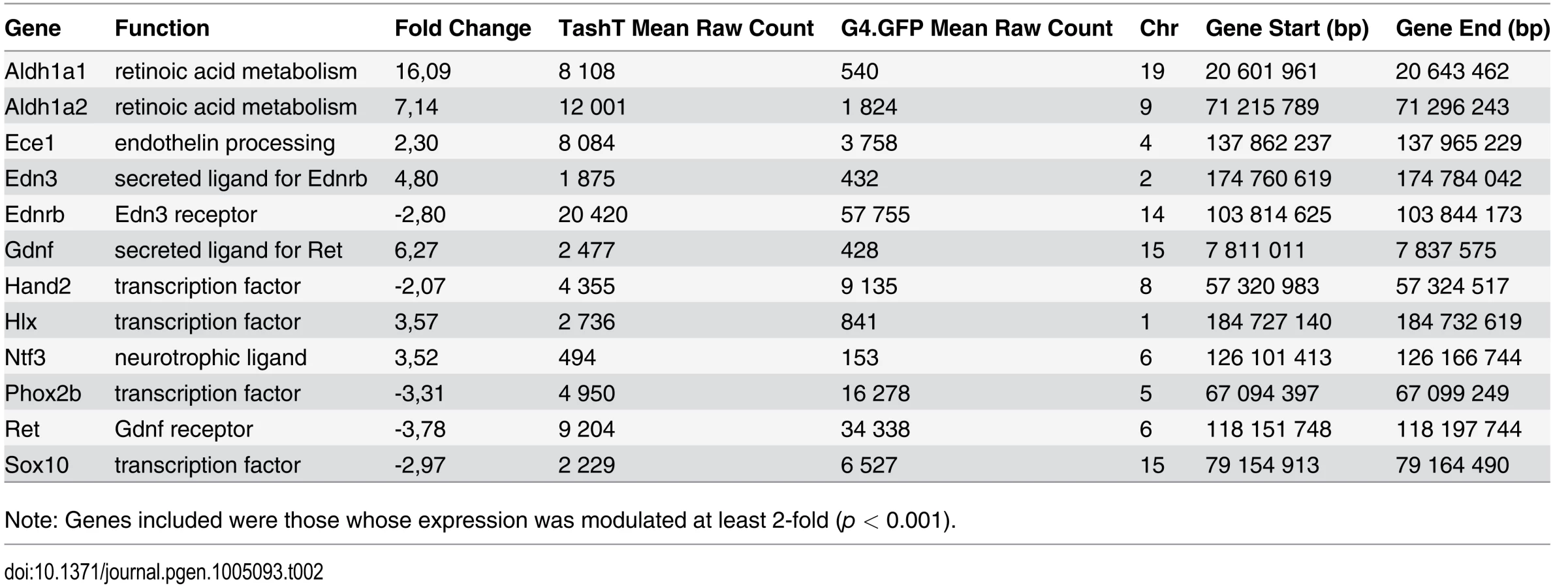
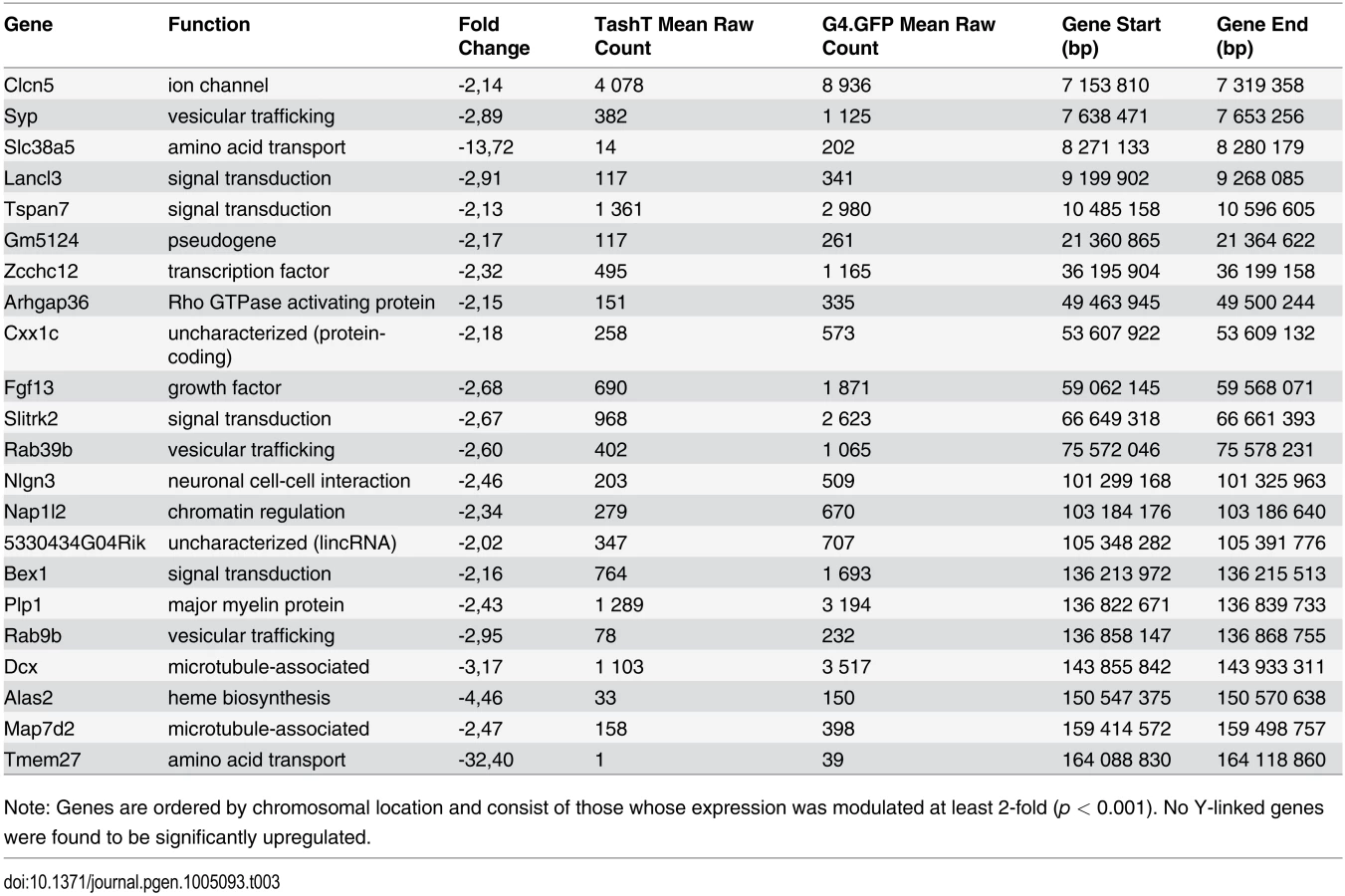
Analysis of RNAseq data revealed that over 1200 coding and non-coding genes are differentially expressed in a significant manner (≥2-fold and p <0.001) in TashTTg/Tg eNCC, among which upregulated and downregulated genes are equally represented (S1 Dataset and S10c Fig). The most deregulated genes (≥4-fold) are listed in Table 1 and this shorter list now indicates a strong enrichment for upregulated genes in TashTTg/Tg eNCC (41 downregulated vs 188 upregulated). Each of these 229 genes was manually assigned to a category based on function and/or localization of their gene product. Assembling these categories in “super-categories” reveals that TashT affected genes are mostly involved in the control of cell signaling (categories: Ligand-receptor and Signal transduction) and gene expression (category: Transcription factor) as well as in the composition of, and interaction with, the cell microenvironment (categories: Extracellular matrix, Cell adhesion as well as Channel and transmembrane transport). Especially notable examples within each of these super-categories include, respectively, genes encoding Gdnf and Edn3 ligands, many Hox transcription factors as well as various Collagen members. Another category worth mentioning is the Metabolic pathway which notably contains many players of retinoid signaling (Aldh1a1, Aldh1a2, Aldh1a7 and Rdh10). We verified the expression level of selected genes by semi-quantitative RT-PCR. Our selection criteria included genes known as playing a major role in HSCR (Table 2 and S10a Fig) as well as genes located on a sex chromosome whose change in expression could explain the observed male bias (i.e. upregulated on chromosome Y or downregulated on chromosome X) (Table 3 and S10b Fig). All genes tested followed the trend set by the RNAseq data.
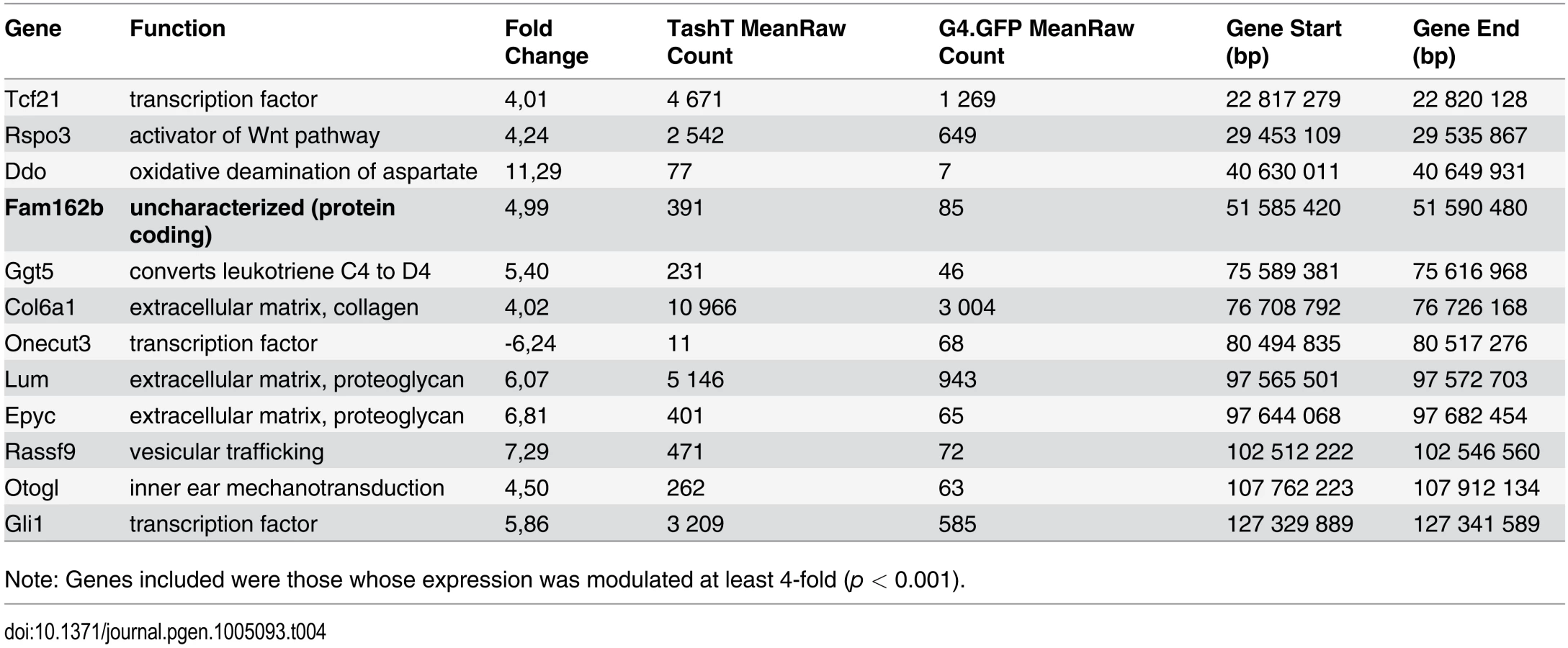
Given that intra-chromosomal regulatory chromatin contacts are much more prevalent than inter-chromosomal ones [26], the selection criteria for identification of the TashT causative gene was its location relative to the repressive elements disrupted by the transgene insertion. We therefore focused on the handful of upregulated genes located on chromosome 10 (Table 4) in our search for a gene directly regulated by the conserved elements. The closest candidate, Fam162b, is ~3.6 Mb telomeric to the conserved elements (Fig. 3b) and its overexpression in TashTTg/Tg eNCC was validated via semi-quantitative RT-PCR (S11a Fig). Note that Fam162b mRNA can also be detected in eNCC FACS-sorted from control embryos, indicating its low level expression in normal eNCC populations (see S1 Dataset). Analyses of Fam162b open-reading frame sequences using ExPASy (www.expasy.org) and Uniprot (www.uniprot.org) resources suggest that this uncharacterized gene encodes a single pass transmembrane protein localized to mitochondria.
Overexpression of Fam162b may explain the TashT ENS defect
We subsequently verified that the conserved silencer elements near the transgene insertion site normally interact with the Fam162b locus using chromosome conformation capture (3C) techniques [27]. These analyses first revealed that such interaction can be detected in wild-type whole embryonic intestines using different primer pairs (Fig. 4a,b). We also found that this interaction is cell type-specific as it is detected in NCC-derived Neuro-2a cells but not in undifferentiated P19 cells (Fig. 4c), the same murine embryonic cell lines used in our luciferase assays (Fig. 3c). Importantly, we further found that this interaction is lost in TashTTg/Tg embryonic gut tissues (Fig. 4b). Therefore, these results strongly suggest that the TashT transgenic insertion disrupts intra-chromosomal contacts that normally repress Fam162b expression in NCC (Fig. 4d).
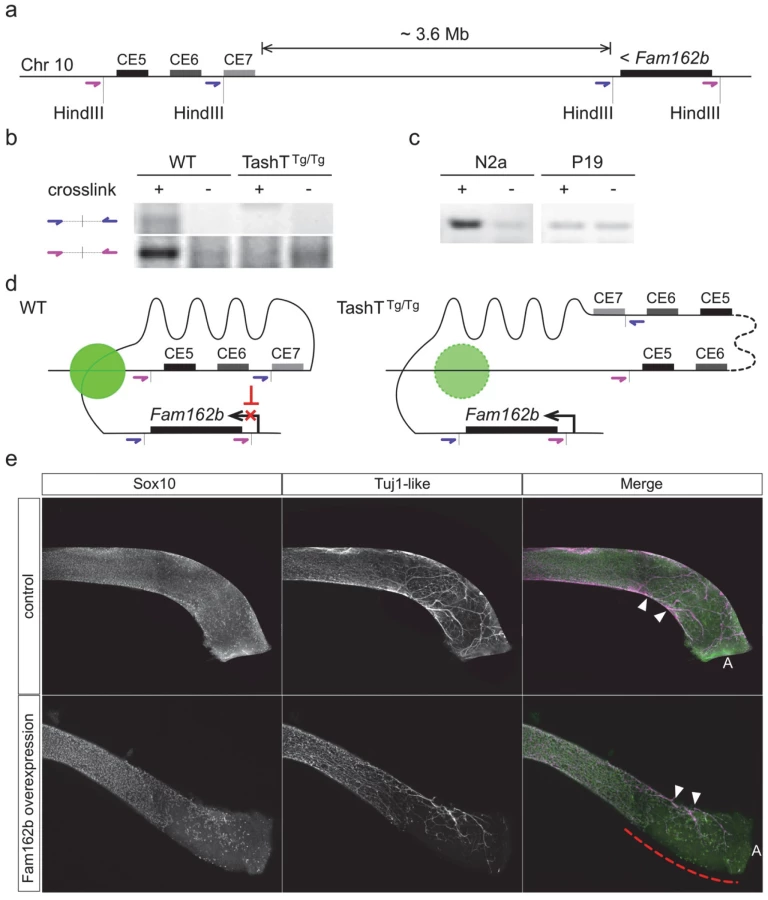
To independently validate the candidacy of Fam162b as being involved in the TashT ENS defect, we generated transgenic e15.5 embryos specifically overexpressing Fam162b in NCC and analyzed the impact on ENS formation using specific markers. The transgenic construct consisted of a bicistronic cassette containing Fam162b and eGFP coding sequences driven by a previously described Sox10 enhancer (U3, also known as MCS4) fused to the Hsp68 minimal promoter [28,29]. Western blotting confirmed that the cloned Fam162b sequences express a protein of expected size (S11b Fig). Using fluorescence from eGFP as a surrogate marker for transgene expression, we obtained a total of three Fam162b transgenic embryos (all females) exhibiting variable levels of transgene expression. In contrast to littermate controls for which bowel tissues were fully colonized, all three transgenic embryos displayed incomplete colonization of the distal hindgut by vagal-derived eNCC and, as a result, this region was found to only contain scattered Sox10-positive eNCC of presumably sacral origin in a way similar to what is observed in TashTTg/Tg tissues (compare Fig. 4e with Fig. 2a). In addition to having used a different genetic background for these experiments (B6C3), we believe that the fact that all Fam162b overexpressers were female likely explains, at least in part, the more modest effect observed in comparison to TashTTg/Tg embryos. Although we cannot currently exclude the possibility that other gene(s) might also be primary target(s) of the TashT mutation and might thus also contribute to the TashTTg/Tg phenotype, these transgenesis data support a role for Fam162b overexpression in TashTTg/Tg pathogenesis.
Discussion
We have identified and characterized a novel mouse model for aganglionic megacolon, called TashT, allowing us to describe the eNCC transcriptome during development and to propose two novel genetic loci implicated in the pathogenesis of megacolon—one that is protein coding, the other involving evolutionary conserved non-coding sequences—as well as an ultra-long-range interaction between these loci. In addition, this mouse model allows us to speculate on the mechanistic nature of the hitherto unexplained male bias of the megacolon phenotype observed in humans, as well as its variable penetrance.
The transcriptional signature of TashTTg/Tg eNCC: deregulation of Gdnf/Ret and Edn3/Ednrb signaling
To the best of our knowledge, this study is the first to report a transcriptome analysis of sorted eNCC. Previous screens for genes expressed in eNCC were performed on whole embryonic intestines using DNA microarrays and based on the differential expression between normal and Ret-null aneural tissues [30,31]. In addition to analyzing eNCC directly, we took full advantage of the RNAseq technology and included non-coding RNA in our analyses. This resulted in a much more extensive list of genes known to be expressed in eNCC, from a few hundred to several thousand (S1 and S2 Datasets).
Importantly, the transcriptional signature of TashTTg/Tg eNCC not only highlighted Fam162b as a potential causative gene, but also provided mechanistic insights into the identified cell migration defect. In this regard, it is noteworthy that several modulated transcripts in TashTTg/Tg eNCC encode components of the extracellular matrix (ECM) (Table 1 and S10c Fig). NCC have been suggested to modify their extracellular environment during, or perhaps as a requirement for, migration [32,33]. One possibility is thus that the modulated ECM in TashTTg/Tg embryonic intestines is less permissive to cell migration [34,35]. Another, not mutually exclusive possibility is that the reduced sensitivity of TashTTg/Tg eNCC to growth factors/chemoattractants normally present in the intestinal ECM underlies the migration defect. In agreement with this, we have found that TashTTg/Tg eNCC have lost their ability to respond to exogenous GDNF and EDN3 in explant assays, with eNCC appearing unable to distinguish between bowel tissue and collagen gel (Fig. 2d). This latter outcome is most likely due to the surprising robust overexpression of both Gdnf and Edn3 by TashTTg/Tg eNCC (Table 2 and S10a Fig). Indeed, given that Gdnf and Edn3 are both normally heavily secreted from the surrounding mesenchyme, additional oversecretion from eNCC is expected to disturb the dosage of ligands these cells normally encounter and/or to disrupt any gradient that might be present. Our RNAseq data also suggest that the lack of responsiveness to GDNF and EDN3 might be due to an overabundance-induced negative feedback on their cognate receptor. This hypothesis is supported by the fact that expression of both Ret and Ednrb is reduced in TashTTg/Tg eNCC (Table 2 and S10a Fig). Interestingly, in the case of Gdnf/Ret signaling, this hypothesis is further supported by the observed overexpression of Lrig1 and Lrig3 in TashTTg/Tg eNCC (S1 Dataset). These genes encode functionally-redundant transmembrane proteins [36] which, as specifically demonstrated for Lrig1 in neuronal cells, can be induced by Gdnf at the transcriptional level and then physically interact with Ret in order to reduce Gdnf binding and tyrosine kinase activity [37]. Regardless of the exact underlying mechanism, a lack of responsiveness to EDN3 might well be responsible for the slower migration of TashTTg/Tg eNCC (Figs2b-c and S4), as inhibition of Ednrb signaling has been recently shown to primarily affect the speed of eNCC migration [38].
Long-range repression of Fam162b expression
Most of the known and characterized long-range acting regulatory elements do not interact with their nearest promoter but bypass several intervening genes in order to reach their target promoter [39]. Long-range enhancer-promoter interactions are also thought to be more commonly involved in the regulation of tissue-specific genes [40]. Moreover, most studies of intra-chromosomal long-range interactions involve loci up to several hundred kb away from each other, though ultra-long-range events (several Mb) between enhancer and promoter are not uncommon [27,40,41]. Our 3C data are in accordance with these observations and indicate that the interaction between the conserved elements near the TashT transgene insertion site and the Fam162b gene ~3.6 Mb away (Fig. 4b) falls in the ultra-long-range category. Little is known of the spatiotemporal pattern of Fam162b expression. The evidence to date is in agreement with an expression in neural derivatives: it is weakly expressed in the mouse olfactory bulb (Allen Brain Atlas, RP_051012_01_G06) and expressed in the frontonasal prominence of mouse embryos, proximal to the oral cavity [42]—a tissue heavily populated by cranial NCC. These observations are consistent with the idea that Fam162b is poised for active transcription in neural and/or neural crest cells but kept in check through a repressive mechanism.
Using luciferase assays, we demonstrated that a subset of the highly conserved elements near the TashT transgene insertion site has a robust negative regulatory function on transcription in a cell type-independent manner (Fig. 3c and S9 Fig). Given the forced juxtaposition of regulatory elements with the proximal promoter in such assays, the absence of a cell type-specific activity in our analysis thus points to a chromatin conformation-dependent mechanism conferring specificity in vivo. As supported by our 3C data (Fig. 4c), we suggest that an ultra-long-range chromatin loop maintains Fam162b expression at a basal level in a subset of neural-derived cells, including eNCC. Further investigations into the regulation of Fam162b expression will be necessary to confirm and expand this hypothesis. The biological function of the Fam162b gene product is also currently unknown. Characterization of this function will clearly be facilitated by the wealth of information obtained from the RNAseq data as well as by the observed cell migration defect.
The source of the male bias
Robust correlation between extent of aganglionosis and expressivity of the megacolon phenotype in TashTTg/Tg animals allowed us to identify the minimal distance of myenteric innervation necessary for successful movement of luminal content across the colon in FVB/n mice. This critical region (~80% of colon length; Fig. 1e) represents a threshold level beneath which intestinal blockage occurs systematically, and in a sex-independent manner. The fact that the mean length of the ganglionated region of TashTTg/Tg males ends in this critical region while females typically show a more extensive ENS explains the apparent contradiction between the male bias in megacolon expressivity and the near complete penetrance of distal aganglionosis in both sexes. A common defect of eNCC colonization, slightly exaggerated in males, is thus the source of the observed sex bias of the megacolon phenotype in TashTTg/Tg animals. In this regard, it is noteworthy that a similar link between extent of aganglionosis and megacolon expressivity has been previously described in mice bearing the Ednrbs-l allele [43] as well as in Ret+/-::Ednrbs/s compound mutants [14]. Interestingly, although only a very modest male sex bias in megacolon expressivity was reported in this latter case (~1.5:1), the correlations made with extent of aganglionosis are in agreement with the threshold level revealed by our study. Indeed, full penetrance of megacolon in Ret+/-::Ednrbs/s males was correlated with a mean length of the ganglionated region clearly beneath the threshold (59% of colon length) whereas partial penetrance of megacolon in Ret+/-::Ednrbs/s females was correlated with a mean length of the ganglionated region much closer to the threshold (72%) [14]. However, as evidenced by the fact that Sox10Dom mutants on a C57BL/6J—C3HeB/FeJ mixed background display a shorter aganglionic zone leading to megacolon (~10%), it should also be noted that position of the threshold level may vary as a function of the genetic background [44].
The aganglionic megacolon of TashTTg/Tg animals share striking similarities with both the variable penetrance and male sex bias of short-segment HSCR, the most common form of the disease (~80% of cases). The threshold level identified with the TashT line is also in accordance with the fact that virtually no sex bias is observed in long-segment HSCR. Our analysis of the TashT line thus provides useful insights into the ontogeny of aganglionic colon and the origin of the sex bias, and shows that, though perhaps suffering from chronic constipation, TashTTg/Tg mice are nevertheless able to pass intestinal material when more than 4/5 of their colon is innervated.
Apart from Ret+/-::Ednrbs/s compound mutants, it is interesting to note that a male bias in the extent of aganglionosis—but not megacolon expressivity—has also been reported in other mouse and/or rat models and in each case implicated a mutation in either Ret or Ednrb [13,45,46]. Taken together with our data showing deregulated Ret and Ednrb signaling in TashTTg/Tg animals as well as with the previous description of a RET non-coding mutation that is twice as frequently transmitted in boys than in girls [47], these observations strongly suggest that both pathways are involved in the regulation of expression of a still undefined sex chromosome-linked gene with critical function in the developing ENS. We reasoned that exaggerated defects in males could arise from overexpression of male-specific genetic material (upregulated genes on Y chromosome) or from a deficiency in the expression of genes present as single alleles (downregulated genes on X chromosome). Another interpretation of this second possibility is that females are protected through biallelic expression of some X chromosome genes, provided they escape X-inactivation [48–51]. Analysis of our transcriptome dataset revealed that expression of Y-linked genes in eNCC is limited to a group of four genes (Kdm5d, Eif2s3y, Uty and Ddx3y) (S2 Dataset). Of these clustered genes—also known to be expressed in the developing brain [52,53]–, Ddx3y is the only one that approaches significant upregulation in TashTTg/Tg eNCC (1.5-fold; edgeR p = 0.0011; DESeq p = 0.0055). In marked contrast, multiple X-linked genes were found to be significantly downregulated in TashTTg/Tg eNCC, including Dcx—a previously suggested potential HSCR susceptibility locus (Table 3) [30]. As X-inactivation escapees tend to be found in clusters [48], other interesting candidates include a group of genes—in the vicinity of Dcx—that contains Bex1 and the Plp1-Rab9b gene pair (Table 3). It is noteworthy that the candidacy of these genes is also supported by human cases with Xq22 microdeletions encompassing them. Indeed, while such cases are assumed to be embryonic lethal in males, female patients suffer from Pelizaeus-Merzbacher-like disease with symptoms of gastrointestinal motility problems including constipation [54].
Materials and Methods
Animals
Work with mice was performed in accordance with the guidelines of the Canadian Council on Animal Care (CCAC) and approved by the relevant institutional committee (Comité institutionnel de protection des animaux; CIPA reference #650) of University of Quebec at Montreal (UQAM). Mice were euthanized by gradual-fill carbon dioxide (CO2) gas preceded by isoflurane anesthesia. TashT transgenic mice and Fam162b transgenic embryos were generated via standard pronuclear microinjection [55], using embryos derived from FVB/n albino and B6C3 mice, respectively. For the TashT line, two transgenes were co-injected at equimolar ratio: a Tyrosinase minigene to allow visual identification of transgenic animals via rescue of pigmentation [15] and a pSRYp[1.6kb]-YFP construct that provides fluorescent marking of migrating NCC in the developing embryo [16]. The previously described Gata4p[5kb]-GFP line (G4-GFP) was used as wild-type control [17]. Mice were mated overnight and noon on the day a vaginal plug was observed was designated as embryonic day (e) 0.5. For Fam162b transient transgenics, a transgene carrying the PCR-amplified Fam162b open reading frame under the control of the Hsp68 minimal promoter [56] and a NCC-specific Sox10 enhancer (U3, also known as MCS4) [28,29] was used. In order to provide a positive control for transgene expression, an IRES-GFP cassette (pIRES2-EGFP, Clontech) was also included immediately downstream of the Fam162b ORF, creating a bicistronic message. Fifteen days after microinjection, foster mothers were sacrificed, embryos were collected, individually analyzed for GFP and immunostained for Sox10 and βIII-Tubulin (see immunofluorescence section below).
Fluorescence in situ hybridization (FISH) of mouse chromosomes
Antisense and sense digoxigenin-labelled RNA probes were synthesized using a DIG transcription kit (Roche). Mouse spleen lymphocytes were collected and metaphase slides prepared using standard cytogenetic protocols [57,58]. Slides were aged at room temperature for 7 days, and then GTG banded, again using standard protocols [55]. Slides were scanned at 100X using a Nikon Eclipse E800 microscope, and representative metaphases were photographed at 1000X using a Nikon DXM1200 digital camera and SimplePCI software. Slides were de-stained using CitriSolv (Fisher) for 10–15 minutes, fixed for 1 minute in a 1% paraformaldehyde (PFA) solution in 2X SSC then dehydrated in graded ethanol washes and stored for future use in 100% ethanol. Slides were air dried then incubated in a denaturation solution for 5 minutes at 73°C, then again dehydrated in graded ethanol washes and stored in 100% ethanol. Probe was generated using a digoxigenin-labeled Tyr minigene DNA fragment [57]. Just prior to adding the denatured FISH probe, slides were air dried. Five to 10 μl of denatured probe was added per slide, which was then incubated overnight at 37° under a coverslip in a humidified dark atmosphere. Detection was performed using a rhodamine-conjugated anti-DIG antibody (Roche), following the manufacturer’s instructions. Slides were counterstained with DAPI (Sigma) and antifade (P-phenylenediamine; Sigma). Previously photographed G-banded metaphases were re-located using epifluorescence, rephotographed, and compared to FISH images.
Fluorescence-activated cell sorting (FACS)
Intestines were dissected from e12.5 G4-GFP and TashT embryos without their posterior end (prospective cecum and colon) because, in contrast to more anterior regions in which only eNCC are fluorescently-labelled (see S1b Fig), TashT intestines also contain high amounts of YFP-positive mesenchymal cells that are not derived from NCC in these regions (see Figs. 2a and S4). A MoFlo XDP (Beckman Coulter) cell sorter was used to collect GFP- or YFP-positive single viable cells from the remaining dissociated intestinal tissue. Dissociation was carried out at 37°C in EMEM containing collagenase (0.4 mg/ml; Sigma C2674), dispase II (1.3 mg/ml; Life Technologies 17105–041) and DNAse I (0.5 mg/ml; Sigma DN25).
High throughput genome and transcriptome sequencing
Whole genome and transcriptome library generation and sequencing was performed by McGill University and Génome Québec Innovation Centre using the HiSeq 2000 platform (Illumina). Fifty to 150 million paired-end sequences (100 bp length) were obtained from 300–500 bp library inserts, resulting in an overall 30x coverage for genome reads. Sequences were filtered based on quality and mapped onto the Mus musculus reference genome (mm9 for genomic DNA, mm10 for RNA). For the transcriptome, total RNA was rRNA-depleted before making the libraries. Three libraries were generated for each cell population, though only two were of sufficient quality for subsequent bioinformatic analysis. DESeq and edgeR differential gene expression analyses (adjusted p-value < 0.001), as well as a minimum 2-fold expression difference, were taken into account to determine significantly deregulated messages between FACS-recovered eNCC from control (G4-GFP) and TashTTg/Tg embryos. Gene Ontology analyses were performed using GOToolBox, and at least 2-fold enriched/depleted categories were selected from a hypergeometric test with a Benjamini-Hochberg corrected p-value threshold of 0.01 (http://genome.crg.es/GOToolBox).
Immunohistochemistry (IHC)
Dissected postnatal intestines were fixed in 4% PFA overnight at 4°C and embedded in paraffin. Sections (7 μm) were stained by immunohistochemistry according to standard techniques. Briefly, the paraffin was removed from slide mounted sections by washing with xylene and ethanol, and sections were treated for antigen retrieval with boiling sodium citrate pH 6.0 for 10 min. Slides were blocked for 1 h in blocking solution (1% bovine serum albumin, 1% milk, in Tris-buffered saline pH 7.5), then incubated overnight at 4°C with mouse anti-βIII-Tubulin (Abcam ab78078, 1:200) primary antibody. Following several Tris-buffered saline (TBS) pH7.5 washes, sections were incubated for 2h at room temperature (RT) in alkaline phosphate-conjugated anti-mouse secondary antibody (Abcam ab97043, 1:200). After two washes in TBS pH 7.5 and one at pH 9.5, the staining was revealed with a nitro blue tetrazolium (NBT, 500μg/ml) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, 187.5μg/ml) solution (Roche Applied Science) for 10 to 20 minutes. The reaction was stopped with a solution of TBS pH7.5 containing EDTA (20mM) and the slides were mounted with glycerol mounting medium (DAKO).
Immunofluorescence (IF)
For embryonic tissues, freshly dissected intestines were fixed 1 hour at RT with 4% PFA in PBS. Alternatively, whole embryos were fixed overnight at 4°C and the intestines dissected afterwards. For adult tissues, whole intestines were fixed in 4% PFA overnight at 4°C, cut longitudinally along the mesentery and washed in PBS. The outermost muscle layers were then stripped from the mucosa/submucosa. Fixed tissues were dehydrated in methanol. After rehydration, the intestines were incubated 2 hours at RT in blocking solution (10% fetal bovine serum, 0.1% Triton-X100 in PBS). Tissues were incubated with primary antibodies overnight at 4°C. The antibodies used were: mouse anti-βIII-Tubulin (1:200; Abcam ab78078), rabbit anti-S100β (1:500; Dako Z0311), goat anti-Sox10 (1:100, Santa Cruz Biotech. sc-17342) and rabbit anti-Ki67 (1:1000; Abcam ab15580). Secondary antibodies Alexa Fluor 594- or Alexa Fluor 647-conjugated anti-goat, -mouse or -rabbit (1:500, Jackson Immunoresearch) were incubated for 2 hours at RT and counterstained with DAPI. All antibodies were diluted with blocking solution. For the TUNEL assay, tissues were permeabilized 20 min at 37°C in 0.3% Triton-X100, 0.1% sodium citrate in PBS 1x, then stained in a 1:9 mix of enzyme solution:label solution from the in situ cell death detection kit, TMR red (Roche Applied Science 12156792910) 1h at 37°C.
Acetylcholinesterase staining
The whole colon (from cecum to anus) was dissected from adults and neonates. It was cut longitudinally along the mesentary (adult tissues only), rinsed, pinned flat and fixed in 4% PFA O/N at 4°C. Staining was performed on tissues as previously described [59]. Following the staining procedure, muscle strips were prepared as described above (adult tissues only).
Ex vivo time-lapse imaging of eNCC
Live imaging of eNCC was performed using a suspended culture technique adapted from Nishiyama et al., 2012 [3]. The abdomen of TashT e11.0 embryos was opened and the surrounding tissues trimmed just enough to expose the developing intestine. The embryo was placed on a small nitrocellulose filter (Millipore GSWP01300) soaked in PBS, and the extra tissues surrounding the intestine were slightly pressed onto the membrane. The PBS was then blotted off before being replaced with DMEM/F12 media (containing 10% FBS and antibiotics). The filter was flipped on top of a DMEM/F12-filled 2 mm-wide trough in a 1% agarose film covering the round glass bottom of a 35 mm culture dish (Greiner Bio One 627860), so that the intestine would float in media without touching either the agarose or the glass. The dish was incubated at 37°C, 5% CO2 during 6 hours, while 250μm-thick stacks were acquired with a 10x objective and a Nikon A1R confocal unit. Cell morphology viewed by YFP fluorescence allowed us to label the center of the cell located at the tip of a chain of migrating eNCC at each timeframe. Five to 6 chain tip cells were tracked from at least 3 intestines of each genotype. Speed and directionality were calculated from this dataset, with the orientation of the mesentery as a reference angle. We cannot totally exclude the possibility that more than one cell was included per chain tip measurement as individual eNCC at the wavefront sometimes exchange places with one another by a leapfrogging process [38].
Ex vivo eNCC migration assays
Ex vivo cell migration assays were performed as previously described [23]. Collagen gels containing or not GDNF (10 ng/ml, Cedarlane CLCYT305–2) and/or EDN3 (250 ng/ml, Sigma E9137) were prepared at least 1 hour before use and kept at 37°C in a CO2 (5%) incubator. Vibratome transverse sections (200 μm) of embryonic small intestines were put down on the collagen gels for 3 days at 37°C with 5% CO2 to let cells migrate into the gel. Bowel sections were removed and cells within the gels were fixed with 4% PFA for 1 hour at RT before being stained with DAPI (Sigma-Aldrich) to detect cell nuclei. All images were taken at 70X magnification with a Leica M205FA fluorescence stereomicroscope as described below.
Image processing and cell counting
YFP expression in TashT tissues or in migrating cells was visualized using a Leica M205FA fluorescence stereomicroscope. IHC slides were observed using a DM2000 Leica upright microscope. Images were acquired with a Leica DFC495 digital camera and Leica Application Suite (LAS) software (Leica microsystems). IF of stained intestines were examined using an inverted Nikon TI microscope. Images were acquired with a Nikon A1 confocal unit and NIS-Element AR4 software, using standard excitation and emission filters for visualizing DAPI, YFP, Alexa Fluor 594 and 647, as well as spectral imaging coupled with linear unmixing in order to distinguish between YFP and GFP fluorescence. All images were processed with ImageJ software [60]. Image J was also used for cell counting with the analyze particles function, or with the cell counter manual function.
Luciferase reporter assays
Constrained elements around the TashT insertion site were identified using Ensembl’s 35 eutherian mammals multiple alignment track (EPO_LOW_COVERAGE) on the NCBIm37 mouse genome version (www.ensembl.org). Seven regions named CE1 to CE7 (ranging between 600 to 1400bp) and containing one or multiple constrained elements were amplified by PCR (oligo sequences available upon request), cloned in the pGEM-T vector (Promega) and validated by sequencing. Luciferase reporter constructs were generated by subcloning each PCR fragment, in both the sense and antisense orientation, into a modified pXP2 vector containing the 109bp Thymidine kinase minimal promoter [61]. Neuro-2a neuroblastoma cells were propagated in EMEM supplemented with 10% FBS whereas P19 embryocarcinoma cells were propagated in alpha-MEM supplemented with 2.5% FBS and 7.5% CBS. Cos7 and NIH 3T3 cells were propagated in DMEM supplemented with 10% FBS. Transfections in 24-well plates and luciferase assays were performed in triplicate at least three times as previously described [62].
Western blots
Western blots using whole cell extracts of transfected Cos7 cells were performed as previously described [63]. Cells were transfected with a CMVp-driven expression vector for the same Fam162b-IRES-eGFP bicistronic cassette used to produce transgenic embryos and protein expression was assessed using the following primary antibodies: rabbit anti-Fam162b (1:1000; Abcam ab122309), rabbit anti-GFP (1:5000; Abcam ab290) and rabbit anti-Gapdh (1:2500, Santa Cruz Biotech. sc-25778).
Semi-quantitative RT-PCR
Total RNA was extracted using the RNAeasy Plus purification mini kit (QIAGEN) on FACS-sorted eNCC. The OneStep RT-PCR kit (QIAGEN) was then used on 100 ng of RNA with primers specific to the desired target (sequences available upon request). PCR consisted of 25–30–35 or 30–35–40 cycles of: 20 seconds at 95°C, 30 seconds at 62°C and 30 seconds at 72°C. Amplicons were resolved on a 2% agarose gel and quantified using the densitometry tools of ImageJ. The expression level of the housekeeping gene Gapdh was used for normalization.
Chromosome conformation capture (3C)
3C was performed as previously described [27] with minor modifications. Starting material was either 1x108 cells (for Neuro-2a and P19 cell lines), as recommended, or ~1x106 cells (for e12.5 intestinal material), in which case reaction volumes were divided by a factor of 20. Only one cycle of phenol, then phenol/chloroform extraction was performed. Glycogen (0.05 mg/ml) was added as a co-precipitant prior to ethanol precipitation. For each PCR reaction, 20 ng (BAC library), 50 ng (embryonic intestines libraries) or 100 ng (cell lines libraries) of library DNA was used. Sequencing of the amplicon confirmed the identity of the chimeric fragment amplified.
Statistics
Data are presented as mean ± standard deviation, with the number of experiments (n) included in the figure and/or legend. Quantification data were subjected to Student's t-test for statistical significance, except for directionality circular data which were compared through an ANOVA. Differences were considered statistically significant when the p value was less than 0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Bergeron KF, Silversides DW, Pilon N (2013) The developmental genetics of Hirschsprung's disease. Clin Genet 83: 15–22. doi: 10.1111/cge.12032 23043324
2. Anderson RB, Newgreen DF, Young HM (2006) Neural crest and the development of the enteric nervous system. Adv Exp Med Biol 589: 181–196. 17076282
3. Nishiyama C, Uesaka T, Manabe T, Yonekura Y, Nagasawa T, et al. (2012) Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nat Neurosci 15: 1211–1218. doi: 10.1038/nn.3184 22902718
4. Burns AJ, Champeval D, Le Douarin NM (2000) Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Dev Biol 219: 30–43. 10677253
5. Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, et al. (2008) Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 45: 1–14. 17965226
6. Alves MM, Sribudiani Y, Brouwer RW, Amiel J, Antinolo G, et al. (2013) Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev Biol 382: 320–329. doi: 10.1016/j.ydbio.2013.05.019 23707863
7. Obermayr F, Hotta R, Enomoto H, Young HM (2013) Development and developmental disorders of the enteric nervous system. Nat Rev Gastroenterol Hepatol 10: 43–57. doi: 10.1038/nrgastro.2012.234 23229326
8. Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, et al. (1994) Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 79: 1267–1276. 8001159
9. Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, et al. (1994) Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79: 1277–1285. 8001160
10. Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, et al. (1996) Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382: 70–73. 8657306
11. Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis V, et al. (2006) Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development 133: 4507–4516. 17050626
12. Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367: 380–383. 8114940
13. Uesaka T, Nagashimada M, Yonemura S, Enomoto H (2008) Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J Clin Invest 118: 1890–1898. doi: 10.1172/JCI34425 18414682
14. McCallion AS, Stames E, Conlon RA, Chakravarti A (2003) Phenotype variation in two-locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and Ednrb. Proc Natl Acad Sci U S A 100: 1826–1831. 12574515
15. Methot D, Reudelhuber TL, Silversides DW (1995) Evaluation of tyrosinase minigene co-injection as a marker for genetic manipulations in transgenic mice. Nucleic Acids Res 23: 4551–4556. 8524641
16. Boyer A, Pilon N, Raiwet DL, Lussier JG, Silversides DW (2006) Human and pig SRY 5' flanking sequences can direct reporter transgene expression to the genital ridge and to migrating neural crest cells. Dev Dyn 235: 623–632. 16411204
17. Pilon N, Raiwet D, Viger RS, Silversides DW (2008) Novel pre- and post-gastrulation expression of Gata4 within cells of the inner cell mass and migratory neural crest cells. Dev Dyn 237: 1133–1143. doi: 10.1002/dvdy.21496 18351674
18. Young HM, Bergner AJ, Anderson RB, Enomoto H, Milbrandt J, et al. (2004) Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Dev Biol 270: 455–473. 15183726
19. Barlow A, de Graaff E, Pachnis V (2003) Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron 40: 905–916. 14659090
20. Nagy N, Goldstein AM (2006) Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Dev Biol 293: 203–217. 16519884
21. Young HM, Hearn CJ, Farlie PG, Canty AJ, Thomas PQ, et al. (2001) GDNF is a chemoattractant for enteric neural cells. Dev Biol 229: 503–516. 11150245
22. Mwizerwa O, Das P, Nagy N, Akbareian SE, Mably JD, et al. (2011) Gdnf is mitogenic, neurotrophic, and chemoattractive to enteric neural crest cells in the embryonic colon. Dev Dyn 240: 1402–1411. doi: 10.1002/dvdy.22630 21465624
23. Bergeron KF, Cardinal T, Pilon N (2013) A quantitative cell migration assay for murine enteric neural progenitors. J Vis Exp: e50709. doi: 10.3791/50709 24084298
24. Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO (2003) GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 130: 2187–2198. 12668632
25. Ngan ES, Garcia-Barcelo MM, Yip BH, Poon HC, Lau ST, et al. (2011) Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J Clin Invest 121: 3467–3478. doi: 10.1172/JCI43737 21841314
26. Wang Z, Cao R, Taylor K, Briley A, Caldwell C, et al. (2013) The properties of genome conformation and spatial gene interaction and regulation networks of normal and malignant human cell types. PLoS One 8: e58793. doi: 10.1371/journal.pone.0058793 23536826
27. Naumova N, Smith EM, Zhan Y, Dekker J (2012) Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods 58: 192–203. doi: 10.1016/j.ymeth.2012.07.022 22903059
28. Antonellis A, Huynh JL, Lee-Lin SQ, Vinton RM, Renaud G, et al. (2008) Identification of neural crest and glial enhancers at the mouse Sox10 locus through transgenesis in zebrafish. PLoS Genet 4: e1000174. doi: 10.1371/journal.pgen.1000174 18773071
29. Werner T, Hammer A, Wahlbuhl M, Bosl MR, Wegner M (2007) Multiple conserved regulatory elements with overlapping functions determine Sox10 expression in mouse embryogenesis. Nucleic Acids Res 35: 6526–6538. 17897962
30. Heanue TA, Pachnis V (2006) Expression profiling the developing mammalian enteric nervous system identifies marker and candidate Hirschsprung disease genes. Proc Natl Acad Sci U S A 103: 6919–6924. 16632597
31. Vohra BP, Tsuji K, Nagashimada M, Uesaka T, Wind D, et al. (2006) Differential gene expression and functional analysis implicate novel mechanisms in enteric nervous system precursor migration and neuritogenesis. Dev Biol 298: 259–271. 16904662
32. Akbareian SE, Nagy N, Steiger CE, Mably JD, Miller SA, et al. (2013) Enteric neural crest-derived cells promote their migration by modifying their microenvironment through tenascin-C production. Dev Biol 382: 446–456. doi: 10.1016/j.ydbio.2013.08.006 23958436
33. Brauer PR, Bolender DL, Markwald RR (1985) The distribution and spatial organization of the extracellular matrix encountered by mesencephalic neural crest cells. Anat Rec 211: 57–68. 3985379
34. Druckenbrod NR, Epstein ML (2009) Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development 136: 3195–3203. doi: 10.1242/dev.031302 19700623
35. Hotta R, Anderson RB, Kobayashi K, Newgreen DF, Young HM (2010) Effects of tissue age, presence of neurones and endothelin-3 on the ability of enteric neurone precursors to colonize recipient gut: implications for cell-based therapies. Neurogastroenterol Motil 22: 331–e386. doi: 10.1111/j.1365-2982.2009.01411.x 19775251
36. Del Rio T, Nishitani AM, Yu WM, Goodrich LV (2013) In vivo analysis of Lrig genes reveals redundant and independent functions in the inner ear. PLoS Genet 9: e1003824. doi: 10.1371/journal.pgen.1003824 24086156
37. Ledda F, Bieraugel O, Fard SS, Vilar M, Paratcha G (2008) Lrig1 is an endogenous inhibitor of Ret receptor tyrosine kinase activation, downstream signaling, and biological responses to GDNF. J Neurosci 28: 39–49. doi: 10.1523/JNEUROSCI.2196-07.2008 18171921
38. Young HM, Bergner AJ, Simpson MJ, McKeown SJ, Hao MM, et al. (2014) Colonizing while migrating: how do individual enteric neural crest cells behave? BMC Biol 12: 23. doi: 10.1186/1741-7007-12-23 24670214
39. Sanyal A, Lajoie BR, Jain G, Dekker J (2012) The long-range interaction landscape of gene promoters. Nature 489: 109–113. doi: 10.1038/nature11279 22955621
40. Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, et al. (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148: 84–98. doi: 10.1016/j.cell.2011.12.014 22265404
41. Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM (2013) Chromosomal contact permits transcription between coregulated genes. Cell 155: 606–620. doi: 10.1016/j.cell.2013.09.051 24243018
42. Feng W, Leach SM, Tipney H, Phang T, Geraci M, et al. (2009) Spatial and temporal analysis of gene expression during growth and fusion of the mouse facial prominences. PLoS One 4: e8066. doi: 10.1371/journal.pone.0008066 20016822
43. Ro S, Hwang SJ, Muto M, Jewett WK, Spencer NJ (2006) Anatomic modifications in the enteric nervous system of piebald mice and physiological consequences to colonic motor activity. Am J Physiol Gastrointest Liver Physiol 290: G710–718. 16339294
44. Cantrell VA, Owens SE, Chandler RL, Airey DC, Bradley KM, et al. (2004) Interactions between Sox10 and EdnrB modulate penetrance and severity of aganglionosis in the Sox10Dom mouse model of Hirschsprung disease. Hum Mol Genet 13: 2289–2301. 15294878
45. Dang R, Torigoe D, Suzuki S, Kikkawa Y, Moritoh K, et al. (2011) Genetic background strongly modifies the severity of symptoms of Hirschsprung disease, but not hearing loss in rats carrying Ednrb(sl) mutations. PLoS ONE 6: e24086. doi: 10.1371/journal.pone.0024086 21915282
46. Vohra BP, Planer W, Armon J, Fu M, Jain S, et al. (2007) Reduced endothelin converting enzyme-1 and endothelin-3 mRNA in the developing bowel of male mice may increase expressivity and penetrance of Hirschsprung disease-like distal intestinal aganglionosis. Dev Dyn 236: 106–117. 17131407
47. Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, et al. (2005) A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature 434: 857–863. 15829955
48. Berletch JB, Yang F, Xu J, Carrel L, Disteche CM (2011) Genes that escape from X inactivation. Hum Genet 130: 237–245. doi: 10.1007/s00439-011-1011-z 21614513
49. Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434: 400–404. 15772666
50. Deng X, Berletch JB, Nguyen DK, Disteche CM (2014) X chromosome regulation: diverse patterns in development, tissues and disease. Nat Rev Genet 15: 367–378. doi: 10.1038/nrg3687 24733023
51. Huynh KD, Lee JT (2003) Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 426: 857–862. 14661031
52. Wolstenholme JT, Rissman EF, Bekiranov S (2013) Sexual differentiation in the developing mouse brain: contributions of sex chromosome genes. Genes Brain Behav 12: 166–180. doi: 10.1111/gbb.12010 23210685
53. Xu J, Burgoyne PS, Arnold AP (2002) Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet 11: 1409–1419. 12023983
54. Yamamoto T, Wilsdon A, Joss S, Isidor B, Erlandsson A, et al. (2014) An emerging phenotype of Xq22 microdeletions in females with severe intellectual disability, hypotonia and behavioral abnormalities. J Hum Genet 59: 300–306. doi: 10.1038/jhg.2014.21 24646727
55. Nagy A, Gertsenstein M, Vintersten K, Behringer R (2003) Manipulating the mouse embryo, A laboratory manual, 3rd Edition. Cold spring Harbor, New-York: Cold Spring Harbor Laboratory Press. doi: 10.1007/978-1-60327-019-9_13 19504073
56. Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, et al. (1989) Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105: 707–714. 2557196
57. Lu W, Phillips CL, Killen PD, Hlaing T, Harrison WR, et al. (1999) Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics 61: 113–124. 10534397
58. Matsuda Y, Chapman VM (1995) Application of fluorescence in situ hybridization in genome analysis of the mouse. Electrophoresis 16: 261–272. 7774567
59. Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, et al. (1998) GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 21: 317–324. 9728913
60. Abramoff MD, Magalhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics International 11: 36–42.
61. Nordeen SK (1988) Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques 6: 454–458. 2908509
62. Sanchez-Ferras O, Coutaud B, Djavanbakht Samani T, Tremblay I, Souchkova O, et al. (2012) Caudal-related homeobox (Cdx) protein-dependent integration of canonical Wnt signaling on paired-box 3 (Pax3) neural crest enhancer. J Biol Chem 287: 16623–16635. doi: 10.1074/jbc.M112.356394 22457346
63. Sanchez-Ferras O, Bernas G, Laberge-Perrault E, Pilon N (2014) Induction and dorsal restriction of Paired-box 3 (Pax3) gene expression in the caudal neuroectoderm is mediated by integration of multiple pathways on a short neural crest enhancer. Biochim Biophys Acta 1839: 546–558. doi: 10.1016/j.bbagrm.2014.04.023 24815547
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 3
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs